Методы исследования строения молекул
Содержание
Введение
. Эксперементальные методы
.1 Рентгеноэлектронная спектроскопия
1.2 Ифракрасная спектроскопия
1.3 Дифракционные методы
. Теоретические методы
.1 Полуэмпирические методы
.2 Неэмпирические методы
.3 Кванто-механические методы
.4 Метод Хюккеля
Заключение
Список использованных источников
ВВЕДЕНИЕ
В современной органической химии
большое значение имеют различные физические методы исследования. Их можно
разделить на две группы. К первой группе относятся методы, позволяющие получать
различные сведения о строении и физических свойствах вещества, не производя в
нем никаких химических изменений. Из методов этой группы, пожалуй, наибольшее
применение получила спектроскопия в широком диапазоне областей спектра - от не
слишком жестких рентгеновских лучей до радиоволн не очень большой длины. Ко
второй группе относятся методы, в которых используются физические воздействия,
вызывающие химические изменения в молекулах. В последние годы к ранее
применявшимся широкоизвестным физическим средствам воздействия на реакционную
способность молекулы прибавились и новые. Среди них особое значение имеют
воздействия жестких рентгеновских лучей и потоков частиц больших энергий,
получаемых в атомных реактора
Целью данной курсовой работы является - узнать о
методах исследований строения молекул.
Задача курсовой работы:
выяснить виды методов и изучить их.
1.
ЭКСПЕРЕМЕНТАЛЬНЫЕ МЕТОДЫ
1.1 Рентгеноэлектронная
спектроскопия
Рисуник 1- Схема электронного
спектрометра: 1-источник излучения; 2-образец; 3- анализатор; 4-детектор;
5-экран для защиты от магнитного поля
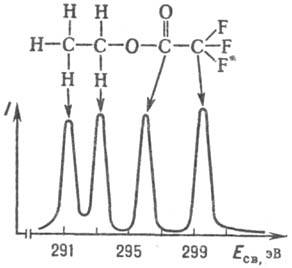
Рисунок 2- Рентгеноэлектронный
спектр Сls этилтрифторацетата
РЭС позволяет
исследовать все элементы, кроме Н, при содержании их в образце ~ 10-5
г (пределы обнаружения элемента с помощью РЭС 10-7-10-9 г).
Относительное содержание элемента может составлять доли процента. Образцы могут
быть твердыми, жидкими или газообразными. Величина Eсв электрона
<#"606051.files/image003.gif">
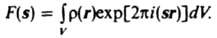
По такой же формуле
рассчитывают и атомный фактор, при этом описывает распределение рассеивающей
плотности внутри атома.
Значения атомного фактора специфичны для каждого вида излучения. Рентгеновские
лучи рассеиваются электронными оболочками атомов. Соответствующий атомный
фактор численно равен числу электронов в атоме, если выражен в названии электронных единицах, т. е. в
относительных единицах амплитуды рассеяния рентгеновского излучения одним
свободном электроне. Рассеяние электронов
определяется электростатическим потенциалом атома. Атомный фактор для электрона связан
соотношением:
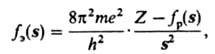
исследование молекула спектроскопия
дифракционный квантовый
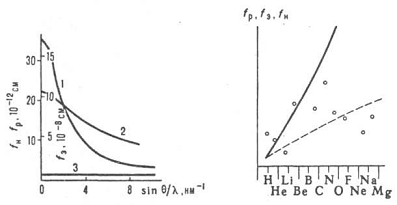
Рисунок 2-
Зависимость абсолютных значений атомных факторов рентгеновских лучей (1), электронов
(2) и нейтронов (3)
от угла рассеяния
Рисунок 3-
Относительная зависимость усредненных по углу атомных факторов рентгеновских
лучей (сплошная линия), электронов
(штриховая)и нейтронов от атомного номера Z
При точных расчетах
рассматривают отклонения распределения электронной плотности или потенциала атомов
от сферической симметрии и название атомно-температурный фактор, учитывающий
влияние тепловых колебаний атомов на
рассеяние. Для излучения помимо рассеяния на электронных оболочках атомов существует роль может играть резонансное рассеяние на
ядрах. Фактор рассеяния fм зависит от волновых векторов и векторов поляризации падающей и рассеянной волн. Интенсивность I(s) рассеяния
объектом пропорциональна квадрату модуля амплитуды: I(s)~|F(s)|2.
