Фенилкетонурия: диагностика и лечение
Содержание
Введение
. Фенилкетонурия как
результат нарушения метаболизма фенилаланина и схожие заболевания
. Диагностика фенилкетонурии
. Лечение фенилкетонурии
Заключение
Список использованной
литературы
Введение
Фенилкетонурия является редким, но тяжелым заболеванием, которое приводит
к нарушению умственного развития человека в раннем возрасте. До 50-х гг.
прошлого века она оставалась малоизученной и не поддавалась лечению. Впервые
заболевание было описано в 1934 г. и постепенно с развитием медицины и, в
частности биохимии и генетики, у учёных сформировалось необходимое
представление о причинах, механизме развития, а, следовательно, и о способах
лечения фенилкетонурии. Тысячи детей были спасены от слабоумия, серьезных
поражений печени и почек, а также летального исхода, который нередко влекло это
заболевание.
Однако, на сегодняшний день некоторые вопросы остаются открытыми:
например до какого возраста больной фенилкетонурией должен придерживаться
диеты, возможны ли новые более эффективные методы ранней диагностики болезни, а
также альтернативные методы лечения?
Все они могут и должны решаться с использованием комплексного подхода, на
основе знаний и опыта медицины, генетики и биохимии. Биохимия занимает важное
место и является неотъемлемой составляющей в системе медицинских наук. Изучение
биохимических процессов человека, животных, растений оказывает содействие
общему прогрессу научных знаний и развитию здравоохранения. Органические
соединения является звеном, что скрепляет представления о живой и неживой
природе в единую цепь. На основе знаний об этих соединениях и процессах
раскрываются тайны развития в материальном мире от простейших молекулярных форм
к полимерным с присущими им функциями, вплоть до функций в живом организме. Все
это определяет актуальность выбора темы работы.
В нашей реферативной работе мы рассмотрим биохимическую природу
фенилкетонурии, распространенные, а также альтернативные методы диагностики и
лечения заболевания в РФ и мире.
. Фенилкетонурия как результат нарушения метаболизма фенилаланина
и схожие заболевания
Фенилкетонурия (ФКУ) («фенил» - от слова фенилаланин, названия
аминокислоты, «кетон» - от слова кетоны, названия химических соединений, «урия»
- выделение продуктов обмена с мочой) - это наследственная болезнь, обусловленная
дефектом гена фермента фенилаланингидроксилазы (ФФГ), что находится на длинном
плече 12 хромосомы (12q 22-24). Дети, родившиеся с фенилкетонурией, не способны
метаболизировать (перерабатывать) фенилаланин (часть протеина), который из-за
этого накапливается в крови. Такое патологически высокое количество
фенилаланина (жизненно необходимой аминокислоты) препятствует нормальному
развитию мозга. При условии отсутствия лечения, оно приводит к умственной
отсталости. Фенилкетонурия - это наследственное заболевание, которое
характеризуется главным образом поражением нервной системы.
человек из 50-70 людей является носителем гена фенилкетонурии. Хотя эта
болезнь не очень распространена (1 ребёнок на каждых 8-13 тысяч новорождённых),
она требует больших затрата для борьбы с ней. В США, например, каждый год
тратится несколько миллионов долларов на лечение больных детей. В РФ на один
выявленный случай заболевания тратится более полумиллиона рублей, не говоря уже
о терапии. Болезнь также сопряжена с моральными и этическими проблемами.
Фенилаланин - одна из 20 аминокислот, которые входят в состав белковой
молекулы (рисунок 1). Принимает участие в биохимических процессах формирования
протеинов и кодируется определённым геном ДНК. Кетоны выступают продуктами
обмена данной аминокислоты.
В норме фенилаланин превращается в тирозин с помощью фермента
гидроксилазы фенилаланина (ФФГ). Для этого преобразования необходим
тетрагидробиоптерин (ВН4) (рис. 2). Он требуется также для биохимических
процессов в ходе синтеза нейромедиаторов - допамина, норэпинефрина, и
серотонина. В норме уровень фенилаланина в крови отвечает 180 mmol/l (3 mg/dl)
или меньше.
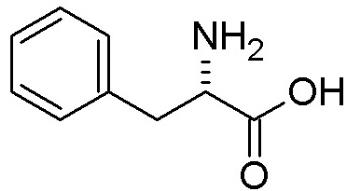
Рисунок 1 Формула фенилаланина - 2-амино-3-фенил-пропановой кислоты
Если же не происходит превращение в тирозин, то помимо повышения уровня
фенилаланина, образовываются побочные продукты обмена: фенилпируват,
фениллактат, фенилацетат и фенилацетилглутамин (рис. 3), которые появляются в
моче больного ребёнка, благодаря чему болезнь и получила своё название.
