Методы анализа антибиотиков
Содержание
Введение
1. Валидация методики количественного
определения антибиотиков
2.
Собственное исследование
Заключение
Список
использованной литературы
Введение
Проблема борьбы с инфекционными заболеваниями не теряет
своей актуальности. Несмотря на значительные успехи в области микробиологии и
антибактериальной химиотерапии, ситуация усложняется увеличением количества
микроорганизмов, резистентных к большому числу противомикробных лекарственных
средств. По данным ВОЗ ежегодно на земном шаре переносит инфекционные
заболевания более 1 миллиарда человек. На долю этих болезней приходится около
25% всех смертей в мире(*). В Республике Беларусь ежегодно регистрируется 2-2,5
миллиона случаев инфекционных заболеваний, в том числе вызванных бактериями.
Одними из наиболее опасных считаются острые инфекционные заболевания
дыхательных путей. Особое значение на сегодняшний день во многих странах
приобретает, в частности, проблема туберкулеза. В Республике Беларусь
показатель заболеваемости туберкулезом составил в 2012 году 39,6 человек на 100
тысяч населения - это на 3,9% ниже по сравнению с 2011 годом. Тем не менее
ситуация с туберкулезом в Республике остается напряженной ввиду появления
лекарственно устойчивых форм, а также групп риска по данному заболеванию (мигранты,
ВИЧ-инфицированные, социально неблагополучные контингенты населения).
ВОЗ рекомендована стратегия борьбы с туберкулезом,
известная под названием Directly Observed Treatment Strategy (DOTS). В основу
стратегии DOTS положены пять основных элементов, в отсутствии каждого из
которых она остается малоэффективной.
Неотъемлемой частью программы является создание надежной
системы поставки высококачественных противотуберкулезных лекарственных
препаратов для учреждений здравоохранения, которые обеспечивали бы непрерывность
процесса лечения больных туберкулезом. В настоящее время противотуберкулезные
препараты делят на препараты 1-ого Со (основного) ряда (изониазид,
этамбутол, рифампицин, стрептомицин и другие), используемые для лечения
обычного (чувствительного) туберкулеза и 2-ого (резервного) ряда (канамицин,
циклосерин, капреомицин), применяемые при наличии резистентности возбудителя к
препаратам первого ряда.
Высокое качество противотуберкулезных лекарственных средств
является гарантией успеха в терапии туберкулеза. Одним из показателей качества
средств антимикробной терапии, в том числе используемых при лечении
туберкулеза, является количественное содержание или активность действующего
вещества. Поэтому разработка методики, обеспечиваюшей стабильное достоверное
определение этого показателя, является актуальной задачей.
1. Валидация методики количественного определения
антибиотиков
Общие сведения о лекарственном средстве
Капреомицин.
Антибиотик капреомицин был выделен в 1960 году Herr и
соавторами из штамма Streptomyces capreolus. Это полипептидный антибиотик,
представляющий собой комплекс из 4-х микробиологически активных компонентов,
структура которых полностью не установлена. Смесь капреомицинов IA, IB, IIA,
IIB имеет примерное процентное отношение 25:67:3:6.
Капреомицин - белое вещество. Растворим в воде с
образованием бесцветного раствора. Практически нерастворим в большинстве
органических растворителей. Применяется в форме капреомицина сульфата с
содержанием активного вещества от 700 мкг/мг до 1050 мкг/мг. Капреомицин
избирательно активен в отношении Mycobacterium tuberculosis, локализующихся
вне- и внутриклеточно. Ингибирует синтез белка в бактериальной клетке,
оказывает бактериостатическое действие. Капреомицин применяется при лечении
легочных форм туберкулеза, вызванных чувствительными к нему штаммами
микобактерий, в том случае, если препараты 1-ого ряда неэффективны или не могут
применяться из-за токсического действия, или присутствуют устойчивые к их
действию микроорганизмы.
Фармакологические и клинические исследования капреомицина
были начаты ещё в 60-ых годах XX века.
В Национальном медицинском исследовательском центре
(Denver, Col, USA) было проведено исследование, целью которого было
проанализировать деятельность всех доступных противотуберкулезных препаратов в
бульонной культуре in vitro в анаэробных условиях. В результате только
капреомицин оказался бактерицидным в этих условиях, его деятельность оказалась
на том же уровне, что и у метронидозола, единственного препарата, известного на
тот момент эффективностью в отношении бактерии туберкулеза в анаэробных
условиях. Этот вывод был подтвержден в 6-ти экспериментах с 3-мя штаммами
Mycobacterium tuberculosis.
