|
Препарат
|
Растворители и
пределы растворимости
|
|
Вода
|
Этанол
|
Метиленхлорид
|
|
1 г
|
1 мл Не
растворился
|
1 мл Не
растворился
|
1 мл Не
растоврился
|
|
100 мг
|
1 мл Не
растворился
|
1 мл Не
растворился
|
1 мл Не
растворился
|
|
100 мг
|
10 мл Не
растворился
|
10 мл
Практически растворился
|
10 мл
Практически растворился
|
|
10 мг
|
10 мл
Практически растворился
|
10 мл
растворился
|
10 мл
растворился
|
Делаем вывод о том что бромгексин очень мало растворим в воде
и мало растворим в этаноле и метиленхлориде.
3. Объясните значение показателя
прозрачность и степень мутности растворов. Проведите определение прозрачности
раствора сульфадиметоксина по методикам ГФ XII: «Раствор 0,4 г субстанции в 10
мл 1 М раствора натрия гидроксида должен быть прозрачным или выдерживать
сравнение с эталоном I»
Прозрачность и степень мутности - есть показатели
доброкачественности жидких лекарственных форм и растворителей.
Прозрачность раствора
Определяют путем сравнения раствора вещества с растворителем.
Испытание проводят при освещении электрической лампой матового стекла мощностью
40 Вт при вертикальном расположении пробирок (рис. 1).
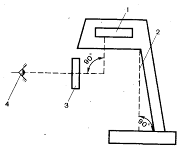
Рис. 1. 1 - источник света; 2 - экран; 3 - зона контроля; 4 -
глаза
Раствор считают прозрачным, если при его рассмотрении
невооруженным глазом не наблюдается присутствие нерастворенных частиц, кроме
единичных волокон. Сравнение проводят с растворителем, взятым для приготовления
жидкостей или эталонами мутности.
Эталонами для определения степени мутности служат взвеси из
гидразина сульфата и гексаметилентетрамина, приготовление которых подробно
описано в общей ФС 42-0051-07 «Определение прозрачности и степени мутности
жидкостей».
Определение степени мутности производят в компараторе.
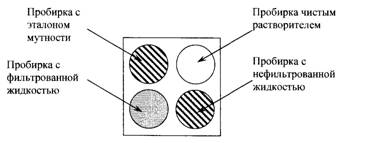
Часть испытуемого раствора в растворителе фильтруют через
бумажный фильтр; в компараторе помещают рядом пробирки с фильтрованным и
нефильтрованным раствором; позади пробирки с нефильтрованным раствором ставят
пробирку с с чистым растворителем, позади пробирки с фильтрованным раствором
помещают последовательно пробирки с соответствующими эталонами мутности до
появления мути, сходной с мутью нефильтрованного раствора. Пробирки
просматривают при подсвечивании электрической лампой 40 Вт.
Приготовление эталона мутности.
Эталонами служат взвеси из гидразина сульфата и
гексаметилентетрамина.
Приготовление раствора гидразина сульфата. 0,50 г. гидразина
сульфата помещают в мерную колбу вместимостью 50 мл, растворяют в 40 мл воды,
доводят объем раствора водой до метки и перемешивают. Раствор выдерживают в
течение 4-6 ч. Приготовление раствора гексаметилентетрамина. 3,00 г.
гексаметилентетрамина растворяют в 30,0 мл воды. Приготовление исходного
эталона. К 25,0 мл раствора гидразина сульфата прибавляют 25,0 мл раствора
гексаметилентетрамина, перемешивают и оставляют на 24 ч.
Приготовление основного эталона. 15,0 мл исходного эталона
помещают в мерную колбу вместимостью 1 л, доводят объем жидкости водой до метки
и перемешивают.
Приготовление эталона сравнения I. 5 мл основного эталона
помещают в мерную колбу вместимостью 100 мл, доводят объем жидкости водой до
метки и перемешивают.
Раствор 0,4 г сульфадиметокисна в 10 мл 1 М гидроксида натрия
и эталон помещают в две пробирки и сравнивают в проходящем свете. Если
прозрачность не меньше чем у эталонного раствора то делают вывод о том что
раствор выдерживает испытания.
4.
Объясните значение показателя цветность растворов. Проведите определение
цветности раствора азитромицина по методикам ГФ XII: «Раствор, полученный в
испытании на Прозрачность раствора, должен быть бесцветным или выдерживать
сравнение с эталоном B9»
Окраску жидкостей определяют визуально путем сравнения с
соответствующими эталонами. Цветность является условно принятой количественной
характеристикой для жидкостей, имеющих незначительную окраску. Цвет - это
восприятие или субъективная реакция наблюдателя на объективный раздражитель в
виде энергии, излучаемой в видимой части спектра и охватывающей диапазон длин
волн от 400 до 700 нм. Для визуальной оценки окраски жидкостей в зависимости от
интенсивности в области коричневых, желтых и красных цветов используют один из
двух методов, описанных в статье.
Цветность раствора
Для приготовления эталонных растворов цветности используют
четыре исходных раствора: А - 6% раствор хлорида кобальта (СоCl2 х
6Н2О); Б - 0,49% раствор дихромата калия (К2Сr2О7);
В - 6% раствор сульфата меди (II) (СuSО4 х 5Н2О); Г -
4,5% раствор хлорида железа (III) (FеСl3 х 6Н2О).
Смешивая исходные растворы в определенных соотношениях, указанных в ФС
«Определение окраски жидкости», получают 4 основных раствора, из которых путем
разбавления их раствором серной кислоты (0,1 моль/л) получают 28 растворов
цветности. Эталон В (коричневый)
Метод 1. Испытания проводят в одинаковых пробирках из
бесцветного, прозрачного, нейтрального стекла с внутренним диаметром около 12
мм, используя равные объемы - 2,0 мл раствора азитромицина и эталона сравнения.
Сравнивают окраску в дневном отраженном свете, горизонтально (перпендикулярно
оси пробирок) матово-белом фоне. Окраска расвтора диазепама не должна быть
интенсивнее указанного эталона.
По методу 2 используют такие же пробирки но сравнение окраски
проводят наблюдением 40 мм слоя раствора и эталона в дневном отраженном свете.
лекарственный дистилляция растворимость
5. Приведите характеристику определения воды
методом дистилляции по ГФ XI. Проведите математическое моделирование проведения
потери в массе при высушивании рибоксина, если масса бюкса составляет 19,9178
г. Методика определения по ГФ XII: «Около 1 г (точная навеска) субстанции сушат
при температуре от 100 до 105 ºС до постоянной массы.
Потеря в массе не должна превышать 1,0%»
Метод дистилляции основан на измерении объема воды,
отогнанной из испытуемого препарата. При этом к измельченному препарату (обычно
растительное сырье) добавляют органический растворитель и нагревают до кипения.