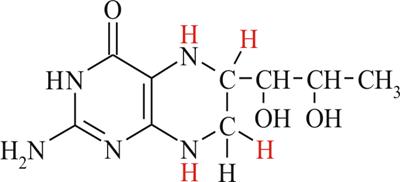
Рисунок 2 Схема тетрагидробиоптерина (ВН4)
При этом различают три типа заболевания: классическое, вариантные и
материнское. В случае классической фенилкетонурии имеет место мутация при
аутосомно-рецессивном наследовании, когда падает активность фермента печени,
почек и поджелудочной железы феналаланина-4-монооксегеназы, обеспечивающей
образование тирозина.
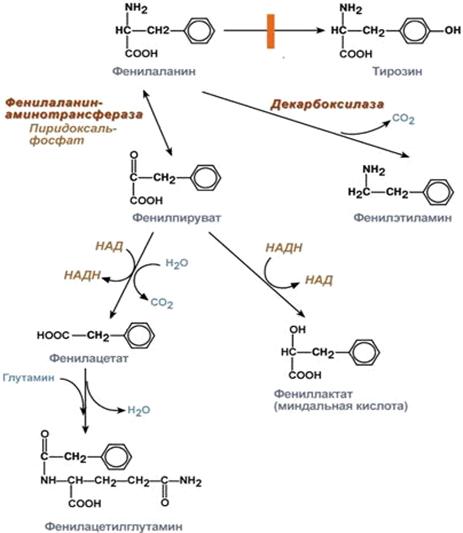
Рисунок 3 Превращение фенилаланина при фенилкетонурии
Первая вариантная Фенилкетонурия характеризуется подобной мутацией
независимого фермента дигидробиоптерин редуктазы, что принимает участие в
восстановлении активной формы тетрагидробиоптерина, являющегося кофактором
гидроксилаз фенилаланина и триптофана. Следствием этого является также
нарушение превращения фенилаланина в тирозин. При втором подтипе вариантого
заболевания недостаток тетрагидробиоптерина (ВН4) возникает из-за
недостаточности 6-пирувоил-тетрагидроптеринсинтазы.
Материнская Фенилкетонурия обнаруживается, когда женщина, больная
фенилкетонурией не соблюдает специальную диету, в т.ч. во время беременности.
Различные формы заболевания характеризуются различным патогенезом,
клинической картиной и способами лечения. Так классическая ФКУ лечится
преимущественно диетой, а первые симптомы появляться на первом году жизни
ребенка. Самым характерным из них является неприятный запах мочи. При
вариантных или атипичной ФКУ (также связанной с дефицитом тетрагидробиоптерина)
симптомы появляются в первые шесть месяцев жизни ребёнка, требуется
заместительная терапия, направленная на изменение метаболизма аминокислот.
Симптомы болезни, такие как рвота, судороги, потливость и пр., связаны с
накоплением производных аминокислоты в различных органах (рис. 4)
Кроме ФКУ существует несколько иных причин повышения уровня фенилаланина
в крови (гиперфенилаланинемия) (табл. 1).
Поэтому биохимическое отклонение организма нуждается в детальном
исследовании при диагностике. Прежде чем устанавливать конкретный диагноз для
грудного ребёнка с гиперфенилаланинемией и назначать лечение должен быть
определён тип заболевания. Ниже представлен перечень причин
гиперфенилаланинемии и биохимические показатели для их дифференциации.
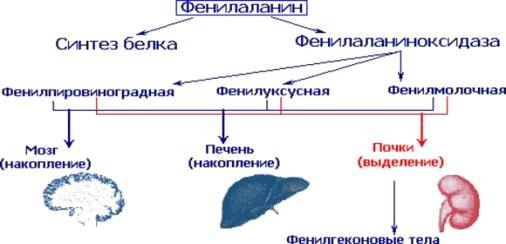
Рисунок 4 Распределение производных фенилаланина при фенилкетонурии
Таблица 1 Причины повышения фенилаланина в крови
|
Уровень фенилаланина в крови (mmol/l)
|
Уровень тирозина в крови (mmol/l)
|
Другие аминокислоты
|
Уровень птеринов (метаболитов тетрагидро-биоптерина) в моче
|
|
ФКУ
|
>1200
|
< 250
|
норма
|
норма
|
|
ФКУ средней тяжести
|
600 -1.200
|
< 250
|
норма
|
норма
|
|
Рост фенилаланина без ФКУ
|
180 - 600
|
< 250
|
норма
|
норма
|
|
Дефицит циклогидролазы
|
180 -1.200
|
< 250
|
норма
|
аномальный
|
|
Тирозенемия др.
|
180 -600
|
> 250
|
Избыток метионина
|
норма
|
Дефицит активности ФФГ приводит к повышению уровня фенилаланина в крови.
