Комплексные соединения
1. Комплексные соединения
Начало классической теории валентности положено
немецким химиком Ф. Кекуле. Одно из ключевых её положений - постоянство
валентности элементов и насыщаемость химической связи. Классическая теория
позволила описать и предсказать множество химических фактов и явлений,
свойственных простым бинарным молекулам, как Н2О, NH3, SO3, HCl, CuCl2, SiCl4
(их химическую активность, порядок расположения атомов и т.д.), но она не
смогла объяснить механизм образования, свойства и строение более обширного и
разнообразного по составу класса сложных соединений высшего порядка
(комплексных), являющихся часто продуктами сочетания простых молекул с уже
реализованными химическими связями: AlCl3 ∙ 6H2O, Fe(CN)2 ∙ 4KCN,
Zn(OH)2 ∙ 2KOH, AgCl ∙ 2NH3. Согласно классической теории,
двухвалентная медь, например, должна существовать в виде солей типа CuSO4,
CuCl2, Cu(NO3)2, однако в действительности помимо бледно-голубых кристаллов
CuSO4, с валентной схемой
известны ярко-синие кристаллы комплексной соли
[Cu(NH3)4]SO4, структура которых не находит объяснения в рамках общепринятых
представлений о валентности.
Наиболее удачно строение и свойства комплексных
соединений объясняет координационная теория швейцарского химика А. Вернера,
предложенная в 1893 году.
2. Основные положения
координационной теории
Подобно многим имеющим большое значение теориям,
координационная теория исключительно проста. Её главный постулат сводится к
нескольким моментам:
помимо главных валентностей у атомов имеются
побочные валентности, проявляющие себя при некоторых реакциях;
насыщение главных валентностей лежит в основе
образования простых соединений первого порядка;
насыщение побочных валентностей лежит в основе
образования соединений высшего порядка, комплексных.
А. Вернер писал: «Даже если, судя по числу
валентностей, соединительная способность нескольких атомов исчерпана, они все
же, в большинстве случаев, могут принимать участие далее в построении сложных
молекул с образованием определенных атомных связей. Эту возможность нужно усматривать
в том факте, что, наряду со связями сродства, называемыми главной валентностью,
между атомами существуют еще другие связи, называемые побочными
валентностями…».
В момент опубликования теория Вернера носила
формальный характер и не рассматривала вопрос о природе сил, обуславливающих
формирование химической связи и механизм возникновения сложных молекул.
Развитая в дальнейшем учениками, она стала основой современных воззрений о
построении комплексных соединений. Согласно им, в молекуле любого комплексного
соединения один из ионов, обычно положительно заряженный, занимает центральное
место и называется комплексообразователем или центральным ионом. Вокруг него, в
непосредственной близости, расположено (координировано) некоторое число
противоположно заряженных ионов или электронейтральных молекул, называемых
лигандами. Комплексообразователь с окружающими его лигандами образуют
внутреннюю координационную сферу комплексного соединения - комплексный ион -
[MLn], где М - комплексообразователь; L - окружающие комплексообразователь
лиганды; n- их количество.
Для комплекса двухвалентной меди [Cu(NH3)4]SO4
роль комплексообразователя выполняют ионы меди, электронейтральные молекулы
аммиака - лиганды. Вместе ионы Cu2+ с молекулами NH3 формируют комплексный ион
[Cu(NH3)4]2+. Остальные ионы, не разместившиеся во внутренней сфере, находятся
на более далеком расстоянии от центрального иона, составляя внешнюю
координационную сферу. Общая запись формулы комплексного соединения имеет вид:
[MLn]Xm, где в квадратные скобки заключаются частицы внутренней сферы, Х -
внешнесферные частицы (молекулы или ионы). Для комплекса [Cu(NH3)4]SO4
внешнесферные частицы - ионы SO42-.
В большинстве комплексных соединений
комплексообразователями служат ионы переходных металлов (d-элементов), хотя ими
могут быть неметаллы в положительной (В3+, Si4+, Р5+, S6+, I7+), реже в
отрицательной (I-, S2- и др.) степени окисления: K[BF4], Cs[IСl8],
(NH4)2[SiF4]. Комплексообразователями могут выступать и нейтральные атомы.
Нейтральными комплексообразователями являются атомы многих элементов побочных
подгрупп V, VI, VII, VIII групп периодической системы. Так, к примеру,
построены молекулы карбонилов металлов Cr(CO)6, Mn2(CO)10, Fe3(CO)12, Ni(CO)4.
В
роли лигандов фигурируют простые (Cl-, Br-, I-, OH) и сложные (CO32-, S2O32-,
SCN-, NO2-) ионы, полярные и легко поляризующиеся в электрическом поле
комплексообразователя нейтральные молекулы как неорганической (H2O, NH3, NO,
CO), так и органической (NH2CH2CH2NH2, C2H5NH2, C3H5N) природы.
Молекула
комплексного соединения в целом электронейтральна. Заряд комплексного иона
определяется алгебраической суммой зарядов комплексообразователя и лигандов.
Нейтральные молекулы вклада в заряженность иона не вносят:
2+ 6∙1- 3+ 0 0 1- 0 0
[Fe(CN)6]4-,
[Cr(H2O)(NH3)4OH]2+,
[Ni(CO)4]0
.
По знаку заряда комплексного иона различают:
катионные ([Со(NH3)6]3+, [Al(H2O)5OH]2+); анионные ([Al(OH)4]-, [Fe(CN)6]4-) и
нейтральные ([Ni(CO)4]0, [Co(NH3)3Cl3]0) комплексы. Последние не содержат
внешней сферы и не являются электролитами.
Центральный атом комплексного соединения
координирует вокруг себя лиганды внутренней сферы, геометрически правильно
располагая их в пространстве. Число лигандов, определенным образом
расположенных в пространстве и непосредственно связанных с центральным ионом,
называют координационным числом этого иона или координационной валентностью.
Более строго: координационное число - это число электронодонорных центров
лигандов (атомов и π-связей),
непосредственно взаимодействующих с комплексообразователем. Для комплексных
соединений с моноцентровыми лигандами координационное число равно числу
лигандов (в комплексных ионах [Cu(NH3)4]2+ координационное число меди - 4, в
[Fe(CN)6]3+ координационное число железа - 6). В случае полицентровых - числу
таких лигандов, умноженному на число электронодонорных центров лиганда (в
[Сo(SO4)3]3- координационное число кобальта - 6).
В химии комплексных соединений координационное
число играет примерно ту же роль, если лиганд ион, что и валентность в
соединениях первого порядка, варьируя от 1 до 12. Обычными координационными
числами, встречающимися более чем у 95% комплексов, являются 4 и 6.
Координационное число не является неизменной
величиной. Его максимальное значение определяется, главным образом,
соотношением сил отталкивания между лигандами, возникающими по мере накопления
их в координационной сфере, и уравновешивающими эти силы силами притяжения
лигандов к центральному иону. Когда внесение следующего лиганда приводит к
такому возрастанию сил отталкивания, которое уже не компенсируется притяжением
лигандов к комплексообразователю, координирование прекращается. Следовательно,
основными факторами, влияющими на координационное число, являются плотность
заряда комплексообразователя, а также размер и заряд (или полярность) лигандов.
В некоторых случаях размер лиганда оказывается
намного больше размера центрального иона. Это будет вести к ограничению
координационного числа центрального иона, поскольку делает физически
невозможным присоединение к нему большого числа лигандов. Показано, что, если
отношение радиусов центрального иона (r) и лигандов (r/) меньше 0,155,
максимальное координационное число не может превышать двух (рис. 1.1). Однако и
в тех случаях, когда отношение радиусов очень мало, основным фактором остается
баланс сил притяжения лигандов к комплексообразователю и сил отталкивания между
самими лигандами.
