Современное состояние и перспективы использования двойных лекарств в качестве лекарственных средств
МИНИСТЕРСТВО ЗДРАВООХРАНЕНИЯ
РЕСПУБЛИКИ БЕЛАРУСЬ
Учреждение образования
БЕЛОРУССКИЙ ГОСУДАРСТВЕННЫЙ
МЕДИЦИНСКИЙ УНИВЕРСИТЕТ
Фармацевтический факультет
Кафедра фармацевтической технологии и
химии
Современное состояние и перспективы
использования двойных лекарств в качестве лекарственных средств
Липская Маргарита Анатольевна
Минск, 2014
Оглавление
Перечень условных обозначений и сокращений
Введение
. Идентичные и неидентичные двойные лекарства
. Гомодимеры и симметричные лиганды
.1 Симметрия в природе
.2 Гомодимеры как лиганды рецепторов
.3 Гомодимеры как ингибиторы ферментов
.4 Гомодимеры как лиганды ДНК
.5 Гомодимеры фармакологического интереса
. Гетеродимеры и лиганды двойного действия
.1 Гибридные молекулы как лиганды двух различных рецепторов
.2 Гибридные молекулы как ингибиторы ферментов
.3 Гибридные молекулы, действующие на один рецептор и фермент
.4 Другие примеры лекарств двойного действия
. Способ закрепления идентичных и неидентичных двойных
лекарств к ассиметричному центру связывания
.1 Идентичные и неидентичные двойные лекарства,
взаимодействующие с двумя смежными центрами связывания, расположенными на одной
макромолекуле
.1.1 Ингибиторы ацетилхолинэстеразы
.1.2 Бисубстратные ингибиторы
.2 Идентичные двойные лекарства, взаимодействующие с двумя
подобными сайтами связывания, расположенными на различных мономерах одной
макромолекулы
.2.1 Ингибиторы сиртуина
.2.2 Ингибиторы глутатион S-трансферазы
.2.3 Лиганды рецепторов, связанных с G-белком
.3 Идентичные и неидентичные двойные лекарства,
взаимодействующие с двумя различными сайтами связывания, расположенными на
различных макромолекулах
.3.1 Лиганды GPCR (гетеродимер)
Заключение
Список использованных источников
Перечень условных обозначений и сокращений
CОХ -
циклооксигеназа- рецепторы, сопряженные с G-белком
GST -
глутатион-S-трансфераза
LO -
липоксигеназа
PAF -
фактор активации тромбоцитов- рецепторы, активируемые пероксисомными
пролифератами
PR -
обратная транскриптаза
SAR -
анализ зависимости структура - активность
АПФ - ангиотензин превращающий фермент
АХЭ - ацетилхолинэстераза
ЛС - лекарственное средство
НЭП - нейтральная эндопептидаза
Введение
В стратегии дизайна препаратов прошлых 20 лет ведущим было традиционное
понятие: одна болезнь - одна мишень - один подход. Благодаря достижениям
биоорганической и бионеорганической химии, молекулярной биологии (особенно
благодаря установлению структур некоторых рецепторов и ферментов методом
рентгеноструктурного анализа), была идентифицирована структура биологических
мишеней, ответственных за болезнь, что привело к дизайну мощных и селективных
лигандов или ингибиторов. Но в большинстве случаев, в основе болезни лежат
многократные и сложные системы, где больше чем одна биологическая мишень,
которые должны быть смодулированы. Некоторые исследования показали, что
одновременное и умеренное блокирование или активация нескольких мишеней - более
действенное, чем использование селективных и мощных препаратов. Поэтому,
создание молекул, действующих на многочисленные биологические мишени, и
способных их модулировать, стало инновационным подходом в дизайне ЛС.
Совсем недавно в литературе появилось множество терминов таких как
“двойной, димерный, дуальный, гибрид, смешанный или многократный” соединенных с
терминами “лиганды, ингибиторы, активаторы, модуляторы или антагонисты”.
Лекарства, содержащие две фармакофорные группы, объединенные ковалентно в одну
молекулу, названы двойными лекарствами.
Двойные лекарства могут быть идентичными (представляют собой комбинацию
двух одинаковых составляющих) и неидентичными (имеющими в качестве составляющих
различные структуры). В частности, возможно конструирование сложных «бинарных»
структур, содержащих в своем составе несколько функционально значимых частей
молекулы. При конструировании двойных лекарств возможны самые различные
комбинации, значительно улучшающие активность и фармакокинетические свойства
ЛС. Например, если известен фермент, разрушающий ЛС в организме, то возможно
конструирование бинарной молекулы, содержащей в своей структуре как фрагмент этого
ЛС, так и фрагмент ингибитора данного фермента. При расщеплении данной молекулы
в организме ингибирование фермента приведет к пролонгированию действия данного
ЛС.
Целью данной курсовой работы является изучение подхода создания двойных
лекарств, преимуществ данного типа модификации в улучшении активности и
фармакокинетических свойств ЛС, оценка современного состояния, а также
перспектив дальнейшего использования двойных лекарств в качестве ЛС.
1. Идентичные и неидентичные двойные лекарства
Соединение двух одинаковых фармакологических объектов приведет к
образованию “идентичного двойного лекарства”, которое является эквивалентом
производного гомодимера. Соединение, где объединены в одну молекулу два
различных фармакологических объекта, называют “неидентичным двойным лекарством”
или гетеродимером. Первая стратегия конструирования - процесс
дублирования/димеризации активного или главного компонента. Цель этого подхода
- производство более мощного и/или более селективного препарата по сравнению с
единичным объектом. Вторая стратегия состоит из соединения двух различных
фармакофоров. В этом случае новое соединение будет обладать всеми
фармокологическими действиями (схема 1.1). Этот подход обладает большим
преимуществом, когда два целевых фермента или рецептора включены в одну болезнь
или расстройство. Лекарство гетеродимер окажет влияние, действуя одновременно
на две биологические мишени [1].
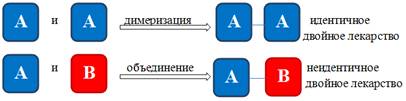
Схема 1.1 - идентичные и неидентичные двойные лекарства
Соединение идентичных или неидентичных фармакофоров можно
классифицировать по способам связи, используемым между двумя объектами.
Объединение может быть достигнуто посредством соединения или другим способом
(простая связь) или способом наложения. Группа связей может быть полимерной
цепью (обычно метиленовая цепь), ароматическим или гетероароматическим кольцом,
а в некоторых случаях неароматическим циклом. Фармакофоры могут быть перекрыты,
когда имеется общая структурная цепь (кольцо или химическая группа),
идентифицированная в двух различных лекарствах. Дублирование аспирина привело к
идентичному двойному лекарству диаспирину, соединенного без связей. Такрин,
ингибитор ацетилхолинэстеразы, был соединен с помощью связей (полиметиленовая
цепь), в результате чего был образован дитакрина. Салициловая кислота и
структура парацетамола перекрываются (способ наложения), образуя неидентичное
двойное лекарство ацетаминосалол (рисунок 1.1) [2].
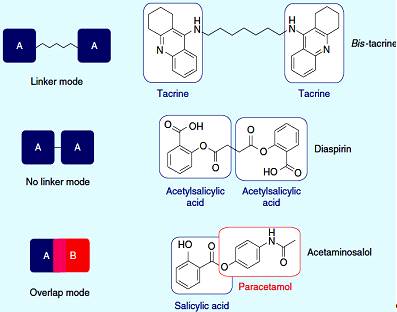
Рисунок 1.1 - Способы соединения двойных лекарств
Неидентичные двойные лекарства также называют лекарствами двойного
действия или гибридами из-за различного фармакологического результата, который
дают две представленные фармакофорные связи. Конструкцию лекарств двойного
действия, названную симбиотическим подходом, можно понять соответственно по
двум стратегиям (схема 1.2). Первая стратегия объединение двух отобранных
неидентичных фармакофоров в гибридную молекулу как иллюстрирование
сульфаниламидной производной. Ассоциативный синтез хлоробензасульфонамида с
производной индола через метиленовые связи приводит к образованию двойного β
- блокатора и
мочегонного агента. Вторая стратегия начинается с соединения-лидера, который,
как уже известно, оказывает оба действия. Рациональная оптимизация приведет к
лекарству двойного действия [1].
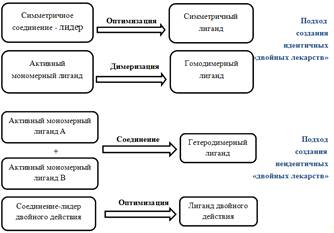
Схема 1.2 - Конструкция двойных лекарств
2. Гомодимеры и симметричные лиганды
Димеризация биологически активной молекулы является альтернативным
подходом в процессе оптимизации соединения-лидера. Обычно дублирование
фармакофора приводит к эквивалентной или более активной производной, которая
показывает различную селективность и фармакокинетические свойства.
