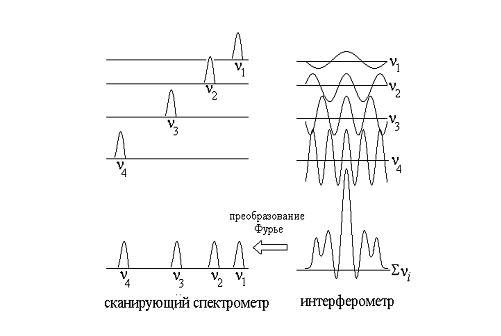
Исследование строения органических соединений с помощью физических методов
Министерство
образования и науки Российской Федерации
ИРКУТСКИЙ
ГОСУДАРСТВЕННЫЙ ТЕХНИЧЕСКИЙ УНИВЕРСИТЕТ
Кафедра
химической технологии
Исследование
строения органических соединений с помощью физических методов
Вариант
№ 8
ПОЯСНИТЕЛЬНАЯ
ЗАПИСКА
к
курсовой работе по дисциплине
«Теоретические
и экспериментальные методы исследования в химии»
.00
.00.00 ПЗ
Иркутск
2013 г
СОДЕРЖАНИЕ
ВВЕДЕНИЕ
1 Инфракрасная спектроскопия
1.1 Физико-химические основы метода ИК-спектроскопии
1.1.1 Оптическая спектроскопия. Инфракрасная спектроскопия (ИК) и
спектроскопия комбинационного рассеяния (КР)
1.1.2 Структура атомных и молекулярных спектров. Вращательные и
колебательные спектры
1.1.3 Колебания многоатомных молекул
1.2 Спектроскопия с преобразованием Фурье1
1.2.1 Физические основы спектроскопии с преобразованием Фурье
1.2.2 Принципиальная схема интерферометра Майкельсона
1.2.3 Регистрация спектров
1.2.4 Некоторые особенности и преимущества спектроскопии с
преобразованием Фурье
2 Явление ядерного магнитного резонанса
2.1 Протонный магнитный резонанс (ПМР), или ЯМР 1Н27
2.1.1 Химический сдвиг
2.1.2 Спин-спиновое взаимодействие
2.2 Спектроскопия ядерного магнитного резонанса ядер 13С
2.2.1 Основные особенности спектроскопии на ядрах 13С
3.
Идентификация органического соединения
3.1 Расшифровка спектров
ЗАКЛЮЧЕНИЕ
СПИСОК ИСПОЛЬЗОВАННЫХ ИСТОЧИКОВ
ВВЕДЕНИЕ
Всякий раз, когда химик синтезирует или выделяет
новое, неизвестное ранее органическое соединение, возникает необходимость в
выяснении его строения. Для этой цели новое соединение подвергают
спектроскопическому анализу, чаще всего при помощи четырех методов, а именно
электронной и инфракрасной спектроскопии (ИК - спектроскопии),
масс-спектрометрии и спектроскопии ядерного магнитного резонанса (ЯМР -
спектроскопия). Полученные в результате такого многостороннего анализа данные
обычно позволяют предложить для неизвестного соединения по меньшей мере
ковалентную структуру, а часто дают возможность сделать определенные выводы и о
его относительной стереохимии.
При спектрометрическом изучении органического
соединения обычно применяется следующая стратегия. Сначала масс -
спектрометрически определяют его элементарный состав (брутто - формулу) и
предварительно отмечают характер его фрагментации. Затем с помощью инфракрасной
спектроскопии определяют природу функциональных групп, а электронный спектр
позволяет выяснить, сопряжены ли эти функциональные группы или нет. Далее по
данным спектроскопии ЯМР 1Н, 13С, анализируя окружение атомов водорода и
углерода в изучаемой молекуле, выбирают одну или несколько наиболее вероятных
структур. Наконец, полученные каждым методом данные сопоставляют друг с другом
и перепроверяют с тем, чтобы убедиться, что они не противоречат найденному
решению задачи.
Конечно, в любой реальной ситуации химик уже
имеет те или иные сведения о природе химической реакции, в результате которой
образует, новое соединение, и таким образом число теоретически возможных
структур резко сокращается. Кроме того, часто задачу выяснения строения можно
решить путем удачного подбора только одного или двух из указанных
спектроскопических методов, иногда подтвердив полученные данные простым
химическим тестом или посредством определения температуры плавления или
кипения. В более сложных случаях необходима полная информация, и тогда химик
вынужден постоянно проверять соответствие друг другу данных, получаемых с
помощью различных методов. Подобный сравнительный критический анализ данных это
ключ к успешному решению задачи, а для химика одним из источников высшего
удовлетворения является тот момент, когда все разрозненные части головоломки
неожиданно складываются в однозначное решение трудной задачи выяснения
структуры.
С практической точки зрения важно обращать
внимание на условие регистрации и особенности интерпретации спектров.
Существенным моментом, например, является вычитание из спектра Пиков,
отвечающих растворителю, или намеренное расширение шкалы при записи сигналов
вне обычного рабочего диапазона прибора.
1.
Инфракрасная спектроскопия
Инфракрасная спектроскопия (ИК - спектроскопия)
используется в различных областях науки, и в каждой из них придается этому
термину различный смысл. Для химика-аналитика это удобный метод решения многих
задач. Физику ИК-спектроскопия представляется методом исследования
энергетических уровней в полупроводниках или определения межатомных расстояний
в молекулах. Она может быть также полезна и при измерении температуры пламени
ракетного двигателя. Для химика-органика это метод идентификации органических
соединений, позволяющий выявлять функциональные группы в молекулах и следить за
ходом химических реакций. Для биолога ИК-спектроскопия - перспективный метод
изучения транспорта биологически активных веществ в живой ткани, ключ к
структуре многих естественных антибиотиков и путь познания строения клетки.
Физику-химику метод позволяет приблизиться к пониманию механизма гетерогенного
катализа и кинетики сложных реакций. Он служит дополнительным источником
информации при расшифровке структуры кристаллов. В этих и многих других
областях знания ИК-спектроскопия служит исследователям мощным средством
изучения тайн вещества.
ИК-спектр поглощения, вероятно, уникальное в
своем роде физическое свойство. Не существует двух соединений, за исключением
оптических изомеров, с различающимися структурами, но одинаковыми ИК-
спектрами. В некоторых случаях, таких, как полимеры с близким молекулярным
весом, различия могут быть практически не заметны, но они всегда есть. В
большинстве случаев ИК-спектр является „отпечатком пальцев" молекулы,
который легко отличим от спектров других молекул.
Кроме того что поглощение характеристично для
отдельных групп атомов, его интенсивность прямо пропорциональна их
концентрации. Таким образом, измерение интенсивности поглощения дает после
простых вычислений количество данного компонента в образце.
По своим возможностям метод почти универсален.
Образцы могут быть жидкими, твердыми или газообразными. Они могут быть
органическими или» неорганическими, хотя неорганические вещества иногда не дают
хорошо выраженных спектров. В обычных условиях для ИК-излучения прозрачны
только одноатомные газы и неполярные молекулы (Ne, Не, 02, N2, Н2).
Другое ограничение заключается в том, что такой
распространенный растворитель, как вода, имеет в ИК- области очень сильное
поглощение и, кроме того, растворяет окна кювет, в качестве которых используют
пластинки из кристаллов солей. Метод ИК- спектроскопии обычно не очень
чувствителен к примесям, если они не превышают 1 %. Это, конечно, может быть
как благом, так и бедствием, все зависит от точки зрения и решаемой проблемы.
Подобным же образом может огорчить и тот факт, что положения характеристических
полос поглощения для многих групп различны при переходе от одной молекулы к
другой, но это подтверждает индивидуальность спектра поглощения и дает больше
для понимания структуры молекулы, чем если бы полосы были неизменны.