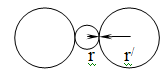
Рис. 1.1
Существует статистическое правило, связывающее
заряд центрального иона (степень окисления) и координационное число, согласно
которому (табл. 1) характерные координационные числа чаще всего превышают заряд
центрального иона в два раза (жирным шрифтом выделены наиболее часто
встречающиеся). Приведенные значения чисел соответствуют максимальному
насыщению координационной сферы лигандами и свойственны
координационно-насыщенным соединениям. В растворах не всегда соблюдаются
необходимые для этого условия и тогда образуются координационно-ненасыщенные
комплексы, с меньшими координационными числами. Т.е., не являясь постоянной
величиной, координационное число комплексообразователя, помимо основных (заряд,
размер частиц), зависит еще от ряда сопутствующих факторов, среди которых
наибольшее значение имеют концентрация исходных компонентов и температура
раствора.
Таблица 1 - Координационные
числа некоторых комплексообразователей
|
Ионы
|
Степень
окисления комплексообразователя
|
Характерное
координационное число
|
|
Au+,
Ag+, Cu+
|
+1
|
2
|
|
Cu2+, Hg2+, Pb2+, Pt2+, Pd2+, Zn2+, Ni2+, Co2+
|
+2
|
4;
6
|
|
Fe3+, Al3+, Cr3+, Co3+, Ni3+
|
+3
|
6;
4
|
|
Mo4+,
W4+
|
+4
|
8
|
Число атомов, предоставляемых лигандами для
образования координационных связей с комплексообразователем, может быть
различным. В большинстве случаев каждый отдельный лиганд присоединяется к
центральному иону через один атом (М - L), занимая во внутренней координационной
сфере одно место. Координационная ёмкость (число мест) или дентатность такого
лиганда равна единице. Лиганды, предоставляющие для связывания с центральным
ионом один атом и занимающие во внутренней сфере одно место, называются
монодентатными. К монодентатным относятся все одноатомные лиганды: анионы
галогенов; анионы CN-, SCN-, OH-; молекулы NH3, H2O, NO, CO и др. Примерами
комплексов с монодентатными лигандами служат [Fe(CN)6]3-, [CrCl6]3-:
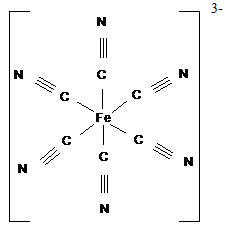
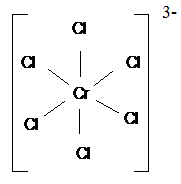
Иные лиганды используют в
комплексообразовании два или большее число атомов, занимая в координационной
сфере центрального иона два или большее число мест. Такие лиганды называются
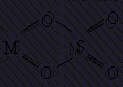
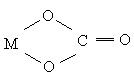
би- и полидентатными. Бидентатным является оксалат-анион С2О42-, формирующий с
катионом-комплексообразователя две связи за счёт двух кислородных атомов:
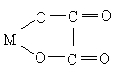 ,
,
а также этилендиамин, сульфатный,
карбонатный и другие анионы:
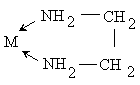 ,
,  ,
, 
При этом один и тот же лиганд, в
зависимости от характера связывания, может быть и моно- и бидентатным:

Примером тетрадентатного лиганда
является двухзарядный анион этилендиаминтетрауксусной кислоты (ЭДТА):
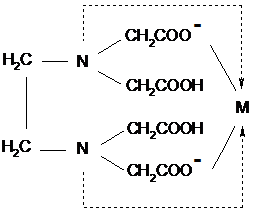
(на этих и последующих схемах
обычные ковалентные связи, образованные неспаренными электронами, обозначены
черточками, а возникающие по донорно-акцепторному механизму - стрелками). Таким
образом, дентатность - это число донорных центров лиганда, участвующего в
координации.
3. Природа химической связи в
комплексном ионе
Механизм образования комплексных
соединений и, прежде всего, природа химической связи комплексообразователя и
лигандов в современной химии описываются исходя из электростатических и
квантовомеханических представлений, ставших основой трех более или менее
отличных друг от друга теорий. Хронологически и по увеличению сложности первой
считается электростатическая теория с её более совершенной модификацией -
теорией кристаллического поля, второй - метод валентных связей и третьей -
метод молекулярных орбиталей. Сочетание последней теории с теорией
кристаллического поля привело к интенсивному развитию нового подхода,
известного под названием теории поля лигандов.
Согласно этой теории, образование
многих комплексных соединений в первом приближении можно объяснить
электростатическим притяжением между центральным катионом металла и
противоположно заряженными анионами или полярными молекулами лигандов. При
достаточном их сближении в пространстве в формирующемся комплексном ионе наряду
с силами притяжения начинают действовать силы электростатического отталкивания,
особенно между одноименно заряженными или, в случае полярных молекул, одинаково
ориентированными лигандами. В результате возникает правильное геометрическое
расположение лигандов и образуется устойчивая структура, с минимальной
потенциальной энергией. Количественные расчеты В. Косселя и А. Магнуса
(1916-1922 г.г.) подтверждают правомочность такой модели, показывают прямую
зависимость координационного числа от заряда центрального иона. Однако простая
электростатическая теория не в состоянии объяснить своеобразие реакционной
способности комплексных соединений, их магнитные свойства, окраску,
избирательность (специфичность) комплексообразования, поскольку не принимает во
внимание природу центрального иона и лигандов, особенности строения их
электронных оболочек, взаимное влияние ионов друг на друга. Более полно и точно
образование химических связей, свойства и организацию комплексных соединений
объясняют теории, основанные на квантовомеханических представлениях о строении
атомов и молекул: метод валентных связей и метод молекулярных орбиталей.
Метод валентных связей
Метод валентных связей к комплексным соединениям
впервые применил американский химик Л. Полинг в 30-х годах прошлого столетия.
Полинг исходил их следующих допущений:
для образования ковалентных связей с орбиталями
лиганда центральный атом или ион металла должен содержать некоторое число
пустых (вакантных) орбиталей, определяющих его координационную валентность;
ковалентные связи образуются перекрыванием
вакантных орбиталей атома (иона) металла с заполненными орбиталями донорного
иона (лиганда). Донорный ион обязан иметь для этого, по крайней мере, одну
неподеленную пару электронов.
Формирующиеся в комплексах σ-связи
являются двухэлектронными и двухцентровыми, а образование их протекает по
донорно-акцепторному механизму; чем больше
электронные облака перекрывают друг друга, тем более прочной является возникающая
химическая связь. Для удовлетворения этого критерия необходимо, чтобы исходные
атомные орбитали были гибридизованы и формировали новую систему перекрывающихся
эквивалентных орбиталей с определенной направленностью в пространстве.
Рассмотрим с позиции метода валентных связей
образование химических связей между комплексообразователем и лигандами на
конкретных примерах. В комплексной соли K2[NiCl4] конфигурация валентной
оболочки иона никеля Ni2+ в основном состоянии описывается формулой 3d84s04p0 и
может быть представлена в виде диаграммы заполнения ячеек:
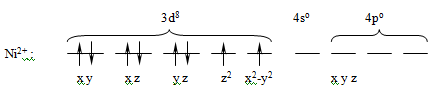
Согласно диаграммы, Ni2+ имеет четыре вакантные
орбитали: 4s, 4px, 4py, 4pz. Ионы хлора на внешней оболочке содержат свободные
неподеленные пары электронов -  . Под действием
электрического поля ионов хлора ион никеля (II) из основного переходит в
возбужденное состояние с гибридизацией одной 4s и трех 4p-орбиталей:
. Под действием
электрического поля ионов хлора ион никеля (II) из основного переходит в
возбужденное состояние с гибридизацией одной 4s и трех 4p-орбиталей:
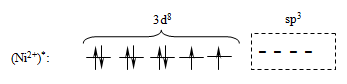
Последующее перекрывание вакантных sp3-атомных
орбиталей возбужденного иона Ni2+ c двухэлектронными sp3-атомными орбиталями
ионов хлора приводит к обобщению четырех неподеленных электронных пар хлора и
формированию комплексного иона [NiCl4]2-, с тетраэдрической ориентацией лигандов
(тетраэдрическая координация):
Возникают четыре равноценные двухэлектронные σ-связи
Ni2+ - Cl-, аналогичные по свойствам одинарным σ-связям
в простых бинарных соединениях (рис. 2):
Рис. 2 -
Комплексный ион [NiCl4]2-
Суммарную реакцию образования комплексного иона
можно записать:
Ni2+ + 4 Сl- [NiCl4]2-.