Экспериментально можно определить лишь модули |F(s)|, а для построения функции
рассеивающей плотности (r) необходимо знать также фазы (s) для каждого s. Тем
не менее теория дифракционных методов позволяет по измеренным I(s) получить
функцию (r), т. е. определить структуру веществ. При этом лучшие результаты
получают при исследовании кристаллов. Структурный
анализ. Монокристалл представляет собой строго упорядоченную систему, поэтому
при дифракции образуются лишь дискретные рассеянные пучки, для которых вектор рассеяния равен вектору
обратной решетки .
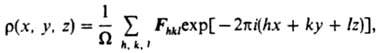
Для построения
функции ( х, у, z )по экспериментально определяемым величинам применяют метод проб и ошибок, построение и анализ функции межатомных
расстояний, метод изоморфных замещений, прямые методы определения фаз. Обработка
экспериментальных данных на ЭВМ позволяет восстанавливать структуру в виде карт
распределения рассеивающей плотности. Структуры кристаллов изучают с помощью рентгеновского структурного
анализа. Этим методом определено более 100 тысяч
структур кристаллов.
Для неорганических кристаллов с применением различных методов уточнения (учет поправок
на поглощение, анизотропию
атомно-температурного фактора и т. д.) удается восстановить функцию с
разрешением до 0.05
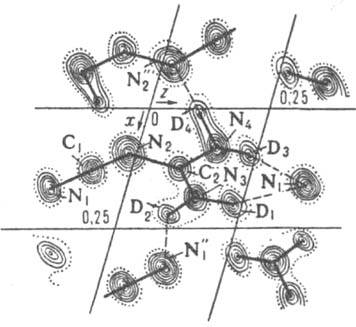
Рисунок 4- Проекция ядерной
плотности кристаллической структуры
Это позволяет определять анизотерапию тепловых колебаний атомов,
особенности распределения электронов,
обусловленные химической связью, и т. д. С помощью рентгеноструктурного анализа
удается расшифровывать атомные структуры кристаллов белков, молекулы которых содержат тысячи атомов. Дифракция рентгеновских лучей используется также для
изучения дефектов в кристаллах (в рентгеновской топографии), исследования
приповерхностных слоев (в рентгеновской спектрометрии), качественного и
количественного определения фазового состава поликристаллических материалов. Электронография как метод изучения структуры кристаллов имеет след. особенности: 1) взаимодействие вещества с электронами намного сильнее, чем с рентгеновскими лучами, поэтому
дифракция происходит в тонких слоях вещества толщиной 1-100 нм; 2) fэ
зависит от атомного ядра слабее, чем fр,
что позволяет проще определять положение легких атомов в присутствии тяжелых; Структурная электронография широко применяется для исследования тонкодисперсных
объектов, а также для изучения разного рода текстур (глинистые минералы, пленки полупроводников и
т. п.). Дифракция электронов низких энергий
(10-300 эВ, 0.1-0.4 нм) - эффективный метод исследования
поверхностей кристаллов: расположения атомов, характера их тепловых колебаний и т. д. Электронная микроскопия
восстанавливает изображение объекта по дифракционной
картине и позволяет изучать структуру кристаллов
с разрешением 0.2-0.5 нм. Источниками нейтронов
для структурного анализа служат ядерные
реакторы на быстрых нейтронах, а также
импульсные реакторы. Спектр пучка нейтронов,
выходящих из канала реактора, непрерывен вследствие максвелловского
распределения нейтронов по скоростям (его
максимум при 100°С соответствует длине волны 0.13 нм).
Монохроматизацию пучка осуществляют
разными способами - с помощью кристаллов-монохроматоров и др. Нейтронографию используется, как правило, для уточнения и дополнения
рентгеноструктурных данных. Отсутствие монотонной зависимости fи от
атомного номера позволяет достаточно точно определять
положение легких атомов. Кроме того, изотопы одного в того же элемента могут иметь сильно различающиеся
значения fи (так, fи углеводорода 3.74.1013
см, у дейтерия 6.67.1013 см). Это
дает возможность изучать расположение изотопов
и получать дополнит. сведения о структуре путем изотопного замещения .
Исследование магнитного взаимодействия. нейтронов с магнитнами моментами атомов дает
информацию о спинах магнитного атомов. Мёссбауэровское -излучение отличается чрезвычайно малой
шириной линии - 108 эВ (тогда как ширина линии характеристических
излучения рентгеновских трубок . 1 эВ). Это обусловливает высокую временную и
пространств. согласованность резонансного ядерного рассеяния, что позволяет, в
частности, изучать магнитное поле и градиент электрического поля на ядрах.
