Процессы гидрирования и дегидрирования органических соединений
Содержание
Введение
1. Понятие о катализаторах
2. Процесс гидрирования
непредельных соединений
2.1 Традиционные процессы и
катализаторы гидрирования
2.2 Современные тенденции в
разработке и использовании новых
катализаторов гидрирования
2.3 Катализаторы на основе
благородных металлов
2.4 Процесс гидрирования
3. Процесс дегидрирования
органических веществ
3.1 Разновидности
дегидрирующего действия катализаторов
3.2 Современные представления
о природе активной поверхности
катализаторов дегидрирования
3.3 Процесс дегидрирования
Заключение
Список литературы
Введение
катализатор
дегидрирование органический
Каталитическое гидрирование включает большую группу реакций присоединения
водорода по ненасыщенным связям С=С, С=О, C≡N, С≡С
и т.д., а также реакции гидрогенолиза (разрыва и восстановления) связей С-С,
С-О, С-S и др.
Кроме гетерогенных катализаторов, применяющихся в крупнотоннажных
процессах, используют также гомогенные металлокомплексные и гидридные
катализаторы (преимущественно в тонком органическом синтезе). Расширяется
применение энантиоселективных катализаторов гидрирования, в основном
металлокомплексных соединений никеля, кобальта и других металлов с оптически
активными лигандами.
Наиболее широкое распространение получили нанесенные никелевые
катализаторы гидрирования, за ними идут катализаторы на основе благородных
металлов. В процессах гидрооблагораживания и гидродесульфуризации широко
применяются бинарные Ni-Мо-,
Ni-W-, Со-Мо- и Со-W-
катализаторы. Новыми перспективными катализаторами гидрирования являются
карбиды металлов [1].
К реакциям каталитического дегидрирования способны углеводороды различных
гомологических рядов (парафины, нафтены, олефины и циклоолефины,
алкилароматические углеводороды). В результате реакции отщепляется водород и
образуются более ненасыщенные соединения или происходит циклизация
(дегидроциклизация). Катализаторами дегидрирования являются различные металлы и
оксиды. Основные побочные реакции, протекающие при каталитическом
дегидрировании: крекинг, изомеризация, коксообразование. Каталитическое
дегидрирование является одним из важнейших промышленных процессов переработки
углеводородного сырья [2].
Цель работы: рассмотреть процессы гидрирования и дегидрирования
органических соединений, а так же природу катализаторов и механизмы их действия.
1. Понятие о
катализаторах
Катализатором называется вещество, которое своим присутствием в небольшом
количестве в смеси веществ способствует химической реакции между ними
(определение Берцелиуса, 1836) или увеличивает скорость реакции (определение
Оствальда, 1894). Второе определение основано на предположении, что реакция
протекает и в отсутствие катализатора, но с очень малой, возможно неизмеримой,
скоростью. Действительно, известны многочисленные реакции, протекающие в
отсутствие катализатора, но в присутствии катализатора их скорость резко
возрастает. Известно также большое число реакций, которые идут только в
присутствии определенного катализатора. В некоторых случаях одни и те же
реагирующие вещества при замене катализатора образуют другие продукты реакции.
Надо полагать, что в последних случаях катализатор играет существенную роль,
поскольку влияет на ход реакции, так что старое определение Берцелиуса, верно,
по крайней мере, в отдельных случаях.
Следует подчеркнуть, что катализатором всегда является вещество. Было бы
неправильно говорить, например, о каталитическом действии света.
Когда говорят, что катализатор не принимает участия в реакции,
подразумевают, что после реакции он находится качественно и количественно в том
же состоянии, что и до реакции. Поэтому катализатор не включают в уравнение
химической реакции. Кроме того, нет необходимости использовать его в каком-либо
определенном стехиометрическом соотношении с реагентами. Иногда достаточно
ничтожно малого количества катализатора, чтобы вызвать реакцию.
Катализаторы ускоряют или вызывают только термодинамически возможные
реакции, т.е. реакции, протекающие самопроизвольно с уменьшением свободной
энергии в сторону установления равновесия.
Изучение реакций, протекающих как в присутствии, так и в отсутствие
катализаторов, раскрыло важную сторону механизма их действия, которое
заключается в том, что катализаторы уменьшают энергию активации реакций.
Например, известно, что энергия активации гомогенной реакции в газовой фазе -
разложения йодистого водорода 2HI → H2 + I2 - равна 44,3 ккал/моль. Энергия активации этой реакции,
проводится в присутствии металлического золота в качестве катализатора
(гетерогенный катализ), равна 25 ккал/моль, а в присутствии платины - только 14
ккал/моль.
Различают две большие группы каталитических реакций: гомогенный и
гетерогенный катализ. В первой группе реакций катализатор и субстрат образуют
одну фазу, обычно жидкую. Во второй группе реакций катализатор является
твердым, а субстрат - жидким или газообразным. Третья группа каталитических
реакций - ферментативные реакции - охватывает процессы, исключительно важные
для животных и растительных организмов. Участвующие в этих реакциях
катализаторы - ферменты - являются органическими соединениями (белки) с
огромными молекулами (макромолекулы), вырабатываемыми живыми клетками.
Гомогенный катализ. Катализаторами огромного большинства реакций этой
группы являются кислоты и основания. В настоящее время в результате
обстоятельных кинетических исследований установлено, что в реакциях,
катализируемых кислотами, субстрат ведёт себя как основание, а промежуточный
продукт является его сопряженной кислотой. В реакциях, катализируемых
основаниями, субстрат ведет себя как кислота, отдавая катализатору протон.
После каждой элементарной реакции катализатор - кислота или основание -
регенерируется. Большинство этих реакций относится к области органической
химии.
Ферментативными реакциями являются реакции гидролиза, гидрирования и
дегидрирования (присоединение водорода к молекулам субстрата или отщепление
водорода от его молекул), реакции окисления (перенос электронов), реакции
переноса групп фосфорной кислоты и органических групп. Резко выраженная
специфичность ферментов указывает на то, что между ферментом и молекулой
субстрата образуются химические соединения, благодаря которым последние
активируются (теория Михаэлиса и Ментен, 1913).
Уравнение
Михаэлиса-Ментен: основное уравнение ферментативной
<#"822644.files/image001.gif">
Уравнение
имеет вид:
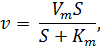
где
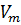 -
максимальная скорость реакции, равная kcatE0;
-
максимальная скорость реакции, равная kcatE0; 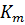 -
константа Михаэлиса, равная концентрации субстрата, при которой скорость
реакции составляет половину от максимальной; S - концентрация
субстрата [3].
-
константа Михаэлиса, равная концентрации субстрата, при которой скорость
реакции составляет половину от максимальной; S - концентрация
субстрата [3].