Химические связи, возникающие при
комплексообразовании за счет неподеленной электронной пары одного из
взаимодействующих атомов (ионов) и свободной орбитали другого атома (иона),
называются донорно-акцепторными координационными связями. Нейтральные молекулы
или ионы, отдающие свои неподеленные пары электронов в «общее пользование»,
называются донорными; атомы - комплексообразователи,
предоставляющие свободные орбитали для размещения этих электронов -
акцепторными. Идентичные
химические связи, с тетраэдрической направленностью sp3-гибридных орбиталей,
возникают в ионе NH4+, при взаимодействии молекул аммиака с протоном H+; ионе
BF4-, при взаимодействии BF3 с анионом F- (рис. 3):
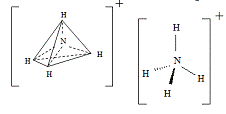
Рис. 3 -
Тетраэдрическая конфигурация атома азота в ионе NH4+
Лиганды отличаются по силе своего воздействия на
комплексообразователи. Объясняется это энергетическим потенциалом лигандов. По
величине энергии лиганды располагаются в следующем порядке (спектрохимический
ряд лигандов):
СО > СN-
> NH2-CH2-CH2-NH2 > NH3 > SCN- > H2O > OH- > F- >Cl- >
Br- > I-
В начале этого ряда находятся лиганды, создающие
наиболее сильное поле, в конце - слабое. Лиганды,
создающие сильное поле, оказывают более значительное влияние на энергетическое
состояние d-подуровня комплексообразователя. В результате меняется
распределение электронов по орбиталям, что будет вести к иному типу
гибридизации, а в итоге к иной структуре и к иным свойствам образующегося
комплексного соединения. Так, если в комплексной соли K2[NiCl4] четыре
вакантные орбитали иона Ni2+ под влиянием слабого поля Cl- находятся в
состоянии sp3-гибридизации, с тетраэдрическим расположением лигандов в
пространстве (рис. 2), то в комплексной соли никеля (II) c цианид-ионами
(K2[Ni(CN)4]) тетраэдрическая конфигурация комплексного иона трансформируется в
плоскостную квадратную (рис. 4). Вызвано это тем, что во втором комплексе, под
влиянием сильного электрического поля цианид-ионов, сначала имеет место
обобщение двух
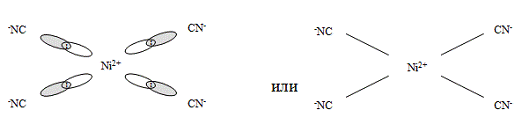
Рис. 4 -
Комплексный ион [Ni(CN)4]2-
Неспаренных
d-электронов Ni2+, которые занимают 3dz2-орбиталь (см. ниже схему), а
освободившаяся 3d x2-y2 - орбиталь, а также 4s- и две 4р-орбитали участвуют
далее в гибридизации с образованием четырех эквивалентных dsp2-орбиталей,
направленных в плоскость квадрата:
В комплексе с цианид-ионами у центрального иона
не остается неспаренных электронов и он не обладает магнитными свойствами
(диамагнитен, μ = 0). При
комплексообразовании с ионами хлора, имеющими слабое поле, два электрона не
спарены. Неспаренные электроны, вращаясь вокруг собственных осей, создают
магнитный момент. Магнитный момент молекул (μ) складывается
из магнитных моментов неспаренных электронов. Если молекула содержит
неспаренные электроны, то μ≠0
и вещество парамагнитно, т.е. втягивается в магнитное поле. Когда неспаренных
электронов нет и μ = 0, вещество
диамагнитно и слабо выталкивается из магнитного поля. Наличие двух неспаренных
электронов у атома Ni2+ в комплексе K2[NiCl4] обуславливает его парамагнетизм.
Достоинство метода валентных связей в том, что
уже многие десятилетия он дает химикам простую, наглядную модель связывания
атомов, успешно объясняет определенные значения координационных чисел и
геометрические формы комплексных частиц. Правильно описываются с позиций метода
и различия в магнитных свойствах - диамагнитизм и парамагнетизм. Однако
основанный на ряде допущений, он не в состоянии объяснить спектры поглощения и
строение некоторых, особенно открытых в последнее время, комплексов, например,
«сэндвичевых». Кроме того, взаимодействие центрального атома (иона) с лигандами
не всегда сводится к элементарному акту передачи электронов лигандом. Известны
лиганды, сами способные принимать электроны металла на свои вакантные орбитали,
например, на свободные d-орбитали в молекуле PF3 или в ионе SnCl3-. Сомнительно
также, чтобы на центральном ионе могла осуществляться концентрация столь
высокого отрицательного заряда, который бы создавался, если бы каждый лиганд
стал отдавать ему свою пару электронов.
Метод молекулярных орбиталей
Среди различных подходов к объяснению
образования комплексного иона наиболее общий дает метод молекулярных орбиталей.
Впервые к комплексным соединениям он был применен Ван-Флеком в 1935 г. При
описании координационной связи в комплексном ионе в методе молекулярных
орбиталей учитываются не только вакантные атомные орбитали
иона-комплексообразователя, что характерно для метода валентных связей, но и
орбитали, координирующихся вокруг комплексообразователя n лигандов.
Формирование молекулярных орбиталей в комплексных соединениях осуществляется по
тому же принципу, что и в простых бинарных молекулах. Отличие заключается в
том, что в комплексном ионе молекулярные орбитали многоцентровые и
делокализованные, как, например, в молекуле бензола или содержащих циклические
ядра «сэндвичевых» комплексах - дибензолхроме [Cr(C6H6)2] и ферроцене
[Fe(C5H5)2]. В последних примерах атомы металлов находятся между циклическими
органическими структурами, энергия которых равномерно распределена
(делокализована) по обоим циклам, в результате чего центральный атом в равной
степени взаимосвязан со всеми атомами комплекса.
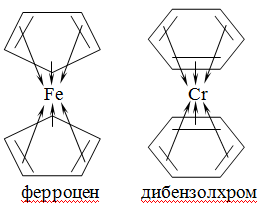
Рис. 5
Комплексные соединения известны практически для
всех элементов таблицы Менделеева. f-элементам свойственна наивысшая
комплексообразующая способность, но актиноиды и лантаноиды короткоживущие
элементы. Поэтому среди стабильных элементов d-элементы, имеющие свободные
орбитали на внешнем уровне и незавершенные предвнешние уровни, обладают
наиболее высокой способностью к комплексообразованию, а их комплексы наиболее
прочны. Среди комплексов d-элементов для жизнедеятельности человека наиболее
важны природные соединения Mn, Fe, Co, Cu, Zn, Mo. Амфотерные р-элементы: Al,
Sn, Pb и др., имеющие три р-орбитали, более слабые комплексообразователи.
Комплексообразующая способность s-элементов самая малая, - более чем на порядок
ниже, чем у p-элементов. Биогенные s-элементы Na, K, Ca, Mg могут образовывать
непрочные комплексы только с лигандами определенной структуры. Таким образом, в
блоках периодической системы способность к комплексообразованию уменьшается в
следующем порядке: f > d > p >> s.
Различия в комплексообразующей способности
прослеживаются не только между элементами отдельных s, p, d, f-семейств
периодической системы, но и между элементами одного семейства. Среди
многообразия факторов, влияющих на комплексообразование, можно выделить
несколько основных, прямо связанных с природой и строением иона
комплексообразователя.