Ингибирование фермента будет улучшено, используя гомодимеры ингибиторы.
.1 Симметрия в природе
Природа древний производитель структур с высокой степенью симметрии,
которая позволяет уменьшать информацию и сложность уровней. Естественная
симметрия наблюдается в макромолекулах (олигомеры) таких как ВИЧ, гемоглобин и
инсулин. Соединение мономеров и гексамеров инсулина в присутствии цинка,
приводит к образованию макромолекулярного комплекса с высокой степенью
симметрии (симметрия C3). ДНК, посредством симметричной двухцепочечной
структуры, определяет морфологию и функции клетки. Эти хорошо организованные
высокомолекулярные системы, представляют собой сайты связывания для небольших
молекул, включая воду и ионы. Естественные симметрические структуры обычно
представляют ось симметрии C2, например алкалоид лобеланин (для лечение
наркотической зависимости), спартеин (лечение болезни Грейвса) или
изохондодендрин, и антикоагулянты дикумарол и госсупол. Известны некоторые
примеры симметрических структур C3. Валиномицин, циклический пептидо-лактонный
антибиотик, селективный K - транспортер. Он состоит из циклического тримера,
содержащего l-валин, d - α-гидроксисовалериановую кислоту, d-валин и l - лактат
[3].
2.2 Гомодимеры как лиганды рецепторов
Идентичные двойные лекарства показали большую способность к
потенцированию и / или селективность к рецептору, как лиганды рецептора, по
сравнению с их составляющим единичным звеном. Несколько биогенных аминов
(катехоламины), четвертичные аммониевые соли (ацетилхолин), и пептиды
(ангиотензин, эндотелин) принадлежат к известному классу - двойных рецепторов
G-белка. Они представлены одной субъединицей с семью трансмембранных
сегментами, три внеклеточных и три внутриклеточных петли, которые соединены с
G-белками. Дублирование лекарств в пределах этой серии лигандов было эффективно
в нескольких случаях. Пиперидин - основы димерного лиганды были подготовлены, и
была оценена их способность блокировать транспорт дофамина (DA) и серотонина
(5-HT). Два единичных транс - пиперидина были связаны пентаметиленовой цепью в
гомодимерный лиганд. Процесс димеризации показал увеличение влияния на
деятельность транспортера DA (DAT) и по 5-HT транспортеру (SERT) [4]. Некоторые
селективные антагонисты β1 адренорецепторов были разработаны дублированием
известного окспренолола. Фармакофор фенолоксипропаноламин был димеризован с
помощью метиленовой цепи, для образования симметричного окспренолола. В
зависимости от длины связывающей цепи, могут быть получены селективные β2 или β1 антагонисты. Метоктрамин, селективный антагонист M2,
является полезным для характеристики рецепторов АХ мускаринового типа.
Селективность к М2 - рецепторам была улучшена, путем модификации
фрагмента метоктрамина. Замена 2-месоксибензильной группы, гидрофобным
фрагментом в пирензепине, приводит к образованию очень мощного антагониста M2,
тогда как пирензепин известен как селективный антагонист M1 -
рецепторов. Димеризация фармакофора пирензепина приводит к новому антагонисту с
различной селективностью. Недавно, исследования показали, что длина
полиметиленовой цепи производного метоктрамина полиамина является очень важной,
при преобразовании антагониста мускариновых рецепторов в селективный антагонист
никотиновых рецепторов. Таким образом, по общему образцу, были разработаны
симметрические двойные лекарства, действующие как антагонисты различных
рецепторов [5].
2.3 Гомодимеры как ингибиторы ферментов
Симметрическое расположение в ферментах гомодимеров или тетрамеров
определяет активный центр фермента симметричного вида. Таким образом,
симметричные ингибиторы будут соответствовать связывающему участку фермента.
Обратная транскриптаза ВИЧ (ВИЧ RT) и протеаза (PR ВИЧ) важны для созревания и
производства инфекционных вирусных частиц. Комбинированное ингибирование ВИЧ
транскриптазы и протеазы способно снизить вирусную нагрузку в крови пациентов
[6]. Ферменты, которые являются соответственно гетеро - и гомодимерами для ВИЧ
транскриптазы и протеазы, хорошо охарактеризованы: больше чем 170 структур
протеазы ВИЧ и ее комплексов с различными ингибиторами были решены методами
кристаллографии белка. Симметричная природа протеазы ВИЧ использовалась в
поиске новых лекарств против ВИЧ, которые воплотят предсказанную особенность
активного сайта. Разработка ингибиторов протеаз ВИЧ привела к симметричным
структурам, которые можно разделить на две группы: (a) псевдосимметричные
структуры, такие как производные А74,704 и L700,417, которые содержат
асимметричные атомы в непосредственной близости к ингибитору двойной оси; (b)
полностью C2-симметричные, как ингибиторы циклической мочевины и производные
диола (рисунок 2.1) [7].
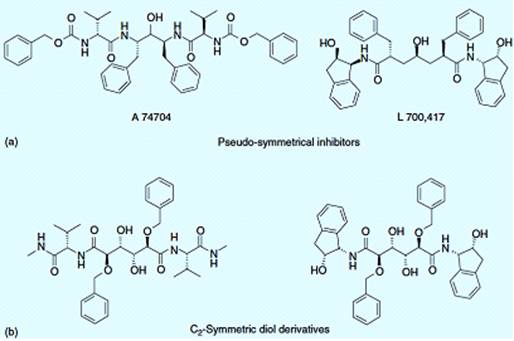
Рисунок 2.1 - Ингибиторы обратной транскриптазы и протеазы ВИЧ
2.4 Гомодимеры как лиганды ДНК
Молекула ДНК является основным объектом многих противоопухолевых агентов.
Маленькие молекулы, которые связываются с ДНК интеркалированием, требуют для
эффективного связывания полициклических систем в своей структуре. Из-за
симметрического расположения двухцепочечной спирали, симметрия найдена в
структуре лигандов ДНК. Пентамидин и диамидинобензимидазол (рисунок 2.2)
связываются в малой бороздке ДНК и показывают более высокое сродство к
областям, богатым основаниями. Соединения с четным числом метиленовых связок
бензольного кольца имидазола, имеют более высокое сродство к ДНК, чем те же с
нечетным числом метиленов. Моно-интеркалятором DACA, был димеризован с помощью
аминополиметиленовой цепи смешанный ингибитор топоизомеразы I/II с
цитостатической активностью на линии опухолевых клеток [8].
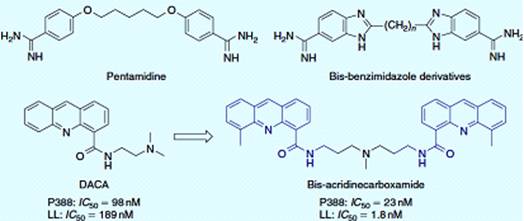
Рисунок 2.2 - ДНК лиганды
Поиск катионных холинергических агентов привел к многочисленным двойным
лекарствам. Димеры четвертичных аммониевых солей гексаметония и декаметония
являются мощными блокаторами в ганглиях и в нейромышечном синапсе
соответственно (рисунок 2.4). Другие нейромышечные блокаторы, такие как
сукцинил и себацил дихолин можно рассматривать как двойные лекарства
ацетилхолина.
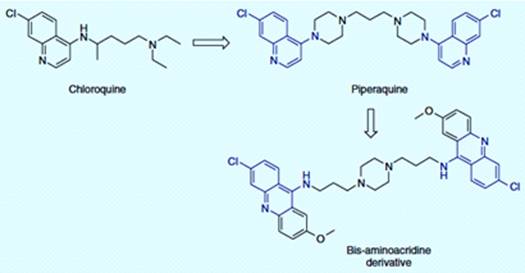
Рисунок 2.3 - Двойные лекарства аминоакридина
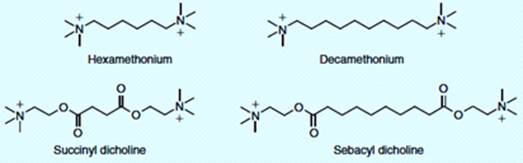
Рисунок 2.4 - Холинергические двойные лекарства
3. Гетеродимеры и лиганды двойного действия
Гетеродимеры и лекарства двойного действия оказывают свое двойное
действие на две различные биологические цели. Это могут быть рецепторы,
ферменты или их комбинация. Объединение двух физиологических эффектов
предназначено для получения синергический эффекта (возрастания эффективности) в
лечении болезни или расстройства. Гибридные молекулы можно получить в
результате объединения двух компонентов различной активности (ассоциативных
синтез) или из соединения с двойным действием. В первом случае, два фармакофора
связаны и идентифицированы, тогда как во втором случае часто возникает
трудность в определении химической части молекулы, которая обладает
биологической активностью. В последнем случае, конструкция лекарства двойного
действия часто основана на структурных модификациях одного из двух
фармакофоров, путем включения важных элементов другого фармакофора.