Гетерогенный
катализ имеет широкое и важное применение в промышленности. Гетерогенный
катализ особенно часто используется в реакциях окисления (пример: 2SO2 +
O2 → 2SO3), в реакциях восстановления
или гидрирования (пример: N2 + ЗН2 → 2NH3), в реакциях
дегидрирования (осуществляемых в нефтехимической промышленности с
использованием в качестве катализаторов платины, оксиды хрома или сульфида
молибдена) и в реакциях разрыва молекул углеводородов нефти (крекинг) в
присутствии катализаторов кислотного характера (оксиды алюминия, алюмосиликаты).
Твердые
катализаторы тем активнее, чем больше их поверхность, т.е. чем они больше
измельчены, и чем ниже температура их получения и последующей обработки. Так,
катализаторы из металлического никеля, используемые в реакциях гидрирования,
представляют собой пирофорные порошки, полученные восстановлением оксида никеля
NiO водородом примерно при 300 °С. При нагревании
примерно до 700 °С эти катализаторы полностью теряют активность, поскольку
атомы на их поверхности перестраиваются в более устойчивое состояние. Можно
сделать вывод, что в реакции принимает участие не вся поверхность катализатора,
а только определенные активные центры (Тейлор, 1926).
Давно
известно, что химическая реакция при гетерогенном катализе происходит во
внешнем слое молекул, адсорбированных на поверхности катализатора (Лэнгмюр,
1916) или точнее на активных центрах катализатора. Различают два типа
адсорбции: адсорбцию вандерваальсовыми силами при низкой температуре и
активированную адсорбцию, или хемосорбцию. Последний процесс, единственно важный
при гетерогенном катализе, состоит в образовании химических соединений между
молекулами реагентов и поверхностными атомами катализатора. Теплоты,
выделяющиеся при вандерваальсовой адсорбции, малы (около 4 ккал/моль для таких
газов, как O2, СО и N2 на Сr2O3 при -183 °С), но достигают значения обычных теплот
реакции при хемосорбции (50 ккал/моль для O2 на Сr2O3
при 0°С). Вандерваальсова адсорбция осуществляется мгновенно, тогда как
хемосорбция происходит медленно, но ее скорость возрастает с температурой.
В
обычной каталитической реакции продукты реакции десорбируются с катализатора и
диффундируют в массу газа (или жидкости), а их место тотчас же занимают
молекулы реагентов, которые повторяют тот же элементарный процесс. Таким
образом, один активный центр поверхности катализатора превращает большое
количество молекул реагента. Некоторые вещества, например сероводород H2S,
фосфин Н3Р и различные соединения азота, прочно закрепляются на поверхности
катализаторов (металлов), блокируя активные центры и тем самым замедляя
каталитический процесс (отравление катализаторов) [4].
2. Процесс
гидрирования непредельных соединений
Гидрирование или гидрогенизация (от позднелатинского hydrogenium -
водород), деструктивная гидрогенизация, - совокупность химических процессов,
происходящих при воздействии водорода на органическое вещество. В
топливоперерабатывающей промышленности гидрогенизацию применяют для получения
из твёрдых горючих ископаемых (угли, сланцы), а также низкосернистых нефтей и
тяжёлых нефтяных остатков моторного горючего, смазочных масел и химических
продуктов. Гидрогенизация твёрдого топлива является универсальным методом
получения из него синтетического жидкого топлива. Также важный резерв для
замены сырой нефти горючими сланцами, битумами, углями [5,6].
Развитие исследований в области гидрогенизации относится к 1897-1900 гг.,
когда П. Сабатье (Франция) и Н.Д. Зелинский (Россия) со своими учениками
разработали основы гидрогенизации катализа органических соединений. Влияние
давления водорода на ускорение реакций гидрогенизации органических соединений
было установлено вначале XX в.
В.Н. Ипатьевым. Промышленное применение гидрогенизации твёрдого топлива впервые
было получено в 30-40-х гг., в Германии. Перед 2-й мировой войной (1939-1945)
установки по гидрогенизации угля и угольных смол работали также в
Великобритании, Италии, Корее; в СССР были построены два опытных завода. В
послевоенный период в основе переработки нефти сырья применяли гидрогенизацию.
Начиная с 60-х гг. ведутся работы по гидрогенизации твёрдого топлива с целью
создания экономически эффективных процессов производства синтетических жидких
топлив.
В СССР был разработан процесс гидрогенизации угля для получения моторного
горючего, котельного топлива и химикатов. Процесс осуществляется при температуре
420-430 °С, давлении водорода 10 Па, в присутствии активных катализаторов,
растворителя и органических добавок-ингибиторов реакций радикальной
полимеризации. В зависимости от исходного сырья выход жидких продуктов 85-95%
(технологическая схема процесса дана на рис.1.).
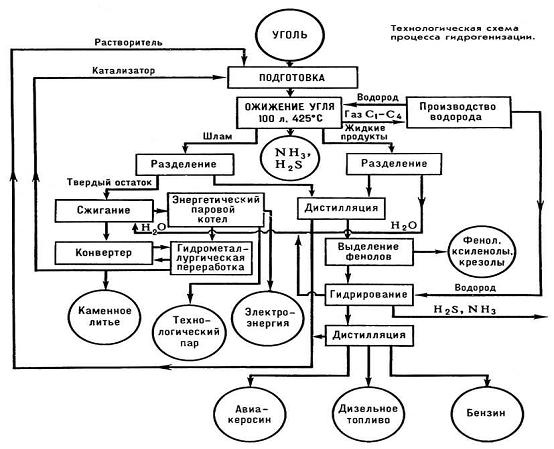
Рис.1. Процесс гидрогенизации угля для получения моторного горючего,
котельного топлива и химикатов.
В США, Великобритании, ФРГ разрабатывается ряд процессов по
гидрогенизации угля с катализатором и без катализатора, под давлением водорода
1-7 и 15-30 Па, температура 400-500 °С, а также экстракции угля растворителями
с последующей гидрогенизации экстрактов. Исследования по гидрогенизации
твёрдого топлива и тяжёлых нефти остатков ведутся в Японии, Индии, Австралии,
Польше и др. [3,5].
2.1
Традиционные процессы и катализаторы гидрирования
В первом промышленном процессе гидрирования использовался никелевый
катализатор, открытый Сабатье в 1897 г. Позже были разработаны другие известные
катализаторы гидрирования: металлы VI-VIII групп, в том числе металлы
платиновой группы, на носителях и в составе сложных оксидных систем, а также
скелетные металлические катализаторы.
Результаты исследования Ипатьевым в начале XX в. каталитических реакций
гидрирования под давлением послужили основой для создания процессов, широко
используемых в настоящее время в промышленности. Был разработан ряд
специфических катализаторов гидроочистки, получаемых пропиткой или соосаждением
(Аl-Со-Мо-, Ni-Мо-, сульфидные Ni-W-S- катализаторы и т.д.), которые устойчивы к каталитическим
ядам, содержащимся в нефтяном сырье (S-, N-, О- содержащие соединения).