Спектроскопия в ИК- области встречается с рядом
специфических трудностей. В силу того что излучение невидимо, юстировку
оптических деталей нельзя проводить визуально. Энергия, с которой приходится
иметь дело, крайне мала и уменьшается с увеличением длины волны.
Из-за низкой энергии сигнал приемника не намного
выше уровня шума, возникающего в результате хаотического теплового движения
электронов в контуре приемника. Более того, поскольку все части спектрометра
теплые (по сравнению с абсолютным нулем), они излучают энергию в ИК-области и
на детектор попадает значительное количество паразитной энергии, которая должна
быть отделена от полезного сигнала. Кто-то сказал, что ИК-спектрометрию грубо
можно сравнить с фотографированием эмиссионного спектра на раскаленном добела
спектрографе.
.1 Физико-химические основы метода
ИК-спектроскопии
1.1.1 Оптическая спектроскопия. Инфракрасная
спектроскопия (ИК) и спектроскопия комбинационного рассеяния (КР)
Спектроскопическими методами анализа называются
методы, основанные на взаимодействии вещества с электромагнитным излучением.
Одним из важнейших понятий, используемых в спектроскопии, является понятие
спектра. Спектр - это последовательность квантов энергии электромагнитных
колебаний, поглощенных, выделившихся или рассеянны при переходах атомов или
молекул из одних энергетических состояний в другие.

Рис. 1 Области электромагнитного спектра
Диапазон электромагнитного излучения
простирается от наиболее длинноволнового излучения - радиоволн с длинами волн
более 0,1 см - до наиболее высокоэнергетического γ-
излучения с длинами волн порядка 10-11 м (рис. 1).
Отдельные области электромагнитного спектра
перекрываются. Следует отметить, что область электромагнитного спектра, которая
воспринимается человеческим глазом, весьма незначительна по сравнению со всем
его диапазоном. Характер процессов, протекающих при взаимодействии излучения с
веществом, различен в разных спектральных областях. В связи с чем,
спектроскопические методы анализа классифицируют по длине волны (энергии)
используемого излучения. В то же время, оптическая спектроскопия подразделяется
и по изучаемым объектам: на атомную и молекулярную. При помощи атомной
спектроскопии можно проводить качественный и количественный анализ элементного
состава вещества, т.к. для каждого элемента характерен свой уникальный набор
энергий и интенсивностей переходов между электронными уровнями в атоме. Из
данных молекулярной спектроскопии можно извлекать данные об электронной
структуре молекул и твердых тел, а также информацию об их молекулярной
структуре. Так, методы колебательной спектроскопии, включающие инфракрасную
(ИК) спектроскопию и спектроскопию комбинационного рассеяния (КР), позволяют
наблюдать колебания связей в веществе. Наборы полос в ИК и как и отпечатки
пальцев человека. По этим спектрам вещество может быть идентифицировано, если
его колебательный спектр уже известен. Кроме того, по ИК и КР спектрам
определяют симметрию и структуру неизученных молекул. Частоты основных
колебаний, находимые из спектров необходимы для расчета термодинамических
свойств веществ. Измерение интенсивности полос в спектрах позволяет проводить
количественный анализ, изучать химические равновесия и кинетику химических
реакций, контролировать ход технологических процессов.
В результате взаимодействия потока излучения с
веществом интенсивность потока (I0) уменьшается вследствие процессов поглощения
(на величину IA), отражения (IR) и рассеяния (IS). Связь между этими величинами
и интенсивностью потока I, прошедшего через вещество,
Выражается следующим соотношением:
I0=I+IA+IR+IS (1)
Методы, основанные на взаимодействии вещества с
излучением ИК- области спектра являются абсорбционными, т.е. основанными на
явлении поглощения излучения. Эмиссионные методы в этой области спектра не
используют ввиду трудностей получения и регистрации спектров испускания. В ИК-
области для характеристики энергии фотонов чаще всего используют величину,
называемую волновым числом:
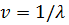 (2)
(2)
Ее размерность - см-1, т.е. это число длин волн,
укладывающихся на отрезке 1 см.
Волновое число прямо пропорционально энергии:
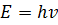 (3)
(3)
В ИК- спектроскопии спектр поглощения (или
пропускания) представляют в
Координатах оптическая плотность (или
интенсивность пропускания) - волновое число.
.1.2 Структура атомных и молекулярных спектров.
Вращательные и колебательные спектры
Для атомов характерны дискретные спектры,
состоящие из отдельных спектральных линий - линейчатые спектры. Количество
спектральных линий в них растет по мере увеличения числа электронов на внешних
электронных оболочках. Спектры молекул в радиочастотном диапазоне и дальней ИК-
области имеют линейчатый характер, а в средней и ближней зонах ИК-, УФ- и
видимой областях наблюдаются полосатые спектры.
Появление полос в молекулярных спектрах связано
с существованием в молекуле трех видов движения: электронного, колебательного и
вращательного. Энергию молекулы E можно приближенно представить в виде суммы
электронной Ee, колебательной Ev и вращательной Er энергий:
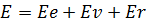 (4)
(4)
Эти виды энергии различаются весьма существенно
Ee » Ev » Er . Каждая из входящих в выражение (4) энергий квантуется, т.е. ей
соответствует определенный набор дискретных энергетических уровней (рис. 2).
Для простоты на ней изображены лишь два электронных уровня Ee . Каждому
электронному уровню отвечает свой набор колебательных уровней Ev , а каждому
колебательному уровню - свой набор вращательных уровней Er . При изменении
энергии электронов у молекулы одновременно изменяются колебательная и
вращательная энергии, и вместо электронных наблюдаются электронно -
колебательно-вращательные переходы. Частоты спектральных линий, отвечающие этим
переходам, определяются выражением
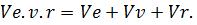
Поскольку число таких линий весьма велико, то
электронно-колебательно-вращательный спектр, обычно называемый электронным,
принимает вид широких перекрывающихся полос. Электронные спектры испускания и
поглощения наблюдают в интервале 50-2500 нм (УФ, видимая и ближняя ИК-
области). По этой же причине полосатую структуру имеют и колебательные спектры
(30-40 ·10-2 см-1, средняя и дальняя зоны ИК-области).
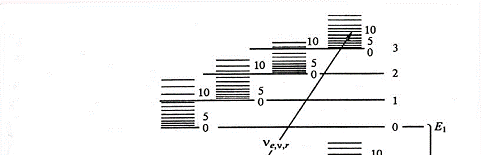
Рис. 2 Схема энергетических уровней двухатомных
молекул
.1.3 Колебания многоатомных молекул
Всевозможные положения молекул в трехмерном
пространстве сводятся к поступательному, вращательному и колебательному
движению.
Молекула, состоящая из N атомов, имеет всего 3N
степеней свободы движения. Эти степени свободы распределяются между видами
движения по-разному в зависимости от того, является молекула линейной или нет.
Для молекул обоих типов существует по 3 поступательных степени свободы, а число
вращательных степеней свободы для нелинейных молекул равно 3, а для линейных -
2. Таким образом, на долю колебательных степеней свободы (рис. 3) приходятся:
N-5 степеней свободы для линейных молекул,
N-6 степеней свободы для нелинейных молекул.
Основные типы колебаний молекулы называются
нормальными колебаниями. Более строго, нормальными колебаниями называются такие
колебания, которые происходят независимо друг от друга. Это означает, что при
возбуждении нормального колебания не происходит никакой передачи энергии для
возбуждения других колебаний. В случае нормальных колебаний атомы колеблются в
одной фазе и с одинаковой частотой.