3.1 Гибридные молекулы как лиганды двух различных рецепторов
обладают физическими, биохимическими и структурными сходствами. Таким
образом, селективность к биогенным аминам, таким как норадреналин (NA), (5-HT)
серотонин, дофамин (DA) и гистамин (H) зависит от типичного взаимодействия и
дополнительных обязательных взаимодействий. Поскольку фармакофоры всех этих
лигандов подобны, контроль за их селективностью является важной проблемой химии
лекарств. Таким образом, представляет интерес синтез гибридных лекарств,
которые связывают различные участки GPCRs как агонисты, антагонисты или
агонист-антагонист.
В результате поиска антипсихотических средств без побочных эффектов (например,
экстрапирамидные побочные эффекты), антагонисты 5-HT2 рецепторов,
такие как ританзерин показали уменьшение побочных симптомов шизофрении. Таким
образом, было предложено, что комбинированное введение антагониста 5-HT2
и антагониста D2 рецепторов может быть эффективным в лечении
шизофрении. Исследование соединенного γ-карболина, показало что мощное
сродство к 5-HT2 и умеренное сродство к D2 рецепторам,
было достигнуто и привело к созданию соединения с равносильным и наномолярным
сродством к обоим рецепторам [10]. Эффективное антипсихотическое средство
Зипрасидон было также разработано из производного нафтилпиперазина с умеренным
антагонизмом к 5-HT2А и D2 рецепторам. Положительные
инотропные препараты, такие как армродипин были разработаны путем объединения в
одной молекуле антагониста гистамина H1 и агониста H2.
Объединение слабого частичного агониста H2 со слабым антагонистом H1
привело к образованию гетеродимера, который является очень мощным агонистом H2
(100-кратное увеличение) и антагонистом H1, в 10 раз более
активного, чем мономерный фармакофор.
Антигистаминный препарат Лоратадин является слабым антагонистом PAF.
Учитывая физиологическое значение PAF при астме, это представляет потенциальный
терапевтический интерес, чтобы противодействовать одной молекулой действию
обоих медиаторов. Была достигнута оптимизация этого лекарства двойного
действия, что привело к повышению антагонизма к PAF и подобному антагонизму к H1.
Тромбоксан А2 (TXA2) также является условием развития
астмы. Поэтому, были приложены усилия, к разработке антагонистов рецептора TXA2.
Концепция симбиотического подхода привела к недавнему открытию производных
дибензоксепина. Противоаллергическое средство и антагонист H1
KW-4994, также обладает слабым антагонизмом к TXA2. Успешные модификации
привели к созданию двойного антагониста TXA2 и H1, такого
как KF 15766 (рисунок 3.1) [11].
Октапептидный гормон ангиотензин II участвует в сокращении гладких мышц
кровеносных сосудов и в секреции других эндогенных веществ. Т. к. типы
рецепторов ангиотензина II (АТ1 и АТ2) находятся в
различных количествах во многих тканях и органах, двойные антагонисты могут
быть эффективными фармакологическими средствами.
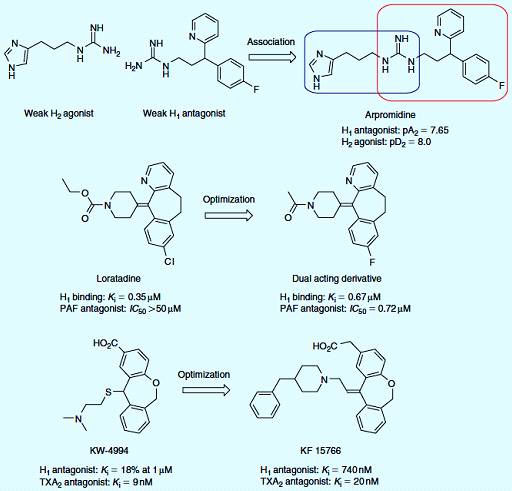
Рисунок 3.1 - Гистаминэргические гибридные лекарства
Начиная с лозартана, селективный антагонист АТ1, и PD 123317, селективный антагонист АТ2,
были разработаны двойные неселективные антагонисты АТ1 и АТ2
рецепторов. Структуры лозартана и PD 123317 были объединены с помощью общих структур (синим цветом), и новые
подмостки были оптимизированы традиционным исследованием SAR, которое привело к
производной ВIВS 222 [12]. Мощный и активный антагонист АТ1 L-159,093,
был модифицирован для увеличения сродства к АТ2 рецепторам. После
оптимизации, был получен мощный и умеренный антагонист АТ1/АТ2
L-159,689 (рисунок 3.2) [13].
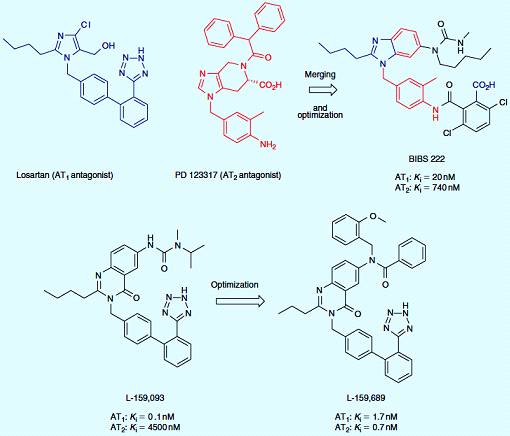
Рисунок 3.2 - Лиганды АТ1/АТ2 рецепторов
Недавние исследования на животных показали, что одновременная блокада АТ1
и АТ2 рецепторов должна быть довольно эффективной в лечении
гипертонии и сердечно-сосудистых заболеваний, таких как сердечная
недостаточность. Бифенилсульфонамид BMS-193884 разработанный как мощный
селективный антагонист ЕТА разделен той же дифенильной структурой
что и ирбесартан, антагонист АТ1. Путем слияния ключевых структурных
элементов этих двух производных, ученые Bristol-Myers Squibb получили гибрид,
который был оптимизирован, для получения производной DARA 7. DARA 7 показал
сбалансированную активность на АТ1 и ЕТА рецепторы и
падение повышенного кровяного давления у крыс (рисунок 3.3) [14].
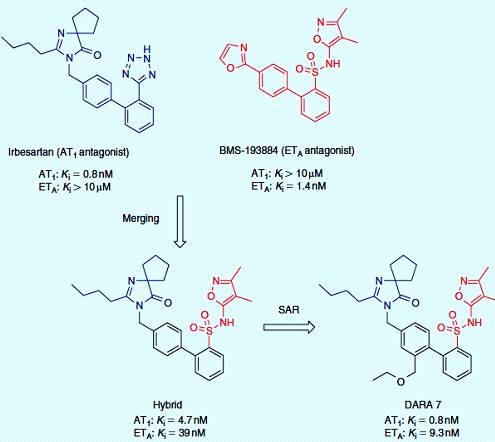
Рисунок 3.3 - Двойные лиганды ангиотензина и ЕТА рецепторов
Лекарства двойного действия также могут быть получены в результате
комбинации лигандов, принадлежащих к абсолютно различным фармакофорам.
Активация субстанции P (SP) и
аденозиновых рецепторов (А1) приводит к тому же эффекту (например,
гипотония и обезболивание), что представляет терапевтический интерес к
объединению в одном соединении этих свойств. Производная ксантина, антагонист
аденозиновых А1 рецепторов, был связан с пентапептидным концевым
фрагментом субстанции с получением конъюгата с подобным сродством к А1
и SP рецепторам (рисунок 3.4). Связывание было достигнуто посредством
аминокислот [15].
Рецепторы, активируемые пероксисомными пролифератами (PPARs) относятся к
ядерным рецепторам, которые включают рецепторы для стероидных, ретиноидых
гормонов и гормонов щитовидной железы.