Для иона комплексообразователя, имеющего электронную
конфигурацию инертного газа (s2 или ns2np6 - щелочные и щелочно-земельные
металлы; многие элементы побочных подгрупп 3-8 групп, в высшей степени
окисления), псевдоинертного ((n-1)d10 - Cu+, Ag+, Au+, Zn2+, Cd2+, Ga3+, In3+,
Sn4+, Pb4+, Sb5+, Bi5+) или псевдоинертного газа плюс два электрона
((n-1)d10ns2 - Ga+, In+, Sn2+, Pb2+, Sb3+, Bi3+) комплексообразующая
способность зависит главным образом от эффективного ионного радиуса и
эффективного заряда ядра атома. Если отношение заряда катиона к его радиусу в
кристалле принять за φ - ионный
потенциал, то можно показать, что способность к образованию комплексных
соединений ионами металлов с небольшими или многозарядными ионными и
полидентатными лигандами увеличивается по мере возрастания ионного потенциала.
Наблюдаемая последовательность для ионов с одним и тем же зарядом подтверждает
такое предположение: Li+ > Na+ > K+ > Rb+ > Cs+; Mg2+ > Ca2+
> Sr2+ > Ba2+ > Ra2+; Al3+ > Sc3+ > Y3+ > La3+. Аналогичная
закономерность свойственна ионам с различными зарядами, но примерно одинакового
размера: Y3+ > Ca2+ > Na+; La3+ > Sr2+ > K+.
Несоответствие возникает при сравнении близких
по размеру и заряду представителей одной и той же группы периодической системы,
например: K и Cu+, Ca и Cd, Sc и Ga3+. Элементы побочной подгруппы всегда более
сильные комплексообразователи, по сравнению с элементами главной, и причина
этого кроется в различии электронных конфигураций. Ионы элементов главной
подгруппы имеют конфигурацию инертного газа ns2np6, а ионы побочной -
псевдоинертного (n-1)d10, либо псевдоинертного плюс два электрона (n-1)d10ns2.
Последние конфигурации в меньшей степени экранируют заряд ядра положительного
одноатомного иона, из-за чего эффективный заряд ядра для ионов элементов
побочной подгруппы в действительности выше, чем для ионов элементов главной.
Например, для иона Cu+ он больше, чем для иона K+.
Значение эффективного заряда можно найти по
величинам энергий ионизации. Так, хотя Na+ и Cu+ почти одинаковы по размеру
(0,95 и 0,93 Ǻ), энергия ионизации для Na+ равна 118,5 ккал/моль, а для
Cu+ - 178 ккал/моль. Это различие связано с наличием у иона Cu+ большего
сродства к электрону, что приводит к большему притяжению электронов лиганда к
меди, чем к иону натрия.
Для ионов металлов с конфигурацией инертного
газа, как s, p так и d-элементов, комплексообразование не является характерным.
Несравненно более высокую комплексообразующую способность проявляют ионы
переходных металлов, когда d-орбитали незаполнены - (n-1)d1-9. Это означает,
что для формирования донорно-акцепторных связей, образования устойчивых
комплексных соединений принципиальное значение имеет не только величина φ
потенциала
катиона, но и наличие, а также число свободных орбиталей на d-подуровне,
которые могут быть «заселены» неподеленными парами электронов лиганда.
Значительное влияние на процесс
комплексообразования оказывает расщепляемость незаполненного d-подуровня под
воздействием силового поля лиганда. Лиганды, окружая центральный ион, способны
возбуждать его, изменяя взаимную ориентацию d-орбиталей. При этом, испытывая
влияние лигандов, наиболее сильному возбуждению подвергаются ближайшие к иону
dx2-y2 и dz2 орбитали, меньшему - удаленные - dxy, dxz и dyz. В результате
ранее единый энергетический подуровень из пяти d-орбиталей центрального иона
расщепляется на два разных энергетических подуровня:
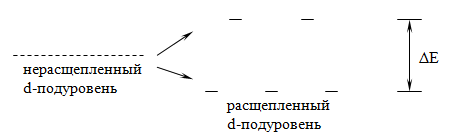
Рассмотрим это на примере иона железа Fe2+. Его
электронная конфигурация описывается формулой 3d6. В свободном ионе
распределение электронов подчиняется правилу Хунда: ↑ ↓ ↑ ↑
↑ ↑ . В присутствии лиганда с сильным энергетическим полем (CO,
CN-) d-подуровень расщепляется и электроны с орбиталей, обладающих большим
запасом энергии, перейдут на орбитали с меньшим:
Возникает синглентное состояние центрального
атома (отсутствие неспаренных электронов). Выравнивание энергии двух свободных
3d-орбиталей и энергий одной 4s- и трех 4p-орбиталей способствует более легкому
их смешиванию (d2sp3-гибридизация) с формированием устойчивого октаэдрического
комплекса. Рассмотренный феномен имеет существенное значение, так как
обусловливает важнейшую биологическую функцию - транспорт кислорода
гемоглобином, в состав которого входит двухвалентное железо.
Для s- и p-элементов эффект расщепления
невозможен, что снижает комплексообразующую способность ионов этих металлов.
4. Пространственное строение и
изомерия комплексных соединений
Ориентация связей металл-лиганд определяет
пространственную конфигурацию комплексных молекул а, следовательно, возможность
существования их в виде изомеров, соединений с одинаковым составом и
молекулярной массой, но отличающихся строением, физическими и химическими
свойствами. У комплексных молекул с одинаковыми лигандами изомеры отсутствуют.
Отталкиваясь за счёт электростатических сил, идентичные по природе лиганды, как
правило, симметрично располагаются в пространстве вокруг центрального атома,
где все положения лигандов эквивалентны. Возникает определенная устойчивая
геометрическая конфигурация комплексного соединения. Для наиболее часто
встречающихся комплексов с координационными числами 2, 4 и 6 свойственны
следующие геометрические конфигурации:
|
Координационное
число
|
Геометрическая
конфигурация
|
|
2
|
L
----------- M ---------- L - линейная Одинаковые лиганды находятся по разные
стороны от комплексообразователя, с углом 1800. Наблюдается у лекарственного
препарата золота (I) хризолана Na3[Au(S2O3)2], используемого при лечении
ревматоидного артрита: [O3S2 - Au - S2O3]3-. Числу 2 отвечает
sp-гибридизация.
|
|
4
|
-
тетраэдрическая По аналогии с катионом аммония (NH4+) лиганды в таких
комплексных соединениях располагаются в вершинах тетраэдра. Чаще свойственна
комплексообразователям со спаренными d-электронами и sp3-гибридизацией
вакантных орбиталей. Проявляется у комплексной соли цинка [Zn(NH3)4]Cl2, с
электронной формулой валентной оболочки катиона 3d104s04p0.
|
|
4
|
-
плоская квадратная Характерна для [Cu(NH3)4]SO4 - комплекса, у которого из-за
наличия неспаренного d-электрона у иона Cu2+ тетраэдрическое расположение
лигандов сменяется на плоское квадратное. Наиболее часто встречаемый тип
гибридизации dsp2.
|
|
6
|
-
октаэдрическая, схематически можно изобразить так: Самая многочисленная
группа комплексных соединений: Na2[SiF6], K2[PtCl6], H4[Mn(CN)6] и т.д.,
образуемых элементами V, VI и VII периодов таблицы Менделеева. Орбитали dz2 и
dx2-y2 в эктаэдрических комплексах направлены прямо к лигандам, обеспечивая в
итоге формирование гибридных d2sp3-орбиталей.
|
При наличии двух или большего числа неоднородных
лигандов эквивалентность расположения их во внутренней сфере
комплексообразователя нарушается и возможно появление комплексных изомеров,
различающихся по цвету, растворимости, реакционной способности, дипольному
моменту, способам получения. Для комплексных соединений свойственны следующие
типы изомерии: геометрическая, сольватная (в водных растворах гидратная),
ионизационная, координационная и оптическая (зеркальная).