При совместном присутствии органического растворителя и воды, перегонка
происходит при температуре более низкой, чем у каждой из жидкостей. В качестве
органических растворителей рекомендованы ксилол и толуол. Погрешность метода
велика вследствие задержки капель воды на стенках холодильника и приемника, а
также за счет образования эмульсии в пограничном слое двух жидкостей. Для
повышения точности определений рекомендуется использование достаточно больших
масс препаратов - 10-20 г.
Потеря в массе при высушивании. Около 1,0 г (точная
навеска) рибоксина сушат при температуре 100-105
ºС до
постоянной массы. Потеря в массе не должна превышать 1%.
Проведем моделирование.
Подготовить бюкс: помыть, высушить, довести до постоянной
массы. Доведенным до постоянной массы считают в том случае, если последующее
взвешивание отличается от предыдущего не более чем на 0,0005 г.бюкса
19,9178 г.
. На весах взвешать около 1 г рибоксина.вещества+бюкса
= 21,0122 г.
. Сушат до постоянной массы. По истечении времени бюкс
достать и поместить в эксикатор для охлаждения. Взвесить.вещества+бюкса
= 21,0030 г.
. Рассчитать массу препарата до высушивания:
вещества= m вещества+бюкса - mбюкса =
21,0122-19,9178 = 1,0944 г.
. Рассчитать массу препарата после высушивания:
. Рассчитать процентное содержание воды:
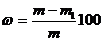 , (%)
, (%)
где m - масса лекарственного препарата до высушивания, m1
- масса после высушивания, ω - массовая доля воды в препарате, %.
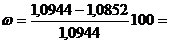 0,84%
0,84%
Соответствует требованиям.
6. Укажите, какие виды золы определяют по ГФ
XII. Промоделируйте определение общей золы для листьев брусники, если масса
тигля составляет 54,9898 г., масса навески - около 1 г
Определяют общую золу (ОФС 42-0055-07) и сульфатную золу (ОФС
42-0056-07) а так же золу растворимую в хлороводородной кислоте.
Проведем моделирование.
Содержание общей золы позволяет судить о минеральном
остатке, связанном с наличием неорганических веществ в растительном объекте, а
также с содержанием в нем примесей, попавших в сырье при сборе и сушке. При
определении в лекарственных формах, например, в таблетках, общая зола отражает
содержание талька, аэросила или двуокиси титана, используемых в качестве
наполнителей и вспомогательных веществ. Количество общей золы зависит от
специфики исследуемого сырья, фазы вегетации, времени и способа сбора, условий
сушки. Наиболее часто в состав общей золы входят K, Na, Mg, Ca, Fe, C, Si, P,
S, Cl в виде солей или оксидов, реже и в меньших количествах - Al, Cu, Mn и др
Определение сульфатной золы выявляет загрязненность
органических лекарственных веществ катионами металлов (Fe, Cu, Zn, Pb, Mn, As,
Cr и др.) Предварительная минерализация повышает чувствительность обнаружения
примесей катионов за счет относительного увеличения содержания примеси в
единице массы. В зависимости от условий прокаливания одни и те же вещества
могут образовывать различные по химическому составу остатки. Так, соли
органических кислот превращаются в карбонаты или оксиды. Галогениды, в
частности хлориды, могут частично улетучиваться. Оксиды некоторых металлов в
присутствии органических соединений могут восстанавливаться до свободных
элементов. Одновременно при минерализации разрушаются возможные связи катионов
примесей с анализируемым соединением, возникшие вследствие соле- и
комплексообразования, т.к. они часто образуют гораздо более прочные связи, чем
с реактивами, применяемыми для обнаружения примесей.
Фарфоровый тигель для сжигания прокаливают в муфельной печи
при 300оС до постоянной массы. Для предотвращения потери вещества
при сгорании, сжигание следует вести при минимальной температуре. При этом
лучше использовать тигель с крышкой.
Когда обуглившееся вещество в основном сгорит, тигель
помещают в муфельную печь и прокаливают в течение часа. После этого тигель
охлаждают, помещают в эксикатор над каким либо водоотнимающим средством на 30
мин.
Охлажденный тигель взвешивают на аналитических весах и
повторяют прокаливание в течение часа. Если при повторном взвешивании разница в
массе не превысит 0,0005 г., то считают, что достигнута постоянная масса и
прокаливание прекращают. При большем расхождении процесс повторяют.
Подготовить бюкс: помыть, высушить, довести до постоянной
массы. Доведенным до постоянной массы считают в том случае, если последующее
взвешивание отличается от предыдущего не более чем на 0,0005 г.бюкса
54,9898 г.
. На весах отвешать необходимое количество исследуемого
вещества.вещества+бюкса = 56,1000 г.
. По завершении процедуры тигль помещают в эксикатор для
охлаждения. Взвесить.вещества+бюкса = 55,1200 г.
. Рассчитать массу препарата до обработки:
вещества= m вещества+бюкса - mбюкса =
56,1000-54,9898 = 1,1102 г.
. Рассчитать массу золы после прокаливания:
золы= m зола+бюкса - mбюкса =
55,1200-54,9898 = 0,1302 г.
. Рассчитать процентное содержание золы:
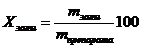 , (%)
, (%)
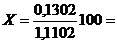 11,72%
11,72%
7.
Укажите общие замечания для испытания на чистоту и допустимые пределы
примесей по ГФ XII. В воде очищенной примеси хлоридов определяют по методике:
«10 мл воды не должны давать реакции на хлориды». Укажите последовательность
определения, химизм реакции, применяемые реактивы
Определение примесей в лекарственных средствах и оценку их
содержания проводят путем сравнения с эталонными растворами, устанавливающими
предел содержания данной примеси, после проведения реакции.
Общие замечания
. Вода и все реактивы должны быть свободны от ионов, на
содержание которых проводят испытания.
. Пробирки, в которых проводят наблюдения, должны быть
бесцветными и одинакового диаметра (около 1,5 см, если не указано иначе).
. Если не указано иначе, навески для приготовления эталонных
растворов отвешивают с точностью до 0,001 г.
. Наблюдения мути и опалесценции растворов проводят в
проходящем свете
на темном фоне, а окраски - по оси пробирок при дневном
отраженном свете на матово-белом фоне.
. Прибавление реактивов к испытуемому и эталонному растворам
должно проводиться одновременно и в одинаковых количествах.
. В случае, когда в соответствующей фармакопейной статье
указано, что в данной концентрации раствора не должно обнаруживаться той или
иной примеси, поступают следующим образом. К 10 мл испытуемого раствора
прибавляют применяемые для каждой реакции реактивы, указанные в методике, кроме
основного реактива, открывающего данную примесь. Затем раствор делят на две
равные части: к одной из них прибавляют основной реактив и оба раствора
сравнивают между собой. Между ними не должно быть заметной разницы.