Степень повышения зависит от степени энзимной недостаточности. Классическая
форма ФКУ, которая в Соединенных Штатах и Европе случается с частотой 1:13.000
(1:500 и 1:10000 по регионам РФ), обычно дает при обследовании такие
показатели:
) концентрация фенилаланина в крови 1200 mmol/l (20 mg/dl) и выше у
новорожденных при грудном вскармливании или употреблении смесей;
) концентрация тирозина в крови меньше 250 mmol/l (5 mg/dl);
) проба на птерин в моче, который указывает на недостаточность ВН4, в
норме.
При классической форме ФКУ активность ФФГ печени приближается к нулю. При
мягких формах ФКУ (атипичная ФКУ) наблюдается уровень фенилаланина в крови в
пределах 600-1200 mmol/l (10-20 mg/dl), нормальный или низкий уровень тирозина,
нормальная проба на птерин в моче. На этом фоне проявляется незначительная
резидуальная (остаточная) активность ФФГ.
Приблизительно 20-50% грудных детей, у которых была выявлена
гиперфенилаланинемия во время скрининга новорожденных (обязательного
обследования в первые дни жизни), имели стойкую мягко проявляющуюся, не
связанную с ФКУ гиперфенилаланинемию с уровнем фенилаланина в крови 180-600
mmol/l (3-10 mg/dl) и стойкой активностью ФФГ до 10%. Гиперфенилаланинемия
считалась не вредной, но, основываясь на результатах последних исследований,
были выдвинуты предположения, что стойкое повышение фенилаланина в крови выше
360 mmol/l (6mg/dl) может снижать показатель умственного развития ІQ. Другие
исследования, наоборот, показывают, что стойкое повышение фенилаланина в крови
до уровня 600 mmol/l (10mg/dl) не вызывает никаких проблем. Существует
расхождение во взглядах на то, назначать ли ограничивающую диету грудным детям
с уровнем фенилаланина 360-600 mmol/l (6-10 mg/dl). В некоторых случаях
диетическое лечение назначается детям с уровнем фенилаланина выше 480 mmol/l
(8mg/dl).
В целом именно нарушение синтеза и переработки ВН4 приводят к
возникновению гиперфенилаланинемии. Важно отметить, что дети с этим отклонением
страдают от прогрессирующей, часто в тяжелой форме, неврологической деградации,
если им по ошибке ставят диагноз классической ФКУ и лечат только диетой со
сниженным содержимым фенилаланина. Недостаточность ВН4 ограничивает
преобразование фенилаланина в тирозин, а связанна с деятельностью ЦНС по
превращению тирозина в ДОФА (рис. 5) и триптофана - в 5-гидрокситриптофан (рис.
6). Таким образом, эти нарушения приводят к повышению фенилаланина и нарушению
в синтезе нейромедиаторов, таких как меланин, мелатонин, серотонин. Редко встречаются
случаи, когда недостаточность ВН4 является только периферийной и не влияет на
синтез медиаторов в ЦНС, где их уровень остается в норме. За последние годы в
некоторых странах был налажен сбор данных по диагностике и лечению этого
редчайшего заболевания (1:1.000.000 или 1% от всех новорожденных с неонатальною
гиперфенилаланинемией). Лечение предусматривает введение дополнительного ВН4 и
медикаментов, которые повышают уровень нейромедиаторов.
Гиперфенилаланинемия, выявленная во время благодаря скринингу новорожденных,
может иметь временный характер. Это, как правило, относительно невысокое
повышение фенилаланина, наблюдающееся в первые месяцы жизни и не нуждающееся в
специальной терапии. В некоторых случаях гиперфенилаланинемия является
вторичной как следствие временной гипертиреоз - анемии. Описаны случаи более
тяжелых форм временной гиперфенилаланинемии, при которых сначала возникала
необходимость в лечении (уровень 480-840 mmol/l или 8-18 mg/dl). Со временем
выздоровление наступало само по себе в процессе роста ребёнка.