Геометрическая или пространственная изомерия
заключается в том, что комплекс, включающий неодинаковые лиганды, может
существовать в двух или нескольких различных формах в зависимости от
пространственного размещения лигандов вокруг комплексообразователя. К
геометрической изомерии в первую очередь относится цис-транс-изомерия,
проявляющаяся в основном в плоско-квадратных и октаэдрических комплексах:
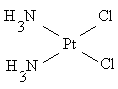

цис (оранжево-желтый) транс
(светло-желтый)
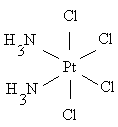
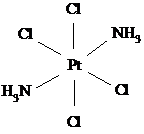
цис (оранжевый) транс (желтый)
Различие в структуре обусловливает
различную физиологическую активность таких соединений. Например, малотоксичный
плоско-квадратный цис-изомер платины обладает противоопухолевой активностью и
нашёл применение в качестве лекарственного препарата; транс-изомер значительно
более токсичен без какого-либо противоопухолевого действия.
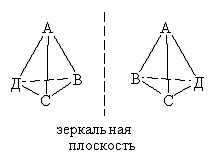
Рис. 6
Оптическая изомерия. Наблюдается у
комплексных соединений, структура которых не имеет элементов симметрии. В
простейшем случае это тетраэдрический комплекс, с четырьмя различными
лигандами, для которого существуют два изомера (см. схему), не совмещающиеся в
пространстве. Такие изомеры называют оптическими антиподами. Один из оптических
изомеров способен вращать плоскость поляризованного света влево (L-изомер),
второй - вправо (D-изомер).
Другие виды изомерии вызваны
различным положением и связью лигандов внутренней и внешней координационных
сфер. Так, гидратная изомерия имеет место при переходе воды из внутренней во
внешнюю сферу. Например, для хлорида хрома (III) известны три модификации:
|
Структура
комплекса
|
Окраска
кристаллов
|
|
[Cr(H2O)6]Cl3 [Cr(H2O)5Cl]Cl2 ∙ H2O
[Cr(H2O)4Cl2]Cl ∙ 2H2O
|
сине-серая
светло-зеленая темно-зеленая
|
Ионизационная изомерия обусловлена различным
распределением неодинаковых по своей природе ионов между внутренней и внешней координационными
сферами, что приводит к изменению степени диссоциации комплексного соединения.
Например: [Co(NH3)5Br]SO4 - красно-фиолетовый; [Co(NH3)4SO4]Br - красный, в
большей степени диссоциирующий комплекс.
Координационная изомерия связана с переходом
лигандов от одного комплексообразователя к другому в соединениях, образованных
комплексными катионами и комплексными анионами: [Co(NH3)6] ∙ [Cr(CN)6] и
[Co(CN)6] ∙ [Cr(NH3)6].
5. Классификация комплексных
соединений
Существует несколько способов классификации
комплексных соединений, основанных на разных признаках. Наиболее
распространённой является классификация по природе лигандов. В зависимости от
природы лигандов различают:
аммиакаты (лиганды - молекулы NH3):
[Cu(NH3)4]SO4; [Co(NH3)6]Cl3; [Pt(NH3)6]Cl4.
Если роль лигандов выполняют молекулы аминов:
этилендиамин - NH2CH2CH2NH2 (обозначаемый как En), метиламин - CH3NH2, этиламин
- C2H5NH2 и др. комплексы называют аминатами;
аквакомплексы (лиганды - молекулы Н2О):
[Cr(H2O)6]Cl3; [Al(H2O)6]Cl3; [Cu(H2O)4](NO3)2. В кристаллическом состоянии
некоторые из аквакомплексов удерживают кристаллизационную воду: [Cu(H2O)4]SO4 ∙
H2O. Кристаллизационная вода не входит в состав внутренней сферы, она связана
менее прочно, чем координированная, и легче отщепляется при нагревании;
ацидокомплексы (лиганды - анионы различных
кислот). К ним относятся:
комплексы типа двойных солей: K2[PtCl4],
K4[Fe(CN)6], которые можно представить как продукты сочетания двух солей: PtCl2
∙ 2KCl, Fe(CN)2 ∙ 4KCN;
комплексные кислоты: H2[SiF6];
гидроксокомплексы (лиганды - ионы ОН-):
Na2[Sn(OH)4], Na2[Sn(OH)6];
переходные ряды комплексов. Включают комплексы с
различными по природе лигандами. Запишем переходный ряд между аммиакатами и
ацидокомплексами платины (II): [Pt(NH3)4]Cl2; [Pt(NH3)3Cl]Cl; [Pt(NH3)2Cl2];
K[Pt(NH3)Cl3]; K2[PtCl4];
циклические или хелатные комплексные соединения.
Содержат би- или полидентатные лиганды, связанные с центральным атомом
несколькими связями. Примерами могут служить оксалатный комплекс железа (III) -
[Fe(C2O4)3]3- и этилендиаминовый комплекс платины (IV) - [PtEn3]4+:

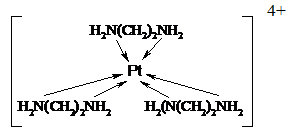
В группу хелатов входят и
внутрикомплексные соединения, в которых центральный атом, включаясь в состав цикла,
образует ковалентные связи с лигандами разными способами: донорно-акцепторным и
за счёт неспаренных атомных электронов. В организме такие комплексы характерны
для ионов Cu или Fe с аминокислотами, например, меди с глицином в глицинате
меди:
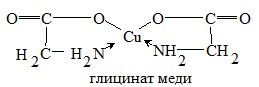
Рис. 8
где две связи комплекса
донорно-акцепторные, возникающие за счёт донорно-акцепторного взаимодействия
неподелённых пар электронов двух аминогрупп кислоты  со
свободными орбиталями возбуждённого иона меди, две другие - обычные ионные,
получаемые замещением в карбоксильных группах ионов Н+ на ионы Cu2+.
со
свободными орбиталями возбуждённого иона меди, две другие - обычные ионные,
получаемые замещением в карбоксильных группах ионов Н+ на ионы Cu2+.
Перечисленные комплексные соединения
относятся к одноядерным, содержащим один центральный атом. Помимо них
существуют полиядерные, в структуре которых одновременно присутствуют два или
несколько центральных атома-комплексообразователя. К полиядерным комплексным
соединениям относятся:
комплексы с мостиковыми лигандами,
где каждые два центральных атома соединены одним, двумя или тремя лигандами
одновременно:
[Sb2F7]- 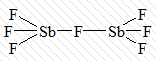
[Al2Cl6] 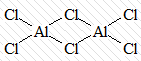
[Co2(OH)2(NH3)8]4+ 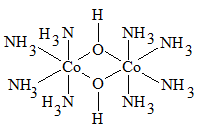
кластерные соединения, в которых центральные
атомы связаны между собой непосредственно:
[(CO)5Mn - Mn(CO)5] 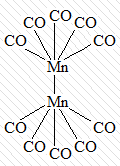
[Re2H2Cl8]2- 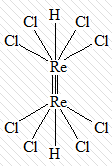
В большинстве случаев в кластерных комплексах
комплексообразователи объединены помимо прочных одинарных или кратных связей
ещё и мостиковыми лигандами:
|
[Ru3O12]12-
|
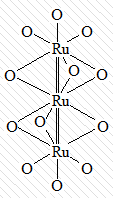
|
- изополисоединения - полиядерные соединения с
комплексными анионами, в которых комплексообразователями выступают атомы одного
и того же элемента, а в качестве лигандов (в том числе и мостиковых) -
оксид-ионы О2-. Наиболее распространенными из комплексов этого типа являются
изополикислоты и их соли: Н4Р2О7, K2Cr2O7 и др., с пространственным
расположением атомов:

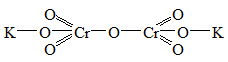
- гетерополисоединения - полиядерные соединения,
в комплексных ионах которых содержатся центральные атомы разных элементов, а в
качестве лигандов (в том числе и мостиковых) выступают оксид-ионы О2-. К таким
соединениям преимущественно относятся гетерополикислоты с общей формулой
Н8-n[MnM12VIO40], где n - степень окисления В+3, Si+4, Ge+4, Ti+4, P+5; MVI -
Mo или W. Например: Н4[GeMo12O40], H3[PW12O40], H5[BMo12O40].