Для изучения бактериостатической активности капреомицина, а
также эффективности его при туберкулезной инфекции, вызванной как
чувствительными, так и устойчивыми к антибиотикам группы стрептомицина
штаммами, в Центральном научно-исследовательском институте туберкулеза (ЦНИИТ)
были селекционированы клонированные штаммы, чувствительные и устойчивые к
различным концентрациям стрептомицина, канамицина, флоримицина (виомицина) и
капреомицина[8]. Опыты in vitro проводили на жидкой среде Школьниковой методом
серийных разведений. Была установлена невысокая бактериостатическая активность
капреомицина по отношению к исходному чувствительному штамму микобактерий
H37Rv. Минимальная подавляющая концентрация (МПК) капреомицина составляла 2
мкг/мл. Она не повышалась при действии капреомицина на штаммы, обладающие
различной степенью устойчивости к стрептомицину. МПК стрептомицина при действии
на штаммы, устойчивые к различным концентрациям капреомицина, по сравнению с
действием его на чувствительный штамм почти не увеличивалась и составляла 0,5
мкг/мл. Таким образом, не было выявлено снижения активности стрептомицина по
отношению к капреомициноустойчивым штаммам и активности капреомицина по
отношению к стрептомициноустойчивым штаммам. При действии капреомицина на
штаммы, устойчивые к 250 мкг/мл канамицина, наблюдалось снижение
бактериостатической активности капреомицина по сравнению с действием его на
чувствительные штаммы в 8-16 раз. МПК капреомицина при действии его на штаммы,
устойчивые к 500 и 1000 мкг/мл канамицина, еще больше увеличивалась и
составляла 120 мкг/мл. При действии канамицина на капреомициноустойчивые штаммы
наблюдалось четкое снижение его активности по сравнению с действием на
чувствительные штаммы. При невысокой: устойчивости штаммов к капреоминину МПК
канамицина повышалась в 5-10 раз, при действии же канамицина на
высокоустойчивые к капреомицину штаммы его активность снижалась более чем в 100
раз. В целом наблюдалось незначительное снижение активности капреомицина по
отношению к высокоустойчивым к канамицину штаммам и резкое снижение активности
канамицина по отношению к штаммам, высокоустойчивым к капреомицину. При
рассмотрении взаимоотношений между капреомицином и флоримицином наблюдались
двустороннее снижение чувствительности для штаммов, обладающих невысокой
устойчивостью к антибиотикам, и двусторонняя перекрестная устойчивость для
штаммов, обладающих высокой устойчивостью к данным антибиотикам.
При действии капреомицина на экспериментальную
туберкулезную инфекцию у мышей, вызванную клонированным и чувствительным
штаммом RV, выявилась низкая эффективность капреомицина по сравнению с другими
препаратами этой группы (индекс эффективности равнялся 5%).
Суть испытания состоит в измерении температуры тела кроликов после
введения им в ушную вену испытуемых стерильных жидкостей. Испытания проводят на
трех кроликах, масса тела которых не отличается более чем на 0,5 кг. Жидкость
считают непирогенной, если сумма повышения температуры у трех кроликов не
превышает 1,4°С. Если эта сумма находится в пределах 1,5-2,2°С, испытание
повторяют на пяти кроликах, а если превышает 2,2°С, то жидкость считается пиро
генной.
Испытание пирогенности на кроликах отличается определенными трудностями.
Поэтому во многие фармакопеи мира (США, Великобритании, Китая и др.) для
определения пирогенности ЛС включен так называемый Л АЛ-тест (определение
бактериальных эндотоксинов). В его основе лежит способность лизата амебоцитов
(клеток крови) мечехвоста специфически реагировать с эндотоксинами
грамотрицательных бактерий (липосахаридами). В результате взаимодействия
эндотоксина и лизата появляется помутнение прозрачной реакционной смеси или
происходит образование твердого геля, что служит подтверждением присутствия
эндотоксина. Сырьем для производства ЛАЛ-реагента служит кровь мечехвостов -
морских животных, обитающих у берегов Северной Америки, Японии, Китая,
Вьетнама.
Л АЛ-гест высокоспецифичен по отношению к эндотоксинам грамотрицательных
бактерий. Его чувствительность во много раз выше, чем у фармакопейного теста на
кроликах, а области применения значительно шире. ЛАЛ-тест применим в
производственном (постадийном) контроле содержания эндотоксинов в инъекционных
ЛФ, поскольку дает возможность получения результатов в течение 1-2 часов и
одновременного испытания большого количества образцов. Кроме того, этот тест
обеспечивает надежность и воспроизводимость получения результатов, сочетающихся
с простотой используемой методики. Реактив для ЛАЛ-теста предсталяет собой
сублимационно высушенный лизат, который готов к использованию после разведения
его апирогенной водой. Учитывая преимущества ЛАЛ-теста, подготовлен проект ОФС
"Определение содержания бактериальных эндотоксинов" для включения в
очередное издание ГФ РФ.