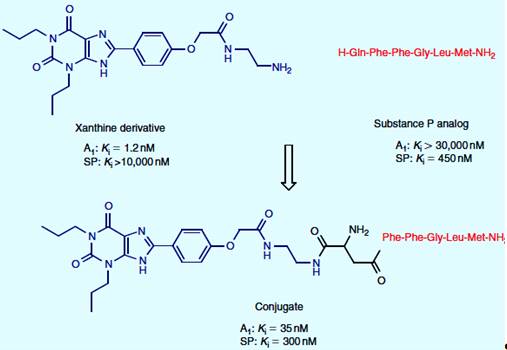
Рисунок 3.4 - Субстанция Р и гибридный лиганд аденозина
Демонстрация того, что PPARα и PPARγ рецепторы, которые являются
посредниками фибратов (гиполипидемическая активность) и глитазона (повышает
чувствительность к инсулину), привела к разработке нового поколения лекарств
двойного действия. Двойные антагонисты PPAR α/γ хорошо подходят для лечения
гипергликемии и профилактики сердечно-сосудистых заболеваний при сахарном
диабете 2 типа. Производные арилоксазола (рисунок 3.5) являются типичным
примером комбинации фенофибриновой кислоты и производной тиазолидинона
Фенофибриновая кислота, антагонист PPARα, была объединена с селективным PPARγ
производным
тиазолидинона; гибридное производное получено замещением фенильной группы, для
увеличения силы. Дальнейшие исследования SAR привели к разработке более мощного
антагониста двойного действия PPARα и PPARγ, такого как мураглитазар. С другой
стороны, оптимизация производных α - замещенной фенилпропановой кислоты
приводит к получению двойных агонистов PPAR α/PPAR δ
[16].
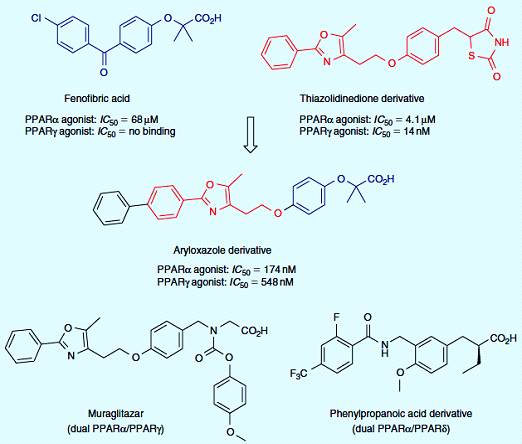
Рисунок 3.5 - Двойные агонисты PPAR
3.2 Гибридные молекулы как ингибиторы ферментов
Как и рецепторы, ферментативные системы можно разделить на классы
ферментов, и каждый тип ферментов представлен несколькими изоформами. Таким
образом, в фармакологических и терапевтических целях, это может представлять
интерес, для объединения в одной молекуле структурных ингибиторов двух
различных изоферментов, двух ферментов, принадлежащих к одному классу или двух
ферментов, ингибиторы которых обладают сходными фармакофорными свойствами.
Ингибиторы циклооксигеназы (СОХ-2) и 5-липооксигеназы (5-LO), ферменты,
участвующие в биосинтезе простагландинов и лейкотриенов, изучаются как
нестероидные противовоспалительные средства с повышенным уровнем безопасности.
Соединения двойного действия с двойным ингибирующим действием, используются для
лечения пациентов, страдающих от артрита и других заболеваний. Тиазолон CI-1004
(рисунок 3.6) был идентифицирован как аналогичный двойной ингибитор COX/5-LO.
По сравнению с соединением KME-4, этот препарат двойного действия
неульцерогенный, водорастворимый и при пероральном введении проявляет
противовоспалительные свойства. В последнее время, было достигнуто соединение
фармакофоров селективных ингибиторов СОХ-2 и 5-LO. Новое сопряженное производное
показало мощное ингибирование COX-2/5-LO и высокую селективность к СОХ-2 [17].
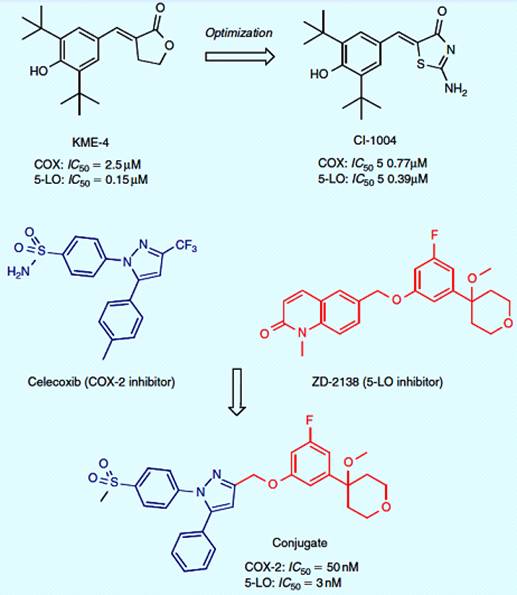
Рисунок 3.6 - Двойной ингибитор СОХ/5-LO
Инактивация эндогенного опиоидного пептида энкефалина является одной из
физиологических функций нейтральных эндопептидаз (НЭП). Было предположено, что
одновременное ингибирование ангиотензин превращающего фермента (АПФ) и НЭП
может быть выгодным для лечения застойной сердечной недостаточности или
гипертензии. Тиорфан, известный ингибитор НЭП, имеет двойные ингибирующие
АПФ/НЭП свойства, но как ингибитор НЭП он в сотни раз мощнее, чем как ингибитор
АПФ (рисунок 3.7). Жесткий бензазепинон был разработан как PheLeu миметик и
показал мощное двойное ингибирование НЭП/АПФ. Исследования в области изучения
ингибиторов двойного ряда привели к проектированию бициклического тиазепинона
омапатрилата. Это соединение показало равносильное двойное ингибирование
НЭП/АПФ и продемонстрировало хорошие результаты в снижении давления у животных.
Омапатрилат был разработан для лечения гипертонии и застойной сердечной
недостаточности [18].
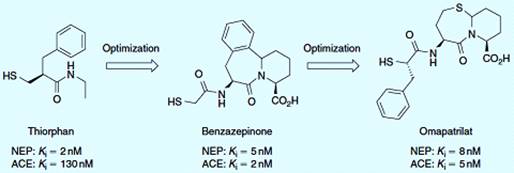
Рисунок 3.7 - Двойные ингибиторы НЭП/АПФ
Нуклеозидные ингибиторы обратной транскриптазы (NRTIs), такие как
зидовудин или зальцитабин показали высокую эффективность, когда они
используются в сочетании с ингибиторами PR ВИЧ. Из-за увеличения
неблагоприятных побочных эффектов, исследование было сосредоточено на
ненуклеозидных ингибиторах обратной транскриптазы (NNRTIs),
неконкурентоспособных ингибиторах сайта связывания. Комбинированная терапия
NRTI-NNRTI проявит синергическую активность и имеет большую эффективность.
Таким образом, соединения, содержащие как нуклеозидные, так и ненуклеозидные
ингибиторы (рисунок 3.8), были разработаны и показали микромолярную анти-ВИЧ
активность. Производная гетеродимер не показала синергетический эффект,
предполагается, что отдельные фармакофоры связываются не одновременно [19].
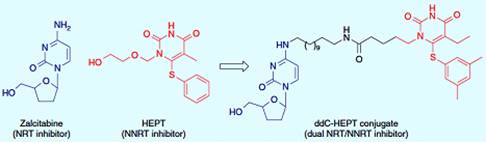
Рисунок 3.8 - Гетеродимер ингибитор ВИЧ
Назначение ингибиторов ацетилхолинэстеразы (АХЭ) представляет
перспективный подход для лечения болезни Альцгеймера, так как была
продемонстрирована связь между этим ферментом и образование амилоида. Мощный и
селективный ингибитор АХЭ был разработан в результате слияния двух различных
ингибиторов АХЭ гуперзина A и такрина (рисунок 3.9). Гибридная производная
показала улучшенную ингибирующую активность с умеренной селективностью. Лиганды
двойного взаимодействия с галантамином были получены при помощи различных
метиленовых связей из фрагмента фталимида и производной галантамина, ингибитора
активного центра. Биохимическое исследование кристаллографической структуры АХЭ
позволило четко идентифицировать два связывающих участка на ферменте: активный
центр, расположенный у основания глубокого и узкого ущелья, и периферический
сайт, расположенный при открытии ущелья. Комбинация фрагмента фталимида,
который, как известно, взаимодействует с периферическим сайтом, с производной
галантамина (ингибитор активного центра), привела к соединению, которое
связывается и с активным и с периферическим центрами [20].