Объем производства катализаторов для получения органических соединений не
очень велик. К числу крупнотоннажных относятся промышленные процессы
гидрирования жиров, 2-этилгексеналя, ацетилена (для удаления его из этилена),
бензола в циклогексан в производстве капролактама и некоторые другие.
Расширяется применение катализаторов гидрирования при производстве лекарств и
физиологически активных соединений. Важными показателями для катализаторов этой
категории являются высокая селективность, длительный срок службы, соответствие
экологическим требованиям. В основном такими характеристиками обладают
металлические (Ni, Pt, Pd, Rh, Ru) катализаторы на носителях,
медно-хромовые и никель-хромовые соосажденные катализаторы.
На примере гидрирования бензола в циклогексан можно проследить тенденцию
к использованию того или иного катализатора в зависимости от того, насколько
жесткие требования предъявляются к чистоте конечного продукта.
С начала века гидрирование бензола традиционно осуществляли на никеле
(никель Ренея, пропиточные катализаторы) и платиновых металлах на носителях. В
ранних промышленных процессах, проводившихся при повышенных температуре и давлении,
наряду с основной протекали и побочные реакции: крекинг с образованием n-гексана, изомеризация циклогексана в
метилциклопентан, изомеризация и крекинг с выделением более легких
углеводородов.
Наилучшие результаты при получении высокочистого циклогексана
обеспечивает двухстадийная схема: на первой стадии используют нанесенный Ni-катализатор (30-45% Ni/Аl2О3), а на второй - Pt-катализатор на носителе (0,2-1,0% Pt/Аl2О3). Чистота
циклогексана достигает при этом 99,57-99,88%.
Большое внимание в нашей стране уделяли соосажденным никель-хромовым
катализаторам, обеспечивающим получение высокочистого циклогексана в трубчатых
реакторах в несколько стадий при строгом соблюдении технологии. Катализаторы
готовили осаждением азотнокислых солей Ni и Cr
раствором соды при комнатной температуре. Затем следовали фильтрация, промывка
осадка от нитрат-ионов, сушка, прокаливание, таблетирование и активация в токе
водорода. Содержание Ni
составляло 48-63%, а Сr2О3 -
37-52%. Недостатками этого и других способов получения Ni-Cr-катализаторов
являются многостадийность, громоздкость фильтровального оборудования, большие
объемы сточных вод на стадии промывки и т.д.
В связи с этим был предложен бессточный метод получения Ni-Сr-катализаторов, включающий стадию смешения растворов Ni(NO3)2 и (NH4)2Gr2O7 с последующими сушкой и прокаливанием смеси солей путем ее
распыления на нагретую насадку из инертного материала в режиме кипящего слоя
при 310-340 °С, таблетированием и восстановлением [1].
2.2
Современные тенденции в разработке и использовании новых кат
ализаторов
гидрирования
В 1980-х годах была проведена серия исследований, направленных на
создание никелевых катализаторов гидрирования на носителях, не уступающих по
активности и другим параметрам скелетным катализаторам. Такими свойствами
обладает, например, катализатор Еurо-Ni-1 (состава 25% Ni/SiO2), разработанный большой группой исследователей из
разных стран во главе с Кененом. Этот катализатор готовят методом гомогенного
осаждения, при котором часть никеля образует на носителе гидросиликат.
Гомогенное осаждение Ni
ведут в присутствии мочевины на суспендированном в водной среде
микросферическом силикагеле, который получают пиролизом SiCI4 в пламени. В присутствии SiО2 рН
водного раствора соли никеля Ni(NО3)2. 6Н2О не превышает 5,3-5,5,
тогда как в его отсутствие при осаждении гидроксида никеля значение рН
достигает 5,8 и остается постоянным.
На сильное взаимодействие Ni и
SiО2 в процессе приготовления катализатора указывает трудность восстановления
последнего, особенно по сравнению с ненанесенным гидроксидом никеля. Для
полного восстановления катализатора Еurо-Ni-1 требуются температуры выше 400 °С,
вплоть до 600 °С.
При нанесении Ni на SiО2
развитие поверхности может происходить за счет образования промежуточного
гидросиликата никеля. Однако это зависит от условий приготовления, соотношения Ni : SiО2 и реакционной способности
SiО2. Наряду с Euro-Ni-1 в последние годы с помощью новых
технологий были приготовлены катализаторы Ni/SiO2 с меньшим
содержанием металла (10-15%) для гидрирования ацетона.
Предприняты и другие попытки увеличить удельную поверхность Ni на носителе при уменьшении его
содержания до 4-10%. Для этого использовали как метод пропитки, так и
металлоорганические комплексы, модифицирующие поверхность носителей SiО2 и Аl2О3, в органических растворителях.
Большой интерес в 1980-х годах в России вызвали так называемые
«сорбционные» катализаторы. Метод их получения, заключающийся в связывании
металлоорганических комплексов поверхностью носителя, достаточно сложен и
дорог.
Для гидроочистки прямогонных бензинов применяют в основном соосажденные Al-Co-Мо-катализаторы, способные удалять серу. Гидрирование
ароматических углеводородов осуществляется на них лишь частично. Такие
катализаторы вообще недостаточно активны в реакциях гидрирования ароматических
и ненасыщенных соединений. Для этой цели в некоторых случаях более пригодны Ni-W-Al- либо Ni-Мо-Al-катализаторы.
В последнее время большое внимание уделяют новым видам Со-Мо-, Ni-Мо- и Ni-W- катализаторов
на носителях, а также катализаторам на основе благородных металлов.
Сероустойчивые катализаторы широко используют как в одно-, двухстадийных
процессах гидрирования ароматических веществ. Установлено, что катализатор
Ni-W/Аl2О3 отравляется серой более
интенсивно, чем Ni-Мо/Аl2О3. Поэтому двухступенчатый процесс
гидроочистки организуют таким образом, что на первой ступени (в основном,
гидрирование сернистых соединений) применяют Ni-Мо-, а на второй ступени («догидрирование») -
низкопроцентный Ni-W-катализатор (например: 6%Ni + 20% WОз/Аl2Оз).
С конца 1980-х годов разработкой новых Ni-W- катализаторов
на носителях занимается ряд фирм, ориентированных на гидроочистку моторных
топлив. Недостатком предложенных катализаторов является значительное содержание
в них WО3; кроме того, для осуществления на этих катализаторах эффективной
гидроочистки требуется применение высоких давлений (до 10 МПа) и температур (до
360 °С).
Таким образом, в настоящее время наиболее привлекательными и дешевыми
катализаторами гидрирования ненасыщенных углеводородов (включая ароматические
соединения) являются модифицированные оксидами вольфрама или молибдена
никельсодержащие катализаторы на носителях [1].