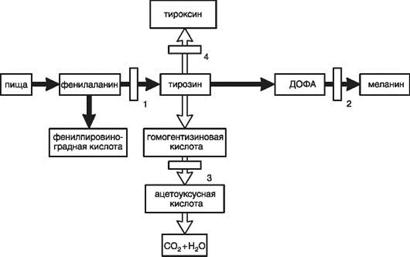
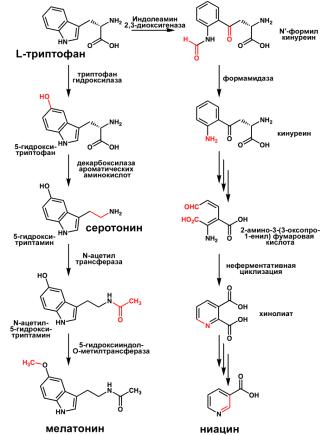
Рисунок 6 Синтез нейромедиаторов из триптофана
Гиперфенилаланинемия часто случается при тирозинемии I-го типа. Это
симптом вторичного характера. Очевидно, он возникает вследствие тяжелого
метаболического поражения печени. Одновременное определение уровней
фенилаланина и тирозина дает возможность определить наличие тирозинемии или
ФКУ, как причины гиперфенилаланинемии.
Галактоземия и другие заболевания, которые вызывают дисфункцию печени,
также могут вызывать повышение фенилаланина в крови вторичного характера. В
таких случаях гиперфенилаланинемия сопровождается повышением в крови уровня
метионина и тирозина. Чтобы проявить такие вторичные причины
гиперфенилаланинемии, необходимо провести дальнейшее обследование ребенка и
узнать его клинический фенотип.
Вследствие нарушения обмена фенилаланин накапливается в крови и тканях и
становится «ядом» для растущего организма. Дети, которые родились с
фенилкетонурией, чаше всего, на протяжении первых месяцев жизни кажутся вполне
здоровыми. Но, если лечение не начато, они через 3-5 месяцев теряют
приобретенные навыки, интерес к окружению. А когда им исполняется год, они уже
имеют умственную отсталость.
Дети с ФКУ также обладают пониженным мышечным тонусом, часто являются
раздражительными. Их тело и моча имеют характерный «мышиный» запах или запах
плесени. Кожа у них довольно сухая, наблюдаются частые дерматиты. В некоторых
случаях возникают судороги, характерные «кивки». Как правило, больные дети
хорошо развиваются физически, имеют чрезмерный вес и более светлый цвет волос,
чем их родственники.
Фенилкетонурия является, как мы уже знаем, наследственной болезнью. Она
проявляется лишь тогда, когда оба родителя имеют ген болезни и передают его
ребенку. Такой тип наследования называется аутосомно - рецессивным. Если в
одной клетке есть один нормальный ген и один ген фенилкетонурии, то такой
человек называется носителем. Он не имеет никаких проявлений болезни, но может
передать болезненный ген своим потомкам (рисунок 7).
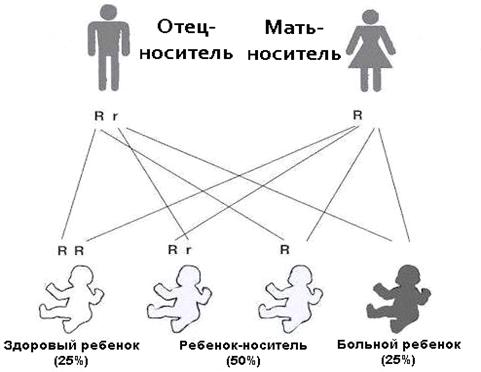
Рисунок 7 Передача аутосомно-рецессивных признаков от родителей с геном
фенилкетонурии
Если оба родители являются носителями, то существует 25% шансов, что
каждый из них передаст дефектный ген ребенку, вызывая, таким образом, рождение
больного ребенка. Есть также 25% шансов, что они передадут здоровые гены и
ребенок родится абсолютно здоровым. И, в конце концов, есть 50% шансов, что у
ребенка будет один нормальный ген и один ген фенилкетонурии. В этом случае
ребенок будет носителем, как и его родители.
. Диагностика фенилкетонурии
С помощью специального тестирования крови (нужно лишь несколько капель,
которые высушивают на фильтровальной бумаге) можно диагностировать болезнь,
когда ребенку всего 5-6 дней от роду. Бумага представляет собой медицинскую
тест-бумагу, на которой сухое пятно крови подвергается радиоимунному анализу.
Искомые стабильные и схожие с ними маркированные радионуклиды со специальными
связывающими системами находятся при помощи радиоспектрометра. В данном случае
проба крови анализируются на уровень фенилаланина, и определяется причина его
повышения, если таковое имеется.