6. Номенклатура комплексных молекул
Названия комплексных соединений составляются по
определенным правилам. Согласно рекомендаций IUPAC, вначале указывают состав
внутренней координационной сферы. Во внутренней сфере в первую очередь называют
анионы, прибавляя к их латинскому названию окончание «о». Например: Cl- -
хлоро, CN- - циано, SO32- - сульфито, Н- - гидридо, ОН- - гидроксо и т.д. Далее
называют нейтральные молекулы и, прежде всего, молекулы аммиака и его
производные. Причём для координированного аммиака используют тривиальный термин
- аммин, для воды - аква, для СО - карбонил, для NO - нитрозил. Число лигандов
указывают греческими числительными: 1 - моно (часто не приводится), 2 - ди, 3 -
три, 4 - тетра, 5 - пента, 6 - гекса.
Затем называют комплексообразователь. Если
центральный атом формирует анионный комплекс, то употребляют латинское название
элемента, прибавляя к корню окончание «ат», с указанием в скобках степени его
окисленности. Если центральный атом входит в состав катиона, используют русское
название элемента и в скобках указывают его степень окисленности. В случае
неэлектролитов степень окисленности центрального атома не отмечается, так как
она вытекает, исходя из электронейтральности комплекса.
После названия внутренней сферы называют
внешнюю: кислотные остатки или катионы. Число кислотных остатков, а также
катионов определяется валентностью комплексного иона и в названии не
отображается. Катионы внешней сферы называются в родительном падеже.
анионные комплексы:[Al(OH)4] -
тетрагидроксоалюминат (III) калия;[Fe(CN)6] - гексацианоферрат (II) натрия;
(NH4)2[Pt(OH)2Cl4] -
тетрахлородигидроксоплатинат (IV) аммония.
катионные:
[Al(H2O)6]Cl3 - хлорид гексаакваалюминия (III);
[Zn(NH3)4]Cl2 - хлорид тетраамминцинка (II);
[CoCl(NH3)5]Cl2 - хлорид пентаамминхлорокобальта
(III).
нейтральные:
[Pt(NH3)2Cl2] - дихлородиамминплатина;
[Cr(H2O)3F3] - трифторотриаквахром;
[Co(NH3)3(NO2)2Cl] -
хлородинитритотриамминкобальт.
7. Диссоциация в растворах
Внутренняя и внешняя сферы комплексного
соединения сильно различаются по устойчивости. Частицы внешней сферы
удерживаются комплексным ионом в основном электростатическими силами и легко
отщепляются в водных растворах. Эта диссоциация называется первичной и
протекает по типу диссоциации сильных электролитов.
Лиганды
внутренней сферы связаны центральным атомом намного прочнее (ковалентными
связями) и при растворении отщепляются незначительно. Обратимый распад
внутренней сферы носит название вторичной диссоциации. Диссоциацию комплекса
[Ag(NH3)2]Cl можно записать следующим образом:
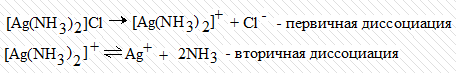
Диссоциация комплексного иона
характеризуется наличием равновесия между комплексной частицей, центральным
ионом и лигандами. Как и в случае слабых электролитов, она подчинена закону
действия масс и может быть описана соответствующей константой равновесия,
называемой константой нестойкости комплексного иона:
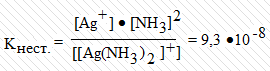
Константы нестойкости комплексных
ионов весьма различны (табл. 1.2) и характеризуют термодинамическую
устойчивость комплексного иона, которая зависит от прочности связи центрального
атома с лигандами. Мерой этой прочности является энергия разрыва
донорно-акцепторной связи. Чем ниже значение константы нестойкости, тем прочнее
и устойчивее комплекс.
Таблица 2 - Константы
нестойкости некоторых комплексных ионов при 25ºС
|
Реакция
|
Кнест.
|
|
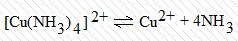 8,5
∙ 10-13 8,5
∙ 10-13
|
|
|
 9,3
∙ 10-8 9,3
∙ 10-8
|
|
|
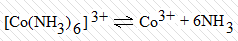 2
∙ 10-34 2
∙ 10-34
|
|
|
 5
∙ 10-31 5
∙ 10-31
|
|
|
 1
∙ 10-20 1
∙ 10-20
|
|
|
 3
∙ 10-42 3
∙ 10-42
|
|
В последнее время вместо константы нестойкости
часто используют обратную ей величину, называемую константой устойчивости. Для
иона [Ag(NH3)2]+ константа устойчивости равна:
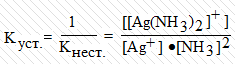 .
.
Диссоциация комплексного иона не
всегда заканчивается только конечными продуктами. В действительности в
растворах имеет место ступенчатый процесс, аналогичный процессу у слабых
электролитов, где каждая ступень диссоциации характеризуется своей константой
нестойкости:
ступень
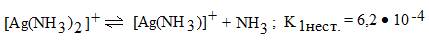 ,
,
2 ступень
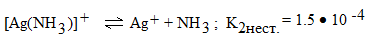 .
.
Причем всегда К1нест. > К2нест.
> К3нест. >
Общая константа нестойкости равна
произведению констант всех ступеней диссоциации комплексного иона:
Кнест. [Ag(NH3)2] + = К1 ∙ К2
= 6,2 ∙ 10-4 ∙ 1,5 ∙ 10-4 = 9,3 ∙ 10- 8.
С помощью Кнест. или Куст. можно предсказать
течение реакций образования и распада комплексных ионов. При сильном различии
констант нестойкости реакция пойдет в сторону формирования иона с меньшей
константой нестойкости или, что равноценно, с большей константой устойчивости.
То же относится и к реакциям распада. Например, если на ион [Ag(NH3)2]+ с
Кнест. = 9,3 ∙ 10-8 подействовать сильной кислотой, то аммиакат серебра
разрушается с образованием ионов Ag+ и NH4+, с константой для NH4+ = 6 ∙
10-10:
 .
.
В то же время комплексный ион
[Pt(NH3)4]2+, с константой нестойкости намного более низкой (5 ∙ 10-34),
чем у комплексного иона серебра, сохраняет стабильность при комнатной
температуре даже в концентрированной соляной кислоте.
По значениям констант диссоциации, с
использованием изотермы реакции, нетрудно рассчитать стандартную энергию
Гиббса: ΔGº
= RTlnКнест.,
которая является энергетической характеристикой процесса комплексообразования.
У ряда комплексных соединений
константа нестойкости чрезвычайно велика и в водных растворах они практически
полностью распадаются на составляющие их ионы. В первую очередь это относится к
соединениям, для которых более принятым считается название двойные соли:
K2[CuCl4] ≡ CuCl2 ∙ 2KCl. В воде равновесие 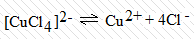 настолько
смещено вправо, что комплексных ионов [CuCl4]2- почти не остается. Содержание
их возрастает по мере концентрирования растоворов. Это означает, что чёткой
границы между комплексными и двойными солями, а также кристаллогидратами,
которые можно отнести к двойным солям (KCl MgCl2 ∙6H2O; (NH4)2SO4∙FeSO4∙6H2O
и др.), не существует. Двойная соль отличается от комплексной лишь степенью
диссоциации: у комплексной она незначительная, у двойной - практически полная.
настолько
смещено вправо, что комплексных ионов [CuCl4]2- почти не остается. Содержание
их возрастает по мере концентрирования растоворов. Это означает, что чёткой
границы между комплексными и двойными солями, а также кристаллогидратами,
которые можно отнести к двойным солям (KCl MgCl2 ∙6H2O; (NH4)2SO4∙FeSO4∙6H2O
и др.), не существует. Двойная соль отличается от комплексной лишь степенью
диссоциации: у комплексной она незначительная, у двойной - практически полная.