Испытания основаны способности ионов серебра образовывать с
хлоридами, в зависимости от их содержания, белый творожистый осадок, белую муть
или опалесценцию, не исчезающие от прибавления азотной кислоты:
+ + Cl- → AgCl↓
Осадок хлорида серебра легко растворим в аммиаке. При этом
образуется бесцветный комплекс хлорида диаммиаката серебра:
+ 2NH3 → [Ag(NH3)2] Cl
Выполнение испытания
Для приготовления эталонного раствора хлорид-ионов, навеску
прокаленного хлорида натрия массой 0,0660 г. растворяют водой в мерной колбе на
100 мл и доводят раствор до метки (раствор А). 0,5 мл раствора А разбавляют
водой до 100 мл (раствор Б). Этот раствор является эталонным и содержит 2 мкг
иона С1- в 1 мл.
В качестве объекта испытаний выбрана вода очищенная. К 10 мл
испытуемой воды и одновременно к 10 мл эталонного раствора прибавляют по 0,5 мл
разведенной азотной кислоты и по 0,5 мл 0,02 М раствора нитрата серебра,
перемешивают и через 5 мин. сравнивают интенсивность появившейся опалесценции в
проходящем свете на темном фоне. При этом исследуемый и эталонный растворы
должны быть в одинаковых пробирках.
Вода очищенная не должна давать опалесценции или мути.
8.
В препарате магния сульфат примеси железа определяют 3 методом по методике:
«Раствор 1,5 г субстанции в 10 мл воды должен выдерживать испытание на железо
(не более 0,002% в субстанции)». Укажите последовательность определения, химизм
реакции, применяемые реактивы
Метод III. Испытание основано на способности тиоцианат-онов с
солями трехвалентного железа давать ярко-красные комплексы:
3+ + 6CNS- → [Fe(CNS)6]3-
Эталонный раствор содержит 3 мкг иона Fe3+ в 1 мл.
Готовится эталон растворением навески железоаммонийных квасцов. Для определения
примеси железа в солях магния к испытуемому раствору предварительно добавляют
0,5 мл раствора хлорида аммония.
Определение проводят следующим образом: к 10 мл раствора 1,5
г сульфата магния и 10 мл стандартного раствора, добавляют 2 мл раствора
хлористоводородной кислоты и 1,5 мл 15% раствора тиоцианата аммония и через 5
мин сравнивают.
Приготовление эталонного раствора соли железа (III):
Готовят 0,1% раствор железо(III) - иона, для чего
рассчитанное количество железоаммонийных квасцов растворяют в воде в мерной
колбе вместимостью 100 мл, добавляют 1 мл хлороводородной кислоты и доводят
объем раствора водой до метки (раствор А). 15 мл раствора А помещают в мерную
колбу вместимостью 500 мл и доводят объем раствора водой до метки (раствор Б).
10 мл раствора Б помещают в мерную колбу вместимостью 100 мл и доводят объем
раствора водой до метки (раствор В). Этот раствор содержит 0,003 мг (3 мкг)
ионов железа(III) в 1 мл.
Сравнивают 10 мл эталона В (30 мкг ионов железа, что
соответствует 0,002% от 1,5 г сульфата магния) с 10 мл раствора сульфата магния
после добавления реактивов. Интенсивность окраски не должна превышать
интенсивности окраски эталонного раствора.
9. Титрованные растворы, их назначение.
Требования к исходным веществам. Способы определения концентрации и поправочных
коэффициентов титрованных растворов. Опишите порядок приготовления, определения
концентрации и разбавления 200 мл титрованного раствора 0,1 М натрия
тиосульфата с поправочным коэффициентом 1,03 по требованиям ГФ XII
Титрованный раствор (титрант, рабочий
раствор) - это раствор с точно известной концентрацией. Готовят их либо
растворением точной навески вещества, либо из фиксаналов. Исходные вещества,
для приготовления титрованных растворов должны соответствовать следующим
требованиям:
• высокая чистота,
• строгая стехиометричность состава,
• устойчивость на воздухе,
• отсутствие гигроскопической влаги,
• большая молярная масса эквивалента,
• доступность,
• нетоксичность.
Для приготовленных титрованных растворов вычисляют
поправочный коэффициент (К) к молярности, представляющий собой отношение
реально полученной концентрации титрованного раствора к теоретически
заданной.
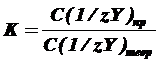
Коэффициент К должен находиться в пределах от 0,98 до 1,02. При
отклонении величины К от указанных пределов растворы необходимо соответственно
укрепить или разбавить, на основании следующих расчетов.
В случае разбавления раствора из величины К вычитают
единицу и полученную разность умножают на 1000. Результат умножения
соответствует количеству воды в мл, которое следует прибавить к каждому литру
разбавляемого раствора.
= (К-1) 1000
В случае укрепления из единицы вычитают К и разность
умножают на количество г исходного вещества, взятого для приготовления 1 л
раствора.
а= (1 - К) а
Полученное количество добавляют на каждый литр укрепляемого
раствора. После этого раствор тщательно перемешивают. При определении
поправочного коэффициента к титру производят не менее трех титрований.
Если результаты титрования отличаются друг от друга не более чем на 0,05 мл,
берут среднее арифметическое из полученных данных. Если же расхождения между
отдельными титрованиями превышают указанную величину, титрование необходимо
повторить до получения требуемого результата. Чтобы ошибка при титровании не
превышала ~0,1%, на титрование нужно расходовать не менее 20 - 30 мл раствора.
Приготовление раствора 0,1 М тиосульфата натрия. Молярная масса тиосульфата
натрия равна 158 г./моль. Для приготовления 200 мл 0,1 М раствора берут навеску
3,16 г. тиосульфата и растворяют в 200 мл воды.
Титруют 0,05М йода, используя 1 мл раствора крахмала,
прибавленного в конце титрования, как индикатор. 1 мл 0,05М йода соответствует
24,82 мг Na2S2O3.5H2O.
I2 + KI ® K[I3][I3]
+ 2Na2S2O3 ® KI + 2NaI + Na2S4O6
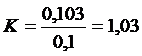
Требуется разбавление раствора V = (К-1) 1000 = 0,03*1000 =
30 мл воды сссследует добавить к 1000 мл раствора, так как взято 200 мл то
нужно добавить 6 мл воды.
10. Микробиологическая чистота. Значение
показателя. Характеристика методов определения. Примеры лекарственных средств,
для анализа которых используется данный метод
Нестерильные лекарственные средства (субстанции, различные
формы препаратов - таблетки, капсулы, гранулы, растворы, суспензии, сиропы,
мази, суппозитории и др., а также вспомогательные вещества) могут быть
контаминированы микроорганизмами. В них допускается наличие лимитированного
количества микроорганизмов, при отсутствии определенных видов бактерий,
представляющих опасность для здоровья человека.