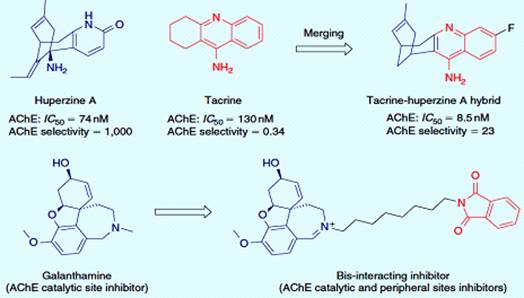
Рисунок 3.9 - Гибридный ингибитор АХЭ
В дополнение к дефициту холинергической нейротрансмиссии, наблюдаемом в
н. э. и коррелированном с когнитивными нарушениями функций, пациенты в наше
время также страдают от депрессии и беспокойства. Для лечения этих симптомов
используются ингибиторы транспортеров серотонина (SERT). Создание двойных
ингибиторов (АХЭ и SERT) было достигнуто, для получения лучшего
терапевтического эффекта. После гибридизации ривастигмина, ингибитора АХЭ, и
флуоксетина, мощного ингибитора SERT, был создан новый двойной ингибитор АХЭ и
SERT (рисунок 3.10). После исследований SAR, было получено соединение ((R) -
аналог) с мощным ингибирующим действием на оба фермента [21].
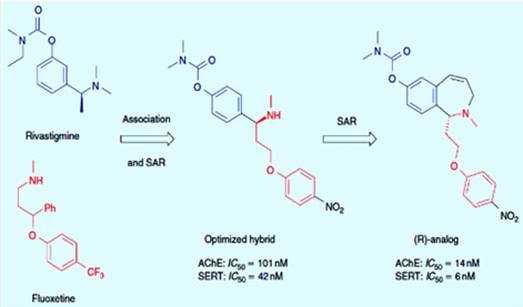
Рисунок 3.10 - Двойной ингибитор АХЭ/SERT
3.3 Гибридные молекулы, действующие на один рецептор и
фермент
Гибридные молекулы, действующие одновременно на рецептор и на фермент,
могут привести к мощным синергетическим эффектам. Рассмотрим на примере
производных взаимодействующих с TXA2, мощным индуктором агрегации
тромбоцитов и сокращения гладких мышц кровеносных сосудов. Ингибирование TXA2
синтазы (TxS) и селективная блокада TXA2 рецептора (TxR)
рассматривается в качестве альтернативной терапевтической стратегии по
предотвращению тромбического действия TXA2. Таким образом, ингибитор
TxS и антагонист TxR были объединены в одну молекулу, такие как самиксогрел,
ингибитор синтазы, и далтробан, антагонист TXA2 рецептора (рисунок
3.11). Поскольку самиксогрел показал умеренные плазменные уровни после
перорального приема (низкая растворимость в водном растворе), то он был
оптимизирован в производную гуанидина тербогрел, который проявляет более мощные
антитромбические эффекты in vivo [22].
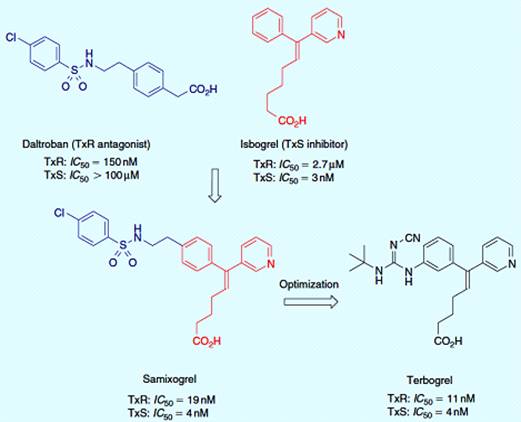
Рисунок 3.11 - Гибридное лекарство ТХА2
Основываясь на физиологической гипотезе, обе мишени могут принадлежать к
различным системам. Дигидропиридины, такие как нифедипин, как известно,
являются антагонистами кальциевых рецепторов кальция (Ca2+). Эти
препараты обычно используются для лечения пациентов с сердечно-сосудистыми
заболеваниями (гипертония, инфаркт миокарда). Сочетание антагониста Ca2+ и
ингибитора TxS может повысить терапевтическую эффективность для конкретных
патологий, где, и повышен синтез TXA2 и наблюдается клеточная
перегрузка кальцием. Имидазольный фрагмент дазоксибена, ингибитора TxS, был
слит с основной структурой нифедипина, что привело к образованию FEC 24265
(рисунок 3.12). Гибридная производная показала более благоприятный
фармакологический эффект in vivo. Фактор активации тромбоцитов (PAF)
участвует в процессе воспаления и связан с патологиями, такими как ишемия,
тромбоз и астма. Объединение антагониста PAF и ингибитора TxS обеспечило бы
благоприятное терапевтическое лечение. Ридогрел, мощный ингибитор TxS, был
непосредственно связан с антагонистом PAF Е-6123. Гетеродимерный лиганд показал
двойное равносильное сродство и ингибирующую способность к PAF и TxS,
выраженную активность после перорального приема [23].
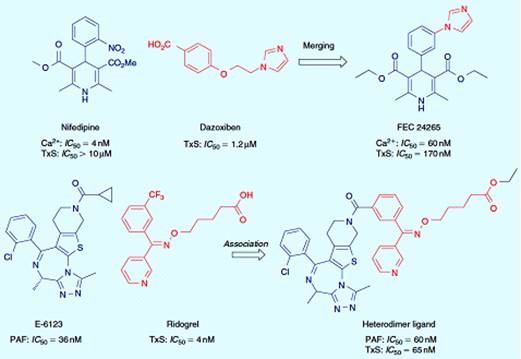
Рисунок 3.12 - Гибридные лекарства с синергическим эффектом
Новые антигипертензивные средства, обладающие β
- блокирующими и
ингибирующими ангиотензин превращающий фермент (АПФ) свойствами представляют
благоприятный подход для лечения повышенного кровяного давления. Гибридизация β
- блокатора пиндолола и
ингибитора АПФ энелаприла привела к производному BW-A575C, который обладает
двумя действиями (рисунок 3.13). Эти двойные лекарства, β
- блокатор/ингибитор
АПФ, терапевтически выгодны при лечении гипертонии и застойной сердечной
недостаточности [24].
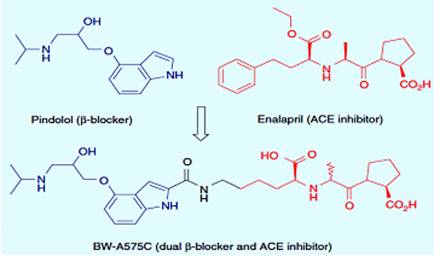
Рисунок 3.13 - Двойной ингибитор АПФ/β - блокатор
3.4 Другие примеры лекарств двойного действия
Комбинированное лечение необходимо при длительном лечении гипертонической
болезни. β - блокирующие и мочегонные свойства в одной молекуле
представляют большой интерес при гипертонии напряжения. Описано мало попыток
синтезировать гибридные молекулы, объединяя структуры антагонистов β-АР и мочегонных средств (рисунок
3.14). Гибридный сульфонамид был получен, путем связывания β
- блокатор производного
пропранолола с 2-хлорбензол сульфамидным фрагментом мефрузида [25].
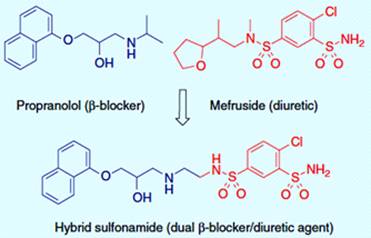
Рисунок 3.14 - Гипотензивные гибридные лекарства
Антибактериальные лекарства двойного действия были разработаны, связывая
хинолоны (рисунок 3.15), такие как ципрофлоксатин, в цефотаксим. Гибридный
лиганд был оптимизирован и продемонстрировал мощную активность против широкого
спектра грамположительных и грамотрицательных бактерий.
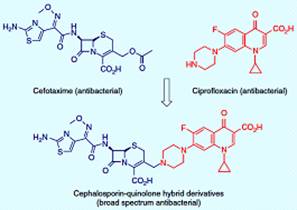
Рисунок 3.15 - Антибактериальные гибридные лекарства
В результате поиска новых эффективных антидепрессантов, были разработаны
лекарства двойного действия с селективным ингибированием серотониновых
рецепторов (SSR) и антагонизмом 5-HT1А рецептора (рисунок 3.16).
Комбинация ингибитора SSR дулоксетина и производных арилпиперазина, которые,
как известно, имеют высокое сродство к 5-HT1А рецепторам, привела к
образованию гибридов бензотиофенпиперазина, класса потенциальных
антидепрессантов с двойным механизмом действия [26].