2.3
Катализаторы на основе благородных металлов
Никельсодержащие катализаторы дешевле катализаторов на основе благородных
металлов (Pt, Pd, Ru, Rh, Ir, Os). Однако в
целом ряде промышленных процессов гидрирования предпочтение отдается последним.
Это обусловлено их высокой каталитической активностью, позволяющей использовать
более низкие температуры и давления. Срок жизни таких катализаторов нередко
исчисляется годами. Особенно большой интерес вызывают Pd-, Pt-, Ru- и Rh- катализаторы на носителях и биметаллические системы.
Применение специальных носителей позволило снизить содержание благородного
металла до 0,01-0,005% и тем самым удешевить катализатор, сохранив его
качество.
Применение современных физико-химических методов исследования дает
возможность изучать механизм действия этих катализаторов, оптимизировать их
состав, используя различные носители и модификаторы, разрабатывать способы
регулирования дисперсности металла на поверхности носителя. Некоторые
катализаторы на основе благородных металлов устойчивы к отравлению S- и N- содержащими соединениями.
Гидрирование ненасыщенных углеводородов.
В некоторых случаях в качестве катализаторов гидрооблагораживания
моторных топлив (деароматизации, гидрирования олефинов) предпочитают применять
благородные металлы, а не никель. Катализаторы на основе благородных металлов
более активны и меньше отравляются сернистыми соединениями. Платина и палладий,
нанесенные на Аl2O3, легко дезактивируются при
концентрации серы менее 1·10-4%, однако их устойчивость к сере растет при
использовании таких кислотных носителей, как цеолиты. В частности, на Pd/LaY даже при давлении H2S 63 кПа, т.е. в
условиях, при которых весь палладий должен был бы превратиться в PdS, 98% продуктов гидрокрекинга
додекана составляют алканы, а активность и селективность катализатора
сохраняются неизменными. Столь необычное поведение благородного металла
объясняют эффектом SMSI или
изменением электронного состояния палладия при взаимодействии с кислотными
центрами цеолита.
Платиновые катализаторы получают ионным обменом или пропиткой носителя
раствором соли Pt(NH3)4CI; последняя связывается с кислотным центром цеолита ≡Аl-O-. Далее следует важная стадия образования центров 2(≡Аl-O-)Pt2+, которые
формируются при прокаливании на воздухе с последующим восстановлением
водородом. При этом платина входит в состав кластеров типа m(≡Аl-O-H)••• Ptn. Кластеры находятся в тесном контакте с сильными
кислотными центрами, что делает возможным переход электронов от благородного
металла и приводит к формированию электронодефицитных частиц металла (Ptδ+), определяющих особые свойства
катализатора.
Чаще всего для гидрирования применяют палладиевые катализаторы на
носителях. В табл. 1 приведены результаты исследования реакций гидрирования СO, альдегидов и ненасыщенных
углеводородов на катализаторах Pd/TiO2, полученных различными способами и
отличающихся размерами частиц и величиной активной поверхности металла.
Как видно из табл. 1, скорости гидрирования СO и низкотемпературного
гидрирования пропионового альдегида в спирт возрастают с увеличением
дисперсности Pd. Однако при гидрировании двойных связей C=C в этилене,
бутадиене и акролеине наблюдается обратный порядок изменения активности, что
особенно заметно в реакции гидрирования сопряженных связей бутадиена.
Средняя активность (число оборотов катализаторов в секунду) различных
катализаторов Pt/TiO2 в реакциях гидрирования CO, альдегидов и олефинов
|
Характеристики катализатора
|
Средняя активность
катализатора при гидрировании
|
|
[Pd], %
|
Sуд, м2 r -1
|
D, Нм
|
H : Pd
|
CO (метании-рование)
|
CO в CH3OH (230 ºС)
|
связи C=O в
Пропионо-вомальде-гиде (80 ºС)
|
C2H4 (80 ºС)
|
связи C=C в
акролеине (80 ºС)
|
C4H6 (80 ºС)
|
|
5,5
|
207
|
4,5-6,5
|
0,32-0,67
|
7,3·10 -4
|
4,3·10 -4
|
0,16
|
380
|
0,09
|
150
|
|
5,2
|
9
|
186
|
0,17
|
2,8·10 -4
|
4,0·10 -5
|
4,5·10 -3
|
500
|
-
|
680
|
|
4,4
|
9
|
295
|
0,067
|
5,5·10 -4
|
0
|
4,5·10 -3
|
580
|
0,4
|
1,05·10 -3
|
Связь между каталитическими свойствами нанесенного палладия и его
дисперсностью активно обсуждается в литературе. В одних работах это явление
объясняют изменением электронного состояния Pd в результате SMSI, в других -
участием в реакции не только металла, но и носителя. Представленные в табл. 1
три Pd-катализатора на одном и том же носителе (TiO2) проявляют различную
активность и селективность. Поэтому причину различий следует искать в свойствах
частиц самого палладия: в изменении их размеров, формы, электронной структуры и
характера взаимодействия с гидрируемыми молекулами [1].
.4 Процесс
гидрирования
Реакцию гидрирования ненасыщенных соединений водородом можно
рассматривать как реакцию восстановления. При этом атомы углерода кратной связи
восстанавливаются, а молекулярный водород окисляется. Присоединение водорода к
алкенам происходит только в присутствии катализаторов:
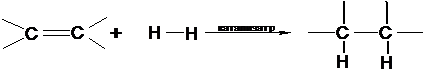
В
качестве катализаторов применяют тонкоизмельченные металлы - платину, палладий,
никель. Наиболее часто используются никель Ренея и катализатор Адамса. Никель
Ренея получают обработкой никель-алюминиевого сплава гидроксидом натрия, в
результате чего получают тонко измельченный никель, насыщенный водородом.
Катализатор Адамса - это платиновая чернь, получаемая восстановлением оксида
платины(IV) водородом непосредственно в процессе реакции.
Палладий для увеличения поверхности наносят на инертный материал - уголь. Все
эти катализаторы не растворяются в органических растворителях.
Водород
и алкен адсорбируются на большой поверхности тонкоизмельченного металла, где и
происходит реакция. Оба атома водород присоединяются с одной стороны π-связи, т.е. процесс идет как син-присоединение:

Молекула
ненасыщенного соединения может содержать другие функциональные группы,
способные к восстановлению. Во многих случаях удается подобрать условия, при
которых происходит селективное восстановление двойной связи. В приведенных ниже
примерах восстановление не затрагивает бензольное кольцо и карбонильную группу:
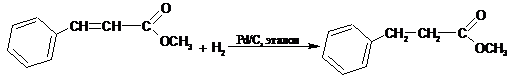
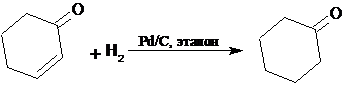
Гидрирование
является экзотермической реакцией. Значения теплот гидрирования дают ценную
информацию об относительной устойчивости ненасыщенных соединений. На основании
этих данных было установлено, что чем более замещенным является алкен, тем он
термодинамически стабильнее [7,8].