Несимметричные движения атомов приводят к более
сложным колебаниям. Каждое колебание атомов в молекуле может быть представлено
как линейная комбинация нескольких нормальных колебаний.
С точки зрения формы колебаний различают:
валентные колебания (ν),
которые
происходят в направлении химических связей и при которых изменяются межатомные
расстояния;
деформационные колебания (δ
), при которых изменяются валентные углы, а межатомные расстояния остаются
постоянными.
При поглощении инфракрасного излучения
возбуждаются только те колебания, которые связаны с изменением дипольного
момента молекулы. Все колебания, в процессе которых дипольный момент не
изменяется, в ИК-спектрах не проявляются.
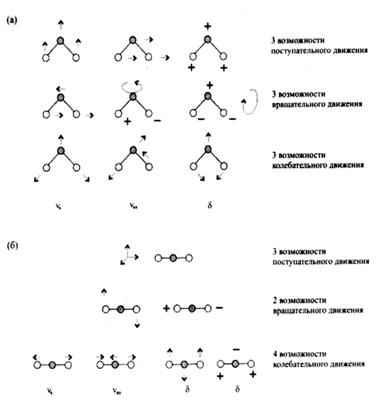
Рис. 3 Различные возможности движения
трехатомных молекул
а) Молекула H2O (нелинейная). б) Молекула CO2
(линейная)
В экспериментально полученных колебательных
спектрах число полос часто не совпадает с теоретическим. Как правило, в
экспериментальных спектрах полос меньше ввиду того, что не все возможные
колебания возбуждаются, а некоторые из них являются вырожденными.
Экспериментальный спектр может быть и более богат полосами по сравнению с
теоретическим из-за наличия обертонов и сложных колебаний. Частоты сложных
колебаний равны линейным комбинациям частот различных валентных и
деформационных колебаний.
.2 Спектроскопия с преобразованием Фурье
Как известно, основное назначение ИК
спектрофотометров заключается в получении колебательного спектра исследуемого
соединения. К концу 20 века были разработаны различные конструкции спектральных
приборов. ИК спектрофотометры, в которых информация о поочередно, вырезаемых
щелью спектральных интервалах регистрируется последовательно во времени,
называют сканирующими. По мере сканирования каждого такого спектрального
интервала, ширина которого определяется спектральной шириной щели, энергия
излучения воспринимается одноканальным приемником. Приборы с пространственным
разделением, использующие многоканальные приемники, в средней ИК - области, в
отличие от видимой, практически не применяются. Примером многоканального
прибора для видимой области служит спектрограф, регистрирующий спектр излучения
на фотопластинку. Многоканальные спектрометры - это такие приборы, в которых
приемник одновременно получает много сигналов, соответствующих различным
участкам спектра. Эти сигналы дешифруются таким образом, что дают информацию о
каждом отдельном спектральном элементе. Так как ИК области присуща низкая
энергия, то конструирование ИК - спектрофотометров направлено на максимальное
увеличение проходящей через прибор и попадающей на приёмник энергии. Со своей
стороны пользователь должен помнить об этом ограничении и выбирать, особенно,
при количественных измерениях оптимальные с точки зрения энергетики режимы
работы и методы пробоподготовки.
В ходе классического спектроскопического
эксперимента входящее в призменный или решеточный монохроматор
полихроматическое излучение (белый свет) разделяется на бесконечное число
монохроматических пучков. Спектр получается путем пространственного разделения
выходящих из призмы пучков с различными длинами волн. Дифракционная решетка
работает подобным же образом, за исключением того, что число пучков равно числу
штрихов решетки и для каждой длины волны на выходе получается больше одного
максимума. Различные порядки спектра, которые перекрываются, необходимо
разделять. Разрешение, достигаемое в спектрометре, определяется шириной щели,
задающей полосу длин волн, которая попадает на фотоприемник и порядком спектра.
В 1970-е годы классические сканирующие спектрометры начали вытесняться
приборами нового поколения - на основе интерферометров с использованием
преобразования Фурье, и к настоящему времени этот процесс завершился.
.2.1 Физические основы спектроскопии с
преобразованием Фурье
Все ИК - спектрофотометры независимо от
конструкции имеют общие элементы: источник излучения, оптическую систему,
приемник, систему усиления сигнала.
Источники излучения. Идеальным источником для ИК
- спектроскопии был бы монохроматический излучатель высокой интенсивности,
непрерывно перестраиваемый в широком частотном интервале. Несмотря на то, что
существуют лазеры с перестраиваемой частотой, в настоящее время наиболее
распространенными являются нагреваемые до температуры 1200 - 1400 К источники с
широкой областью излучения: глобар (карбид кремния), штифт Нернста (оксиды
циркония, тория, иттрия), нихромовая спираль, платиновая проволока с
керамическим покрытием. В дальней ИК области используется излучение стенок
ртутной лампы низкого давления. Излучательная способность тепловых источников
подчиняется закону Планка для излучения абсолютно черного тела. Ведутся
исследования по применению терагерцового излучения (субмиллиметровый диапазон)
в спектроскопии.
Оптические системы. Назначение оптической
системы - направлять излучение источника по нужному пути с минимальными
потерями. Использование отражательных зеркал с наружным покрытием (напыленный
алюминий, просветляющие покрытия) позволяет избежать хроматической аберрации.
Отражательная оптика может иметь плоские, сферические, параболические,
эллиптические или тороидальные поверхности. Разработано большое число типов
оптических систем спектрофотометров. Классические схемы спектральных приборов
рассмотрены в специальной литературе
Приемники излучения. Приемники ИК излучения
делятся на две группы: тепловые и фотоэлектронные. Первая группа включает
термоэлементы (термопары), болометры (сопротивления с большим температурным
коэффициентом), пневматические приемники, пироэлектрические приёмники.
Пироэлектрические детекторы (на основе триглицинсульфата (NH2 CH2 COOH)3 H2 SO4
) используются в интерферометрах из-за их высокой чувствительности в широкой ИК
области.
В основе работы фотоэлектронных
полупроводниковых приемников, к которым относятся фоторезисторы и фотодиоды,
лежит явление внутреннего фотоэффекта. В ближнем ИК диапазоне наиболее
распространены фотодиоды на основе германия и твёрдого раствора InGaAs. В
среднем ИК диапазоне применяются охлаждаемые жидким азотом фотодиоды на основе
твёрдого раствора HgCdTe (MCT Mercury-Cadmium-Tellurium). Полупроводниковые
детекторы для работы в низкочастотной области требуют охлаждения до низких
(азотных или гелиевых) температур. Ширина запрещённой зоны определяет
длинноволновую границу чувствительности фотоэлектронных приёмников.