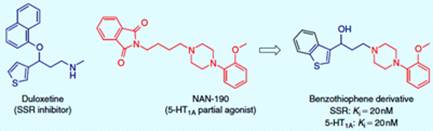
Рисунок 3.16 - Гибридный антидепрессант
4. Способ закрепления идентичных и неидентичных двойных лекарств к
ассиметричному центру связывания
.1 Идентичные и неидентичные двойные лекарства, взаимодействующие с двумя
смежными центрами связывания, расположенными на одной макромолекуле
.1.1 Ингибиторы ацетилхолинэстеразы
Трехмерная структура АХЭ из электрического органа ската (Torpedo
californica) показала, что активный центр расположен у основания глубокого и
узкого ущелья (20 Å), выровненный кольцами нескольких ароматических
аминокислот. На “анионном подместе” активного центра, моделированием было
предположено, что четвертичная аминогруппа ацетилхолина связывается с индольной
боковой цепью сохраненного остатка, Trp84, как было продемонстрировано в
дальнейшем для нескольких комплексов с АХЭ. Комплексы АХЭ с дичетвертичными
лигандами (дэкаментоний и BW284C51) привели к назначению Trp279 как основного
элемента второго сайта связывания, в верхней части ущелья активного центра,
названное “периферическим сайтом связывания, в 14 Å
от активного центра.
Эти структурные назначения были подтверждены большим числом биохимических
исследований, включая сайт направленный мутагенез, которые подтвердил важность
остатков ароматических аминокислот в АХЭ. Чтобы улучшить силу и селективность
препарата, была применена стратегия двойных лекарств к созданию ингибитора
двойного действия на центр АХЭ. Несколько идентичных димеров такрина, например
ди(5)-такрин и ди(7)-такрин, были синтезированы и оценены после того, как
вычислительные исследования показали слабое сродство такрина к остаткам периферического
сайта связывания (Trp279 и Tyr70) в сочетании с высоким сродством такрина к
анионному каталитическому сайту (Trp84 и Phe330).
Увеличенная ингибирующая активность двухвалентных молекул такрина была
пояснена закреплением к двойному центру. Определение кристаллических структур
ди(5) - такрина и ди(7)-такрина (рисунок 4.1) дало структурное основание для
наблюдения, что ди(7)-такрин - оптимальный ингибитор. Он формирует
благоприятные взаимодействия укладки типа сэндвича в обоих центрах с Trp84 (анионный
центр) и Trp279 (периферический сайт связывания) с минимальной перестройкой
белка. Ди(5)-такрин, который является менее мощным, имеет только одностороннее
благоприятное взаимодействие в периферическом сайте и вызывает конформационные
изменения в белковой цепи возле ацил-связывающего кармана. Структурные
изменения в ферменте, наблюдаемые в комплексе ди(5)-такрин/АХЭ показали, что
выбор оптимальной длины связывающей цепи оказывает существенное влияние на
ингибирующую активность.
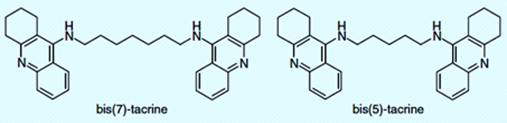
Рисунок 4.1 - Молекулярные структуры сокристаллизующихся ингибиторов АХЭ
Эти результаты подчеркивают проблему текущих, основанных на структуре,
подходов дизайна препарата, где не приняты во внимание представления о
структуре белка и конформационные изменения. Помимо такрина, также известны
другие ингибиторы “анионного центра” АХЭ, которые используются в разработке
двойных лекарств АХЭ. Развитие идентичных и неидентичных двойных лекарств
контролируется, состыковывая исследования, которые указали на сродство
гуперзина и такрина к каталитическим и периферическим сайтам связывания АХЭ.
Исследования моделирования также показали полезные гидрофобные эффекты,
переданные алкиленовой связывающей цепью периферическому центру лиганда.
Дальнейшее биохимическое исследование АХЭ показало, что периферический сайт
связывания в устье ущелья, участвовал в образовании β-амилоидного пептида, отвечающего за
нейродегенеративный процесс в наше время. Эта особенность АХЭ инициировала
разработку двойного ингибитора сайта связывания, в надежде на увеличение силы
ингибирования АХЭ и защиту нейронов от Аβ (amyloid beta) токсичности. Увеличение сродства к АХЭ двойных
(-)-галантамина, такрина, гипиридона, производных гуперзина, и гетеродимерных
лигандов такрин/пропидиум стало стимулом в разработке ингибиторов, связывающих
двойной сайт (рисунок 4.2). (-)-ди(10)-гупиридон проявляет ингибирующую
активность 2.4 нМ (Torpedo
californica АХЭ), которая является более чем в 200 раз выше по сравнению с
мономерным ингибитором АХЭ гуперзином A [27].
Рисунок 4.2 - Молекулярные структуры последовательно кристаллизующихся
ингибиторов АХЭ
4.1.2 Бисубстратные ингибиторы
Другая стратегия использует симметрические и асимметричные двухвалентные
лиганды, предназначенные для связывания в кофакторе и сайтах связывания
фермента, таким образом, оказывая конкурентное ингибирование. Этот тип лигандов
называют бисубстратными ингибиторами. Бисубстратная концепция привела к
разработке соединений с мощными лечебными свойствами. Мупироцин (псевдомониевая
кислота А) является фемптомолярным (10-15 моль) ингибитором
бактериальной изолейцил-тРНК-синтетазы и одним из наиболее широко используемых
антибиотиков. Были также обнаружены другие асимметричные бисубстратные
ингибиторы для GNC5 - связанной N-ацетилтрансферазы
и протеинкиназы.
Юнг и др. применили этот подход, для разработки ингибиторов для фермента
протеин - аргинин - метилтрансферазы (PRMT1). Был получен идентичный ингибитор
PRMT1, который блокирует оба сайта связывания, кофактор S-аденозилметионин
(SAM) и основание (гистон), одновременно (рисунок 4.3). Хотя сайт связывания
асимметричен, симметричный лиганд проявляет благоприятное взаимодействие как с
основанием (гистоном) так и с кофактором сайта связывания [28].
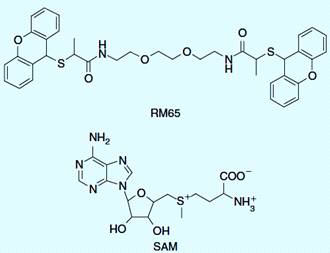
Рисунок 4.3 - Молекулярная структура бисубстратного ингибитора PRMT1 и SAM
.2 Идентичные двойные лекарства, взаимодействующие с двумя подобными
сайтами связывания, расположенными на различных мономерах одной макромолекулы
.2.1 Ингибиторы сиртуина- зависимые деацетилазы гистонов (сиртуины)
являются ферментами, которые расщепляют ацетильные группы остатков лизина в
гистонах, а также и в других белках. Реверсивное ацетилирование является важным
фактором в регулировании деятельности таких белков. Мощные селективные
ингибиторы сиртуина - интересные инструменты для исследования биологических
функций этих ферментов и могут в будущем использоваться в качестве лекарств для
лечения рака. Кристаллическая структура сиртуина подтипа Sirt5 в комплексе с
идентичным двойным лекарством сурамином показала, что два белковых мономера
связаны одной молекулой сурамина (рисунок 4.4). , который представляет собой в
растворе мономер, также был обнаружен в виде димера в растворе после
закрепления сурамина, что подтверждается гель - хроматографией. Мономер -
мономер взаимодействие в основном неполярное, нет прямых водородных связей
между двумя мономерами, таким образом, димерная структура Sirt5, главным
образом, стабилизирована самой связанной молекулой сурамина. И в
кристаллической структуре, и в растворе, сурамин действует как связывающее
звено, приводящее к димеризации Sirt5. Также было отмечено одновременное
закрепление одной молекулы сурамина на поверхностях двух мономеров в
кристаллической структуре комплекса сурамин - миотоксин II.
Для сиртуинов, однако, это важное открытие может привести к новому классу
ингибиторов, которые не только специфически связывают активный центр, но также
и выполняют функцию связывающего компонента, таким образом, ограничивая подвижность
и доступность фермента. Чтобы получить более мощные и селективные ингибиторы
сиртуина, была изменена структура сурамина. Было обнаружено, что полученные
производные являются мощными ингибиторами сиртуина, с высокой активностью к
подтипу Sirt1. Интересно, что была найдена не только двойная молекула сурамина,
чтобы блокировать Sirt1 (297 nМ),
но также соединение NF154 (рисунок 4.4) - которое напоминает половину структуры
сурамина - мощный ингибитор Sirt1 (525 nM). Исследования показали, что NF154
аналогично взаимодействует с связывающим карманом сиртуина. Однако перекрестное
связывание двух мономеров сиртуина не возможно (рисунок 4.5) [29].