Для количественного определения микроорганизмов используют методы:
. Чашечный агаровый метод
. Метод мембранной фильтрации
. Метод наиболее вероятностных чисел (НВЧ)
Чашечный агаровый метод.
Для количественного выявления бактерий образец в разведении
1:10 в количестве 1 мл вносят в 2 стерильные чашки Петри, заливают
расплавленным и остуженным до 40-45 0С мясо-пептонным агаром (МПА) в объёме
15-20 мл и быстро перемешивают вращательными движениями. Для выявления грибов
чашки заливают средой Сабуро. После застывания среды чашки помещают в термостат
на 5 суток: МПА при Т350С, среду Сабуро - Т20-250С. После окончания
сроков инкубации считают колонии, выросшие на поверхности питательных сред,
находят среднее арифметическое и умножают на степень разведения образца.
Полученный результат будет соответствовать количеству микроорганизмов в 1 г
(мл) исследуемого ЛС.
Метод мембранной фильтрации.
Данный метод используют для количественного определения
микроорганизмов в жидких, водорастворимых и жиросодержащих ЛС, растворимых в
изопропилмиристате, обладающих или не обладающих антимикробным действием. Для
посева используют мембранные фильтры с диаметром пор не более 0,45 мкм,
способные эффективно задерживать микроорганизмы.
Образец в разведении 1:10 в объёме 10 мл (что соответствует 1
г или 1 мл образца) наносят на фильтр. После фильтрации мембранный фильтр
переносят на чашки с соответствующей средой: МПА и агаром Сабуро. Если
исследуемый препарат обладает выраженным антимикробным действием или в его
состав входит консервант, то фильтр отмывают от лекарственного препарата
стерильным физиологическим раствором или специальными жидкостями, указанными в
фармакопейной статье. После окончания сроков инкубации (5 суток) производят
подсчёт колоний, выросших на фильтре, что будет соответствовать количеству
микроорганизмов в 1 г (мл) исследуемого ЛС.
Метод наиболее вероятных чисел (НВЧ).
Данный метод является менее чувствительным и точным. Его
используют, когда невозможно применить указанные выше методы.
Исследуемый образец готовят в виде раствора, суспензии или
эмульсии в разведениях: 1:10, 1:100 и 1:1000. Каждое разведение в объёме 1 мл
(что соответствует 100 мг, 10 мг и 1 мг образца) вносят в 3 пробирки с жидкой
питательной средой. Каждое разведение, таким образом, представляет ряд из 3-х
пробирок. Среды с посевами инкубирую при Т350С в течение 5 суток.
После окончания сроков инкубации в каждом ряду отмечают
количество пробирок в которых визуально отмечается рост микроорганизмов.
Испытания на микробиологическую чистоту проводят для многих
лекарственных средств, в основном для мазей, глазных капель, сиропов.
11. Метод нитритометрии и применение его в
фармацевтическом анализе. Способы установления конца титрования. Теоретические
основы метода. Преимущества и недостатки различных методов. Приведите примеры
определения лекарственных средств основного, кислотного характера, солей.
Напишите уравнения химических реакций, укажите факторы эквивалентности, титры
титрантов по определяемым веществам, расчетные формулы содержания
Метод основан на реакциях первичных и вторичных ароматических
аминов с нитритом натрия, который используют в качестве титранта.
Первичные ароматические амины образуют с нитритом натрия в
кислой среде диазосоединение:
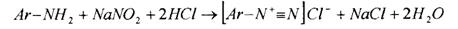
Вторичные ароматические амины в этих условиях образуют
N-нитрозосоединения:
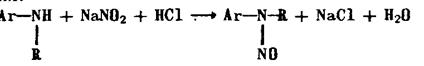
Эквивалентную точку устанавливают с помощью внешних
индикаторов (иодкрахмальная бумага); внутренних индикаторов (тропеолин 00,
нейтральный красный, смесь тропеолина 00 с метиленовым синим) или
потенциометрически. При потенциометрическом титровании индикаторным служит
платиновый электрод, а электродом сравнения хлор серебряный или насыщенный
каломельный.
Нитритометрическое титрование проводят, как правило, при 15 -
200С, в некоторых случаях анализируемый раствор охлаждают до 0 - 50
С. Кроме температуры на результаты нитритометрического титрования оказывают
влияние концентрация хлористоводородной кислоты, природа растворителя и
катализатора. В качестве последнего используют бромид калия. Он оказывает
каталитическое действие на скорость титрования препаратов, повышает четкость
перехода окраски внутренних индикаторов в точке эквивалентности и увеличивает
скачок потенциала на платиновом электроде при потенциометрическом способе
индикации. Нитритометрию применяют в фармацевтическом анализе для определения
сульфаниламидных препаратов (стрептоцид, норсульфазол, этазол, сульгин,
сульфацил-натрий и др.), производных п-аминобензойной кислоты (анестезин,
новокаин, новокаинамид), п-аминосалициловой кислоты (натрия п-аминосалицилат),
представляющих собой первичные ароматические амины, а также вторичные амины
(дикаин). Если первичная ароматическая аминогруппа ацилирована (парацетамол,
стрептоцид растворимый), то обычно вещество предварительно гидролизуют.
Ароматические нитропроизводные (левомицетин) вначале количественно
восстанавливают до первичных аминов, а затем титруют нитритом натрия.
Примеры определений
Количественное
определение парацетомола
Определяют по продукту кислотного гидролиза n-аминофенолу,
используя нитритометрический метод.
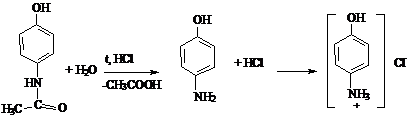
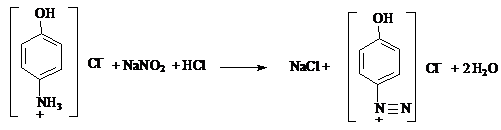
Эквивалентную точку устанавливают потенциометрически или с помощью
йодкрахмальной бумаги, которая синеет от выделившегося йода.
2
+HCI → NaCI +HNO2
KI +HCI → KCI +HI
HNO2 +2HI → I2 + 2NO↑ + 2H2O
Эквивалентную точку при нитритометрическом определении
парацетамола можно также установить со смешанным индикатором, содержащим
0,1%-ный раствор тропеолина 00 и 0,15%-ный раствор метиленового синего.
Фактор эквивалентности 1. Содержание действующего вещества
(массовая доля, %) в навеске или аликвоте ЛС (субстанции, препарата) при прямом
титровании:
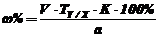
где, V - объем израсходованного титрованного
раствора NaNO2, мл; T Y/X - титр
соответствия, г/мл; К - поправочный коэффициент; а -
объем, мл или масса, г лекарственного средства, отобранные для проведения
анализа.
