Синтез (-)-каиновой кислоты
Белорусский
государственный университет
Химический
факультет
Курсовая
работа
по теме
«Синтез
(-)-каиновой кислоты»
Выполнила студентка
курса 3 группы Демидова Д.Д.
Преподаватель Шклярук Д. Г.
МИНСК 2013
Список сокращений
1. LiHMDS - оксаметилдисилазан
лития.
2. KHMDS
- оксаметилдисилазан калия.
3. NaHMDS
- оксаметилдисилазан натрия.
4. DEPC
- диэтилпирокарбонат.
5. DPPA
- дифенилфосфоразид.
6. Cbz
- карбобензоксильная защитная группа.
7. Boc
- трет-бутилоксикарбонильная защитная группа.
8. Boc2O
- ди-трет-бутилдикарбонат.
9. DBU
- диазабициклоундецен.
10. THF
- тетрагидрофуран.
12. TMSCl
- триметилсилилхлорид.
13. TFA
- трифторуксусная кислота.
14. DMAP
- диметиламинопиридин.
15. DMF
- диметилформамид.
16. DMS
- диметилсульфид.
17. PhNTf2
- N-фенил-бис (трифторметансульфонимид).
Введение
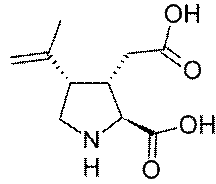
Каиновая кислота является прирордным
пирролидином с функциональными заместителями при С2, С3, С4 в цис и транс
положениях. Эта кислота впервые была получена из морской водоросли с японским
названием «Каинин-соу» (Digenea simplex). Первоначально
каинат употреблялся как глистогонное средство. В настоящее время используется в
нейронаучных экспериментах для исследования нейродегенеративных процессов,
моделирования эпилепсии, болезни Альцгеймера. В 1999 году была прекращена коммерческая
добыча каината из вышеуказанной водоросли. В связи с этим, химики начали искать
синтетические способы получения этого, на первый взгляд простого, продукта. И
даже сейчас, синтез каиновой кислоты является затратным и состоит из
значительного количества стадий. Далее будут рассмотрены некоторые способы
синтеза каиновой кислоты.
Методы синтеза
эпоксид азетидинон карвон синтез
Синтез из (+)-карвона
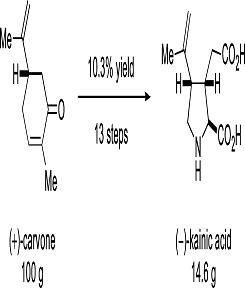
Данный синтез основан на использовании
2-пропенильной группы у природных терпенов (Схема 1).
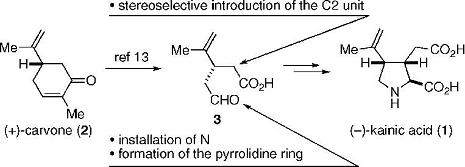
Схема 1: план синтеза (-)-каиновой кислоты
(+)-Карвон является представителем терпенов, и,
как известно, подвергается окислительной деструкции, что, в итоге, даёт
универсальный синтетический промежуточный продукт(3). Предположили, что
продукт(3) имеет пригодные функциональные возможности для стереоселективного
введения заместителя по положению С2, внедрения атома азота, а также для
формирования кольца пирролидина.
Этот синтез начинается с эпоксидирования
(+)-карвона(2), с использованием NaOH(в.)
для получения эпоксида(4) с 89% выходом (Схема 2).
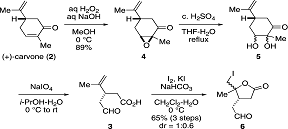
Схема 2: синтез йодолактона(6)
После того как был разработан эффективный способ
получения йодолактона(6), было уделено большее внимание внедрению атома азота,
с последующим стереоселективным алкилированием (Схема 3).
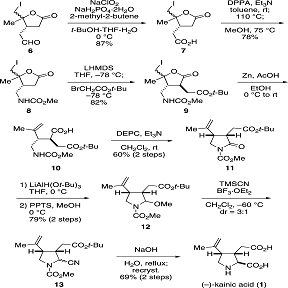
Схема 3: полный синтез (-)-каиновой кислоты(1)
Окислением альдегида(6), с использованием
NaClO2, получают карбоновую кислоту(7) с выходом 87% . Курциусовой
перегруппировкой карбоновой кислоты(7), с использованием DPPA и Et3N , с
последующей обработкой метанолом , получали метилкарбамат(8) с выходом в 78%.