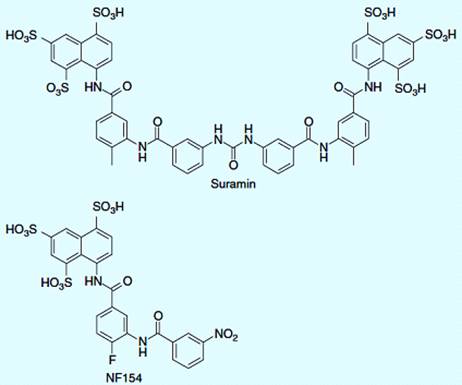
Рисунок 4.4 - Молекулярная структура сурамина и NF 154
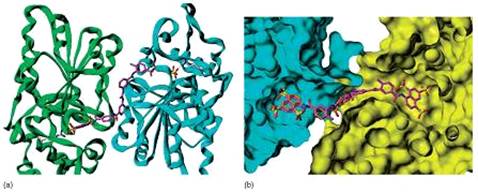
Рисунок 4.5 - (a)
перекрестное связывание двух мономеров Sirt5 (голубой и зеленый) с сурамином (пурпурный).
Оранжевым цветом показаны водородные связи; (b) Связывание сурамина и NF 154
4.2.2 Ингибиторы глутатион S-трансферазы
Глутатион S - трансферазы (GSTs)1 катализируют конъюгацию нуклеофильного
трипептида глутатиона (GSH, γ-Glu-Cys-Gly) в структурно различные гидрофобные
электрофилы. Среди электрофильных субстратов для GSTs алкилирующие агенты,
используемые в химиотерапии рака. GSTs, как известно, повышено продуцируются в
злокачественных тканях, предполагая, что они могут играть определенную роль в
приобретенной резистентности к противоопухолевым агентам. Поэтому, совместное
введение мощных, селективных ингибиторов GST как адъювантов в химиотерапии,
появилось в качестве возможной стратегии восстановления лекарственной
чувствительности резистентных клеток. Геометрия димера GST, с его двумя
идентичными активными центрами на противоположных концах, доступной для растворителя
межсубъединичной расщелины, представляет возможность, проектировать
симметрические двойные лекарства, которые связывают оба активных центра
одновременно.
Имеющаяся кристаллическая структура GST в комплексе с сульфатной
производной антрахинона (рисунок 4.6), использовалась в качестве основы для
разработки новых эффективных ингибиторов. Авторы использовали связанную
молекулу, производную Uniblue А (рисунок 4.7), для создания молекулы двойного
лекарства и чтобы проанализировать свободную энергию связывания [28]. Так как
авторы полагали о близости двух активных центров димера GST и их местоположении
на противоположных концах доступной для растворителя межсубъединичной
расщелина, то предполагалось, что могут быть разработаны двойные ингибиторы,
чтобы одновременно связывать оба активных центра. Соответствующая длина
связывающей цепи, для соединения обоих мономеров основана на исследованиях и
молекулярном моделировании. В то время как мономер производной Uniblue А
показал IC 50 - 5000 nM, двойное лекарство является более чем 100 раз более
активным (IC 50 - 44 nM).
Результаты исследований подтвердили повышенное сродство двойных ингибиторов по
сравнению с мономерными аналогами.
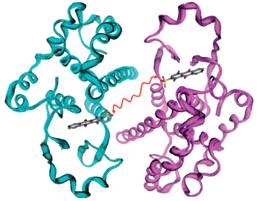
Рисунок 4.6 - Схема комплекса димера GST и сульфатной производной антрахинона. Связывающий
фрагмент между компонентами обозначен красным цветом
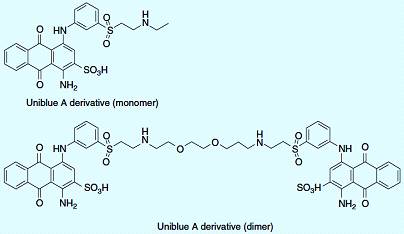
Рисунок 4.7 - Молекулярная структура ингибиторов GST
4.2.3 Лиганды рецепторов, связанных с G-белком
Рецепторы, сопряженные с G-белком (GPCRs) являются мембранными белками,
которые характеризуются общими семью трансмембранными сегментами. GPCRs играют
важную роль во многих биологических процессах. В последние десятилетия, все
больше появилось доказательств, что GPCRs, после активации, димеризуются в
активную форму, а затем оказывают свое биологическое действие. GPCRs находятся
в клеточной мембране или как гомодимеры или как гетеродимеры. Исследования,
показывающие, что гетеродимеризация GPCR может изменить фармакологию рецептора,
вызвали интерес к развитию лекарств, которые селективно нацелены на
гетеродимерные рецепторы. Один из подходов, предназначается для пары GPCRs,
должны быть синтезированы и использованы двойные молекулы, предназначенные для
двух центров связывания рецептора на гомо - или гетеродимере одновременно.
Обоснование использования двойного лекарства к GPCRs выходит из возможности
того, что двухвалентные лиганды способны к связыванию независимого рецептора на
димерном рецепторе, которое приводит к более благоприятным термодинамическим
взаимодействиям, чем связывание двумя одновалентными лигандами. Этот тип
взаимодействия схематично показан на рисунке 4.8.
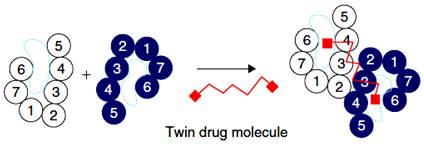
Рисунок 4.8 - Димеризация GPCR и
связывание молекулой двойного лекарства. Двухвалентный лиганд содержит два
обязательных независимых сайта GPCR
Главный прорыв в понимании класса GPCR был достигнут в 2000 году, когда
была открыта кристаллическая структура бычьего родопсина. Структура бычьего
родопсина использовалась различными группами для генерации гомологичных моделей
GPCRs. Эти модели использовались, в руководстве решений конструирования
двухвалентных лигандов, обращенных к двум сайтам связывания GPCR. Portoghese и
др. сообщили о несколько идентичных двойных молекулах с переменной длиной
связывающего фрагмента, предназначенных для исследования фармакодинамики и
особенностей организации опиоидных рецепторов. Подход двойных лекарств, как
было показано, применим и к другим GPCRs. До сих пор нет сведений о двойных
лигандах для рецепторов аденозина, дофамина, гонадотропин-рилизинг гормона,
мелатонина, мускарина, опиоидов, серотонина и вазопрессина [30].
.3 Идентичные и неидентичные двойные лекарства, взаимодействующие с двумя
различными сайтами связывания, расположенными на различных макромолекулах
Лиганды GPCR (гетеродимер). Недавно также было доказано, что многие
мономеры различных GPCRs, способны взаимодействовать друг с другом, в
результате чего формируются гетеродимеры. Все большее число исследований
указывают на роль гетеродимеризации GPCR в модуляции фармакологии рецептора и
предполагают, что гетеродимеры могут представлять собой функциональную единицу.
Таким образом, создание селективных двойных лигандов может быть перспективной
стратегией. Было опубликовано несколько примеров, где селективные
гетеродимерные лиганды, оказывали меньше побочных эффектов из-за большей
селективности. Portoghese и др. синтезировали серию двойных лигандов димерных
опиоидных рецепторов, состоящих из фармакофора антагониста μ
- рецептора и фармакофора
антагониста δ - рецептора, разделенных прокладками переменной длины.
Антиноцицептивная активность двухвалентных лигандов была выше, чем достигнутая
путем совместного введения индивидуальных лигандов опиоидных рецепторов.
Будущий анализ закрепления таких лигандов к гетеродимерам и гомодимерам,
основанный на структуре моделирования GPCR димеров, обеспечит понимание
молекулярных детерминант, необходимых для селективного размещения и/или
активации гетеродимеров [31].
Заключение
Лекарства, объединяющие два фармакофора в одной молекуле, описаны в
многочисленных областях лекарственной химии. Исторически, их структура
подвергалась модификации, и на сегодня рациональный дизайн гомо - и
гетеролигандов основан на знании структуры белка, который содержит сайты связывания.
Дизайн двойных лекарств более сложен, чем дизайн соединений с одним видом
активности. Наряду с множеством успешных примеров двойных лекарств, могут
существовать и некоторые недостатки:
· объединение двух фармакофоров в одной молекуле, может привести к
инертному составу. Для успеха подхода необходимо хорошее знание данных о SAR в пределах каждого фармакофора (типы
связей, гидрофильные и гидрофобные участки). Также важен выбор связывающего
фрагмента (природа, длина).