Испытание на токсичность проводят на белых мышах обоего пола массой 19-21
г. Испытуемый раствор вводят в хвостовую вену пяти мышам и ведут1 наблюдение за
ними в течение 48 час. Если ни одна из подопытных мышей в течение этого срока
не погибнет, то ЛП считается выдержавшим испытание на токсичность. В случае
гибели хотя бы одной мыши испытания повторяют по определенной схеме и делают
окончательное заключение о его токсичности.
Испытаниям на микробиологическую чистоту подвергают не стерилизуемые в
процессе производства ЛП (таблетки, капсулы, гранулы, растворы, экстракты, мази
и др.). Эти испытания имеют своей целью определение состава и количества
имеющейся в ЛФ микрофлоры. При этом устанавливается соответствие нормам,
ограничивающим микробную обсеме- ненность (контаминацию). Испытание включает
количественное определение жизнеспособных бактерий и грибов, выявление
некоторых видов микроорганизмов, кишечной флоры и стафилококков. Испытание выполняют
в асептических условиях в соответствии с требованиями ГФ XI (в. 2, с. 193)
двухслойным агаровым методом в чашках Петри.
Испытание на стерильность основано на доказательстве отсутствия в ЛС
жизнеспособных микроорганизмов любого вида и является одним из важнейших
показателей безопасности ЛС. Этим испытаниям подвергаются все ЛП для
парентерального введения, глазные капли, мази и т.д. Для контроля стерильности
применяют биогликолевую и жидкую среду Са- буро, используя метод прямого посева
на питательные среды. Если ЛС обладает выраженным антимикробным действием или
разлито в емкости более 100 мл, то используют метод мембранной фильтрации.
Валидация - это подтверждение обоснованности выбора метода анализа для
установления норм качества Л С по каждому разделу НД. Она проводится при
подготовке проектов НД на новые ЛС или при последующем пересмотре НД. Ва-
лидации подвергаются аналитические методы, используемые для идентификации ЛВ,
установления содержания в нем различных примесей, количественного определения индивидуальных
ЛВ и содержания их в ЛФ, определения вспомогательных веществ и консервантов.
Валидация метода анализа предполагает оценку его специфичности, линейной
зависимости результатов испытаний, аналитической области методики,
правильности, воспроизводимости результатов, предела обнаружения.
Ревалидация необходима в тех случаях, когда произошли изменения в синтезе
ЛВ, в составе ЛС, в аналитической методике. Параметры аналитического метода,
устанавливаемые при его валидации и ревалидации, рассчитываются в соответствии
с существующими правилами статистической обработки результатов анализа.
Специфичность метода анализа обусловливает его способность достоверно
установить наличие ЛВ в присутствии других компонентов (примесей,
вспомогательных веществ). Оценка специфичности необходима для методов,
используемых при идентификации, определении примесей и количественного
содержания Л В.
Линейная зависимость аналитических сигналов от концентрации ЛВ
устанавливается графически. Оценивается она на основании не менее 5 испытаний,
выполненных с помощью используемой аналитической методики. Параметрами,
подтверждающими линейную зависимость, являются коэффициент регрессии, угол
наклона линии регрессии и остаточная сумма площадей.
Аналитическая область методики охватывает интервал между верхним и нижним
пределами содержания испытуемого вещества, в котором соблюдается линейная
зависимость. При этом данная методика должна обеспечивать определение с
требуемыми воспроизводимостью и точностью. Аналитическая область выражается в тех
же единицах,что и результаты испытаний с помощью данной методики (проценты,
миллионные доли).
Правильность (точность) аналитического метода характеризует близость
результатов, полученных с помощью данной методики, к истинному значению. При
установлении этого параметра для количественного определения субстанций,
примесей могут быть использованы стандартные образцы, другие независимые
методик! ьные смеси, метод добавок.
Правильность оценивается не менее чем на трех повторностях определения
для трех аналитических концентраций в пределах аналитической области.
Воспроизводимость аналитического метода отражает степень совпадений
результатов отдельных испытаний при многократном использовании методики. Она
устанавливается при количественном определении не менее 9 аликвот образца и
выражается в результате статистической обработки по величинам стандартного
отклонения, коэффициента вариации и доверительного интервала.
Межлабораторная воспроизводимость аналитического метода показывает
степень воспроизводимости результатов испытаний, выполненных по разработанной
методике в различных лабораториях на соответствующем оборудовании, разными
аналитиками, в разное время.