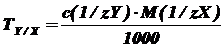
где, с (1/z Y) - молярная концентрация
эквивалента титранта, моль/л; M (1/z X) - молярная масса
эквивалента растворенного вещества, г/моль.
Т = 0,1*151 г/моль/1000 = 0,0151 г./мл (для
парацетамола)
Количественное определение сульфаниламидных
препаратов
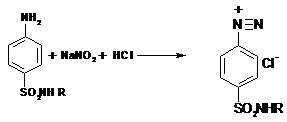
Точку эквивалентности устанавливают с помощью индикаторов:
тропеолин 00, нейтральный красный, смесь тропеолина 00 с метиленовым синим
(внутренние индикаторы), с помощью внешних индикаторов (иодкрахмальная бумага)
или потенциометрически. Этим методом определяют: сульфаниламид, сульфацетамида
натрий, сульфадиметоксин, сульфален.
Фактор эквивалентности 1. Т = 0,1*310 г/моль/1000
= 0,0310 г./мл (для сульфадиметоксина)
Количественное определение п-аминосалицилата
натрия
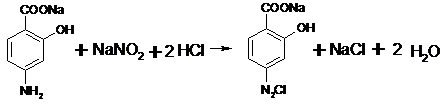
Фактор эквивалентности 1. Т = 0,1*175 г/моль/1000
= 0,0175 г./мл (для п-аминосалицилата натрия)
12. Приведите описание, методы определения
подлинности, чистоты и количественного определения натрия фторида
фармакопейными и нефармакопейными методами. Ответ проиллюстрируйте написанием
химических реакций и расчетных формул. Опишите особенности хранения и
применения лекарственного средства
Определение
подлинности
А. Даёт реакции на натрий.
1 мл раствора NaF, содержащей ион натрия подкисляют
разведенной уксусной кислотой, затем добавляют 0.5 мл раствора
цинк-уранилацетата; образуется желтый кристаллический осадок.
+ Zn(UO2)3(CH3COO)8
+ CH3COOH + 6H2O → NaZn(UO2)3(CH3COO)9.
6H2O↓ + HF
Cмоченный хлороводородной кислотой NaF внесенный в бесцветное
пламя, окрашивает его в желтый цвет.
Б. Даёт реакции на фториды:
К 2 мл раствора добавляют 0.5 мл раствора кальция хлорида.
Образуется белый желатиновый преципитат, который растворяется при добавлении 5
мл раствора железа хлорида.
+ CaCI2 ® CaF2 + 2NaCI-
+ Ca2+ ® CaF2¯
Приблизительно к 4 мг препарата добавляют смесь из 0.1 мл
ализарина С и 0.1 мл раствора циркония нитрата. Встряхивают. Цвет меняется от
красного к жёлтому.
Испытание
на чистоту (определение примесей)
Испытуемый раствор (С). Растворяют 2,5 г препарата в воде без
углекислоты при нагревании и доводят объем раствора до 100 мл тем же растворителем.
Прозрачность раствора. Раствор должен быть
прозрачным и бесцветным.
Хлориды. Разбавляют 10 мл раствора С до 15 мл водой.
Содержание хлорид-ионов не должно превышать 200 мкг/мл (ррm).
Количественное определение.
К 80 мг препарата добавляют смесь из 5 мл уксусного ангидрида
и 20 мл безводной уксусной кислоты и нагревают до растворения. Затем охлаждают
и добавляют 20 мл диоксана. Используя 0,1 мл раствора кристаллического
фиолетового, как индикатора, титруют 0.1М раствором хлорной кислоты до появления
зелёного окрашивания.
+ CH3COOH ® (CH3COONaH)+F-
CH3COOH + HCIO4® CH3COOH2)+CIO4
(CH3COONaH)+F- +
(CH3COOH2)+CIO4 ® 2CH3COOH +
NaCIO4 + HF
Лекарственного вещества в субстанции не менее 98.5% и не
более 100.5% процентов, в пересчёте на абсолютно сухое вещество.
Фактор эквивалентности 1. Содержание действующего вещества
(массовая доля, %) в навеске или аликвоте ЛС (субстанции, препарата) при прямом
титровании:
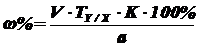
где, V - объем израсходованного титрованного
раствора NaNO2, мл; T Y/X - титр
соответствия, г/мл; К - поправочный коэффициент; а -
объем, мл или масса, г лекарственного средства, отобранные для проведения
анализа.
где, с (1/z Y) - молярная концентрация
эквивалента титранта, моль/л; M (1/z X) - молярная масса
эквивалента растворенного вещества, г/моль.
Т = 0,1*42 г/моль/1000 = 0,0042 г./мл (0.1М
хлорная кислота)
Применение: Фармакологическое действие - восполняющее
дефицит фтора, противокариозное, ингибирующее костную резорбцию.
Ионы фтора стабилизируют кальций в процессе минерализации,
замещая гидроксильную группу в кристаллах апатитов с образованием плохо
растворимого фторапатита (обусловливает плотность твердых тканей). Индуцирует
остеогенез путем стимуляции остеобластов. Уменьшает резорбцию кости, повышает
ее устойчивость к действию остеокластов, увеличивает костную массу.
Способствует пенетрации ионов фтора в эмаль зуба, стимулирует созревание и
обеспечивает прочность эмали, предупреждая развитие кариеса. Оказывает бактерицидное
действие в отношении кариесогенной микрофлоры, уменьшает продукцию ею кислот, в
частности молочной. Остеопороз: первичный (постменопаузальный, пресенильный,
сенильный, идиопатический), стероидный (профилактика и лечение); локальные
остеопатии, профилактика кариеса у детей и взрослых, при содержании в питьевой
воде фторидов ниже 0,6 мг/мл.
Хранение. Хранить в хорошо закрытой таре.
13. Приведите описание, методы определения
подлинности, чистоты и количественного определения железа фумарата фармакопейными
и нефармакопейными методами. Ответ проиллюстрируйте написанием химических
реакций и расчетных формул. Опишите особенности хранения и применения
лекарственного средства
Железа(II) фумарат (Ferrosi fumaras) Железа (II) бутендиоат

Железа
фумарат применяется для лечения железодефицитной анемии. Он представляет собой
порошок от красновато-оранжевого до красно-коричневого цвета, без запаха. Мало
растворим в воде, очень мало - в спирте.
Определение
подлинности
1. ИК-спектрофотометрия (определение фумарат - иона)
. ТСХ - хроматография в тонком слое
. Реакция с резорцином на фумарат-ион.
К препарату добавляют резорцин, серную кислоту и нагревают.
Образуется осадок темно-красного цвета. При разбавлении водой раствор
окрашивается в оранжево-желтый цвет с флюоресценцией
. Реакции на ион железа (II)
А. К 2 мл раствора добавляют 0.5 мл разведенной
хлороводородной кислоты и 1 мл раствора калия феррицианида; образуется синий
осадок.