Ограничения метода - слабая мощность мёссбауэровских источников и обязательное
присутствие в исследуемом кристалле ядер, для
которых наблюдается эффект Мёссбауэра. Структурный анализ некристаллических веществ. Отдельные молекулы в газах, жидкостях и твердых аморфных телах по-разному ориентированы в
пространстве, поэтому определить фазы рассеянных волн, как правило, невозможно.
В этих случаях интенсивность рассеяния обычно представляют с помощью т. наз.
межатомных векторов rjk, которые
соединяют пары различных атомов (j и k) в молекулах: rjk=
rj - rk. Картина рассеяния усредняется по всем
ориентациям:
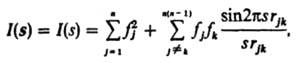
.1
Полуэмпирические методы
Полуэмпирические методы квантовой химии, методы
расчета мол. характеристик или свойств вещества с привлечением
экспериментальных данных. По своей сути полуэмпирические методы аналогичны
неэмпирическим методам решения уравнения Шрёдингера для многоатомных систем,
однако для облегчения расчетов в полуэмпирических методах вводятся дополнит.
упрощения. Как правило, эти упрощения связаны с валентным приближением, т. е.
основаны на описании лишь валентных электронов, а также с пренебрежением
определенными классами молекулярных интегралов в точных уравнениях того
неэмпирического метода, в рамках которого проводится полуэмпирический расчет.
Выбор эмпирических параметров основан на
обобщении опыта неэмпирических расчетов, учете химических представлений о
строении молекул и феноменологических закономерностей. В частности, эти
параметры необходимы для аппроксимации влияния внутренних электронов на
валентные, для задания эффективных потенциалов, создаваемых электронами остова,
и т.п. Использование экспериментальных данных для калибровки эмпирических
параметров позволяет устранить ошибки, обусловленные упомянутыми выше
упрощениями, однако лишь для тех классов молекул, представители которых служат
опорными молекулами, и лишь для тех свойств, по которым параметры определялись.
Наиболее распространены полуэмпирические методы,
основанные на представлениях о мол. орбиталях (см. Молекулярных орбиталей
методы, Орбиталь). В сочетании с ЛКАО-приближением это позволяет выразить
гамильтониан молекулы через интегралы на атомных орбиталях . При построении
полуэмпирических методов в мол. интегралах выделяют произведения орбиталей,
зависящих от координат одного и того же электрона (дифференциальное перекрывание),
и пренебрегают некоторыми классами интегралов. Напр., если нулевыми считаются
все интегралы, содержащие дифференциальное перекрывание cаcb при а . b,
получается т. наз. метод полного пренебрежения дифференциала. перекрыванием
(ППДП, в англ. транскрипции CNDO-complete neglect of differential overlap).
Применяют также частичное или модифицированное частичное пренебрежение
дифференциальное перекрыванием (соотв. ЧПДП или МЧПДП, в английской
транскрипции INDO- intermediate neglect of differential overlap и
MINDO-modified INDO), пренебрежение двухатомным дифференциальное перекрыванием
- ПДДП, или neglect of diatomic differential overlap (NDDO), - модифицирование
пренебрежение двухатомным перекрыванием (МПДП, или modified neglect of diatomic
overlap, MNDO). Как правило, каждый из полуэмпирических методов имеет несколько
вариантов, которые принято указывать в названии метода цифрой или буквой после
косой черты. Напр., методы ППДП/2, МЧПДП/3, МПДП/2 параметризованы для расчетов
равновесной конфигурации ядер молекулы в основном электронном состоянии,
распределения заряда, потенциалов ионизации, энтальпий образования химических
соединений, метод ЧПДП используется для расчета спиновых плотностей. Для
расчета энергий электронного возбуждения применяют спектроскопическую
параметризацию (метод ППДП/С). Распространено также использование в названиях
полуэмпирических методов соответствующих программ для ЭВМ. Напр., один из
расширенных вариантов метода МПДП называют Остинской моделью, как и
соответствующую программу (Austin model, AM). Имеется несколько сотен различных
вариантов полуэмпирических методов, в частности разработаны полуэмпирические
методы, аналогичные конфигурационного взаимодействия методу. При внешних
схожести разных вариантов полуэмпирических методов каждый из них можно
применять для расчета лишь тех свойств, по которым проведена калибровка
эмпирических параметров. В наиб. простых Полуэмпирических расчетах каждая мол.