Последующее депротонирование соединения(8) с 2,5 экв LiHMDS,
с дальнейшим добавлением к трет-бутиловому эфиру бромуксусной кислоты при -78
°C, повлекло за собой получение эфира(9) с выходом 82% . Как и ожидалось,
алкилирование происходит от противоположной стороны заместителя к β
-положению к карбонильной группе, конфигурация в γ
-положении лактона не влияет на селективность при алкилировании . Реакция
восстановительного раскрытия кольца из йодолактона(9) была осуществлена путем
обработки Zn в присутствии уксусной кислоты с получением карбоновой
кислоты(10), которая , при обработке DEPC и триэтиламина при температуре
окружающей среды , подвергся циклизации с получением желаемого цис-замещенного
лактама(11) с 60 % выходом из продукта(9).
Наконец, ввели заместитель по положению С1.
Лактам(11) селективно восстанавливают LiAlH(Ot-Bu)3
при 0 °C, и полученную смесь обрабатывают ППТС в метаноле с получением
соединения(12) с 79% выходом в виде одного изомера. Обработка вещества(12) в
TMSCN и BF3*OEt2 дала аминонитрил(13) в виде смеси диастереомеров (3:1). К
счастью, эпимеризация нежелательного изомера произошла в ходе щелочного
гидролиза. Таким образом, диастереомерная смесь нитрила(13) подаётся к
гидролизу NaOH, затем, после перекристаллизации получают чистую (-)-каиновой
кислоту (1) с 69% выходом. Этот синтетический маршрут позволило получить 14,6 г
(-)-каиновой кислоты (1) из 100 г (ю)-карвон (2) в 13 шагов и 10,3% общего выхода.
Этот синтетический путь позволил получить 14,6 г (-)-каиновой кислоты(1) из 100
г (+)-карвона(2) в 13 стадий и с 10,3% выхода от общего.
Методы синтеза
Синтез из азетидинона
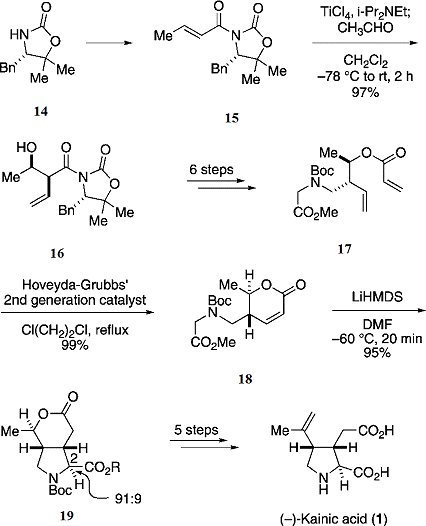
Схема 4: полный синтез (-)-каиновой кислоты(1)
Оксазолидинон Эванса(14) обробатывают LDA
в
ТГФ при -78 °С, это даёт продукт(15). Обработка продукта(15) даёт соединение
альдольной конденсации(16) в виде одного изомера , который преобразуется в
акрилата(17) через шесть стадий. Пирролидиновое кольцо(19) с полностью
замещёнными функциональными группами было получено по внутримолекулярной
реакции Михаэля из ненасыщенного лактона(18) с высокой диастереоселективностью.
Наконец, формирование изопропенила и манипуляции с функциональными группами
привели к получению каиновой кислоты(1). Использование внутримолекулярной
реакции Михаэля оказалось весьма эффективным для формирования полностью
разработанного кольца пирролидина. В этом методе синтеза раскрывается более
удобный способ получения каиновой кислоты с учётом недостатков получения
продукта(17). Это синтез второго поколения позволил получить (-)- каиновую
кислоту в 12 стадий с 14 % выходом из коммерчески доступного и недорогого
азетидинона(25).