FеСl2 + K3[Fe(CN)6]
® 2КСl + KFeFe(CN)6¯
или Fe2+ + 3K+ + [Fe(CN)6]3-
® 2K+ + KFeFe(CN)6¯
Б. К раствору добавляют раствор аммония сульфида; образуется
черный осадок, растворимый в разведенных минеральных кислотах.
2+ + S2- → FeS↓+ 2H+
→ Fe2+ + H2S↑
Оценка чистоты
Допустимыми примесями являются сульфаты, мышьяк, железо
(III), кадмий, хроматы, никель, цинк. Эти элементы определяют методом
атомно-абсорбционной спектрометрии.
Количественное определение
Цериметрический метод:
Титрант - сульфат церия (IV) - Се(SО4)2.
Индикатор - ферроин, происходит изменение окраски из
оранжевой в голубовато-зеленую. Титруют по железу:
2++Ce4+ = Fe3++Ce3+
Фактор эквивалентности 1. Содержание действующего вещества
(массовая доля, %) в навеске или аликвоте ЛС (субстанции, препарата) при прямом
титровании:
где, V - объем израсходованного титрованного
раствора NaNO2, мл; T Y/X - титр
соответствия, г/мл; К - поправочный коэффициент; а -
объем, мл или масса, г лекарственного средства, отобранные для проведения
анализа.
где, с (1/z Y) - молярная концентрация
эквивалента титранта, моль/л; M (1/z X) - молярная масса
эквивалента растворенного вещества, г/моль.
Т = 0,1*170 г/моль/1000 = 0,017 г./мл (0.1М
сульфат церия).
Применение: Фармакологическое действие - гемопоэтическое,
эритропоэтическое, противоанемическое.
Восполняет в организме дефицит железа, необходимого для
синтеза гемоглобина и других глобиновых ферментов.
Характеризуется высокой биодоступностью, поскольку
двухвалентное железо (закисное) легко абсорбируется, а фумаровая кислота
усиливает его всасывание. Cmax достигается через 4 ч после приема. T1/2 - около
12 ч. При железодефицитных анемиях прием 350 мг 1 раз в сутки обеспечивает
минимальную эффективную дозу железа. При этом повышается уровень гемоглобина (и
железа) в сыворотке крови, увеличивается число эритроцитов; все
гематологические и клинические симптомы анемии регрессируют через 3-4 нед
лечения. Железодефицитные анемии: постгеморрагические (меноррагии, хронические
кровопотери в ЖКТ и др.); повышенная потребность в железе (беременность,
лактация, период интенсивного роста и полового созревания, особенно у девочек)
- лечение и профилактика; недостаточное поступление железа с пищей или
нарушения его всасывания (хроническая диарея, глистная инвазия);
железодефицитная анемия у лиц пожилого и старческого возраста (в качестве
пробного лечения).
Хранение: в плотно закрытой ёмкости, в защищённом от света
месте.
14. Приведите описание, методы определения
подлинности, чистоты и количественного определения колларгола фармакопейными и
нефармакопейными методами. Ответ проиллюстрируйте написанием химических реакций
и расчетных формул. Опишите особенности хранения и применения лекарственного
средства
Колларгол представляет собой зеленовато-черные или
синевато-черные пластинки с металлическим блеском. Растворяется в воде с
образованием коллоидного раствора (1:50), который при разбавлении водой
(1:2000) имеет коричневый или красновато-бурый оттенок. Колларгол представляет
собой 70-75% золь серебра (Ag), в котором коллоидные частицы стабилизированы
гидролизатом казеина (основного белка молока всех млекопитающих).
Реакции
подлинности
1. После минерализации образца путем озоления остаток
обрабатывают кислотой азотной, при этом серебро, связанное с белком, переводят
в ионную форму. Далее проводят реакции на ион серебра:
+ 2HNO3 ® Ag NO3 + NO2 + H2O+
+ Cl- ® AgCl¯
белый
. Белок обнаруживают по специфическому запаху жженого рога
(жженой шерсти) при сгорании препарата.
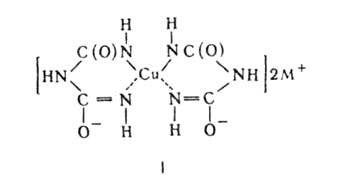
. Белок можно обнаружить биуретовой реакцией, которая впервые
была описана в 19 веке для простого соединения - биурета: NH2-CO-NH-CO-NH2.
После предварительного кислотного гидролиза препарата пептиды дают фиолетовые
комплексы при добавлении раствора CuSO4, например:
Оценка чистоты
Недопустимыми примесями являются посторонние соединения
серебра. Колларгол также не должен давать щелочную реакцию (по фенолфталеину),
и не должен содержать нерастворимых в воде веществ и продуктов разложения
белка, которые обнаруживают по возникновению запаха аммиака при нагревании
раствора с раствором натрия гидроксида.
Количественное определение
Сначала коллоидные препараты разрушают кипячением в смеси
концентрированной серной и азотной кислот в колбе КьельдаляЗатем образовавшиеся
ионы серебра определяют тиоцианатометрическим методом:
3 + NH4NCS ® AgNCS¯+NH4NO3
NH4NCS + FeNH4(SO4)2
® (NH4)3[Fe(NCS)6] + 2 (NH4)2SO4
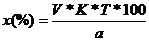
- объем 0,1 н раствора тиоцианата аммония (титрант), мл
К - поправочный коэффициент
Т - титр равный 0,0169 г./мл,
а - объем раствора, взятый для определения, мл
Фактор эквивалентности 1.
Применение: Колларгол используется как
антисептический препарат для лечения гнойных ран, бленнореи, мягкого шанкра,
конъюнктивитов, ринита, аденоидов, воспаления лимфатических узлов, а также
воспалений в мочевом пузыре.
Хранение. В хорошо укупоренных банках оранжевого стекла в
защищённом от света месте.
15. Приведите описание, методы определения
подлинности, чистоты и количественного определения гидроперита фармакопейными и
нефармакопейными методами. Ответ проиллюстрируйте написанием химических реакций
и расчетных формул. Опишите особенности хранения и применения лекарственного
средства
1. Образование надхромовой кислоты (ГФ): к 1 мл препарата
прибавляют 0,2 мл H2SO4 разведенной, 2 мл эфира, 0,2 мл
раствора K2Cr2O7 и взбалтывают, эфирный слой
окрашивается в синий цвет.
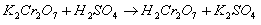
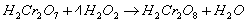
. Взаимодействие с иодидами и бромидами
калия или натрия в кислой среде (ГФ):
KI+H2O2+H2SO4
→ I2+K2SO4+2H2O
KBr+H2O2+H2SO4
→ Br2+K2SO4+2H2O
Выделяющиеся галогены обнаруживаются с
помощью крахмала или иод-крахмальной бумаги по посинению.