орбиталь для валентных электронов определяется как решение одноэлектронного
уравнения Шрёдингера с оператором Гамильтона, содержащим модельный потенциал
(псевдопотенциал) для электрона, находящегося в поле ядер и усредненном поле
всех остальных электронов системы. Такой потенциал задают непосредственно с
помощью элементарных функций или основанных на них интегральных операторов. В
сочетании с ЛКАО-приближением подобный подход позволяет для многих сопряженных
и ароматического мол. систем ограничиться анализом p-электронов (см. Хюккеля
метод), для координационной соединений-пользоваться расчетными методами поля
лигандов теории и кристаллического поля теории и т.п. При изучении
макромолекул, напр. белков, или кристаллических образований нередко пользуются
полуэмпирическими методами, в которых электронное строение не анализируется, а
определяется непосредственно поверхность потенциальной энергии. Энергию системы
приближенно считают суммой парных потенциалов взаимодействия атомов, напр.
потенциалов Морса (Морзе) или Леннард-Джонса (см. Меж молекулярные
взаимодействия). Такие полуэмпирические методы позволяют проводить расчет
равновесной геометрии, конформационных эффектов, энергии изомеризации и т.п.
Нередко парные потенциалы дополняют определенными для отдельных фрагментов
молекулы многочастичными поправками. Полуэмпирические методы такого типа, как
правило, относят к молекулярной механике. В более широком смысле к
полуэмпирическим методам относятся любые методы, в которых определенные
решением обратных задач параметры мол. системы используются для предсказаний
новых экспериментальных данных, построения корреляционных соотношений. В этом
смысле полуэмпирическими методами являются методы оценки реакционной
способности, эффективных зарядов на атомах и т. п. Сочетание полуэмпирического
расчета электронного строения с корреляц. соотношениями позволяет оценивать
биологическую активность различных веществ, скорости химических реакций,
параметры технологических процессов. К полуэмпирическим методам относятся и
некоторые аддитивные схемы, напр. применяемые в химической термодинамике методы
оценки энергии образования как суммы вкладов отдельных фрагментов молекулы.
Интенсивное развитие полуэмпирических методов и неэмпирических методов
квантовой химии делает их важными средствами современные исследования
механизмов хим. превращений, динамики элементарного акта хим. реакции,
моделирования биохимических и технологических процессов. При правильном
использовании (с учетом принципов построения и способов калибровки параметров)
полуэмпирические методы позволяют получить надежную информацию о строении и
свойствах молекул, их превращениях.
2.2Неэмпирические
методы
Принципиально иное направление расчетной
квантовой химии, сыгравшее огромную роль в современном развитии химии в целом,
состоит в полном или частичном отказе от вычисления одноэлектронных (3.18) и
двухэлектронных (3.19)-(3.20) интегралов, фигурирующих в методе ХФ. Вместо
точного оператора Фока используется приближенный, элементы которого получают
эмпирическим путем. Параметры оператора Фока подбирают для каждого атома
(иногда с учетом конкретного окружения) или для пар атомов: они либо являются
фиксированными, либо зависят от расстояния между атомами. При этом часто (но не
обязательно - см. ниже) предполагается, что многоэлектронная волновая функция
является однодетерминантной, базис - минимальным, а атомные орбитали Х; -
симметричными ортогональными комбинациями ОСТ Хг Такие комбинации легко
получить, аппроксимируя исходные АО функциями Слейтера 'Xj (2.41) с
помощью преобразования Полуэмпирические методы работают гораздо быстрее, чем
неэмпирические. Они применимы к большим (часто - к очень большим, например,
биологическим) системам и для некоторых классов соединений дают более точные
результаты. Однако следует понимать, что это достигается за счет специально
подобранных параметров, справедливых лишь в пределах узкого класса соединений.
При переносе на другие соединения те же методы могут дать абсолютно неверные
результаты. Кроме того, параметры часто подбираются таким образом, чтобы
воспроизводить только определенные молекулярные свойства, поэтому придавать физический
смысл отдельным параметрам, используемым в схеме расчета, не следует.
Перечислим основные приближения, используемые в полуэмпирических методах .
. Рассматриваются только валентные
электроны. Считают, что электроны, относящиеся к атомным остовам, лишь
экранируют ядра. Поэтому влияние этих электронов учитывают, рассматривая
взаимодействие валентных электронов с атомными остовами, а не с ядрами, и вводя
энергию отталкивания остовов вместо энергии межъядерного отталкивания.
Поляризацией остовов пренебрегают.