Оптические материалы. Так как обычные оптические
стёкла поглощают среднее и длинноволновое ИК излучение, то в качестве
материалов для изготовления окон кювет и светоделителей используют
монокристаллы различных солей. В спектроскопии внутреннего отражения применяют
материалы с высокими показателями преломления. В таблице 1 представлены
сведения о свойствах некоторых распространённых материалов для ИК.
|
Материал
|
Область
прозрачности, мкм (см-1)
|
Показатель
преломления
|
Примечания
|
|
LiF
|
0,11-7,0
(90910-1429)
|
1,39
|
Чувствителен
к кислотам
|
|
Флюорит
CaF2
|
0,12-9,0
(83333-1111)
|
1,40
|
Нерастворим
в воде. Чувствителен к солям NH4+
|
|
BaF2
|
0,14-12,0
(71430-833)
|
1,45
|
Нерастворим
в воде
|
|
NaCl
|
0,20-16,0
(50000-625)
|
1,52
|
Растворим
в воде, слегка растворим в спиртах
|
|
KBr
|
0,21-25,0
(47620-400)
|
1,53
|
Гигроскопичен,
растворим в воде, спиртах, глицерине
|
|
Si
|
От
1,5 мкм (6667-)
|
3,40
|
Растворим
в щёлочи и HF, длинноволновая граница зависит от чистоты материала
|
|
ZnSe
|
0,5-20
(20000-50)
|
2,42
|
Водостойкий,
раств. в сильных кислотах, разлагается в HNO3
|
|
KRS-5,
(TlI TlBr)
|
0,55-35,0
(18180-286)
|
2,38
|
Мягкий,
раств. в тёплой воде, основаниях, спирте и HNO3. Токсичен!
|
|
Полиэтилен
высокой плотности.
|
20,0-200,0
(500-50)
|
1,52
|
Мягкий,
устойчив к большинству растворителей.
|
Таблица 1. Свойства некоторых оптических
материалов, применяемых в ИК области спектра
В основе оптической схемы типичного
Фурье-спектрометра лежит схема интерферометра Майкельсона, разработаны и другие
системы. Прежде, чем рассматривать работу спектрометра с преобразованием Фурье,
рассмотрим интерферометр, созданный американским физиком Альбертом Абрахамом
Майкельсоном (Нобелевская премия 1907 г.).
.2.2 Принципиальная схема интерферометра
Майкельсона
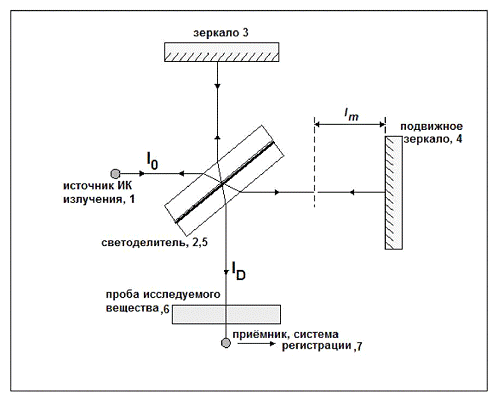
Светоделитель,
Неподвижное зеркало,
Подвижное зеркало,
Компенсатор,
Проба исследуемого вещества,
Детектор ИК-излучения.
Рис. 4 Схема интерферометра Майкельсона.
Свет, идущий от источника излучения 1 (I0)
,разделяется полупрозрачным плоскопараллельным зеркалом - светоделителем 2 на
два когерентных пучка. Материалы, из которых изготавливают светоделитель (и
компенсирующую пластинку 5), подбирают в зависимости от исследуемой области
спектра. Один пучок направляется к неподвижному плоскому зеркалу 3 и отражается
от него на светоделитель, другой идет к плоскому зеркалу 4 и также
возвращается, на светоделителе они соединяются. Эти два когерентных пучка
интерферируют между собой, в результате чего они могут либо усиливать, либо
ослаблять друг друга в зависимости от разности хода между ними. В фокальной
плоскости объектива возникают интерференционные полосы, которые можно наблюдать
визуально или регистрировать каким-либо способом (детектор 7). Зеркало 4
совершает возвратно-поступательное перемещение вдоль луча. Смещение этого зеркала
происходит относительно нулевого положения, в котором оптическая разность хода
в плечах интерферометра равна нулю. Наибольшие смещения зеркала составляют ±1m.
При смещении подвижного зеркала 4 на четверть длины волны светлые полосы в
интерферограмме заменяются на темные и наоборот.
Детектор 7 регистрирует интерферограмму -
зависимость интенсивности выходящего из интерферометра светового потока от
оптической разности хода, которая может бытьразличной от сантиметров до метров.
В интерферограмме содержится полная информация о спектральном составе
излучения, идущего от источника.
Интерферограмма является результатом рабочего
цикла интерферометра - сканирования (“скана”) по оси l от 0 до lm -
одностороннее сканирование, или от - lm до + lm - двухстороннее сканирование.
По мере движения зеркала 4 на приемник попадает световой пучок, интенсивность
которого в случае монохроматического источника меняется по косинусоидальному
закону. Если - I(x) интенсивность света, попадающего на приемник, х - смещение
зеркала 4 в сантиметрах, В(v) - интенсивность источника как функция волнового
числа V в см-1, то интенсивность сигнала для монохроматического источника Vj
изменяется по закону:
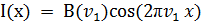
В случае “классического” сканирующего
спектрометра спектр будет состоять из единственной полосы с максимумом 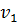 .
(рис. 5).
.
(рис. 5).
Если в источник излучения добавить вторую
частоту v2, то результирующая зависимость в координатах “положение зеркала -
интенсивность” будет представляться в виде суммы двух косинусоидальных волн:
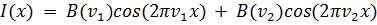
Если добавлять третью, четвертую и т. д. до
бесконечного числа частот (т. е. рассматривать полихроматические источники,
какими являются лампа накаливания или тепловой источник - глобар, то в
приемнике возникает сигнал от суммы косинусоидальных волн - интерферограмма
(1):
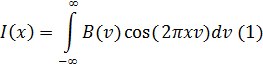
Реальная интерферограмма типичного спектра,
показана на рис. 6. В интерферограмме в закодированном виде содержится вся спе
ктральная информация о попадающем в интерферометр излучении.
Максимальная амплитуда интерферограммы
соответствует одинаковому удалению обоих зеркал от светоделительной пластинки.
В этой точке колебания с частотами для всех длин волн находятся в фазе и
взаимно усиливают друг друга, образуя наиболее интенсивную полосу
интерференции. Это место на интерферограмме называется положением нулевого
порядка интерференции, или же положением стационарной фазы колебании ( рис. 6).
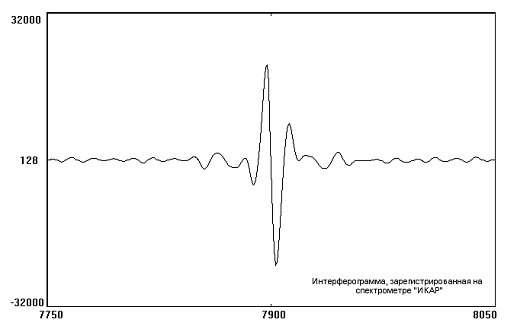
Рис. 6. Типичная интерферограмма
На практике идеального полихроматического
источника излучения не существует, так как имеется собственный спектр излучения
материала, из которого изготовлен источник. Это приводит к появлению в области
крыльев интерферограммы источника дополнительных затухающих колебаний.
Для получения спектра излучения источника нужно
выражение (1) подвергнуть преобразованию Фурье по косинусам. Восстановленный
спектр описывается уравнением следующего вида (2):
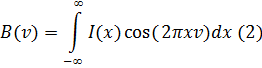
Уравнения (1) и (2) определяют взаимосвязь между
интерферограммой и спектром.
Если в один из каналов интерферометра помещена
поглощающая проба 6 (Рис. 4), то используются сходные рассуждения. При этом из
полихроматического излучения источника вычитается ряд волновых чисел,
соответстваующих полосам поглощения пробы. Результирующая интерферограмма пробы
образуется всеми волновыми числами, за исключением тех, что поглощены.
Преобразования Фурье представляют собой взаимно
обратные интегральные преобразования. В этом смысле можно сказать, что спектр
есть Фурье - образ интерферограммы по косинусам, а интерферограмма есть Фурье -
образ спектра по косинусам.
.2.3 Регистрация спектров
Для получения спектра пробы необходимо
зарегистрировать при одинаковых условиях эталонную интерферограмму (сравнение)
Bref(v) и интерферограмму с пробой Bsample(v), отношение их Фурье-образов даст
спектр пропускания (Т) пробы:
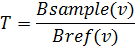
Регистрируемый в отсутствие пробы спектр
называется эталонным или спектром сравнения (reference) (рис.7, 1-верхний).