8. Реакции комплексообразования
Комплексные соединения образуются в
тех же химических реакциях, что и простые соединения первого порядка: в
реакциях соединения, обмена, замещения. Поскольку степени окисления атомов,
входящих в состав реагирующих веществ, в реакциях замещения меняются, их
принято называть реакциями окисления-восстановления.
Реакции соединения:
комплексный соединение
ион молекула
HgI2 + 2KI = K2[HgI4];
Zn(OH)2 +
2NaOH = Na2[Zn(OH)4];+ 4NH3 = [Cu(NH3)4]SO4;+ 5CO = [Fe(CO)5].
- обмена:
FeSO4 + 6KCN
= K4[Fe(CN)6] + K2SO4;+ (NH4)2[Hg(SCN)4] = Zn[Hg(SCN)4] + 2NH4Cl;+ 2Na2C2O4 =
Na2[Co(C2O4)2] + 2NaCl.
- окисления-восстановления:
Zn + 2[Ag(NH3)2]Cl = [Zn(NH3)4]Cl2 + 2Ag;
2K4[Fe(CN)6]
+ Cl2 = 2K3[Fe(CN)6] + 2KCl.
Вероятность комплексообразования простых молекул
в более сложные определяется возможностью формирования термодинамически более
устойчивых соединений:
9. Хелатирование. Его роль в
биологии и медицине
Хелатирование комплексообразователя лигандом,
когда лиганд предоставляет комплексообразователю два и более атома, образуя с
ним циклическую структуру, широко распространено в природе. Центральный атом
такого комплекса оказывается как бы втянутым внутрь лиганда (см. строение глицината
меди) и охвачен связями наподобие клешней рака. Отсюда их название хелатные
(chelate - клешня).
Би- и полидентатные лиганды связываются с ионом
комплексообразователя намного прочнее, чем монодентатные. Определяемая по
величинам констант нестойкости прочность хелатных этилендиаминовых комплексов
[M(En)2]n+ на 8-10 порядков выше, чем прочность комплексных ионов тех же
металлов с аммиаком [M(NH3)4]n+, хотя у тех и у других по четыре однотипных
связи M - N. Объясняется это взаимосвязанностью донорных атомов в пределах би-
и полидентатного комплекса. Чтобы удалить молекулу NH3 из комплекса
[M(NH3)4]n+, требуется разорвать только одну связь M - N. Для удаления
H2NCH2CH2NH2 из комплекса [M(En)2]n+ необходим разрыв уже двух связей,
образованных двумя атомами азота.
Повышенная прочность комплексных соединений с
би- и полидентатными лигандами называется хелатным эффектом. Хелатный эффект
полидентатных лигандов настолько велик, что они могут формировать устойчивые
комплексы даже с ионами щелочных и щелочно-земельных металлов. Хорошо известным
примером является хлорофилл - важнейший природный комплекс магния с порфином:
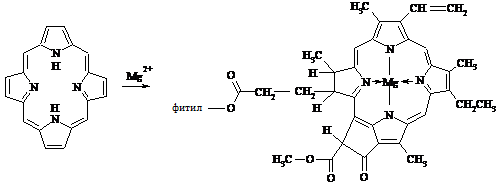
порфин хлорофилл
В молекуле порфина два протона,
связанные с азотом, легко замещаются на атом металла. Еще две связи, двух
оставшихся атомов азота с атомами магния, формируются по донорно-акцепторному
механизму.
Хлорофилл играет ключевую роль в
процессе фотосинтеза, являющемся самой масштабной химической реакцией на Земле.
Хотя при этом утилизируется не более 1% световой энергии, падающей на лист
растения, продукция фотосинтеза (1011 тонн органического вещества в год) в
сотни раз превышает продукцию всего нефтехимического производства, а запасаемая
в растениях энергия во столько же раз превышает энергию сжигаемого
человечеством топлива.
Примерно одинаковую структуру ядра
(порфиновое кольцо) имеет второй природный комплекс - гемоглобин. Разница лишь
в том, что у гемоглобина роль комплексообразователя выполняют катионы
двухвалентного железа. Координационное число ионов Mg2+ и Fe2+ равно 6, поэтому
по вакантным местам присоединяются ещё две молекулы иных веществ. Например, в
гемоглобине по одну сторону хелата присоединяется молекула белка глобина, а по
другую - молекула кислорода, благодаря чему это соединение является
переносчиком кислорода в крови:
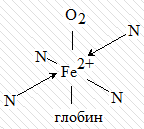
Роль металла в биокомплексах
высокоспецифична: замена его даже на близкий по физико-химическим параметрам
металл приводит к значительной или полной утрате физиологической активности комплекса.
В случае гемоглобина только кобальтовый аналог природного соединения обладает
незначительной способностью связывать и переносить кислород.
Из других значимых в биологическом
отношении комплексных соединений следует выделить витамин В12 и многие металлоферменты
(каталаза, переносчики электронов - цитохромы), дефицит которых у человека
приводит к серьезным заболеваниям.
Хелатный эффект дает ключ к
пониманию состояния катионов, особенно d-элементов, в организме. Все d-элементы
оказываются прочно связанными теми или иными полидентатными биолигандами, как
правило, аминокислотами, полипептидами или циклическими биолигандами -
производными порфина. В свободном виде, и то в виде аквакомплексов:
[Cu(H2O)4]2+, [Fe(H2O)6]3+ и др., эти катионы обнаруживаются лишь в плазме
крови, причем в ничтожных количествах.
Наблюдаемое при хелатировании
снижение констант нестойкости комплексных ионов обусловило использование
полидентатных лигандов (комплексонов) в аналитической химии, токсикологии,
гигиене и т.д. Часто это базируется на разрушении комплексоном менее
устойчивого соединения с переводом его в более прочное комплексное, имеющее
меньшую константу нестойкости. Примером может служить растворение осадка
Cu(OH)2 в NH4OH, при этом образуется комплексная соль:
Cu(OH)2↓ + 4NH4OH =
[Cu(NH3)4](OH)2 + 4H2O.
Связывание гемоглобина молекулами угарного газа
(СО) является аналогичным примером разрушения одного биокомплекса -
оксигемоглобина за счет формирования в 300 раз более прочного другого
биокомплекса - карбоксигемоглобина:
HbO2 + CO HbCO + O2.
Комплексоны так прочно связывают катионы
металлов, что при их добавлении растворяются такие плохо растворимые соли как
сульфаты кальция и бария, оксалаты и карбонаты кальция. Поэтому их применяют
для умягчения воды, для маскировки «лишних» ионов металла при крашении и в
изготовлении цветной плёнки. В химическом анализе явление комплексообразования
позволяет разделять столь близкие по свойствам элементы как лантаноиды и
актиноиды. Некоторые давно используемые комплексоны приведены в таблице 3.
Таблица 3 - Полидентатные
лиганды
|
Название
|
Химическая
формула
|
|
Этилендиаминтетрауксусная
кислота (ЭДТА)
|
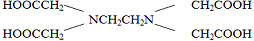
|
|
Диметилглиоксим
|
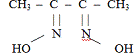
|
|
D-пеницилламин
|
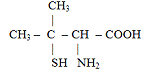
|
|
Дитизон
|
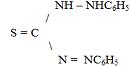
|
|
2,3
- димеркаптопропанол
|
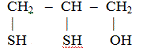
|
|
Унитиол
|
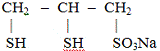
|
|
Аммонийная
соль нитрозофенилгидроксиламина (купферон)
|
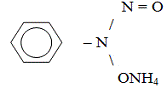
|
10. Хелатотерапия
В организме непрерывно происходит образование и
разрушение биокомплексов из катионов металлов и биолигандов (порфинов,
аминокислот, белков, полинуклеотидов), в состав которых входят донорные атомы
кислорода, азота, серы. Обмен с окружающей средой поддерживает концентрации
этих веществ на постоянном уровне, обеспечивая металло-лигандный гомеостаз.