. В МО учитывают только АО с главным квантовым
числом, соответствующим высшим заселенным электронами орбиталям изолированных
атомов (минимальный базис). Предполагают, что базисные функции образуют набор
ортанормированных атомных орбиталей- ОСТ, ортогонализованных по Лёвдину .
. Для двухэлектронных кулоновских и обменных
интегралов вводят приближение нулевого дифференциального перекрывания (НДП) .
Молекулярной структуре в пределах структурной
области может соответствовать набор модификаций молекулы, сохраняющих
одинаковую систему валентных химических связей при разной пространствеиной
организации ядер. В этом случае глубокий минимум ППЭ дополнительно имеет
несколько неглубоких (эквивалентных или неэквивалентных по энергии) минимумов,
разделенных небольшими потенциальными барьерами. Различные пространствеиные
формы молекулы, преобразующиеся друг в друга в пределах данной структурной
области путем непрерывного изменения координат атомов и функциональных групп
без разрыва или образования химических связей, составляют множество конформаций
молекулы. Набор конформаций, энергии которых меньше намнизшего барьера,
примыкающего к данной структурной области ППЭ, называется конформационным
изомером, или конформером. Конформеры, соответствующие локальным минимумам ППЭ,
называются устойчивыми или стабильными. Таким образом, молекулярную структуру
можно определить как совокупность конформаций молекулы в определенной
структурной области Часто встречающимся в молекулах типом конформационного
перехода является вращение отдельных групп атомов относительно связей: говорят,
что имеет место внутреннее вращение, а различные конформеры называют
вращательными изомерами, или ротамерами. При вращении меняется и электронная
энергия, причем ее значение в процессе такого движения может проходить через
максимум; в этом случае говорят о барьере внутреннего вращения. Последние во
многом обусловлены способностью этих молекул легко адаптировать структуру при
взаимодействии с разными системами. Каждому энергетическому минимуму ППЭ
соответствует пара энантиомеров с одинаковой энергией - правый (R) и левый (S).
Эти пары имеют энергии, отличающиеся всего на 3.8 ккал/моль, однако они
разделены барьером высотой 25.9 ккалjмоль и, следовательно, весьма устойчивы
при отсутствии внешних воздействий. Результаты квантово-химических расчетов
энергий барьеров внутреннего вращения для некоторых молекул и соответствующие
экспериментальные значения. Теоретические и экспериментальные величины барьеров
вращения для связей С-С, С-Р, C-S отличаются всего на 0.1 ккал/моль; для связей
С-0, C-N, C-Si, несмотря на использование базисного набора с включением
поляризационных функций (см. ниже), разница заметно выше. 1'ем не менее, можно
констатировать удовлетворительную точность расчета энергий барьеров внутреннего
вращения методом ХФ.
Подобные расчеты энергий барьеров внутреннего
вращения для простых молекул помимо спектроскопических приложений важны как
критерий качества того или иного расчетного метода. Большого внимания
заслуживает внутреннее вращение в сложных молекулярных системах, например, в
полипептидах и белках, где этот эффект обусловливает мноmе биологически важные
функции этих соединений. Вычисление поверхностей потенциальной энергии для
таких объектов представляет собой сложную задачу как в теоретическом, так и в
практическом плане. Распространенным видом конформационного перехода является
инверсия, такая, какая имеет место в пирамидальных молекулах типа АХ3 (А= N,
Si, Р, As, Sb; Х =Н, Li, F и др.). В этих молекулах атом А может занимать
позиции как выше, так и ниже плоскости, образованной тремя атомами Х. Например,
в молекуле аммиака NH3 метод ХФ дает величину энергетического барьера, равную
23,4 ккал/моль; это неплохо согласуется с экспериментальным значением барьера
инверсии - 24.3 ккал/моль. Если барьеры между минимумами ППЭ сопоставимы с
тепловой энергией молекулы, это приводит к эффекту структурной нежесткости
молекулы; конформационные переходы в таких молекулах происходят постоянно. Для
решения уравнений ХФ применяется метод самосогласованного поля. В процессе
решения оптимизируются только орбитали, занятые электронами, следовательно,
энергии лишь этих орбиталей находят физически обоснованно. Однако метод. ХФ
дает и характеристики свободных орбиталей: такие молекулярные спин-орбитали
называются виртуальными. К сожалению, они описывают возбужденные энергетические
уровни молекулы с погрешностью около 100%, и применять их для трактовки
спектроскопических данных следует с осторожностью - для этого существуют другие
методы. Также как и для атомов, метод ХФ для молекул имеет различные версии, в
зависимости от того, является ли однодетерминантная волновая функция
собственной функцией оператора квадрата полного спина системы S2 или нет. Если
волновая функция построена из пространствеиных орбиталей, занятых парой электронов
с противоположными спинами (молекулы с замкнутыми оболочками), это условие
выполняется, а метод называется ограниченным методом Хартри-Фока ( ОХФ). Если
требование быть собственной функцией оператора на волновую функцию не
накладывается, то каждая молекулярная спин-орбиталь отвечает определенному
спиновому состоянию (а или 13), то есть электроны с противоположными спинами
занимают разные спин-орбитали. Такой метод обычно применяется для молекул с
открытыми оболочками и называется неограниченным методом ХФ (НХФ), или методом
разных орбиталей для разных спинов. Иногда низколежащие энергетические
состояния описывают орбиталями, дважды занятыми электронами, а валентные
состояния описывают однократно занятыми молекулярными спин-орбиталями; этот
метод назьmается ограниченным методом Хартри-Фока для открытых оболочек ( ОХФ-
00). Как и в атомах волновая функция молекул с открытыми оболочками не
соответствует чистому спиновому состоянию, и могут возникать решения, у которых
симметрия волновой функции по спину понижена. Они называются НХФ-нестабильными
решениями.