Предел обнаружения - минимальное содержание анализируемого вещества,
которое можно обнаружить с помощью данной методики (выражается в процентах или
миллионных долях). Устанавливается для химических методов визуально. Для
физико-химических методов устанавливается по минимальной концентрации
испытуемого вещества, которое может быть достоверно обнаружено или рассчитывается
по величине стандартного отклонения и углу наклона калибровочной кривой.
Предел количественного определения - минимальное содержание (в процентах)
анализируемого вещества, которое может быть определено с достаточной точностью
и воспроизводимостью. Устанавливается для любых методов визуально или расчетным
путем подобно установлению предела обнаружения.
Пригодность системы - интегральная часть аналитических методик,
подтверждающая надежность анализа в заданных условиях его проведения.
Стандартными образцами (СО) называют вещества, с которыми сравнивают
испытуемые ЛС при проведении их анализа физико-химическими или биологическими
методами. Условно разделяют СО на химические и биологические, но это не
исключает использования одного и того же из них и для физико-химического, и для
биологического анализа. В ГФ XI, вып. 2 (с. 60) даны определения терминов
"государственные стандартные образцы" (ГСО). "рабочие
стандартные образцы" (PCO) и "стандартные образцы
веществ-свидетелей" (СОВС). Активность, или содержание вещества (%) в ГСО
принимается за 100%, если нет других указаний на этикетке. Выпуск ГСО
осуществляют в соответствии с требованиями ФС, которая разрабатывается
предприятием-разработчиком. В качестве PCO используют образцы серийных Л В,
которые соответствуют требованиям ФС (ФСП). Расчет количественного содержания
ЛВ в ЛФ проводят исходя из фактического содержания его в PCO. В качестве СОВС
используют ГСО, PCO и вещества, специально изготовленные в порядке,
предусмотренном ФС. Применяют СОВС для определения примесей или компонентного
состава испытуемых ЛС.
Ряд особенностей имеют СО на антибиотики. Среди них имеются как
однокомпонентные вещества, так и сложные, состоящие из нескольких компонентов
близкой природы (аминогликозиды, полиены), каждый из которых определяет как
активность и токсичность, так и физико-химические константы ЛС. При разработке
такого СО его активность определяется исходя из количественного содержания в
нем действующего ЛВ. Кроме того, при стандартизации антибиотиков имеется
тенденция к созданию единого стандарта к группе веществ, сходных по химическому
строению.
Аттестацией, хранением и реализацией СО занимается НИИКЛС. Он проводит
экспертизу всех проектов ФС на ГСО, разработанных предприятиями и
организациями, выпускающими эти ГСО. Только после этого они утверждаются МЗ РФ
в установленном порядке.
Номенклатура ГСО отечественного производства включает около 140
наименований. Они используются при выполнении физико-химических и биологических
испытаний более чем 300 ЛС (прежде всего синтетических). Еще более широко
используются PCO, представляющие собой образцы серийных ЛВ. Их применение
отражено в более 700 ФС для испытаний подлинности, чистоты и количественного
определения.
. Собственное исследование
Валидационная оценка аналитических методик
Приказом Министерства здравоохранения и социального развития РФ от 30
октября 2006г. №736 принят Административный регламент Федеральной службы по
надзору в сфере здравоохранения и социального развития, который предусматривает
в структуре регистрационного досье данные о валидации аналитических методик.
Кроме того, в соответствии с ГОСТ Р 52249-2004 "Правила производства и
контроля качества лекарственных средств" (раздел 6) отдел контроля
качества должен обеспечить валидацию методик контроля качества. Однако, в тоже
время, данные документы не регламентируют требования к проведению валидации
аналитических методик и испытания. Следует отметить, что в Российской Федерации
на сегодняшний день нет собственного однозначного нормативного документа
регламентирующего порядок проведения процедуры валидации. Однако, в 2007 году в
Российской Федерации издано "Руководство для предприятий фармацевтической
промышленности (методические рекомендации) ", которое утверждено и введено
в действие решением общего собрания членов Ассоциации российских
фармацевтических производителей, а также рекомендовано к использованию
Федеральной службой по надзору в сфере здравоохранения и социального развития
специалистам, занятым в сфере обращения лекарственных средств. Одна из частей
данного руководства посвящена валидации методик анализа лекарственных средств.
Официальному признанию методики анализа предшествует процедура её
метрологической аттестации. В фармацевтической практике этот процесс называется
валидацией, то есть экспериментальное доказательство того, что методика
пригодна для решения поставленных задач. В зависимости от характера
аналитической методики могут применяться те или иные валидационные
характеристики (табл.1).