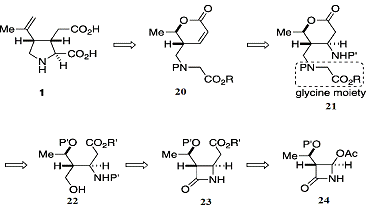
Схема 5: синтез второго поколения
Если к промежуточному соединению(20) добавляется
аминогруппа, то образуется соединение(21), которое может быть легко получено из
азетидинона(25) путём восстановительного раскрытия кольца β-лактама,
с последующей установкой глицина и формирования лактона. Соединение(23) может
быть получено из коммерчески доступного азетидинона, производное(24), которое
было изготовлено в промышленных масштабах, в качестве исходного материала для
антибиотиков на основе β-лактам,
таких как имипенем.
Синтез начался с введения карбоксиметилирующей
группы по [3R(1R, 4R)-4-ацетокси-3-[1 - (трет-бутилдиметилсилилокси)
этил]-2-азетидинона с помощью трет-бутилбромоацетата цинка (Схема 6).
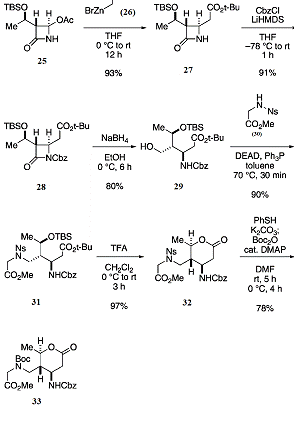
Схема 6: синтез интермедиата(33)
Активация кольца β-лактама
с группой Cbz (карбобензоксильная защитная группа) с последующим
восстановлением с помощью NaBH4 дает аминоспирт(29). Вводится глицин, затем
проводится реакция по Мицунобу с нозил(Ns)-активированным
сложным эфиром(30) для получения вещества(31). При обработке трифторуксусной
кислотой происходит циклизация соединения(31) с последующим образованем
вещества(32). Наконец, происходит переход из нозильной группы в Boc-группу,
что в итоге даёт желаемый продукт(33) с 46% общим выходом из соединения(25).
После удаления группы Cbz из соединения(33)
путём гидрогенолиза, полученный первичный амин обрабатывали 10-кратным молярным
избытком йодистого метила в присутствии карбоната цезия. Затем идёт удаление
группы NH2, вследствие
чего образуется желаемый α,β-ненасыщенный
лактон(18) с 64% выходом (Схема 7).
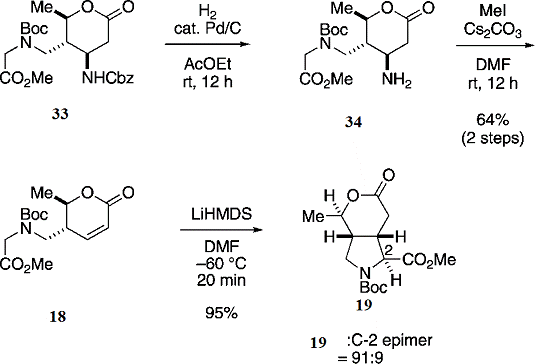
Схема 7: синтез кольца пирролидина
Этот метод синтеза обеспечивает 14% выход
каиновой кислоты в 12 стадий из недорого и доступного азетидинона(25). Данный
синтез существенно лучше, чем синтез первого поколения, представленный на схеме
4.
Методы синтеза
Синтез из транс-4-гидрокси-L-пролина.
Данный синтез основан на реакции Дильса-Альдера.
Реакция Дильса-Альдера из дегидропролина(36) с электроннобогатым диеном(37)
может привести к енону(38) (Схема 8) .
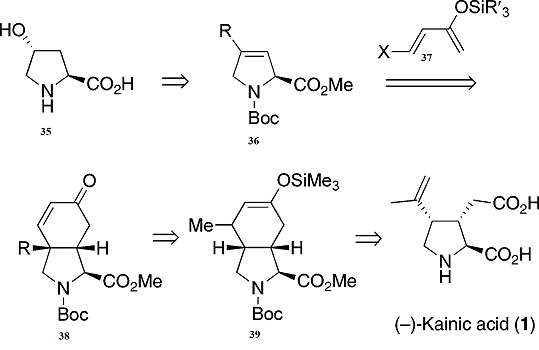
Схема 8: ретросинтетический анализ синтеза
(-)-каиновой кислоты
Этот продукт затем может быть быстро
преобразован в каиновую кислоту через последовательность , которая включала бы
сопряженное добавление метильной группой и захвата енолята для получения(39) ,
с последующим окислительным расщеплением , образование двойной связи , и гидролиза
.