2.3
Квантово-механические методы
Успехи теоретической химии, развитие квантовой
механики создали возможность приближенных количественных расчетов молекул.
Известно два важнейших метода расчета: метод электронных пар, называемый также
методом валентных связей, и метод молекулярных орбит. Первый из этих методов,
разработанный Гейтлером и Лондоном для молекулы водорода, приобрел широкое
распространение в 30-х годах нынешнего столетия. В последние годы все большее
значение приобретает метод молекулярных орбит (Гунд, Э. Хюккель,
Мулликен,Герц-берг, Ленард-Джонс).
В этом приближенном методе расчета состояние
молекулы описывается так называемой волновой функцией ψ,
которая
составляется по определенному правилу из ряда слагаемых:
Сумма этих слагаемых должна учитывать все
возможные комбинации, возникающие в результате попарного связывания атомов
углерода за счет π-электронов.
Для того чтобы облегчить расчет волновой функции
ψ,
отдельные
слагаемые (C1ψ1, C2ψ2 и т. д.)
условно изображаются графически в виде соответствующих валентных схем, которые
используются как вспомогательные средства при математическом расчете. Например,
когда указанным способом рассчитывают молекулу бензола и принимают во внимание
только π-элек-троны,
то таких слагаемых получается пять. Этим слагаемым соответствуют следующие
валентные схемы:
Часто приведенные валентные схемы изображают с
учетом
σ-связей,
например для бензола
Такие валентные схемы называют «невозмущенными
структурами» или «предельными структурами»
Функции ψ1, ψ2,
ψ3 и
т. д. различных предельных структур входят в волновую функцию ψ
с
тем большими коэффициентами (с тем большим весом), чем меньше энергия,
рассчитанная для соответствующей структуры. Электронное состояние,
соответствующее волновой функции ψ, наиболее
устойчиво сравнительно с электронными состояниями, изображаемыми функциями ψ1,
ψ2, ψ3 и т. д.; энергия же состояния,
изображаемого функцией ψ (реальной
молекулы), естественно, является наименьшей сравнительно с энергиями предельных
структур.
При расчете молекулы бензола по методу
электронных пар учитываются пять предельных структур (I-V). Две из них
тождественны классической структурной формуле Кекуле и три-формуле Дьюара. Так
как энергия электронных состояний, соответствующих предельным структурам III,
IV и V, выше, чем для структур I и II, то вклад структур III, IV и V в
смешанную волновую функцию молекулы бензола ψменьше,
чем вклад структур I и II. Поэтому в первом приближении для изображения
распределения электронной плотности в молекуле бензола достаточно двух
эквивалентных структур Кекуле.
Предельные структуры не соответствуют каким-либо
реальным электронным состояниям в невозбужденных молекулах, однако не
исключено, что они могут осуществляться в возбужденном состоянии или в момент
реакции.
Вышеизложенная качественная сторона теории
резонанса совпадает с концепцией мезомерии, несколько ранее развитой Инголдом и
независимо от него Арндтом.
Согласно этой концепции, истинное состояние
молекулы является промежуточным («мезомерным») между состояниями, изображенными
двумя или несколькими «предельными структурами», которые можно написать для
данной молекулы, пользуясь правилами валентности.