Здесь необходимо обратить внимание на то, что Фурье - спектрометр является
однолучевым прибором. По этой причине спектр сравнения представляет собой
спектр излучения теплового источника, на который наложен спектр поглощения
паров атмосферной воды (~3700, 1500-1700 см-1), углекислого газа (~2350 см-1) и
других веществ, возможно находящихся в атмосфере. Те же полосы регистрируются и
в спектре пробы - полистирола (рис.7, 2-средний), однако, при делении одного
спектра на другой в результирующем спектре остаётся только поглощение пробы
(рис.7, 3-нижний), представляющее в данном случае обычный спектр поглощения
полистирола. Можно также получить спектр поглощения пробы в координатах
оптическая плотность (absorbance) - волновое число, что важно при проведении
количественных измерений. Для выполнения указанных преобразований разработано
стандартное программное обеспечение.
В настоящее время производятся Фурье -
спектрометры с быстрым сканированием, точное положение оптических узлов
(скорость перемещения, положение подвижного зеркала и оптическая разность хода)
контролируется в них с помощью гелий-неонового лазера (632,8 нм), а применение
современных технологий позволило их сделать компактными и сравнительно
дешевыми. Возможности этих приборов гораздо шире, чем возможности
“классических” приборов.
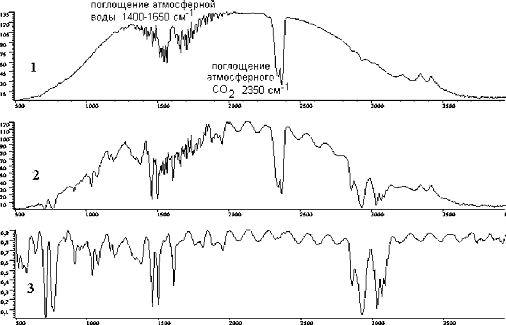
Рис.7.
- эталонный (референтный) спектр,
- спектр пробы,
- спектр пропускания пробы.
.2.4 Некоторые особенности и преимущества
спектроскопии с преобразованием Фурье
В классических спектрофотометрах регистрация
спектра производится во времени при последовательном движении выходящего из
монохроматора спектра по выходной щели. Этот процесс называется сканированием
по волновым числам. Спектрометры с преобразованием Фурье принадлежат к типу
многоканальных приборов, что приводит к значительному снижению энергетических
потерь.
Разрешающая способность является важнейшей
характеристикой как классических так и Фурье-спектрофотометров. Разрешающая
способность обычного сканирующего спектрометра определяется шириной щели и
дифракцией на ней световых волн. В интерферометре предел разрешения равен
обратной величине максимальной разности хода в интерферограмме. Этот вопрос
подробно рассматривается в книгах по оптике. Интерферограмма измеряется от
нулевой разности хода до некоторого максимального значения ху, на котором она
обрывается.
Гармонический анализ позволяет вычислять все
частоты, для которых на интервале от -хi до +х1 укладывается целое число
периодов. Если спектр рассматривается в шкале волновых чисел, то предел
разрешения интерферометра в см-1 можно считать равным 1/2 х1, где х1 - разность
хода выражена в сантиметрах. Ж.Конн использовала в своем интерферометре
разность хода до 200 см и добилась разрешения 0,006 см-1, что на два порядка
превосходит возможности лучших классических спектрофотометров с дифракционными
решетками. В случае серийных Фурье-спектрометров для рутинных измерений
спектров газов приемлемым является разрешение 0,1 см-1. В отличие от
дисперсионных спектральных приборов разрешение постоянно по всему спектральному
диапазону, и его легко менять, изменяя величину хода зеркала.
Высокая точность определения частот.
Фурье-спектрометры не нуждаются во внешних стандартах для градуировки шкалы
волновых чисел. В них используется внутренний стандарт, обычно одночастотный
газовый лазер (для гелий-неонового лазера λл
= 632,8 нм νл = 15804 см-1) для
калибровки разности хода по счету интерференционных полос во время регистрации
интерферограммы. При этом достигается очень высокая точность определения
волновых чисел. Так, в Фурье-спектрометре с криптоновым стандартом была
достигнута точность определения волновых чисел до 10-4 см-1, т. е. более чем на
порядок лучше, чем у самых точных дифракционных приборов.
Широкая область регистрации спектра. За один
проход зеркала (один скан) получается интерферограмма, содержащая информацию о
всем спектральном диапазоне для данного светоделителя. Так, светоделитель на
основе пленки Gе, напыленной на КВг перекрывает область 5000 - 400 см-1, т. е.
всю среднюю ИК-область, в которой проявляются колебательные спектры
органических соединений. Переход в длинноволновую область (менее 400 или 300
см-1) достигается сменой светоделителя, источника излучения и приёмника. Набор
светоделителей из пленок лавсана (майлара) разной толщины позволяет получать
спектры в области до 10 см-1. Низкая энергия источника в этой области может
быть частично скомпенсирована увеличением числа сканирований, что, конечно,
увеличивает время получения спектра. Однако, по сравнению с приборами
классического типа получать длинноволновые ИК-спектры на Фурье-спектрометре
значительно проще. Получение ИК спектров в этой области чрезвычайно важно, так
как там располагаются частоты колебаний кристаллических решеток, связей
металл-углерод, полупроводниковых структур, собственных колебаний водородных
связей.
Малое время регистрации спектра.
Фурье-спектрометр позволяет существенно сократить время регистрации спектра.
Этот выигрыш тем более существен, чем выше необходимое разрешение. Иногда,
особенно при использовании охлаждаемых до температуры жидкого азота детекторов,
имеющих очень низкий уровень собственных шумов, можно получить спектры хорошего
качества от единственного хода зеркала, на что требуется около 1 с. При
регистрации рутинных спектров с разрешением 4 см-1 время одного скана также
составляет около 1 с.
Возможность регистрации слабых сигналов. Высокое
пропускание энергии и возможность улучшения соотношения сигнал/шум путем
накопления интерферограмм позволяет получать спектры удовлетворительного
качества в условиях, когда получение спектра на дифракционном приборе вряд ли
возможно. Например, при малых концентрациях пробы в таблетке из KBr или в
случае сильно рассеивающих излучение образцов. Отношение сигнал/шум для
Фурье-спектрометров имеет величину порядка десятков тысяч.
Отсутствие рассеянного света. Рассеянный свет в
спектрометре очень сильно влияет на точность измерений в дисперсионных
приборах. Устранение его представляет сложную задачу для конструкторов
“классических” приборов. В Фурье-спектрометрах проблемы рассеянного не
существует. Фотометрическая точность современных приборов не хуже 0,1%.
Наличие в приборе встроенной ЭВМ (или
подключение к ПК), позволяет использовать ЭВМ не только для преобразования
Фурье и для управления прибором. К настоящему времени выработан и реализован на
практике минимальный набор требований к возможностям стандартного программного
обеспечения Фурье-спектрометра для хранения и обработки спектров. Некоторые из
них перечислены ниже.
Возможность создавать библиотеки спектров и базы
данных, как на основе собственных результатов, так и с использованием
каталогов. Например, фирма “Bruker” продает в комплекте с Фурье-спектрометрами
каталоги спектров, включающие до 12000 спектров веществ в твердой и жидкой
фазах в области 4000 - 400 см-1 и 7300 спектров газов. Программы поиска
позволяют использовать эти библиотеки в разных целях, например, отыскивать
спектры, близкие к спектру неизвестного вещества, с целью идентификации, или
отбирать спектры по какому-либо признаку или их совокупности. Существуют
специализированные базы данных, например, нефтепродуктов, растворителей,
лекарственных веществ и многие другие.