3. Процесс
дегидрирования органических веществ
Дегидрирование или дегидрогенизация - это химическая реакция отщепления
водорода от молекул органических соединений; одна из фаз процесса биологического
окисления. Осуществляется в присутствие катализаторов или под действием
акцепторов водорода. Каталитическое дегидрогенизация и обратная реакция -
гидрирование - связаны подвижным термодинамическим равновесием. Протеканию
дегидрогенизация способствует повышение температуры и понижение давления.
Осуществляют дегидрогенизация обычно при температуре > 300 °С и давлении
0,1-5 МПа; при необходимости применение более высоких давлений сочетают с
соответствующим повышением температуры. Катализаторы дегидрогенизации - обычно
многокомпонентные системы, содержащие переходные металлы, их оксиды или
сульфиды. При дегидрогенизации молекула реагирующего соединений образует
комплекс с катализатором, распадающийся затем на Н2 и продукт, десорбируемые с
поверхности катализатора [3,5].
Дегидрогенизация парафинов в ароматических соединениях
(дегидроциклизация) - одна из стадий каталитического риформинга; осуществляется
на оксиднохромовом катализаторе при 330-400 °С, давление 0,2-0,8 МПа.
Каталитическая дегидрогенизации используют в промышленности для получения
1,3-бутадиена, изопрена, стирола и др. В СССР в 1984 путем дегидрогенизация
было получено более 2 млн. бутадиена и около 11 млн. стирола. К каталитической
дегидрогенизации относят также так называемая окислительная дегидрогенизации,
протекающее в газовой фазе под действием окислителей (например: О2, SO2, H2O2,
I2, Вr2 и др.). Реакция практически необратима, что способствует увеличению
выхода целевых продуктов. Так, метанол в присутствии серебра (Ag) окисляется О2
воздуха при ≈ 400 °С в формальдегид с выходом около 80%. Окислительное
дегидрогенизация олефинов и алкилбензолов осуществляют в присутствие фосфатов
алюминия, молибдатов висмута, цеолитoв, активированного угля и др.
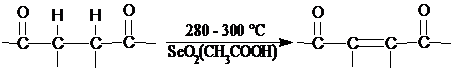
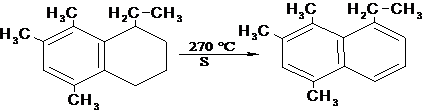
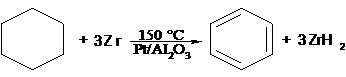
В
качестве акцепторов водорода при дегидрогенизация используют обычно
2,3-дициано-1,4-бензохинон, пероксиды Ni, SeO2, Se, S, а также металлы,
способные образовывать гидриды (Zr, Ti, интерметаллиды и др.). Выше указаны
некоторые примеры [5].
3.1
Разновидности дегидрирующего действия катализаторов
Катализаторы дегидрирования весьма многочисленны и разнообразны по своему
химическому составу. Так, еще по сводке из книги Платэ количество элементов,
упоминаемых в качестве активных компонентов катализаторов ароматизации
парафинов, составляло 51 и включало элементы почти всех групп периодической
системы, а главным образом IV-VI
групп. Число известных катализаторов и способов их приготовления непрерывно
пополняется.
Один и тот же катализатор, если он достаточно активен, способен к
дегидрированию углеводородов различных гомологических рядов и строения. Так, на
алюмохромокалиевых катализаторах были получены достаточно высокие выходы
продуктов дегидрирования низших n-парафинов
С3-С5 и ароматизации n-парафинов
С6-С7, циклогексана, дегидрирования этилбензола, олефинов, разветвленных
парафинов, пятичленных нафтенов, алкилгидроароматических и других
углеводородов.
Недегидрирующие побочные и вторичные реакции.
Как правило, не наблюдается параллелизма между дегидрирующей активностью
катализаторов и активностью их в реакциях крекинга и изомеризации. Это говорит
о том, что реакции изомеризации и крекинга обусловлены не теми активными
центрами, которые ответственны за дегидрирование.
Изомеризация углеводородов на дегидрирующем катализаторе может не только
быть побочной реакцией, но и в некоторых случаях является важной стадией процессов
дегидрирования. Так, ароматизация парафинов с длиной цепи не более пяти атомов
углерода обязательно включает стадию изомеризации.
При отравлении алюмохромового катализатора пиридином или хинолином
уменьшается не только изомеризующая активность, но и выходы «ненормальных»
продуктов дегидроциклизации n-октана
(т- и р-ксилолов), которые образуются через стадию изомеризации n-октана в изооктан или через
промежуточные циклические соединения.
С другой стороны, в условиях риформинга (давление водорода, «бифункциональные»
катализаторы) дегидрирование может быть одной из стадий изомеризации
углеводорода. Так, при контактировании n-гептана с алюмосиликатом или платиной, нанесенной на Аl2O3, изогептаны не образуются; в присутствии же смеси этих
контактов выход изогептанов равен 24%. При этом образуется также толуол.
Изомеризация протекает через промежуточные стадии дегидрирования -
гидрирования:

Дегидрирование
углеводородов, содержащих и не содержащих двойную связь.
Из соотношения продуктов превращения циклогексана в бензол и
метилциклопентан и метилциклопентана в бензол и циклогексан Гензель и сотр.
сделали вывод, что при действии сернистых соединений утрачивается активность
фторированного алюмоплатинового катализатора по отношению к стадии превращения
циклогексена в бензол, но не циклогексана в циклогексен, и, следовательно, что
указанные реакции протекают на разных активных центрах. Подразделение активных
центров катализаторов на такие, которые дегидрируют парафины и олефины.
К выводу о том, что для разграничения активных центров дегидрирования
парафинов и олефинов нет достаточных оснований, приходим и при анализе
экспериментального материала по влиянию химического состава на активность
катализатора. Как правило, все активные катализаторы дегидрирования парафинов
оказываются активными и в дегидрировании олефинов. О том, что это так,
свидетельствуют хотя бы данные о промежуточном образовании олефинов при
ароматизации парафинов, которое в противном случае не могло бы иметь места.
Наблюдается параллелизм, с одной стороны, в относительной реакционной
способности насыщенных и ненасыщенных углеводородов на катализаторах различной
природы, и, с другой стороны, - в соотношении активностей различных
катализаторов по отношению к дегидрированию указанных углеводородов. Так,
реакционная способность углеводородов возрастает с увеличением ненасыщенности
на алюмохромовом, алюмомолибденовом катализаторах, платине, никеле и др.