Кроме этого основного положения теории
мезомерии, к ее аппарату относятся хорошо разработанные представления об
электронных смещениях, в обосновании, интерпретации и опытной проверке которых
важная роль принадлежит Инголду. Согласно Инголду, механизмы электронных
смещений (электронных эффектов) различны в зависимости от того, осуществляется
ли взаимное влияние атомов через цепь простых или сопряженных двойных связей. В
первом случае это - индукционный эффект I (или также статический индукционный
эффект Is), во втором случае - мезомерный эффект М (статический эффект
сопряжения).
В реагирующей молекуле электронное облако может
поляризоваться по индукционному механизму; такое электронное смещение
называется индуктомерным эффектом Id. В молекулах с сопряженными двойными
связями (и в ароматических молекулах) поляризуемость электронного облака в
момент реакции обусловлена электромерным эффектом E (динамический эффект
сопряжения).
Теория резонанса не вызывает никаких
принципиальных возражений, пока речь идет о способах изображения молекул, но
она имеет и большие претензии. Аналогично тому, как в методе электронных, пар
волновая функция описывается линейной комбинацией других волновых функций ψ1,
ψ2, ψ3 и т. д., теория резонанса предлагает
описывать истинную волновую функцию ψмолекулы
в виде линейной комбинации волновых функций предельных структур.
Однако математика не дает критериев для выбора
тех или иных «резонансных структур»: ведь в методе электронных пар волновую
функцию можно представить не только как линейную комбинацию волновых функций ψ1,
ψ2, ψ3 и т. д., но и как линейную
комбинацию любых других функций, подобранных с определенными коэффициентами.
Выбор же предельных структур может быть сделан только на основе химических
соображений и аналогий, т. е. здесь концепция резонанса по существу не дает
ничего нового по сравнению с концепцией мезомерии.
При описании распределения электронной плотности
в молекулах с помощью предельных структур необходимо постоянно иметь в виду,
что отдельно взятые предельные структуры не соответствуют какому-либо реальному
физическому состоянию и что никакого физического явления «электронного
резонанса» не существует.
Из литературы известны многочисленные случаи,
когда сторонники концепции резонанса приписывали резонансу смысл физического
явления и считали, что за определенные свойства веществ ответственны те или
иные отдельные предельные структуры. Возможность возникновения таких ошибочных
представлений заложена во многих пунктах концепции резонанса. Так, когда
говорят о «различных вкладах предельных структур» в реальное состояние
молекулы, легко может возникнуть представление о реальном существовании этих
соотношений. Реальная молекула в концепции резонанса считается «резонансным
гибридом»; этот термин может навести на мысль о якобы реальном взаимодействии
предельных структур, подобно гибридизации атомных орбит.
Неудачен также термин «стабилизация за счет
резонанса», так как стабилизация молекулы не может быть обусловлена
несуществующим резонансом, а представляет собой физическое явление
делокализации электронной плотности, характерное для сопряженных систем.
Целесообразно поэтому это явление называть стабилизацией за счет сопряжения.
Энергия сопряжения (энергия делокализации, или энергия мезомерии) может быть
определена экспериментальным путем, независимо от «энергии резонанса»,
вытекающей из квантово-механических расчетов. Это - разность между энергией,
вычисленной для гипотетической молекулы с формулой, соответствующей одной из
предельных структур, и энергией, найденной экспериментально для реальной
молекулы.
С указанными выше оговорками способ описания
распределения электронной плотности в молекулах с помощью нескольких предельных
структур несомненно может быть использован наряду с двумя другими также весьма
распространенными способами.
2.4
Метод Хюккеля
Хюккеля метод, квантовохимический метод
приближенного расчета энергетических уровней и мол. орбиталей ненасыщенных орг.
соединений. Основан на предположении, согласно которому движение электрона
вблизи атомного ядра в молекуле не зависит от состояний или числа др.
электронов. Это позволяет максимально упростить задачу определения мол.