Возможность, регистрации спектров поглощения и
отражения в любом желаемом виде (А, Т%) и в разных форматах. Изменение
масштабов шкал в сочетании с накоплением позволяет получать спектры в случае
очень слабых сигналов.
Числовое сглаживание спектров уменьшает шумы,
хотя вносит некоторые искажения в спектр и ухудшает эффективное разрешение.
Операция может быть полезна, например, при воспроизведении спектров с сильной
растяжкой по шкале ординат.
Сложение и вычитание спектров проводится в шкале
оптических плотностей. Цифровое вычитание спектров резко расширило возможности
дифференциальной спектроскопии. При вычитании, спектра растворителя из спектра
раствора совсем не обязательно, чтобы спектры были записаны при одинаковой
толщине слоя. Вычитание можно проводить, умножая спектр на любой коэффициент. Возможно
выполнение и других математических операций (дифференцирование, разложение
сложных контуров полос на составляющие функции и др.).
Возможность программирования выполнения
эксперимента. Статистическая обработка спектров и результатов количественных измерений.
В Фурье-спектрометрах используются прецезионные
оптико-механические компоненты, например, наклон подвижного зеркала в процессе
сканирования не должен изменяться больше чем на четверть длины волны.
Спектральный интервал, хотя и достаточный для рутинной работы, ограничен
средней областью (400 - 4000 см-1), из-за неравномерности излучения теплового
источника, близкого к излучению абсолютно чёрного тела. Вблизи пределов этого
интервала резко увеличиваются шумы (ухудшается отношение сигнал/шум). Длинноволновая
(менее 400 или 300 см-1) и ближняя (более 5000 см-1) спектральные области
требуют различных светоделителей, источников излучения и приёмников. Важно
отметить, что для прецезионных работ необходимо вакуумирование или осушка
прибора (что является одним из немногих недостатков Фурье-спектрометров).
2.
Явление ядерного магнитного резонанса
Спектроскопия ядерного магнитного резонанса -
вид спектроскопии, которая регистрирует переходы между магнитными
энергетическими уровнями атомных ядер, вызываемые радиочастотным излучением.
Только ядра со спиновым квантовым числом I, отличным от «0», могут вызывать
сигнал ЯМР, или быть активными в ЯМР.
Спиновое квантовое число ядра определяется
числом протонов и нейтронов в ядре. Существует эмпирическое правило:
) I равно «0» для ядер с четным числом протонов
и нейтронов;
) I равно целым числам (1, 2, 3…) для ядер с
нечетными числами и протонов, и нейтронов;
) I равно полуцелым числам (1/2, 3/2, 5/2 и
т.д.) для ядер с четными числами протонов и нечетными числами нейтронов и
наоборот.
В приложенном магнитном поле с напряженностью Н0
ядро со спиновым числом I может принимать 2I + 1 ориентаций (или занимать 2I +
1 энергетических уровней). Количество энергии, на которое отличаются эти уровни
(разность энергий уровней), возрастает с возрастанием Н0, однако при данном
значении Н0 разность энергий между двумя соседними уровнями есть величина
постоянная.
Разность энергий двух соседних уровней ΔЕ
определяется выражением:
ΔЕ = Н0γ
h/2π
где γ - гиромагнитное
(магнитогирическое) отношение, постоянное для данного изотопа; Н0 -
напряженность внешнего магнитного поля; h - постоянная Планка.
В сущности, эксперимент ЯМР состоит в том, чтобы
сообщить энергию ядру и перевести его с одного энергетического уровня на
другой, более высокий уровень. Поскольку точное значение ΔЕ
зависит от молекулярного окружения возбуждаемого ядра, имеется возможность
связать величину ΔЕ со
строением молекулы и в конечном итоге определить структуру всей молекулы.
К сказанному следует добавить следующее:
сигналы в спектрах ЯМР могут давать только ядра
атомов, обладающих нечетным спиновым числом.
Таким образом, наиболее распространенные изотопы
углерода 12С, кислорода 16О и многие другие, например, дейтерий, являясь
немагнитными, не регистрируются в ЯМР-спектрах.
Из ядер атомов, наиболее часто встречающихся в
органических соединениях, магнитным моментом обладают изотопы 1Н, 13С, 19F,
31P, 15N, 17O.
Спектроскопия ЯМР используется для регистрации
сигналов данных ядер. Наибольшее распространение в исследовании органических
веществ имеет спектроскопия протонного магнитного резонанса (ПМР, ЯМР 1Н) и ЯМР
на ядрах изотопа 13С (ЯМР 13С). Эти виды спектроскопии и будут рассматриваться
в данном пособии.
Теория ядерного магнитного резонанса детально
обсуждена в многочисленных руководствах и монографиях, приведенных в списке
литературы. Мы остановимся на основных моментах, имеющих практическое значение.
Для исследования с помощью ЯМР спектроскопии,
как правило, вещество растворяют в подходящем растворителе (однако ЯМР-анализ
можно проводить и в твердой фазе). Для анализа необходимо ~ 10-20 мг образца.
Приготовленный раствор помещают в ампулу объемом ~ 0.5 мл и диаметром 5 мм.
Выбор растворителя определяется растворимостью
анализируемого вещества и наиболее полным разделением сигналов резонанса
вещества и растворителя, если последний содержит ядра, по которым проводится
регистрация спектра ЯМР. Для проведения анализа удобно использовать
дейтерированные растворители, поскольку дейтерий не дает сигнала в спектре ПМР.
Однако эти растворители содержат остаточные количества протонов, которые дают
сигналы небольшой интенсивности.
Ампулу с образцом помещают между полюсами
сильного магнита. В магнитном поле протоны мгновенно ориентируются в
направлении поля Н0 (подобно маленьким стержневым магнитам). В первый момент
после внесения образца число ядер, ориентированных вдоль поля и против поля,
одинаково (50% на 50%).
Вследствие обмена энергией между системами ядер
(«спинов») и их окружением («решеткой») число ядер на нижнем энергетическом
уровне достаточно быстро возрастает до величины, чуть большей 50%.
Протоны, ориентированные вдоль поля, находятся в
более низком энергетическом состоянии, чем протоны, ориентированные против
магнитного поля.
Мы помним, что в конечном итоге ΔЕ
= hν;
это
означает, что должна существовать такая частота электромагнитного излучения,
которая окажется равной разности энергий между более высоким энергетическим
состоянием ядра (ориентация против Н0) и более низким его состоянием
(ориентация вдоль Н0). Если на ядро воздействовать именно этой частотой, оно
будет взаимодействовать с излучением и изменит свое энергетическое состояние.
Те ядра, которые находились в более высоком энергетическом состоянии, перейдут
на нижний уровень, и наоборот. Однако, поскольку на нижнем энергетическом
уровне существует некоторый избыток ядер, в более высокое энергетическое
состояние перейдет большее число ядер, и в результате взаимодействия ядер с
излучением данной частоты произойдет поглощение электромагнитного излучения.
Именно это поглощение и вызывает сигнал ЯМР.
Точное значение частоты, которая вызывает
переходы между энергетическими уровнями данного ядра, называется резонансной
частотой этого ядра.
Резонанса можно достичь и другим путем: оставляя
частоту постоянной, менять напряженность магнитного поля. Во многих
спектрометрах ЯМР используют генератор фиксированной частоты 200, 300, 400, 500
и даже 800 МГц.
Для существования различия в заселенности
энергетических уровней необходим перенос энергии молекулярного движения на
спины ядер. Различие в заселенности возникает только в том случае, если после
наложения магнитного поля, т.е. с того момента, когда ядра окажутся в магнитном
поле, проходит некоторое время. Это время называется временем спин-решеточной
релаксации.