Нарушение сложившегося равновесия ведет к ряду патологических явлений -
металлоизбыточным и металлодефицитным состояниям. В качестве примера можно
привести неполный перечень заболеваний, связанных с изменением
металло-лигандного баланса только для одного иона - катиона меди. Дефицит этого
элемента в организме вызывает синдром Менкеса, синдром Морфана, болезнь
Вильсона-Коновалова, цирроз печени, эмфизему лёгких, аорто- и артериопатии,
анемии. Избыточное поступление катиона может вести к серии заболеваний самых
разных органов: ревматизму, бронхиальной астме, воспалению почек и печени,
инфаркту миокарда и т.д., называемых гиперкупремиями. Известен и
профессиональный гиперкупреоз - медная лихорадка.
Негативные воздействия чаще всего свойственны
катионам d-металлов, что связано с устойчивостью образуемых ими биокомплексов.
Если константа нестойкости комплексного соединения с таким переходным металлом
ниже, чем константа нестойкости комплекса с каким-либо биокатионом организма,
при его поступлении идёт вытеснение последнего. Это видно из схемы:
 , где
, где
M1L - биокомплекс;- ион поступающего
d-металла.
В результате возможно накопление
чуждого организму комплекса M2L, обладающего токсичным действием. К примеру, из
данных таблицы 1.2 следует, что катионы ртути прочнее связываются цианид
ионами, чем катионы меди, вытесняя их, а также катионы марганца, железа и др.
из относительно нестабильных биокомплексов, что будет вести к токсикозному
состоянию.
Опасность представляет отравление
человека именно теми металлами, которых он в нормальных условиях не содержит
или содержит в незначительных количествах, поэтому поступление даже небольших
их доз приводит к заметному изменению концентрации таких компонентов. Природа
снабдила человеческий организм некоторыми средствами защиты от токсикозов.
Во-первых, ионы металлов, за исключением натрия, калия и кальция, лишь с трудом
попадают в организм через пищеварительный тракт и их поступление с пищей не
обязательно оканчивается отравлениями. Во-вторых, почки освобождают кровь от
металлов, не являющихся необходимыми для жизнедеятельности. Однако в ряде
случаев, в условиях возрастающей антропогенной активности человека, возникает
необходимость в фармакотерапии.
Выведение ионов тяжелых металлов из
организма под действием хелатирующих лигандов называется хелатотерапией.
Чтобы выполнить функцию детоксикантов, вводимые
комплексоны-лиганды должны отвечать ряду требований:
быть нетоксичными;
не подвергаться разложению или какому-либо
изменению в биологической среде;
эффективно связывать ионы-токсиканты, причем
вновь образующиеся соединения должны быть более прочными, чем те, которые
существовали в организме;
не разрушать жизненно необходимые комплексы:
гемоглобин, витамин В12, цитохромы и др.
Одним из первых лекарственных комплексонов,
используемых в таких целях, был 2,3-димеркаптопропанол (британский антилюизит;
табл. 1.3). Близок ему по структуре применяемый в России унитиол. Унитиол
эффективно выводит из организма мышьяк, ртуть, хром, висмут; препарат менее
перспективен при отравлениях свинцом.
С учётом приведенных требований наибольшее
распространение среди комплексонов получили различные соли
этилендиаминтетрауксусной кислоты (ЭДТА), из которых самой доступной является
динатриевая соль, известная как трилон Б. Применение трилона Б показано, к
примеру, при отравлениях соединениями кальция - СаО (негашеная известь),
Са(ОН)2 (гашеная известь), СаС2 (карбид кальция). При этом, связывая ионы
металла, он превращается в тетацин:
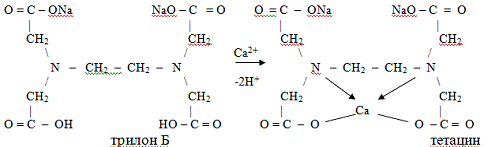
По возрастающей степени устойчивости
образующихся с ЭДТА комплексов металлы располагаются в следующем порядке: Sr,
Mg, Ca, Fe2+, Mn, Co, Zn, Cd, Pb, Cu, Hg, Ni. Это значит, что включая в себя
жизненно важные ионы кальция и не выводя их из организма, тетацин является эффективным
детоксикантом при отравлениях ионами, расположенными правее кальция, так как
катионы этих металлов, замещая центральный ион в комплексе, подвергаются более
прочному хелатированию. Получающийся в реакции тетацина с катионом свинца
комплекс Na2Pb-ЭДТА хорошо растворим в воде и легко удаляется из организма
через почки. Таким образом, тетацин оказался достаточно универсальным
реагентом.
В последнее время разработаны перспективные
хелатообразующие средства, позволяющие избирательно выводить из организма те
или иные ионы. Для удаления ионов железа используют, например, дефероксамин,
для удаления меди - при острых отравлениях и гепатоцеребральной дистрофии -
пеницилламин (табл. 3). Последний менее специфичен как лиганд: помимо ионов
меди, он связывает также ионы ртути, свинца, что усиливает его достоинства, но
также железа и кальция, что может вести к нежелательным последствиям из-за
снижения концентрации этих металлов.
Высокой степенью комплексообразования отличается
фитин - сложный органический препарат. Его получают из конопляных жмыхов. Фитин
полностью защищает животных, токсицированных смертельными дозами свинца. При
этом, в отличие от солей ЭДТА, фитин выводит яд преимущественно через
желудочно-кишечный тракт, а не через почки, снижая нагрузку на этот жизненно
важный орган.
Важную группу лекарственных препаратов
составляют комплексы, в которых ионы-комплексообразователи выполняют роль не
токсикантов, а лекарственных средств. К 2500 году до н.э. восходит история
применения в медицине золота, которое использовали в Китае для лечения проказы.
В настоящее время соединения этого металла употребляют в основном при лечении
ревматоидного артрита, когда наиболее активные в физиологическом отношении
атомы золота, в степени окисления +1, стабилизируются серосодержащими лигандами
гидролитических ферментов, что блокирует разрушающее действие их на суставы.
Инертные, устойчивые хелаты золота обладают
значительной антивирусной активностью. При этом они эффективны даже по
отношению к тем из микроорганизмов, которые нечувствительны к влиянию
антибиотиков. Существенно, что требующиеся для этого концентрации хелатов в
большинстве случаев практически нетоксичны. Поэтому хелаты золота, а также
цинка всё чаще используют для борьбы с вирусными инфекциями кожи и ран.
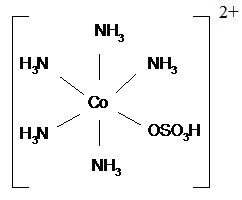
Токсический эффект некоторых комплексов
реализуется при создании противораковых препаратов. Для разрушения раковых
клеток особенно удачным оказалось применение соединений элементов восьмой
группы: никеля, палладия и платины. Анионные комплексы платины (IV) оказывают бактерицидное
действие (например, гексахлороплатинаты (IV)); нейтральные способны
приостанавливать деление, но не рост клеток (например,
цис-тетрахлородиамминплатина (IV)); цис-комплексы платины (II) обладают
противоопухолевыми и лизогенными свойствами (например,
цис-дихлородиамминплатина (II)). Использование некоторых из этих комплексов в
качестве противораковых препаратов приводило к снижению смертности, повышению
числа случаев полного излечения и давало иммунитет к опухолям, вызываемых
канцерогенами и вирусами. При совместном использовании таких комплексов с
другими лекарственными препаратами наблюдался синергический эффект, а вред,
наносимый клеткам нормальных тканей, оказался возместимым. Положительное
действие комплексных соединений в лечении раковых заболеваний, как правило,
вызвано блокированием ими участков ДНК, принимающих участие в передаче
генетической информации.
Список литературы
1.
Болтромеюк
В.В. «Общая химия. Физическая и коллоидная химия»
2011.