Также наиболее активными катализаторами для реакций дегидрирования низших
парафинов в олефины и ароматизации парафинов С≥6 являются промотированные
алюмохромовые катализаторы, причем, как правило, вводимые в катализатор добавки
или изменение способа приготовления или обработки катализатора оказывают
аналогичное влияние (активация, разрабатываемость и т.д.) на реакции, а также
на дегидрирование шестичленных нафтенов (табл. 2).
Таблица 2
Влияние добавок некоторых веществ на свойства катализаторов
дегидрирования
|
Исходные вещества
|
Добавки
|
Углеводороду
|
Влияние на каталитические
свойства
|
|
Cr2O3 - AI2O3
|
Оксиды щелочных металлов
|
Парафины C3-C7
циклогексан, изопентены
|
Повышение активности
|
|
То же
|
CaO, SrO, BaO
|
n-Бутан, n-гептан, циклогексан
|
То же
|
|
»
|
Оксиды Fe
|
n-Бутан, n-гексан
|
Уменьшение активности
|
|
»
|
Оксиды щелочных металлов
|
Изопентан, n-гептан,
циклогекссан
|
Замедление разработки
|
|
»
|
K2O
|
n-Бутан, n-гептан
|
Уменьшение коксообразования
|
|
MoO3 - AI2O3
|
Оксиды K, Rb, Cs, Ce, Nd, HI
|
n-Гексан,
n-гептан, циклогесксан
|
Повышение активности
|
|
Pt - AI2O3
|
Na2O
|
n-Гексан, циклогексан
|
Активность проходит через
максимум
|
|
Pd - AI2O3с добав. HF
|
Оксиды редкоземельных
элементов
|
n-Гексан, циклогексан, метилциклогексан
|
Повышение активности
|
Различия в удельных активностях отдельных металлов по отношению к
реакциям дегидрирования циклогексана, циклогексена и циклогексадиена достигают
нескольких порядков.
Если пренебречь сравнительно небольшими изменениями (в 2-3 раза), то
удельные активности металлов для дегидрирования всех трех углеводородов
располагаются в ряд:
Pd ≈ Pt ≈ Rh ≈ Ir > Ni > Ag ≈ Cu ≈ Co ≈ Fe
Естественно, представление об однотипности активных центров для
дегидрирования насыщенных и ненасыщенных углеводородов не означает того, что
для обеих реакций оптимальными должны быть одни и те же катализаторы.
Индивидуальные особенности углеводородов могут проявляться в побочных
реакциях и вторичных превращениях продуктов. Так, например, способность к
закоксовыванию катализатора у бутадиена значительно больше, чем у бутана или
бутилена. Для уменьшения коксообразования процесс дегидрирования n-бутилена в дивинил проводят в
присутствии водяного пара, а пары воды отравляют алюмохромовые катализаторы.
Поэтому для дегидрирования n-бутилеиов
применяют не алюмохромовый, а другие катализаторы, стойкие по отношению к воде,
например железохромокалиевый.
Разумеется, могут иметь значение и другие факторы, например соотношение
скоростей отдельных элементарных стадий. Так, если лимитирующей стадией
дегидрирования менее реакционно-способного из двух сравниваемых углеводородов
является химическое превращение, то на скорость реакции дегидрирования более
реакционно-способного углеводорода на том же катализаторе будут, возможно,
оказывать влияние и такие факторы, как скорость адсорбции или внутренней
диффузии. Поэтому для обоих углеводородов оптимальные катализаторы должны быть
разными [2].
3.2
Современные представления о природе активной поверхности катализаторов
дегидрирования
Оксиды хрома и некоторые другие оксидные катализаторы.
По современным представлениям, адсорбированный углеводород на оксидах
металлов присоединяется к ионам металла, а не кислорода. Образование связи
между адсорбированным углеводородом и ионом переходного металла происходит за
счет незаполненных d-уровней металла
или путем образования s-d-связи.
Некоторые авторы установили корреляцию между количеством Сr6+, содержащейся в алюмохромовом
катализаторе, и его дегидрирующей активностью, и в связи с этим высказали
предположение, что носителем каталитической активности является содержащийся в
катализаторе шестивалентный хром.
Это предположение, однако, недостаточно обосновано, так как в условиях
реакции восстановление Сr6+ и
других возможных соединений с валентным состоянием хрома выше трех происходит
быстро, а активность алюмохромового катализатора сохраняется продолжительное
время. При регенерации алюмохромового катализатора в токе СO2 высшие оксиды хрома не образуются,
а вместе с тем такие катализаторы не менее активны, чем регенерированные
воздухом.
Ван-Рейн из измерений магнитных моментов и сигналов ЭПР алюмохромовых
катализаторов, подвергавшихся различной окислительно-восстановительной
обработке, сделали вывод, что высшие оксиды хрома восстанавливаются в токе
сухого водорода, т.е. в условиях, благоприятствующих получению активного
алюмохромового катализатора, не только до трех-, но и до двухвалентного хрома
[2]. Соотношение между 2- и 3-валентными оксидами хрома определяется
равновесием:
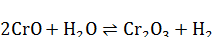
Дальнейшие экспериментальные подтверждения образования двухвалентного
хрома при восстановлении алюмохромовых катализаторов были получены Рубинштейном
[2]. Так, при обработке тщательно восстановленных алюмохромовых катализаторов
водой наблюдается выделение водорода. При обработке Н2O катализатора, восстановленного дейтерием, в газе обнаружен
только протий. Это значит, что водород образуется при разложении воды ионами Сr2+, обладающими восстановительными
свойствами, а не выделяется в результате десорбционного вытеснения водой.
Особенно активной катионной структурой в реакциях с участием водорода на
полупроводниках является d3.
Такую электронную конфигурацию имеет ион хрома в Сr2O3. Согласно Казанскому
и Швецу, активными центрами дегидрирования являются координационно ненасыщенные
ионы Сr+.
В образцах, содержащих адсорбированную воду, все ионы Сr+ находятся в октаэдрической
конфигурации. При отщеплении одной молекулы воды координация понижается до квадратной
пирамиды:
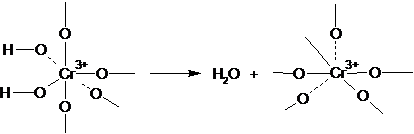
а
отщепление следующих молекул воды может приводить к дальнейшему понижению
координации. Для выяснения, являются ли присутствующие на поверхности
катализаторов, десорбировавших воду при высокой температуре, координационно
ненасыщенные ионы Сr3+ активными центрами дегидрирования, необходимы
дальнейшие исследования.