· гибрид показал ожидаемый фармакологический эффект, но попытка
не удалась, из-за не предсказанных фармакодинамических и токсикологических
побочных действий.
· должна быть тщательно проанализирована сбалансированность
силы лекарственных составляющих. Например, дизайн из агонистов/антагонистов
должен принимать во внимание, что лекарства с антагонистической активностью на
рецепторы должны быть в значительно меньших концентрациях, чем агонисты (нМ и
мкМ соответственно). Подобным образом, при разработке гибридов, состоящих из
лиганда рецептора и ингибитора фермента, должны учитываться кинетические
свойства рассматриваемого фермента.
Однако, из-за большей терапевтической выгоды, поиск селективных лекарств
заменен на дизайн двойных лекарств. В большинстве случаев, одновременное
воздействие идентичных двойных лекарств (симметрично связанных участков)
связано с увеличением силы воздействия. Димер также проявляет большую
селективность, по сравнению с начальным мономером. Объединение в одной молекуле
двух неидентичных фармакофоров приводит к новому соединению, обладающему
свойствами обоих составляющих.
Таким образом, дизайн двойных лекарств и использование их в качестве ЛС
является перспективным и многообещающим подходом в лечении сложных нарушений.
На сегодня уже описаны успешные попытки использования двойных лекарств в
сердечно-сосудистой области, особенно для лечения гипертонии, в исследованиях
ЦНС.
идентичный гомодимер гибридный лекарство
Список использованных источников
1. Contreras J.M., Bourguignon J.J. Identical and
non-identical twin drugs. In The Practice of Medicinal Chemistry (Wermuth, C.
G., Ed.). Academic Press: London, 2003, pp. 251 - 273.
. Homodimeric tacrine congeners as
acetylcholinesterase inhibitors / M.K. Hu [and others] // J. Med. Chem. 2002,
45, 2277 - 2282.
. Neupert-Laves, K., Dobber, M. The crystal structure
of a K+ complex of valinomycin. Helv. Chim. Acta. 1975, 58, 432 - 442.
. Pharmacological and behavioral analysis of the
effects of some bivalent ligand-based monoamine reuptake inhibitors / A.P.
Tamiz [and others] // J. Med. Chem. 2001, 44, 1615 - 1622.
. Structure-activity relationships of
methoctramine-related polyamines as muscular nicotinic receptor noncompetitive
antagonists. 2. Role of polymethylene chain lengths separating amine functions
and of substituents on the terminal nitrogen atoms / M. Rosini [and others] //
J. Med. Chem. 2002, 45, 1860 - 1878.
. De Clerq, E. Toward improved anti-HIV chemotherapy:
therapeutic strategies for intervention with HIV infections. J. Med. Chem.
1995, 38, 2491 - 2517.
. Design and synthesis of new potent C 2 -symmetric
HIV-1 protease inhibitors. Use of the l -mannaric acid as a peptidomimetic
scaffold / M. [and others] // J. Med. Chem. 1998, 41, 3782 - 3792.
. Structure, DNA minor groove binding, and base pair
specificity of alkyl- and aryl-linked bis(amidinobenzimidazoles) and
bis(amidinoindoles) / T.A. Fairley [and others] // J. Med. Chem. 1993, 36, 1746
- 1753.
. Antimalarial, antitrypanosomal, and antileishmanial
activities and cytotoxiciy of bis(9-amino-6-chloro-2-methoxyacridines):
influence of the linker / S. Girault [and others] // J. Med. Chem. 2000, 43,
2646 - 2654.
. Bridged γ -carbolines and
derivatives possessing selective and combined affinity for 5-HT 2 and D 2
receptors / R.E. Mewshaw [and others] // J. Med. Chem. 1993, 36, 1488 - 1495.
. Dibenz[b,e]oxepin derivatives: novel antiallergic
agents possessing thromboxane A 2 and histamine H 1 dual antagonizing activity
1/ E. Ohshima [and others] // J. Med. Chem. 1993, 36, 417 - 420.
. Characterization of BIBS 39 and BIBS 222: two new
non peptide angiotensine II receptor antagonists / J.C. Zhang [and others] //
Eur. J. Pharmacol. 1992, 218, 35 - 41.
. A potent, orally active balanced affinity
angiotensin II AT1 antagonist and AT2 binding inhibitor / S.E. de Laszlo [and
others] // J. Med. Chem. 1993, 36, 3207 - 3210.
. Discovery of N -isoxazolyl biphenylsulfonamides as
potent dual angiotensin II and endothelin A receptor antagonists / N. Murugesan
[and others] // J. Med. Chem. 2002, 45, 3829 - 3835.
. Binary drugs: conjugates of purines and a peptide
that bind to both adenosine and substance P / K.A. Jacobson [and others] // J.
Med. Chem.1987, 30, 1529 - 1532.
. The PPARs: from orphan receptors to drug discovery /
T.M. Willson [and others] // J. Med. Chem. 2000, 43, 527 - 550.
. Synthesis and activity of a new
methoxytetrahydropyran derivative as dual cyclooxygenase-2/5-lipoxygenase
inhibitor / S. Barbey [and others] // Bioorg. Med. Chem. Lett. 2002, 12, 779 -
782.
. Application of a conformationally restricted phe-leu
dipeptide mimetic to the design of a combined inhibitor of angiotensin
I-converting enzyme and neutral endopeptidase 24.11 / G.A. Flynn [and others]
// J. Med. Chem. 1993, 36, 2420 - 2423.
. Synthesis and evaluation of «AZT -HEPT»,
«AZT-pyridinone», and «ddC-HEPT» conjugates as inhibitors of HIV reverse
transcriptase / R. Pontikis [and others] // J. Med. Chem. 2000, 43, 1927 -
1939.
. Potent acetylcholinesterase inhibitors: design,
synthesis, and structure-activity relationships of bis-interacting ligands in
the galanthamine series / A. Mary [and others] // Bioorg. Med. Chem. 1998, 6,
1835 - 1850.
. Van der Schyf C.J., Geldenhuys W.J., Youdim M.B.
Multifunctional drugs with different CNS targets for neuropsychiatric
disorders. J. Neurochem. 2006, 99, 1033 - 1048.
. 6,6-Disubstituted hex-5-enoic acid derivatives as
combined thromboxane A 2 receptor antagonists and synthetase inhibitors / R.
Soyka [and others] // J. Med. Chem. 1994, 37, 26 - 39.
. Approach to dual-acting platelet activating factor
(PAF) receptor antagonist/thromboxane synthase inhibitor (TxSI) based on the
link of PAF antagonists and TxSIs / M. Fujita [and others] // Bioorg. Med.
Chem. Lett. 2002, 12, 341 - 344.
. BW A575C a chemically novel agent with angiotensin
converting enzyme inhibitor and beta-adrenoceptor-blocking properties / G.
Allan [and others] // Br. J. Pharmacol. 1987, 90, 609 - 615.
. Symbiotic approach to drug design:
N-[(4-Chloro-3-sulfamoylbenzamido) ethyl] propanolamine derivatives as
beta-adrenergic blocking agents with diuretic activity / V. Cecchetti [and
others] // Eur. J. Med. Chem. 1991, 26, 381 - 386.
. New 1-aryl-3-(4-arylpiperazin-1-yl)propane
derivatives, with dual action at 5-HT1A serotonin receptors and
serotonin transporter as a new class of antidepressants / J. Martinez-Esparza
[and others] // J. Med. Chem. 2001, 44, 418 - 428.
. Complexes of alkylene-linked tacrine dimers with
Torpedo californica acetylcholinesterase: Binding of Bis5-tacrine produces a
dramatic rearrangement in the active-site gorge / E.H. Rydberg [and others] //
J. Med. Chem. 2006, 49, 5491 - 5500.
. A novel arginine methyltransferase inhibitor with
cellular activity / A. Spannhoff [and others] // Bioorg. Med. Chem. Lett. 2007,
17, 4150 - 4153.
. Structural basis of inhibition of the human NAD+
-dependent deacetylase SIRT5 by suramin / A. Schuetz [and others] // Structure,
2007, 15, 377 - 389.
. Heterodimers of G protein-coupled receptors as novel
and distinct drug targets / R. Rozenfeld [and others] // Drug Discov. Today,
2006, 3, 437 - 443.
. Filizola M., Wang S.X., Weinstein H. Dynamic models
of G-protein coupled receptor dimers: indications of asymmetry in the rhodopsin
dimer from molecular dynamics simulations in a POPC bilayer. J. Comput. Aided
Mol. Des. 2006, 20, 405 - 416.