Также используют биохимический анализ крови (скрининг-тест «пяточка»).
Ошибочно положительные результаты скрининга новорожденных - это временные
отклонения от нормы (например, из-за недоношенности). Поэтому необходимо
проведение повторного обследования.
Существует также тест мочи с трихлорным железом (фенилпируват, вступая в
реакцию, дает зеленую окраску), но он предоставляет менее надежные результаты.
Причина заключается в том, что в первые дни жизни фенилаланин не может
накапливаться на уровнях, которые необходимые для получения положительного
результата теста.
Молекулярная диагностика (диагностика на уровне гена) возможна как для
пренатальной (дородовой), так и постнатальной (послеродовой) диагностики
болезни. В данном случае производится обычный и расширенный поиск частых
мутаций в гене ФАГ (PAH), кодирующим фенилаланингидроксилазу, а также прямая
диагностика тетрагидробиоптерина (ВН4), базирующая на поиске мутаций гена QDRP. Параллельно с этим проходит поиск
мутаций генов PTS, GCH1. Для исследования, как правило, берутся образцы
крови родителей, реже других родственников, иногда слюна.
Обследование ребенка с ФКУ включает установление количественных уровней
аминокислот в плазме и сыворотке крови при особом внимании к фенилаланину и
тирозину, а также сбор проб крови и мочи на анализ птерина. Параллельно с этим
родителей информируют об особенностях ФКУ и подобных заболеваний и
предоставляют консультацию по генетическим вопросам. В некоторых случаях,
особенно когда необходимость введения диеты становится более спешной, чем
представлялось после первых анализов, целесообразно сделать анализ на ФФГ -
генотипа по сыворотке крови, чтобы убедить родителей в точности диагноза и в необходимости
продолжительного медицинского надзора. Определение уровня фенилаланина в крови
каждого из родителей должно быть сделано в начале обследования ребенка. Редко,
но случается так, что у кого-то из родителей повышен уровень фенилаланина.
Раннее начало терапии означает проведение скрининговой программы в
родильных домах. Без четко налаженного скрининга получить хорошие результаты в
лечении ФКУ невозможно. В РФ этот процесс регламентируется такими
нормативно-правовыми актами, как Приказ Минздрава РФ от 25.03.2009 N 133н «Об
организации скрининга в учреждениях государственной и муниципальной систем
здравоохранения», Приказ Минздрава РФ от 30.12.1993 N 316 (ред. от 05.08.2003)
«О дальнейшем развитии медико-генетической службы Министерства здравоохранения Российской
Федерации», Приказ от 16.04.2012 N 366н «Об утверждении Порядка оказания
педиатрической помощи», Приказ от 15.11.2012 N 921н «Об утверждении Порядка
оказания медицинской помощи по профилю «неонатология» и др. Но все мероприятия
опираются на метрологическую базу биохимической науки.
В действующей практике диагностики фенилкетонурии существует ряд ключевых
моментов, продиктованных именно биохимической спецификой, на которые нужно
обратить особое внимание.
Во-первых, очень важно контролировать время забора крови, в противном
случае возможны псевдонегативные результаты. В практике бывали случаи, когда в
медико-генетический центр обращались семьи с ребенком в возрасти 6-8 месяцев с
жалобами на задержку психомоторного развития. При обследовании был выявлен
уровень крови фенилаланин 1200 mmol/l. Оказывалось, что ребенок был
обследован по скрининг-программе и в образце крови, полученном из родильного
дома, фенилаланин не превышал 120 mmol/l (данные анализа были ретроспективно
проверены в центре). В этих случаях причиной низкого уровня фермента у больного
ФКУ ребенка в роддоме стал забор крови на 1-2 день жизни, когда ребенок еще не
получил достаточного количества грудного молока. Ведь метаболизация происходит
после употребления пищи. В литературе также встречаются описания случаев
псевдонегативных результатов; большинство авторов считают, что одиночные случаи
пропуска ФКУ при скрининге возможны именно за счет технических ошибок во время
забора крови. Поэтому на бланке с образцом крови обязательно должны стоять две
даты: родов и забора крови.
Другой необходимый момент при проведении скрининг-программы - это ее
максимальный охват. При отсутствии образца крови ребенка, ее забор
осуществляется по месту проживания или в отделениях стационаров для
новорожденных, если ребенок переводится туда к 4-му дню жизни. Такой учет
новорожденных разрешает обеспечить охват скринингом до 98-99% детей.