Трифлат(41) был подготовлен, чтобы служить в
качестве прямого предшественника дегидропролин производного от продукта(36)
(Схема 9).
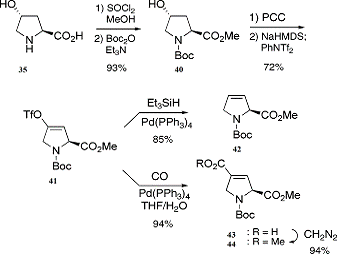
Схема 9: синтез дегидропролинов
Транс-4-гидрокси-L-пролина был преобразован с выходом
93% в N-Вос-производное метилового эфира(40), который был окислен до
соответствующего кетона с выходом 85%. Затем был получен региоселективно
трифталат (41) и с высоким выходом из этого кетона с NaHMDS-PhNTf2.
При обработке продукта(41) Et3SiH
в присутствии Pd-(PPh3)4
приводит к 3,4-дегидропролина(42)
с выходом 85%. Желаемое производное акрилата(44)
с выходом 60% и с энантиомерной частотой в 94% получается при
гидроксикарбонилировании продукта(41).
Как и ожидалось, однако, рацемизации этого соединения происходит довольно
легко: после 10 дней при 20°С, энантиомерная частота была только 20%.
Предположили, что если проводить реакцию при низких давлениях, то степень
рацемизации значительно уменьшится и реакцию можно будет проводить при
комнатной температуре. Омыление продукта(45),
с последующей перекристаллизацией сырого продукта, даёт двухосновную кислоту(46)
с 80% выходом. Эта кислота этерифицируется даизаметаном, что дает енон(47)
с 83% выходом (Схема 10).
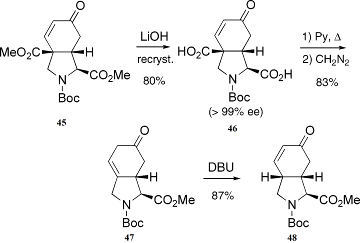
Схема 10:
декарбометокслирование продукат(45)
Трансформация продукта(47)
в сопряженный енон была осуществлена с DBU
в
дихлорметане. После присоединения метильной группы к енону(48),
за счет использования более высокого порядка цианокупрата в
триметилсилилхлориде, получают указанное триметилсилил эфирное производное(49)
в енольной форме, которое было подвергнуто озонолизу. После обработки
продукта(49) диметилсульфидом и диазометаном получается альдегид(50) с 70%
выходом (Схема 11).
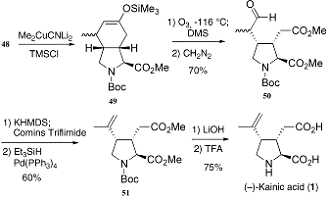
Схема 11: финальные стадии синтеза
Далее происходило преобразование этого
альдегида(50) в олефин(51) с 60% выходом. После омыление соединения(51) и
удаления Вос-группы с помощью TFA получается каиновая кислота с 75%-ным выходом
после перекристаллизации. Синтетически полученный материал был
хроматографически неотличим от аутентичного образца натурального продукта.
Таким образом, была получена (-)-каиновая кислота из 4-гидроксипролина с почти
10% общим выходом.
Заключение
Каждый из этих синтезов состоит из довльно
большого числа стадий (12-14). В среднем общий выход (-)-каиновой кислоты
состовляет 10-13%. Во всех синтезах используются довльно доступные и
сравнительно недорогие реагенты. Все стадии в данных синтезах были довольно
простыми, но всё равно требуется достаточно много времени, чтобы получить
каинат. Думаю в будущем будут найдены новые методы синтеза, которые будут менее
затратными и более быстрыми.
1. High-pressure
Diels-Alder approach to natural kainic acid/
S. K. Pandey// Organic Letters. - 2006. - Vol.8, - No.24, - p.5665-5668
2. Total
Synthesis of (−)-Kainic Acid via Intramolecular Michael Addition: A
Second-Generation Route/ Hiroshi
Sakaguchi// Organic Letters. - 2008. - Vol.10, No.9, - p.1711-1714
. A
Practical Synthesis of (−)-Kainic Acid/
Satoshi Takita// Organic Letters. - 2011. - Vol.13, - No.8, - p.2068-2070