Таблица 1 - Валидационные характеристики основных типов методик
|
Наименование характеристики
|
Типы методик
|
|
Испытание на подлинность
|
Посторонние примеси
|
Количественное определение:
|
|
|
Количественные методики
|
Пределы содержания
|
Основного действующего
вещества, нормируемых компонентов
|
Действующего вещества в
тесте "Растворение"
|
|
Специфичность**
|
Да
|
Да
|
Да
|
Да
|
Да
|
|
Линейность
|
Нет
|
Да
|
Нет
|
Да
|
Да
|
|
Аналитическая область
|
Нет
|
Да
|
*
|
Да
|
|
Правильность
|
Нет
|
Да
|
*
|
Да
|
Да
|
|
Прецизионность: -
повторяемость (сходимость) - внутрила-бораторная воспроизводимость
|
Нет Нет
|
Да Да
|
Нет Нет
|
Да Да
|
Да Нет
|
|
Предел обнаружения
(чувствительность)
|
Нет
|
Нет2
|
Да
|
Нет
|
Нет
|
|
Предел количественного
определения
|
Нет
|
Да
|
Нет
|
Нет
|
Нет
|
|
Робастность (устойчивость)
|
Нет
|
*
|
*
|
*
|
*
|
*
может определяться при необходимости;
**отсутствие специфичности одной аналитической методики может быть
компенсировано использованием другой аналитической методики;
1-необходимо
только для новых методик, не имеющих аналогов;
2-необходимо
в случаях, когда предел обнаружения близок к пределу количественного
определения.
Для проведения анализа по показателям специфичность, линейность
прецензионность и правильность используются растворы модельных смесей
следующего состава:
Раствор модельной смеси А1. Точные навески левомицетина -
субстанции (0,200 г) и таблеточной массы - натрия хлорида (0,200 г) помещают в
мерную колбу вместимостью 100 мл, прибавляют 15 мл 0,1 М раствора кислоты
хлористоводородной, перемешивают, доводят тем же растворителем до метки,
перемешивают.
Раствор модельной смеси А2. Точные навески левомицетина -
субстанции (0,250 г) и таблеточной массы - натрия хлорида (0,150 г) помещают в
мерную колбу вместимостью 100 мл, прибавляют 15 мл 0,1 М раствора кислоты
хлористоводородной, перемешивают, доводят тем же растворителем до метки,
перемешивают.
Модельной смеси А3. Точные навески левомицетина - субстанции
(0,300 г) и таблеточной массы - натрия хлорида (0,100 г) помещают в мерную
колбу вместимостью 100 мл, прибавляют 15 мл 0,1 М раствора кислоты
хлористоводородной, перемешивают, доводят тем же растворителем до метки,
перемешивают.
Так же используется раствор сравнения который приготавливают следующим
образом:
Точную навеску СО левомицетина (0,1г) помещают в мерную колбу
вместимостью 100 мл, растворяют в 0,1 М растворе кислоты хлористоводородной,
доводят 0,1 М раствором кислоты хлористоводородной до метки и перемешивают.
Специфичность
Под специфичностью методики следует понимать способность достоверно
определять анализируемое соединение в присутствии других компонентов образца -
лекарственных веществ, вспомогательных веществ и посторонних прим Установление
специфичности в тестах "Испытание на подлинность".
Для установления специфичности в идентификационных тестах, основанных на
качественных реакциях, следует убедиться как в отсутствии по-ложительного
эффекта реакции на сопутствующие вещества, так и в возможности ингибирования
данной реакции компонентами образца.
При доказательстве подлинности спектрофотометрическим методом необходимо
убедиться в отсутствии светопоглощения у второго компонента, вспомогательных и
сопутствующих веществ.
Установление специфичности в тестах "Количественное
определение"
Методики, используемые для количественного определения также должны
подвергаться валидационной оценке по данному критерию. Спектрофотометрический
метод. При выполнении анализа спектрофото-метрическим методом нужно
подтвердить, что сопутствующие компоненты не мешают определению.
Готовят модельные смеси по следующей методике:
Раствор модельной смеси А1. Точные навески левомицетина -
субстанции (0,200 г) и таблеточной массы - натрия хлорида (0,200 г) помещают в
мерную колбу вместимостью 100 мл, прибавляют 15 мл 0,1 М раствора кислоты
хлористоводородной, перемешивают, доводят тем же растворителем до метки,
перемешивают.
Раствор модельной смеси А2. Точные навески левомицетина -
субстанции (0,250 г) и таблеточной массы - натрия хлорида (0,150 г) помещают в
мерную колбу вместимостью 100 мл, прибавляют 15 мл 0,1 М раствора кислоты
хлористоводородной, перемешивают, доводят тем же растворителем до метки,
перемешивают.