Очень важным является и время, которое занимает верификация диагноза. Все
это должно быть сделано в течение 1-го, 2-х месяцев жизни, в противном случае
программа теряет смысл.
Своеобразным контролем качества проведения массового скрининга
новорожденных является позднее выявление больных ФКУ.
. Лечение фенилкетонурии
С того момента, как обследование показало, что уровень фенилаланина у
ребенка повышен (во многих клиниках это 480 mmol/l или 8mg/dl), вводится диета
с ограниченным содержимым фенилаланина. Если анализы на птерин показали
отклонение от нормы, может возникнуть потребность в дополнительном приеме ВН4 и
медикаментов, которые увеличивают уровень нейромедиаторов.
Принцип лечения фенилкетонурии - специальная диета, т.е. лечебное
питание. Он заключается в ограничении поступления в организм ребенка
фенилаланина с пищей. Если начать диету с 10 - 20-го дня жизни, то это
предотвращает развитие умственной отсталости.
Фенилаланин входит в состав всех белковых продуктов (рисунок 8, таблица 2). Поэтому из рациона
питания больного необходимо исключить те продукты, которые имеют много белка. К
ним относятся мясо, рыба, яйца, молоко и молочные изделия, хлеб.
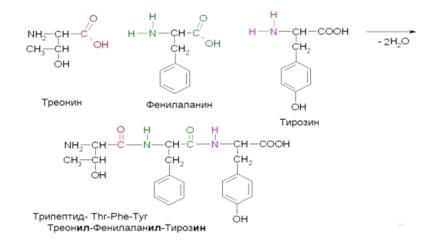
Рисунок 8 Пример соединения фенилаланина с другими аминокислотами и их
остатками в составе белка
Таблица 2 Сравнительное содержание фенилаланин в продуктах питания
|
Название продукта
|
Содержание фенилаланин на 100 г/мл (мг)
|
Общее количество белка на 100 г/мл (мг)
|
|
Хлеб из муки высшего сорта
|
21
|
0,5
|
|
Макароны
|
46
|
1,0
|
|
Сливки
|
93
|
2,0
|
|
Мясной бульон
|
66
|
10,7
|
|
Томатный концентрат
|
121
|
0,6
|
|
Морковный сок
|
19
|
0,93
|
|
Бобы
|
343
|
7,1
|
|
Картофель
|
85
|
1,9
|
|
Яблоко
|
12
|
0,4
|
|
Сахар
|
0
|
0
|
Лечебное меню больного, в основном, составляют продукты растительного
происхождения - овощи, фрукты, ягоды, зелень. В них мало белков и почти
отсутствует фенилаланин (в основном он содержится в бобовых, овсе и зародышах
пшеницы), достаточно жиров, углеводов, а также витаминов, макро-, и
микроэлементов, необходимых для развития организма.
Но организм ребенка не может формироваться при таком дефиците белка.
Поэтому в рацион ребенка добавляют специальные лечебные смеси, в составе
которых содержится уменьшенное количество белка и белок, в котором полностью
удален фенилаланин, такие как «Лофеналак», «Фенил- Фри», «Нофелан» и прочие.
Эти продукты изготавливают либо из смеси кристаллических аминокислот (без
фенилаланина), либо на основе гидролизата-белка, из которого удален
фенилаланин. Гидролиз сывороточных белков может быть осуществлен химическим
(под действием минеральных кислот и щелочей при повышенных температурах) и
ферментативным (с использованием препаратов протеолитических ферментов)
способами. В ходе кислотного гидролиза расщепляются все типы пептидных связей
между различными аминокислотными остатками, и поэтому возможно практически
полное расщепление белка. Однако кислотный гидролиз как технологический процесс
имеет ряд недостатков (частичное или полное разложение ряда аминокислот,
образование меланоидинов, необходимость нейтрализации кислоты).
При щелочном гидролизе белка происходит рацемизация аминокислот, в
результате чего получающийся гидролизат утрачивает биологическую ценность. В
пищевых производствах щелочной гидролиз белка не применяется. Весь процесс
является цепью сложных биохимических и физико-химических реакций (рис. 9).
Расчет белкового компонента пищи для больного ребенка должен производить
врач.