Величина времени спин-решеточной релаксации
имеет важное значение. Если время релаксации мало (у ядер быстрый перенос
энергии), то сигнал ЯМР уширенный. Большое время релаксации, например, у ядер
13С, также затрудняет наблюдение сигналов поглощения: столь важное для
резонанса различие в заселенности уровней при наложении относительно сильного
переменного поля выравнивается быстрее, чем его удается обнаружить (сигнал как
таковой исчезает). Последнее обстоятельство является одной из причин более
низкой чувствительности метода ЯМР 13С по сравнению с ЯМР 1Н.
Основными характеристиками спектров ЯМР
являются:
химический сдвиг,
мультиплетность,
константа спин - спинового взаимодействия;
площадь сигнала резонанса.
Эти характеристики зависят от химического
окружения данного ядра или группы ядер, от числа соседних ядер, обладающих
магнитным моментом, от их относительного расположения, а также от числа
анализируемых ядер в различных структурных фрагментах молекулы.
.1.1 Химический сдвиг
Разность положения сигнала данного протона и
положение сигнала стандарта называется химическим сдвигом данного протона.
В качестве стандарта чаще всего используют
тетраметилсилан (ТМС) Si(CH3)4. Запись ЯМР-спектра проводят таким образом,
чтобы Н0 возрастало слева направо. При этом химический сдвиг сигнала ТМС
принимают за ноль, и регистрируется в наиболее сильном поле (правая часть
спектра).
В практике ЯМР-анализа химический сдвиг выражают
в миллионных долях (м.д.) и обозначают символом «δ». Химические
сдвиги не зависят от рабочей частоты спектрометра:
δ = Δν∙106/ рабочая
частота прибора, (Гц)
Оказалось, что химические сдвиги протонов
органических соединений различных классов лежат в разных областях и, таким
образом, по положению сигнала ЯМР можно определить строение вещества. Ниже
приведены обобщенные области химических сдвигов протонов.
.1.2 Спин-спиновое взаимодействие
Сигналы протона (группы протонов) в спектре
могут быть представлены в виде одиночной линии (такой сигнал называется
«синглет») или в виде групп линий.
Если сигнал представлен в виде двух линий
определенной интенсивности (рис. 8) - сигнал называется «дублет»; в виде трех
линий - «триплет», в виде четырех линий - «квадруплет», или «квартет». Сигнал
может быть представлен группой из шести и более линий, в этом случае говорят о
мультиплете.
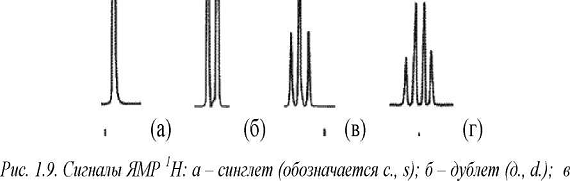
Рис. 8 Сигналы ЯМР 1Н: а - синглет (обозначается
с., s); б - дублет (д., d.); в - триплет (т., t.), г - квадруплет (кв., q.)
Каждая линия любого мультиплета будет отстоять
от соседних линий того же мультиплета на одно и то же число герц.
Химический сдвиг дублета определяется следующим
образом. Находится «центр тяжести» дублета и берется соответствующее значение
м.д. Химический сдвиг триплета, квадруплета и др. мультиплетов указывается
двумя значениями: левое значение - положение крайнего левого компонента
мультиплета, правое значение - положение крайнего правого компонента
мультиплета. Химические сдвиги обычно приводят с двумя знаками после запятой.
Расщепление сигнала протона на компоненты
происходит благодаря спин-спиновым взаимодействиям - взаимодействие спинов
протонов через электронные связи.
Согласно принципу Паули, электроны, связывающие
два ядра, спарены, т.е. их спины антипараллельны. В магнитном поле имеется
определенная тенденция для каждого из ядер спаривать свой спин со спином одного
из связывающих электронов таким образом, чтобы большинство из них было
антипараллельными (соответствует устойчивому состоянию).
Обычно спин-спиновое взаимодействие
распространяется очень слабо не далее трех связей, если только это не
напряженные циклы, мостиковые системы, делокализованные системы (в
ароматических или ненасыщенных структурах).
Спин-спиновое взаимодействие через две связи
называется геминальным.
Спин-спиновое взаимодействие через три связи
называется вицинальным. С- H Геминальное спин-спиновое взаимодействие через 2
связи (2J)C- C -H Вицинальное спин-спиновое взаимодействие через 3 связи (3J)
Возможно ли заранее предсказать вид сигнала
(мультиплетность)?
Да, возможно. Для этого используют следующее
правило.
Если n протонов одной группы (обозначим «А»)
взаимодействуют с n’ протонами другой группы (обозначим «В»), то сигнал
протонов группы «А» будет состоять из n’+1 линий, а сигнал протонов «В» - из n
+1 линий (общее правило 2nI+1, т.к. для протона I = ½,
то
мультиплетность равна n + 1). Таким образом, то или иное расщепление сигнала в
спектре ПМР позволяет определять структуру вещества.
.2 Спектроскопия ядерного магнитного резонанса ядер
13С
Ядро 12С магнитно неактивно (спиновое число
равно 0). Однако ядро 13С, как и протон, имеет спин ½.
Поскольку
природное содержание изотопа 13С составляет только 1.1%, а чувствительность
ядра 13С (большое значение времени релаксации) составляет лишь 1.6% от
чувствительности протона, общая чувствительность метода ЯМР 13С равна ~1/5700
от чувствительности ПМР.
2.2.1 Основные особенности спектроскопии на
ядрах 13С
1. При регистрации спектров ЯМР 13С с полным
подавлением спин-спинового взаимодействия с протонами все сигналы являются
синглетами, если только в молекуле нет других магнитно активных ядер (например,
2Н, 31Р, 19F).
Природное содержание 1Н более 99%, поэтому
каждое ядро 13С связано спин-спиновым взаимодействием с каким-то числом
протонов. В силу этого в спектрах ЯМР 13С без подавления спин-спиновых
взаимодействий с протонами обнаруживаются сложные перекрывающиеся мультиплеты,
которые трудно интерпретировать.
. Сигналы 13С распределены по значительно
большему диапазону химических сдвигов по сравнению с диапазоном для ядер 1Н.
Как и в случае с ЯМР 1Н, в спектрах ЯМР 13С
химические сдвиги выражают в единицах м.д. Диапазон химических сдвигов обычно
составляет 220 м.д. в слабое поле от сигнала ТМС. В силу этого, а также узости
спектральных линий, совпадение химических сдвигов от неэквивалентных атомов
углерода менее вероятно, чем в спектрах ПМР. При этом часто даже спектр смеси
веществ дает ценную информацию. Например, с помощью ЯМР 1Н трудно анализировать
стереоизомеры, в то время как в спектрах ЯМР13С эти изомеры дают различающиеся
сигналы.
. Интенсивности сигналов 13С не коррелируют с
числом ядер углерода.
Снятие спектра в режиме широкополосной
«развязки» от протонов приводит к возникновению ядерного эффекта Оверхаузера
(ЯЭО), который позволяет не только подавить спин-спиновое взаимодействие
13С-1Н, но и существенно (до 3-х раз) увеличить интенсивность сигнала.
В то же время атомы углерода, не имеющие
протонов (четвертичные С-атомы, иногда называют «узловые атомы углерода»), не
показывают усиления за счет ЯЭО и всегда имеют низкую интенсивность. В общем, в
спектроскопии ЯМР 13С интегральные интенсивности сигналов не пропорциональны
числу соответствующих атомов углерода.
. Вследствие более низкой чувствительности
метода ЯМР 13С по сравнению с ПМР для анализа необходимо большее количество
образца (~20 мг).