Крылова,
Козуненко и Кобозев [2] наблюдали максимум содержания стирола в катализаторе
дегидрирования этилбензола и парамагнитной восприимчивости при содержании хрома
в катализаторе ≈ 2%. Максимум удельной восприимчивости при небольшом
содержании хрома связан, по-видимому, с образованием δ-фазы Сr2O3. Что касается активности катализатора, то следует
иметь в виду, что оксид хрома достаточно активна сама по себе, и смешанная
алюмохромовая структура, если бы она играла существенную роль в катализе,
должна была бы быть весьма активной.
Однако
испытанные в работе образцы были мало активнымии, повидимому, обладали
значительной кислотностью. Поэтому наблюдавшееся при увеличении содержания
хрома свыше 2% снижение выходов стирола могло быть связано с образованием
побочных продуктов. Действительно, при дегидрировании менее
реакционно-способных углеводородов - парафинов - не наблюдалось максимума активности
при столь небольших добавках хрома, как 2%.
Буянов
связывает активность железохромовоцинковых катализаторов в реакции
дегидрирования бутиленов с присутствием в нем хромовой шпинели. Однако
значительной дегидрирующей активностью обладают оксид железа или оксид цинка,
не содержащие хрома, не говоря уже о самой оксида хрома. Поэтому в
железохромоцинковых катализаторах активными центрами могут быть, ионы любого
металла, входящего в состав катализатора. Образование же твердых растворов и
смешанных структур скорее способствует стабильности катализатора, препятствуя
рекристаллизации и нежелательным химическим превращениям катализатора, что
может быть особенно важно при дегидрировании бутилена и других олефинов ввиду
жесткости условий эксплуатации этих катализаторов.
Имеющиеся
данные не дают также оснований полагать, что дегидрирующая способность
хромовомагниевых или хромосиликагелевых катализаторов обусловлена образованием
активных смешанных структур. Остается неясной природа активирующего действия
малых количеств воды. Полностью обезвоженные катализаторы дегидрирования
неактивны. При увеличении содержания воды в сырье активность алюмохромовых
катализаторов проходит через максимум.
Можно
предположить, что вода входит в состав активных центров катализатора, оказывая
промотирующее действие. Обратимое отравление алюмохромового катализатора более
значительными ее количествами может быть объяснено блокированием активных
центров [2].
3.3 Процесс
дегидрирования
Отщепление водорода от алканов (дегидрирование) является обратимым
высокотемпературным каталитическим процессом и используется главным образом в
промышленности. Ввиду сложности протекания гетерогенных каталитических реакций
будут рассмотрены только некоторые общие закономерности.
В качестве катализаторов дегидрирования применяют оксиды металлов (Сr2Оэ, Fe2O3, ZnO и др.), а также металлы (Pt, Pd, Ni, Fe); при катализе оксидами температура
процесса 450-650 °С, при катализе металлами - около 300 оС.
Низшие алканы (С2-С4) при дегидрировании превращаются в алкены, а из
бутана, в зависимости от применяемого катализатора и условий реакции, может
получаться и бутадиен-1,3:
 Алканы,
содержащие пять атомов углерода в цепи (но не более), подвергаются
дегидроциклизации с образованием циклопентанового углеводорода:
Алканы,
содержащие пять атомов углерода в цепи (но не более), подвергаются
дегидроциклизации с образованием циклопентанового углеводорода:
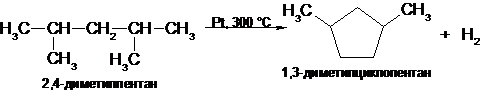
Если
в цепи алкана содержится шесть и более атомов углерода, то образующийся в
подобных условиях циклогексан (или его гомологи) подвергается дальнейшему
дегидрированию с образованием энергетически выгодного ароматического кольца:
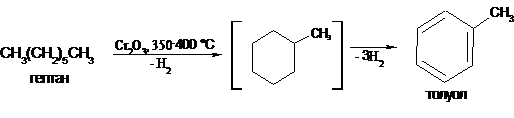
Дегидрирование алканов - реакция, обратная гидрированию ненасыщенных
углеводородов. Положение равновесия определяется температурой и давлением.
Обычно дегидрирование проводят при более высоких температурах, а гидрирование -
при сравнительно низких (до 200 °С), согласно принципу Лe Шателье.
Принцип
Ле Шателье-Брауна (1884 <http://ru.wikipedia.org/wiki/1884>) - если на
систему, находящуюся в устойчивом равновесии, воздействовать извне, изменяя
какое-либо из условий равновесия (температура <http://ru.wikipedia.org/wiki/%D0%A2%D0%B5%D0%BC%D0%BF%D0%B5%D1%80%D0%B0%D1%82%D1%83%D1%80%D0%B0>,
давление
<http://ru.wikipedia.org/wiki/%D0%94%D0%B0%D0%B2%D0%BB%D0%B5%D0%BD%D0%B8%D0%B5>,
концентрация
<http://ru.wikipedia.org/wiki/%D0%9A%D0%BE%D0%BD%D1%86%D0%B5%D0%BD%D1%82%D1%80%D0%B0%D1%86%D0%B8%D1%8F>,
внешнее электромагнитное поле
<http://ru.wikipedia.org/wiki/%D0%AD%D0%BB%D0%B5%D0%BA%D1%82%D1%80%D0%BE%D0%BC%D0%B0%D0%B3%D0%BD%D0%B8%D1%82%D0%BD%D0%BE%D0%B5_%D0%BF%D0%BE%D0%BB%D0%B5>),
то в системе усиливаются процессы, направленные на компенсацию внешнего
воздействия.
Анри
Ле Шателье
<http://ru.wikipedia.org/wiki/%D0%9B%D0%B5_%D0%A8%D0%B0%D1%82%D0%B5%D0%BB%D1%8C%D0%B5,_%D0%90%D0%BD%D1%80%D0%B8_%D0%9B%D1%83%D0%B8>
(Франция <http://ru.wikipedia.org/wiki/%D0%A4%D1%80%D0%B0%D0%BD%D1%86%D0%B8%D1%8F>)
сформулировал этот термодинамический принцип подвижного равновесия, позже
обобщённый Карлом Брауном
<http://ru.wikipedia.org/wiki/%D0%91%D1%80%D0%B0%D1%83%D0%BD,_%D0%9A%D0%B0%D1%80%D0%BB_%D0%A4%D0%B5%D1%80%D0%B4%D0%B8%D0%BD%D0%B0%D0%BD%D0%B4>.
Принцип
устойчивости применим к равновесию любой природы: механическому, тепловому,
химическому, электрическому (эффект Ленца
<http://ru.wikipedia.org/wiki/%D0%9F%D1%80%D0%B0%D0%B2%D0%B8%D0%BB%D0%BE_%D0%9B%D0%B5%D0%BD%D1%86%D0%B0>,
явление Пельтье <http://ru.wikipedia.org/wiki/%D0%AD%D1%84%D1%84%D0%B5%D0%BA%D1%82_%D0%9F%D0%B5%D0%BB%D1%8C%D1%82%D1%8C%D0%B5>)
[3].