фенилкетонурия
биохимический метаболизм лечение
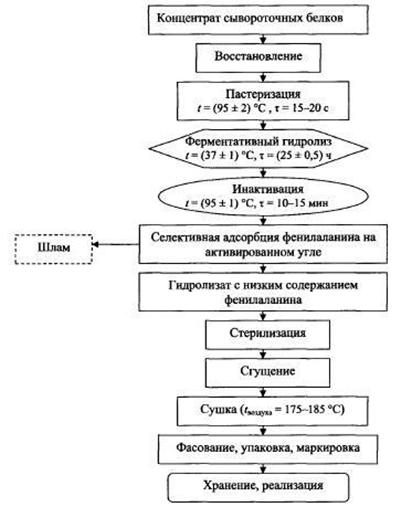
Рисунок 9 Технологическая схема гидролиза сывороточных белков,
используемых в энтеральных смесях для детей больных фенилкетонурией
Всем больным ФКУ назначается диета с применением белковых гидролизатов
или аминокислотных смесей, которые обеспечивают концентрацию фенил-сыворотки
крови до 240 - 360 mmol/l (4-6 mg/dl).
Если ребенок находится на искусственном вскармливании, то натуральным
продуктом для него будет та детская смесь, которую он получает. Состав, как
правило, указан на банке. В дальнейшем коррекция диеты осуществляется с обязательным
определением уровня фенилаланина 1 раз в неделю до 1-го года жизни, а потом 1
раз в месяц до 3-х лет и 1 раз в квартал детям, которые старше 3-х лет.
Практика свидетельствует, что до окончания пубертатного возраста (до 16-18 лет)
диеты нужно придерживаться даже наименее пораженным детям.
На рынке начинают появляться новые формы, альтернативные аминокислотным
добавки порошкообразной формы. В спрессованном виде или в капсулах они занимают
меньше места, и могут употребляться дополнительно или вместо иного препарата во
время путешествий.
ФКУ является незаметной инвалидностью. При нарушении диеты недуг
проявится с новой силой спустя некоторое время.
Количество взрослых молодых людей, которые проходят лечение с раннего
детства, возрастает. Некоторые из них придерживаются диетического питания,
другие - потребляют обычную пищу согласно тем рекомендациям, которые
существовали до середины 80- х гг. Есть также те, кто возвратился к диете после
периодов обычного питания. Взрослые больные, которые избрали диетическое
лечение, отмечают, что их состояние улучшается, когда они удерживают низкий
уровень сывороточного фенилаланина.
Фенилкетонурии не является ограничением для брака, совместного
проживания, семейной жизни. Но молодые люди должны сознавать, что их дети будут
носителями гена ФКУ. Это не означает, что они обязательно заболеют, поскольку
заболевание передается от обоих родителей, как мы рассмотрели ранее.
Правильность диетического лечения ФКУ подтверждается определением уровня
фенилаланина в крови. На фоне диетотерапии он не превышает 240 - 480 mmol/l.
Частота проведения мониторинга крови тоже разная, но британские медики
рекомендуют делать анализы каждую неделю для детей до 4-х лет, каждые 2 недели
для детей 10-ти лет и ежемесячно для детей после 10-ти лет и для взрослых.
Контроль за лечением, физическим и умственным развитием ребенка осуществляют
врач-генетик, детский психиатр, психолог.
В периоды частых инфекционных заболеваний может возникнуть потребность
чаще проверять уровень фенилаланина в сыворотке крови.
Когда женщина с фенилкетонурией беременеет, существует высокий риск
микроцефалии, врожденных недостатков сердца, умственной отсталости у ее
ребенка, так как мозг развивающегося ребенка повреждается ненормальным обменом
веществ матери. Поэтому женщины перед зачатием и беременные с ФКУ должны
придерживаться диеты, если необходимо принимать добавки и следить за уровнем
аминокислоты в крови.
Данные литературы свидетельствуют, что при соблюдении диеты и
компетентном биохимическом контроле для поддержания уровня фенилаланина на
показателях 120-360 mmol/l на протяжении беременности, рождаются здоровые дети.
В настоящее время интенсивно разрабатываются сразу несколько видов
альтернативной терапии ФКУ. Среди них: так называемый метод «больших
нейтральных аминокислот» (large neutral amino acids), энзимотерапия
фенилаланингидроксилазой, фенилаланинаммониалиазой; лечение
тетрагидробиоптерином («Сапроптерин»).
Есть данные об успешном лечении пациентов с умеренной или легкой ФКУ с
применением тетрагидробиоптерина (10-20 мг/кг/сут).
Некоторые зарубежные исследователи доказали, что использование пищевых
гликомакропептидов при ФКУ (с ограниченной дотацией незаменимых кислот) снижает
концентрации фенилаланина в плазме крови и головном мозге, а также способствует
адекватному физическому развитию. Экспериментальным методом лечения ФКУ
является введение гена фенилаланингидроксилазы непосредственно в пораженные
клетки печени. В РФ указанные методы в настоящее время не применяются. Однако,
в нашей стране также идут поиски новых путей борьбы с фенилкетонурией и
улучшения качества жизни больных людей.