Модельной смеси А3. Точные навески левомицетина - субстанции
(0,300 г) и таблеточной массы - натрия хлорида (0,100 г) помещают в мерную
колбу вместимостью 100 мл, прибавляют 15 мл 0,1 М раствора кислоты
хлористоводородной, перемешивают, доводят тем же растворителем до метки,
перемешивают.
В модельной смеси 1 с помощью качественных реакций необходимо доказать
подлинность каждого компонента лекарственного средства.
В модельной смеси 2 с помощью качественных реакций на второй компо-нент
подтверждают отсутствие аналитического эффекта у первого компонента.
В модельной смеси 3 с помощью качественных реакций на первый компонент
подтверждают отсутствие аналитического эффекта у второго компонента.
Необходимо провести определение специфичности одного из ингредиентов в
тесте "Количественное определение" титриметрическим методом.
Для этого используют модельную смесь 2, содержащую только первый
ингредиент, определяемый спектрофотометрическим методом.
Согласно методике проводят титрование навески модельной смеси 2.
Необходимо убедиться, что компонент 1 не титруется в данных условиях.
Проводят определение специфичности одного из ингредиентов в тесте
"Количественное определение" спектрофотометрическим методом. Для
этого используют модельную смесь 3, содержащую только второй ингредиент,
определяемый титриметрическим методом.
Выводы:
Ø В модельной смеси 1 реакции на подлинность компонентов смеси
- левомицетин и натрия хлорид дают положительный результат.
Ø В модельной смеси 2 реакции на натрия хлорид дают
отрицательный результат.
Ø В модельной смеси 3 отсутствие аналитического эффекта у
качественных реакций на левомицетин.
Ø Количественное определение модельной смеси 2 дает понять что
количественно левомицетин не обнаруживается методом аргентометрии.
Ø Количественное определение модельной смеси 3 дает понять что
содержание натрия хлорида нельзя определить с помощью спектрофотометрии.
Основываясь на данных выводах можно сделать заключение о специфичности
данной методики.
Линейная зависимость устанавливается на основании результатов испытаний,
которые пропорциональны концентрации анализируемого вещества в образце в
пределах аналитической методики. Линейность результатов может быть представлена
графически в виде зависимости аналитических сигналов от концентрации вещества
(не менее 5).
Аналитическая методика должна быть охарактеризована следующими
параметрами для подтверждения линейности: коэффициент регрессии, уголнаклона
линии регрессии и остаточная сумма площадей.
Для установления максимума светопоглощения раствора левомицетина снимают
спектр поглощения 0,002% раствора СО в 0,1 М растворе кислоты
хлористоводородной. Приготовление раствора описано ниже при построении
градуировочного графика. Максимум должен находиться при длине волны 278 нм
(например, оптическая плотность Аст=0,295).
Точную навеску СО левомицетина (0,10 г) помещают в мерную колбу
вместимостью 100 мл, прибавляют 15 мл 0,1 М раствора кислоты
хлористоводородной, перемешивают, доводят тем же растворителем до метки,
перемешивают.
В мерные колбы вместимостью 100 мл последовательно вносят 1,0 мл; 2,0 мл;
3,0 мл; 4,0 мл; 5,0 мл раствора А и доводят 0,1 М раствором кислоты
хлористоводородной до метки (получают 0,001%; 0,002%; 0,003%; 0,004% и 0,005%
растворы).
Измеряют оптическую плотность каждого раствора на спектрофотометре при
длине волны 278 нм в кювете с толщиной слоя 10 мм. Раствор сравнения - 0,1 М
раствор кислоты хлористоводородной.
|
V 0,02% р-ра (мл)
|
1
|
2
|
3
|
4
|
5
|
|
С р-ра в %
|
0,02
|
0,04
|
0,06
|
0,08
|
0,1
|
|
Оптическая плотность
|
0,005
|
0,018
|
0,053
|
0,07
|
0,135
|
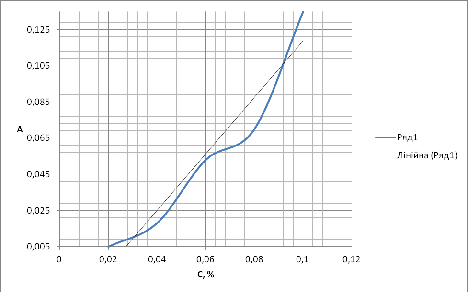
Рисунок Градуировочный график линейной зависимости оптической плотности
от концентрации левомицетина.
Прецензионность методики характеризуется рассеянием результатов
относительно среднего значения. Определение проводят следуя данной методике:
Готовят параллельно по три раствора.
Растворы Б1, Б2, Б3. В три мерные колбы
вместимостью 100 мл переносят по 0,5 мл раствора модельной смеси А2
и доводят 0,1 М раствором кислоты хлористоводородной до метки.