. Для данного дейтерированного растворителя
сигналы ЯМР 1Н и 13С имеют разную мультиплетность.
Образцы для измерений спектров ЯМР 13С обычно
представляют собой растворы в дейтерированных растворителях, для обеспечения
внутреннего стандарта используется добавка ТМС. Необходимо отметить, что для
данного дейтерированного растворителя его сигналы в спектрах ЯМР 1Н и ЯМР 13С
существенно различаются по мультиплетности. Например, небольшое количество
остаточного CHCl3 в дейтерированном коммерческом хлороформе (CDCl3) дает
небольшой синглет около 7.26 м.д. В спектре ЯМР 13С растворитель CDCl3 дает
интенсивный триплет вида 1:1:1 при 77 м.д., который вызван спин-спиновым
взаимодействием ядра 13С с одним ядром дейтерия.
3.
Идентификация органического соединения
Вариант 8
Определить структуру соединения.
Элементный состав, %: C - 68,29; H - 7,32; N -
11,38.
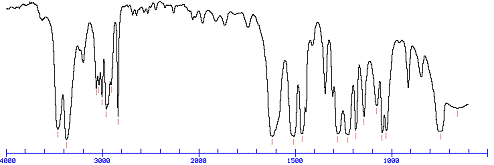
ИК-спектр
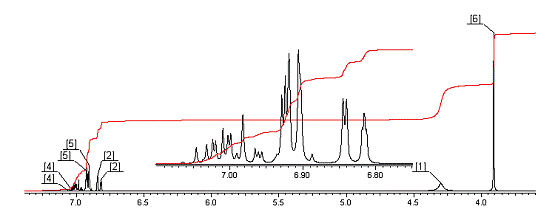
Спектр ЯМР 1Н
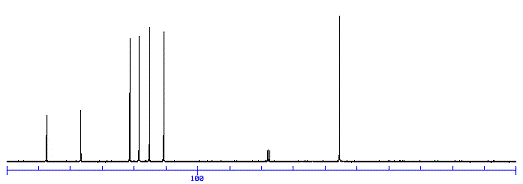
Спектр ЯМР 13С
3.1 Расшифровка спектров
По элементному составу определим брутто-формулу:
В соединении содержатся атомы кислорода 13,01 %.
С : Н : N : О = 68,29/12 : 7,32/1 : 11,38/14
:13,01/16 = 5,69 : 7,32 : 0,81 : 0,81.
Делим на минимальное число из получившихся
значений:
С : Н : N : О = 5,69/0,81 : 7,32/0,81 :
0,81/0,81 : 0,81/0,81 = 7 : 9 : 1: 1.
Тогда брутто-формула соединения С7Н9NO.
Количество двойных связей и/или циклов в
молекуле (формальная непредельность вещества) легко определяется из
брутто-формулы по следующему уравнению:
ФН = n углеродов - (n водородов + галогенов)/2
+(n азотов)/2 +1
ФН=7-9/2+1/2+1=4.
При анализе ИК-спектра видно, что на нем
присутствует интенсивная полосы с частотой 1470 - 1620 см-1, что
свидетельствует о наличии в соединении аренов. Тогда полосы 2820 - 3100 см-1
характерны для метильного радикала (-СН3), при бензольном кольце. Сигналы в
районе 3390-1 и 3450-1 можно отнести к валентным колебаниям амидов (NH2).
Учитывая все выше сказанное, можно написать ряд
возможных структур:

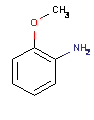
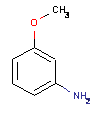
На спектре ЯМР 1Н сигнал с
химическим сдвигом 3,9 м.д. можно отнести к C6H5-O-CH3. Другой сигнал,
мультиплет с химическим сдвигом в диапазоне 6,8 - 7,05 м.д. можно отнести к
сигналам атомов водорода в бензольном кольце.
На спектре ЯМР 13С виден сигнал с
химическим сдвигом 55 м.д. который сигнализирует о присутствии в молекуле
соединения алкильного углерода, связанного с гетероатомом (-O-CH3).
Сигналы химического сдвига в
диапазоне 110-150 м.д., пять сигналов, подтверждают гипотезу о том, что в
молекуле есть углероды аренов. Сигналы делятся на две группы по интенсивности
сигнала, по которым можно судить о расположении радикалов в бензольном кольце.
В результате изучения спектров ЯМР 13С оставляем один вариант:
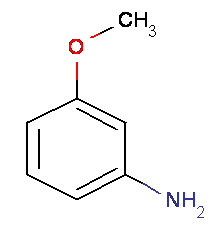
м-АНИЗИДИН
ЗАКЛЮЧЕНИЕ
Исследованное вещество м-АНИЗИДИН.
Экспериментальные данные полностью подтверждают данную структуру.
о-АНИЗИДИН - бесцветная маслянистая жидкость.
На воздухе о-АНИЗИДИН окисляется и приобретает темно-коричневую окраску.
о-АНИЗИДИН плохо растворим в воде,
хорошо в этаноле
<http://www.russian-chemistry.ru/reagents/3477>, эфире <http://www.russian-chemistry.ru/reagents/214>,
ацетоне
<http://www.russian-chemistry.ru/reagents/75>.
о-АНИЗИДИН образует соли с
минеральными кислотами, ацилируются, диазотируются.
В промышленности о-АНИЗИДИН получают
метоксилированием п-нитрохлорбензола с последующим восстановлением
образующегося нитроанизола полисульфидом Na или NaHS при T = 135°С и P = 0,2
МПа, а также под действием Н2 в присутвии никелевых катализатаров.
о-АНИЗИДИН используют для получения
гваякола, азотола, прямых, кислотных и жирорастворимых красителей.
о-АНИЗИДИН раздражает слизистые
оболочки и кожу.
Физические свойства м-АНИЗИДИНА
Температура кипения 251°C
Температура плавления < 0°C
Плотность 1.1 г/см3
Растворимость в воде, г/100 мл при
20°C 2.05
Давление паров, Па при 25°C 0.31
Температура вспышки >112°C
Температура самовоспламенения 515°C
Коэффициент распределения
октанол/вода как lg Pow 0.93
СПИСОК ИСПОЛЬЗОВАННЫХ ИСТОЧИКОВ
органическое соединение спектр
резонанс
Смит
А. Прикладная ИК-спектроскопия. - М.: Мир, 1982. - 328 с.
Накамото
К. ИК-спектры и спектры КР неорганических и координационных соединений. - М.:
Мир, 1991. - 535 с.
Иоффе.
Б.В. Физические методы определения строения органических соединений / Б.В.
Иоффе, Р.Р. Костиков, В.В. Разин. - М.: Высш. шк., 1984. - 336 с.
Володько
Л. В., Комяк А. И., Умрейко Д. С. Ураниловые соединения. Спектры, строение. -
Минск: БГУ, 1981. - 432 с.
Физические
методы исследования в химии. Резонансные и электрооптические методы. - М.:
Высшая школа, 1989. - 288 с.
Казицына
Л.А. Применение УФ-, ИК- и ЯМР-спектроскопии в органической химии / Л.А.
Казицына, Н.Б. Куплетская. - М.: Высш. шк., 1971. - 264 с.
Беккер,
Ю. Спектроскопия / Ю. Беккер; пер. с нем. Л. Н. Казанцевой ; под ред. А. А.
Пупышева, М. В. Поляковой . - М.: Техносфера, 2009. - 527 с.
Вершинин,
Вячеслав Исаакович. Компьютерная идентификация органических соединений / В. И.
Вершинин, Б. Г. Дерендяев, К. С. Лебедев. - М.: Академкнига, 2002. - 196 с.