Заключение
Фенилкетонурия (ФКУ) («фенил» - от слова фенилаланин, названия
аминокислоты, «кетон» - от слова кетоны, названия химических соединений, «урия»
- выделение продуктов обмена с мочой) - это редкое наследственное заболевание,
обусловленное дефектом гена фермента фенилаланингидроксилазы (ФФГ), что
находится на длинном плече 12 хромосомы (12q 22-24). Дети, родившиеся с
фенилкетонурией, не способны метаболизировать (перерабатывать) фенилаланин
(часть протеина), который из-за этого накапливается в крови. Такое
патологически высокое количество фенилаланина препятствует нормальному развитию
мозга. При отсутствии лечения, оно приводит к умственной отсталости, тяжелым
патологиям и даже смерти. Различают три типа заболевания: классическое,
вариантные и материнское. Первые типы связанны с аутосомно-рецессивными
мутациями генов, отвечающих за активность тетрагидробиоптерина (ВН4), что
принимает участие в нормальном превращении фенилаланина в тирозин. Если процесс
превращения нарушен, то повышается уровень фенилаланина в крови и
образовываются побочные продукты обмена: фенилпируват, фениллактат, фенилацетат
и фенилацетилглутамин. Они воздействуют на мозг, печень и почки, в частности в
моче накапливаются фенилмолочная и фенилуксусная кислота, придающая ей
неприятный запах. Это выступает одним из симптомов фенилкетонурии у детей.
Очень важно, чтобы болезнь была выявлена на ранней стадии. Поэтому на
протяжении первых дней жизни проводится обследование - скрининг новорожденных.
При выявлении повышенного уровня фенилаланина и др. веществ устанавливается
причина. Если ею является фенилкетонурия, то ребенку назначается лечение в виде
специальной диеты, а возможно заместительная терапия, направленна на изменение
метаболизма. Для питания детей изготавливаются специальные смести без
фенилаланина.
Список использованной литературы
1. Алексеева
И.В. От самого рождения // НАУКА из первых рук. 2010. Т. 32. № 2. С. 95-96.
. Биохимия
наследуемых нарушений метаболизма. Избранные разделы. Коллектив авторов. Под
ред. Ещенко Н.. - СПб.: Издательский дом Санкт-Петербургского государственного
университета, 2011. - 156 с.
. Гидранович
В.И., Гидранович А.В. Биохимия. - М.: ТетраСистемс, 2012. - 528 с.
. Денисенкова
Е.В., Кузнецова Л.И. Влияние материнской фенилкетонурии на исход беременности и
родов // Вопросы детской диетологии. 2009. Т. 7. № 3. С. 55-58.
. Карпищенко
А.И. Медицинские лабораторные технологии. Руководство по клинической
лабораторной диагностике. В 2 томах. Том 2. - М.: ГЭОТАР-Медиа, 2013. - 790 с.
. Новиков
П.В. Редкие (орфанные) наследственные и врожденные болезни у детей: проблемы и
задачи на современном этапе // Вопросы практической педиатрии. 2011. № 1. С.
34-44.
. Рубан
Э.Д. Генетика человека с основами медицинской генетики. - М.: Феникс, 2014. -
319 с.
. Сарычева
С.Я., Зелинская Д.И. Наследственные болезни обмена веществ, выявляемые методом
неонатального скрининга // Медицинская сестра. 2011. № 8. С. 6-10.
. Северин
С.Е. Биологическая химия. - М.: ГЭОТАР-Медиа, 2013. 624 с.
. Соколова
Н.Г. Настольная книга педиатра. - М.: Феникс, 2012. - 448 с.
. Студеникин
В.М., Боровик Т.Э., Бушуева Т.В. Фенилкетонурия у детей и ее лечение // Лечащий
врач. 2011. № 9. С. 55.
. Табиева
И.С. и др. Опыт мировой и отечественной практики неонатального скрининга на
наследственные заболевания // Педиатрия. Журнал им. Г.Н. Сперанского. 2012. Т.
91. № 1. С. 128-132.
. Тюкавкина
Н.А., Бауков Ю.И,, Зарубян С.Э. Биоорганическая химия. - М.: ГЭОТАР-медиа,
2012. - 416 с.
. Чупак-Белоусов
В.В. Фармацевтическая химия. Книга 2. 4 курс. - М.: Бином, 2012. - 616 с.