Растворы Б4, Б5, Б6. В три мерные колбы
вместимостью 100 мл переносят по 1,0 мл раствора модельной смеси А2
и доводят 0,1 М раствором кислоты хлористоводородной до метки.
Растворы Б7, Б8, Б9. В три мерные колбы
вместимостью 100 мл переносят по 1,5 мл раствора модельной смеси А2
и доводят 0,1 М раствором кислоты хлористоводородной до метки.
Измеряют оптическую плотность каждого из 9 приготовленных растворов на
спектрофотометре при длине волны 278 нм в кювете с толщиной слоя 10 мм. Раствор
сравнения - 0,1 М раствор кислоты хлористоводородной.
Параллельно измеряют оптическую плотность СО 0,002% раствора
левомицетина.
Расчет содержания левомицетина в г/мл проводят по формуле:
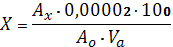 (1),
(1),
где: Ах - оптическая плотность исследуемого раствора; Ао
- оптическая плотность раствора СО левомицетина; 0,00002 - содержание
рибофлавина в г в 1 мл раствора СО. Va - объем аликвоты раствора модельной смеси 2, взятый для
разведения, мл.
|
Xi
|
 -Xi -Xi
|
|
SD=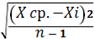 . SD= . SD= 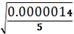 = 0.00053 RSD = = 0.00053 RSD = 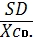 * 100% . RSD = * 100% . RSD = 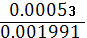 * 100% = 2.7 % * 100% = 2.7 %
|
|
0.002136
|
-0,000145
|
0,00000021
|
|
|
0.00205
|
-0,000059
|
0,000000059
|
|
|
0.00199
|
0,000001
|
0,000001
|
|
|
0.00196
|
0,000031
|
0,0000000096
|
|
|
0.00193
|
0,000061
|
0,000000037
|
|
|
0.00188
|
0,000111
|
0,00000012
|
|
|
=0,001991
|
|
∑=0,000014
|
|
Вывод: данная методика не превышает стандартное отклонение в 3%
следовательно данная методика подходит по показателю прецинзионность.
Правильность
Правильность (точность) аналитического метода характеризует близость
результатов испытаний, полученных данным методом, к истинному значению. При
количественном определении лекарственного вещества этот параметр может быть
установлена путем применения аналитического метода к анализируемому объекту с
использованием стандарта известной степени чистоты или путем сравнение
результатов, полученных предлагаемой аналитической методикой, с результатами,
которые получены другой независимой методикой, правильность которой известна.
В случае количественного определения вещества в лекарственной форме
правильность аналитической методики устанавливается по результатам ее
применения к анализу модельной смеси, включающей все компоненты лекарственной
формы. Правильность методики количественного определения идентифицированных
примесных соединений устанавливается по результатам анализа методом добавок.
При отсутствии образцов примесных соединений или в тех случаях, когда структура
их не установлена, правильность предлагаемой методики их определения должна
быть подтверждена результатами анализадругой аналитической методикой с
охарактеризованной правильностью.
Правильность должна быть оценена на основе не менее 9 определений на
минимум 3 уровнях концентраций в пределе аналитической области (например, 3
повторности определения для 3 аналитических концентраций).
Для проведения анализа методики по данному показателю необходимо
приготовить модельные смеси пропорции которых указаны в таблице 2.
Таблица 2. Приготовление модельных смесей таблеток левомицетина.
|
Модельная смесь
|
1 (А1)
|
2 (А2)
|
3 (А3)
|
|
Навеска левомицетина -
субстанции (г)
|
0,200
|
0,250
|
0,300
|
|
Таблеточная масса - натрия
хлорид (г)
|
0,150
|
0,100
|
|
Средняя масса
|
0,400
|
0,400
|
0,400
|
Растворы модельных смесей приготавливают аналогично методике
специфичность.
Растворы Б1, Б2, Б3. В три мерные колбы
вместимостью 100 мл переносят по 1,0 мл раствора модельной смеси А1
и доводят 0,1 М раствором кислоты хлористоводородной до метки.
Растворы Б4, Б5, Б6. В три мерные колбы
вместимостью 100 мл переносят по 1,0 мл раствора модельной смеси А2
и доводят 0,1 М раствором кислоты хлористоводородной до метки.
Растворы Б7, Б8, Б9. В три мерные колбы
вместимостью 100 мл переносят по 1,0 мл раствора модельной смеси А3
и доводят 0,1 М раствором кислоты хлористоводородной до метки.