Дегидрированию благоприятствует пониженное давление; на практике
используют давление 1-10атм (1атм = 101,3 кПа.), а иногда - ниже атмосферного.
Кроме того, для каждого соединения существует довольно узкий диапазон
температур, в котором протекает прямой или обратный процесс [6,7,8].
Заключение
Роль катализаторов в химическом производстве исключительно велика.
Получение серной кислоты, синтез аммиака, получение из твердого угля жидкого
топлива, переработка нефти и природного газа, получение искусственного каучука,
пластмасс, гидрогенизация жиров - вот далеко не полный перечень важнейших
производств, где применяются катализаторы. Очевидно, поиски новых, все более
совершенных катализаторов будут способствовать повышению производительности
труда и снижению себе стоимости продукции.
Особую роль играют биологические катализаторы - ферменты. При их участии
протекают сложные химические процессы в растительных и животных организмах [9].
Для катализаторов гидрирования наибольшее внимание уделено нанесенным Ni-катализаторам, в последнее время все
шире применяющимся не только в крупнотоннажных процессах, но и в тонком
органическом синтезе. Обсуждены катализаторы на основе благородных металлов (Pt, Pd, Ru, Rh, Ir), нанесенные сульфидные катализаторы гидроочистки и
принципиально новые катализаторы гидрирования, такие как карбиды металлов,
исследование которых быстро развивается в последние годы. Приведены некоторые
примеры использования приемов механохимии для приготовления катализаторов, а
также контактов, обеспечивающих получение экологически чистых моторных топлив.
В настоящее время наибольшее распространение имеют катализаторы,
приведённые в таблице 3.
Таблица 3
Наиболее распространённые процессы и соответствующие катализаторы
|
Процесс
|
Катализатор
|
|
Гидрирование олефинов
|
Нанесенные Ni,
Сu, Pt, Pd, Ru, Rh
|
|
Гидрирование ацетиленовых
Углеводородов
|
Нанесенные Ni,
Pd, Ni - Pd,Со - Pd, Сu - Pd
|
|
Гидрирование ароматических
Углеводородов
|
Нанесенные Ni,
Pd, Rh, Pt, Со - Мо, скелетный Ni
|
|
Гидрооблагораживание
моторных топлив
|
Ni, Ni - Мо, Со - Мо на АI2О3
икзельгуре
|
|
Гидрирование карбонильных
соединений
|
Скелетные и нанесенные Ni
и Со, нанесенные Pd, Rh и Pt, Сu - Сr - О, Сu на кизельгуре
|
|
Гидроочистка
(гидродесульфуризация)
|
Ni - Мо, Ni - W, Со - Мо, Со
- W, Ru на АI2O3 и углероде, карбиды Мо и W
|
|
Гидрирование нитрилов
|
Нанесенные Ni, Pd, Pt
|
|
Гидрирование СО и
СО2(метанирование)
|
Нанесенные Ni,
Си, Со, Fe, Pt, Rh, сетки Pt, карбиды Мо и W
|
Обзор ограничен гетерогенными катализаторами гидрирования, в нем не
рассматриваются многочисленные гомогенные системы, а также скелетные
металлические катализаторы Ренея (Ni, Со, Сu и др.) и соосажденные катализаторы
[1].
Для дегидрирования следует вывод, что вопрос о структуре активных центров
даже для наиболее изученных катализаторов остается спорным.
Носителями каталитической активности в реакциях дегидрирования могут быть
атомы металла (в металлических), атомы или ионы металла (в нанесенных
металлических) и ионы металла в оксидных катализаторах. В состав активных
центров входят также хемосорбированные атомы или молекулы (водород в случае
металлического, вода в случае оксиднохромового катализатора). Дегидрируемая
молекула может быть связана с поверхностью одно-, двух- или многоточечной
адсорбцией с различными типами химической связи (σ-связь или разновидности
координационных связей).
Тем не менее наблюдается лишь небольшое число разновидностей
дегидрирующего действия катализаторов на углеводороды. Имеется достаточно
оснований полагать, что дегидрирование и ароматизация парафинов протекают на
одних и тех же активных центрах, и то же можно сказать относительно парафинов и
олефинов, а также нафтенов (кроме случая непосредственного превращения шестичленных
нафтенов в ароматические углеводороды).
Для всех этих случаев, при которых проявляется аналогия в действии
катализаторов различной природы и превращении углеводородов различных
гомологических рядов и строения, следует предпочесть модель промежуточного
соединения с одно- или двух-, но не с многоточечной адсорбцией, а из
существующих гипотез о структуре активных центров те, которые связывают
активность с самим веществом катализатора и его дисперсностью, а не с теми или
иными соединениями его с носителями. Активный катализатор, как, например,
платина или оксид хрома, сохраняет значительную дегидрирующую способность на
любом носителе различной природы, способном обеспечить стабильность его
дисперсности.
Список литературы
. Крылов
В.О., Навалихина Д.М. Гетерогенные катализаторы гидрирования / В.О Крылов [и
др.] // Журнал «Успехи химии», - М.: 1998. - 67 (7). - С. 587 - 616.
2. Скарченко
В.К. Каталитическое дегидрирование углеводородов / В.К Скарченко [и др.] //
Журнал «Успехи химии» - М.: 1971 - 40 (12). - С. 997 - 1013.
3. Свободная энциклопедия
«Википедия»: [сайт]. - URL: <http://ru.wikipedia.org/wiki>. (дата
обращения 20.09.2012г).
. НеницескуК. Общая
химия / НеницескуК. - перевод с румынского Д.Г. Батыра, И.М. Рейбеля, Х.Ш.
Харитона и А.И. Марина / [под ред. А.В. Аблова]. - М.: Изд-во
«Мир», 1968. - 816 с.
. Словарь Академик / Е.
А. Дембовская, М. А. Меньковский//: [сайт]. - URL: <http://dic.academic.ru>. ( дата обращения 20.09.2012г).
. Химический
энциклопедический словарь / Е.В. Вонский, А.А. Гусев [и др.] / [гл. ред. И.Л.
Кнунянц]. - М.: Сов. Энциклопедия, 1983. - 792 с.
. Органическая химия:
учеб. для вузов: Кн. 1 / В. Л. Белобородов [и др.] / [под ред. Н. А.
Тюкавкиной]. - 2-е изд., стереотип. - М.: Дрофа,200. - 640 с
. Грандбер И. И.
Органическая химия: Учебник для вузов, обучающихся по агроном.спец. - М.:
Изд-во Дрофа, 2001. - 674 с.
. Хомченко Г.П. Пособие
по химии для поступающих в вузы. - М.: РИА «Новая волна»: Изд-во Умеренков,
2008. - 480 с.