. Окисление сульфидов
+4H2O2 = PbSO4+4H2O
Черный сульфид свинца при этом
превращается в белый сульфат.
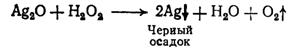
. Пероксид водорода окисляет Fe2+до
Fe3+, которые можно обнаружить по берлинской лазури:
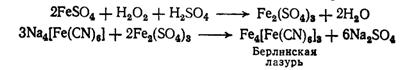
. В кислой среде пероксид водорода
восстанавливает перманганат калия в кислой среде (раствор обесцвечивается):
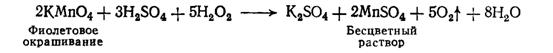
Испытание
на чистоту (определение примесей).
Кислотность. К 10 мл добавляют 20 мл воды и 0,25 мл
раствора метилового красного. Не менее чем 0,05 мл и не более чем 1 мл 0,1М
раствора натрия гидроксида требуется для изменения окраски индикатора.
Органические
стабилизаторы. Встряхивают 20 мл с 10 мл хлороформа и затем отбирают 2 аликвоты,
каждая по 5 мл. Выпаривают хлороформный слой при температуре не выше 250С
и высушивают остаток в эксикаторе. Остаточная масса не должна быть более 5 мг
(250 ppm).
Нелетучий остаток. Помещают 10 мл вещества
в платиновом тигле. Выпаривают раствор досуха на водяной бане и высушивают
остаток при температуре 1000С-1050С. Весовой остаток не
должен быть более 20 мг (2 г/л).
Количественное определение:
. Безындикаторная перманганатометрия: 10 мл раствора помещают
в мерную колбу на 100 мл и доводят водой до метки, к 10 мл полученного раствора
прибавляют 5 мл H2SO4 разведенной и титруют 0,1М KMnO4
до устойчивой розовой окраски:
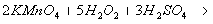
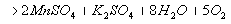
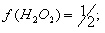
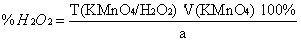
Т = ½*0,1*34/1000 = 0,0017 г./мл (для 0,1 М перманганата калия)
. Косвенная йодометрия:
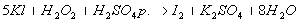
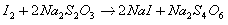
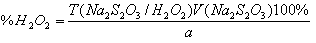
Т = ½*0,1*34/1000 = 0,0017 г./мл (для 0,1 М тиосульфата нтария)
Хранят в темном стекле исключая воздействие света, пыли, тепла,
так как в этих условиях возможно каталитическое разложение перекиси водорода.
Применяют в качестве наружного асептического средства и в составе полосканий.
16. Атомно-адсорбционная спектроскопия. Сущность
метода. Схема прибора. Применение в фармацевтическом анализе. Примеры
лекарственных средств, для анализа которых используется данный метод
Атомно-абсорбционная спектроскопия (ААС) -
спектроскопический метод анализа, основанный на измерении поглощения
электромагнитного излучения оптического диапазона невозбуждёнными свободными
атомами.
При поглощении атомом кванта электромагнитного излучения один
из его электронов переходит на более высокий энергетический уровень - атом
переходит из основного в возбуждённое состояние. Атом способен поглощать только
такое электромагнитное излучение, энергия которого точно равна разности между
энергией возбуждённого и основного состояния. Например, атом Na может поглощать
электромагнитное излучение с длинами волн 589,0 нм (3s →3p), 330,23 нм
(3s → 4p), 285,28 нм (3s → 5p) и некоторыми другими.
Принцип измерения аналитического сигнала в ААС такой же, как
и в других абсорбционных спектроскопических методах анализа. В ААС измеряют
относительную интенсивность двух потоков излучения, один из которых проходит
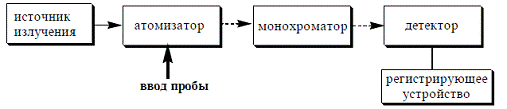
через атомный пар, а другой является потоком сравнения. Принципиальная схема
атомноабсорбционного спектрометра приведена на рисунке.
Используют метод в основном для определения примесей металлов
при испытаниях на чистоту.
Современная медицинская практика характеризуется широким
ассортиментом используемых лекарственных средств. Среди них значительное и
важное значение имеют металлсодержащие препараты, относящиеся к различным
фармакологическим группам. Их специфическая активность определяется прежде
всего наличием в структуре химического элемента (например железа и кобальта в
составе лекарственных средств, стимулирующих кроветворение, висмута - в составе
вяжущих средств с антисептическими свойствами и т.п.). По этой причине
фармакопейные предписания предусматривают определение отдельных элементов в
структуре лекарственных веществ.
При этом анализ металлсодержащих лекарственных средств,
основанный обычно на титриметрических методах недостаточно совершенен, длителен
при проведении отдельных операций, трудоемок, что особенно затрудняет
количественный анализ при малой дозировке вещества в лекарственных формах.
Вместе с тем, наиболее перспективный метод определения металлов -
атомно-абсорбционная спектроскопия, практически не используется в анализе
лекарственных средств отечественного ассортимента.
17. Спектрофотометрия в УФ- и видимой области
спектра. Сущность метода. Схема прибора. Применение в фармацевтическом анализе.
Примеры лекарственных средств, для анализа которых используется данный метод
Спектрофотометрия в УФ- и видимой областях - один из наиболее широко
используемых физико-химических методов в фармацевтическом анализе.
Анализируемые лекарственные вещества должны иметь в структуре молекулы
хромофорные группы (сопряженные связи, ароматическое ядро и др.), обуславливающие
различные электронные переходы в молекулах и поглощение электромагнитного
излучения.
Измерение спектров поглощения растворов анализируемых веществ
в УФ (190 380 нм) и видимой (380 - 780 нм) областях производят с помощью
спектрофотометров различных марок. В качестве растворителей используют
свободные от примесей воду, растворы кислот и щелочей, этанол, хлороформ и
другие органические растворители.
Идентификацию лекарственных веществ можно провести по
удельному показателю поглощения, характеру спектральных кривых в различных
растворителях, положению максимума и минимума светопоглощения или их отношению
при различных длинах волн. Для количественного спектрофотометрического анализа
важен выбор аналитической полосы поглощения. Последняя должна быть свободна от
наложения полос поглощения других компонентов смеси и иметь достаточно высокий
удельный показатель поглощения анализируемого вещества.
Принципиальная схема спектрофотометра показана на рисунке:
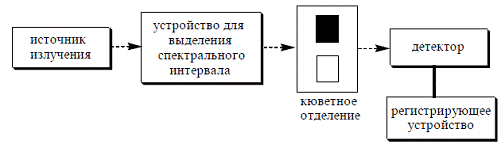
Применяется метод для количественного определения многих
лекарственных веществ согласно основному закону светопоглащения A = e*l*C. И
для идентификации сложных молекул ЛС, таких как алкалоиды, антибиотики.