орбиталей (МО) в представлении линейной комбинацией атомных орбиталей. Метод
предложен Э. Хюккелем в 1931 для расчета электронного строения углеводородов с
сопряженными связями. Считается, что атомы углерода сопряженной системы лежат в
одной плоскости, относительно которой высшие занятые и низшие виртуальные
(свободные) МО (граничные мол. орбитали) антисимметричны, т. е. являются
орбиталями, образованными атомными 2рz-орбиталями (АО) соответствующих атомов
С. Влиянием остальных атомов, напр. Н, или мол. фрагментов с насыщенными
связями пренебрегают. Предполагается, что каждый из М атомов углерода
сопряженной системы вносит в систему один электрон и описывается одной атомной
2рz-орбиталью(k = 1, 2, ..., М). Простая модель электронного строения молекулы,
даваемая Хюккеля методом, позволяет понять многие хим. явления. Например
неполярность альтернантных углеводородов обусловлена тем, что эффективные
заряды на всех атомах углерода равны нулю. Напротив, неальтернантная
конденсированная система 5- и 7-членного циклов (азулен) имеет дипольный момент
ок. 1Д (3.3 x 10-30 Кл x м). В нечетных альтернантных углеводородах
основное энергетическое. состояние отвечает электронной системе, в которой есть
хотя бы одна однократно занятая орбиталь. Можно показать, что энергия этой
орбитали та же, что и в свободном атоме, в связи с чем она наз. несвязывающей
МО. Удаление или добавление электрона изменяет заселенность лишь несвязывающей
орбитали, что влечет появление заряда на некоторых атомах, который
пропорционален квадрату соответствующего коэффициент в разложении несвязывающей
МО по АО. Для определения такой МО применяют простое правило: сумма коэффициент
Ck для всех атомов, соседних с любым данным, должна быть равна нулю. Кроме
того, значения коэффициент должны отвечать дополнит. условию нормировки: Это
приводит к характерному чередованию (альтернированию) зарядов на атомах в мол.
ионах альтернантных углеводородов. В частности, указанное правило объясняет
выделение по хим. свойствам орто- и пара-положений в бензольном ядре по
сравнению с мета-положением. Закономерности, установленные в рамках простого
Хюккеля метода, искажаются при более полном учете всех взаимодействие в
молекуле. Однако обычно влияние множества разнородных дополнит, факторов
(например электронов остова, заместителей, межэлектронного отталкивания и т.
п.) качественно не меняет орбитальную картину электронного распределения.
Поэтому Хюккеля метод часто используют для моделирования сложных механизмов
реакций с участием орг. соединений. При введении в молекулу гетероатомов (N, О,
S, ...) существенными становятся параметры матрицы H, принимаемые для
гетероатома и для атомов углерода. В отличие от случая полиенов, разные типы
атомов или связей описываются разными параметрами или и их соотношение
существенно влияет на вид МО; качество предсказаний, получаемых в рамках
простого Хюккеля метода, как правило, в итоге ухудшается. Простой по своей
идее, наглядный и не требующий сложных вычислений Хюккеля метод является одним
из наиболее распространенных средств создания квантовохимической модели
электронного строения сложных мол. систем. Наиб. эффективно его применение в
тех случаях, когда свойства молекулы определяются в основные топологические
структурой хим. связей, в частности симметрией молекулы. Попытки построить
улучшенные варианты Хюккеля метода в рамках простых молекулярных орбиталей
методов имеют мало смысла, т. к. приводят к методикам расчета, сравнимым по
сложности с более точными методами квантовой химии.
Заключение
В настоящее время создана «целая отрасль науки -
квантовая химия, занимающаяся приложением квантово-механических методов к
химическим проблемам. Однако было бы принципиально ошибочным думать, что все
вопросы строения и реакционной способность органических соединений могут быть
сведены к задачам квантовой механики. Квантовая механика изучает законы
движения электронов и ядер, т. е. законы низшей формы движения, сравнительно с
той, которую изучает химия (движение атомов и молекул), а высшая форма движения
никогда не может быть сведена к низшей. Даже для весьма простых молекул такие
вопросы, как реакционная способность веществ, механизм и кинетика их
превращений, не могут быть изучены только методами квантовой механики. Основой
изучения химической формы движения материи являются химические методы
исследования, и ведущая роль в развитии химии принадлежит теории химического
строения.
Список
использованных источников
1. Минкин,
В.И. Теория строения молекул/ В.И. Минкин. -М.:Высш.шк., 2006- 640с.
. Вилков,
Л.В. Физические методы исследования в химии./ Л.В. Вилков, Ю.А. Пентин. -
М.:Высш.шк., 2005-380с.
. Гардымова,
А.П. Научная электронная библиотека: элементы и устройства вычислительной
техники и систем управления [www.dissercat.com]/
А.П. Гардымова. - 2005.
. Ельяшевич,
М.А. Атомная и молекулярная спектроскопия/ М.А. Ельяшевич, В. Демтредер. -М.:
Мир, 1989-260с.
. Цирельсон,
В.Г. Квантовая химия, молекулы, молекулярные системы и твердые тела -М.:
«БИНОМ» 2010-496с.