Измеряют оптическую плотность каждого из 9 приготовленных растворов на
спектрофотометре при длине волны 278 нм в кювете с толщиной слоя 10 мм. Раствор
сравнения - 0,1 М раствор кислоты хлористоводородной.
Параллельно измеряют оптическую плотность СО 0,002% раствора левомицетина. В мерную колбу вместимостью 100 мл вносят 2,0 мл раствора А и
доводят 0,1 М раствором кислоты хлористоводородной до метки.
мл раствора Б содержит 0,00002 г СО левомицетина.
Расчет содержания левомицетина в процентах (открываемость) проводят по
формуле:
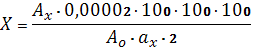 (2),
(2),
где: Ах - оптическая плотность исследуемого раствора;
Ао - оптическая плотность раствора СО левомицетина;
ах -масса навеска исследуемого образца левомицетина;
,00002 - содержание левомицетина в г в 1 мл раствора СО.
Таблица 3
|
Aх
|
ax
|
R(%)
|
(Ri-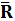 ) )
|
(Ri-)2
|
|
0,385
|
0,206
|
99,86
|
4,96
|
24,6
|
|
0,386
|
0,206
|
94,5
|
-0,04
|
0,0016
|
|
0,386
|
0,206
|
94,5
|
-0,04
|
0,0016
|
|
0,147
|
0,248
|
107,3
|
12,4
|
153,76
|
|
0,152
|
0,248
|
108,2
|
13,3
|
176,89
|
|
0152
|
0,248
|
108,2
|
13,3
|
176,89
|
|
0,257
|
0,294
|
78,8
|
-16,1
|
259,21
|
|
0259
|
0,294
|
82,6
|
-12,3
|
151,29
|
|
0,259
|
0,294
|
82,6
|
-12,3
|
151,29
|
|
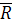 =94,9 =94,9
|
|
|
SD= 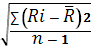 RSD=
RSD= 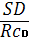 * 100%.
* 100%.
SD= 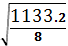 = 11.89
= 11.89 RSD=
RSD= 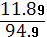 * 100% = 1.3 %
* 100% = 1.3 %
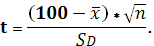
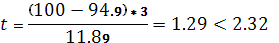
Вывод: полученное значение меньше коэффициента Стьюдента отсюда следует
что данная методика соответствует по показателю правильность.
Заключение
В данной работе изложена суть и важность валидационных оценок, что
позволяет оценить важность этого вида контроля над производством лекарственных
средств.
Так как методики количественного и качественного анализа дают
удовлетворительные результаты только при наличии данного вещества или при
определенных условиях, то аналитические методики по данным показателям
специфичны.
Следуя из того что данная методика не превышает стандартное отклонение в
3% она соответствует по показателю прецинзионность.
Полученное значение меньше коэффициента Стьюдента отсюда следует что
данная методика соответствует по показателю правильность.
Список использованной литературы
валидация антибиотик лекарственный специфичность
1) Атлас по медицинской микробиологии, вирусологии и
иммунологии. Воробьев А.А., Быков А.С. Беликов, В.Г. Фармацевтическая химия.
/В.Г Беликов. В 2 ч.: Ч. 1. Общая фармацевтическая химия; Ч. 2. Специальная
фармацевтическая химия. Учеб для вузов. - Пятигорск. 2003. стр. 333-339.
) Машковский, М.Д. Лекарственные средства: в 2 т./М.Д.
Машковский. - 18-е изд., перераб. и доп.- М.: Новая волна, 2010. - 1234 с.
) М.В. Гаврилин, И.Я. Куль, Н.В. Благоразумная, Л.Н.
Дуккардт, С.П. Сенченко, С.Н. Степанюк, А.Ю.Курегян./Метрологическая аттестация
(валидация) методик анализа лекарственных средств.-Пятигорск 2013.
) Методы анализа лекарств /Н.П. Максютина. - Киев:
Здоровья, 1984. - 224 с.
) Лабораторные работы по фармацевтической химии:
Учебное пособие/В.Г. Беликов. - 2-е изд. перераб. и доп. - Пятигорск, 2003. -
342 с.
) Анализ лекарственных смесей. А.П. Арзамасцев, В.М.
Печенников, Г.М.Родионова и др. Москва компания Спутник+, 2000 год.
) Фармакопейная статья: "Таблетки левомицетина
0,25. Валидационная оценка методик анализа левомицетина". Государственная
фармакопея. СССР. Вып 2 Общие методы анализа.
) Фармакопейная статья "Таблетки".
Государственная Фармакопея СССР. Вып 2 Общие методы анализа.
) Машковский М.Д. Лекарственные средства. издание
пятнадцатое переработанное, исправленное и дополненное, Москва Новая волна 2005
год.