18. Хроматографические методы анализа.
Классификация. Высокоэффективная жидкостная хроматография. Сущность метода.
Применение в фармацевтическом анализе. Примеры лекарственных средств, для
анализа которых используется данный метод
Хроматография - метод разделения и анализа смесей веществ, а
также изучения физико-химических свойств веществ. Основан на распределении
соединений между двумя фазами - неподвижной и подвижной (элюент).
Хроматографические методы применяют для разделения, очистки, идентификации и
количественного определения исследуемых соединений. Качественной
характеристикой во всех видах хроматографии является подвижность определяемого
вещества, которая зависит от его структуры и молекулярной массы, а также от
условий хроматографирования. Отношение пути перемещения вещества к пути
перемещения растворителя есть величина постоянная, обозначаемая Rf. Она
является константой для данных условий разделения и используется для
идентификации лекарственных веществ. Количественной характеристикой служит
интенсивность аналитического сигнала, который в различных методах различен:
например, размеры и интенсивность окраски пятна на пластинке, величина отклика
детектора и др.
Классификация видов хроматографии может быть различной в
зависимости от признака, положенного в основу.
I. По агрегатному состоянию фаз или по характеру
подвижной и неподвижной фазы
. Газо-жидкостная хроматография
. Газо-твёрдофазная или газо-адсорбционная хроматография 3.
Жидкостно-жидкостная хроматография
. Жидкостно-твёрдофазная хроматография
II. По механизму разделения компонентов на
молекулярном уровне. Например,
. Распределительная хроматография. В распределительной
хроматографии разделение происходит за счет разной растворимости веществ в
подвижной фазе (растворителе) и неподвижной фазе, находящейся на поверхности
неподвижного носителя.
. Ионообменная хроматография. Этот вариант хроматографии
позволяет разделять ионы и полярные молекулы, основываясь на ионных
взаимодействиях.
. Адсорбционная хроматография. Этот вид хроматографии основан
на адсорбции исследуемых веществ поверхностью твердой неподвижной фазы.
IIII. По технике выполнения
. Колоночная хроматография. Компоненты разделяют на
колонке-трубке, заполненной хроматографическим сорбентом.
. Капиллярная хроматография. Разновидность колоночной хроматографии;
для разделения используют трубку (капилляр), в которой слой сорбента расположен
только на внутренних стенках колонки, а центральная часть остается свободной.
. Плоскостная хроматография. Для разделения используют
плоский слой сорбента небольшой толщины. Плоскостная хроматография делится на
бумажную хроматографию и хроматографию в тонком слое сорбента. Для бумажной
хроматографии в качестве неподвижной фазы используют специальную
хроматографическую бумагу. В методе хроматографии в тонком слое сорбента
неподвижная фаза представляет собой тонкий слой сорбента (например,
силикагель), закрепленный на инертной подложке (алюминий, стекло и т.д.)
IV. По цели проведения
. Аналитическая хроматография. Целью этого метода является
собственно хроматографическое определение.
. Препаративная хроматография проводится с целью выделения
индивидуальных соединений из смеси в чистом виде.
ВЭЖХ отличается от ГЖХ тем, что подвижной фазой служит не
газ, а жидкость, причем она проходит через колонку, наполненную сорбентом, с
большой скоростью за счет значительного давления, Поэтому ВЭЖХ позволяет
разделять многокомпонентные смеси на индивидуальные вещества высокой степени
чистоты. ВЭЖХ отличается высокой чувствительностью. На разделение 10-15
компонентов затрачивается 20-30 мин. Жидкостный хроматограф включает такие
узлы, как дозатор, насос высокого давления, высокоэффективная колонка, детектор
с регистрирующим устройством. Колонки изготавливают из нержавеющей стали, они
имеют длину 10-25 см. Внутренний диаметр 0,3 - 0,8 см и плотно набиваются
адсорбентом с размером частиц 5 - 10 мкм. В качестве элюента используют
различные углеводороды в сочетании с этанолом. Детектором обычно служит
спектрофотометр с переменной длиной волны (190-900 нм), но существуют также
флуориметрические, электрохимические и другие детекторы.
Идентификацию испытуемых лекарственных веществ проводят по
времени выхода каждого компонента смеси из колонки, которое будет стабильно при
одинаковых условиях проведения эксперимента.
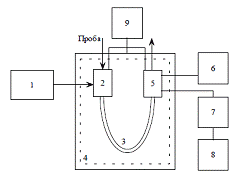
Принципиальная схема газового хроматографа:
- система подготовки газов;
- система дозирования;
- колонка;
- система термостатирования;
- система детектирования; \
- блок питания детектора;
- усилитель сигнала детектора; 8 - регистратор (самописец,
компьютер);
9 - измерители режима
хроматографа (расход газов, стабилизация температур и электрического питания
детекторов)
Благодаря развитию жидкостной тонкослойной и колоночной
хроматографии стало возможным определение свободных аминокислот и нуклеотидов в
биообъектах; исследователи получили инструмент для анализа состава белков и
нуклеиновых кислот, а клиницисты - «аминокислотную» и «нуклеотидную»
диагностику патологических состояний; стало возможным определение различного рода
лекарственных препаратов и их метаболитов в крови и тканях органов больных
экспериментальных животных, следовательно, фармакологи имеют возможность
изучать фармакокинетику, а клиницисты - контролировать эффективность лечения
Литература
1.
Государственная фармакопея РФ XII: Часть 1. - М.: Научный центр экспертизы
средств медицинского применения, 2008. - 704 с.
.
Фармацевтическая химия: учеб. пособие для вузов/ ред. А.П. Арзамасцев. М.:
«ГЭОТАР-Медиа», 2005. - 635 с.
.
Беликов В.Г. Фармацевтическая химия в 2-х частях / Учеб. для фарм. инст. и
фарм. факул. мед. инст. - Ч. 1. - М.: Высшая школа, 1993. - 432 с. Ч. 2. -
Пятигорск, 1996. - 608 с.
.
Руководство к лабораторным занятиям по фармацевтической химии: Учеб. Пособие.
ред. А.П. Арзамасцев. М:.:Медицина, 2004.
.
Лабораторные работы по фармацевтической химии: Учеб. пособие для фарм. инст. и
фарм. факул. мед. инст. / Беликов В.Г., Вергейчик Е.Н., Годяцкий В.Е. и др. -
М.: Высшая школа, 1989. - 375 с.
.
Фармацевтическая химия: руководство к практическим занятиям для студентов,
обучающихся по специальности 060108 фармация / Е.С. Гагарина, А.В. Озерская,
Н.В. Кувачева и др. - Красноярск: типография КрасГМУ, 2009. - 176 с.
.
Дудко В.В., Тихонова Л.А. Анализ лекарственных веществ по функциональным
группам. - Томск, 2004. - 137 с.