Электрохимические методы анализа
МОСКОВСКИЙ ГОСУДАРСТВЕННЫЙ МЕДИЦИНСКИЙ УНИВЕРСИТЕТ
ИМ. СЕЧЕНОВА
кАФЕДРА АНАЛИТИЧЕСКОЙ, ФИЗИЧЕСКОЙ И КОЛЛОИДНОЙ ХИМИИ
РЕФЕРАТ НА ТЕМУ
«ЭЛЕКТРОХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА»
Работа
студентки 18 группы 1 курса
Фармацевтического
факультета
Гончаренко
Светланы Валерьевны
Научный
консультант: Харитонов Ю.Я.
Ведущий
преподаватель: Ершов Ю.А.
Москва
2011г.
1. Общие понятия. Классификация
электрохимических методов анализа
Электрохимические методы основаны на измерении
электрических параметров электрохимических явлений, возникающих в исследуемом
растворе. Такое измерение осуществляют с помощью электрохимической ячейки,
представляющей собой сосуд с исследуемым раствором, в который помещены
электроды. Электрохимические процессы в растворе сопровождаются появлением или
изменением разности потенциалов между электродами или изменение величины тока,
проходящего через раствор.
Электрохимические методы классифицируют в
зависимости от типа явлений, замеряемых в процессе анализа. В общем случае
различают две группы электрохимических методов:
). Методы без наложения постороннего потенциала,
основанные на измерении разности потенциалов, который возникает в
электрохимической ячейке, состоящей из электрода и сосуда с исследуемым
раствором. Эту группу методов называют потенциометрическими. В
потенциометрических методах используют зависимость равновесного потенциала
электродов от концентрации ионов, участвующих в электрохимической реакции на
электродах.
). Методы с наложением постороннего потенциала,
основанные на измерении: а) электрической проводимости растворов -
кондуктометрия; б) количества электричества, прошедшего через раствор -
кулонометрия; в) зависимости величины тока от приложенного потенциала - вольт
амперометрия; г) времени, необходимого для прохождения электрохимической
реакции - хроноэлектрохимические методы (хроновольтамперометрия,
хронокондуктометрия). В методах этой группы на электроды электрохимической
ячейки налагают посторонний потенциал.
Основным элементом приборов для
электрохимического анализа является электрохимическая ячейка. В методах без
наложения постороннего потенциала она представляет собой гальванический
элемент, в котором вследствие протекания химических
окислительно-восстановительных реакций возникает электрический ток. В ячейке
типа гальванического элемента в контакте с анализируемым раствором находятся
два электрода - индикаторный электрод, потенциал которого зависит от
концентрации вещества, и электрод с постоянным потенциалом - электрод
сравнения, относительно которого измеряют потенциал индикаторного электрода.
Измерение разности потенциалов производят специальными приборами -
потенциометрами.
В методах с наложением постороннего потенциала
применяют электрохимическую ячейку, названную так потому, что на электродах
ячейки под действием наложенного потенциала происходит электролиз - окисление
или восстановление вещества. В кондуктометрическом анализе используют
кондуктометрическую ячейку, в которой замеряют электрическую проводимость
раствора. По способу применения электрохимические методы можно классифицировать
на прямые, в которых концентрацию веществ измеряют по показанию прибора, и
электрохимическое титрование, где индикацию точки эквивалентности фиксируют с
помощью электрохимических измерений. В соответствии с этой классификацией различают
потенциометрию и потенциометрическое титрование, кондуктометрию и
кондуктометрическое титрование и т.д.
Классификация по способу применения
электрохимических методов. Различают прямые и косвенные методы.
а) Прямые методы. Измеряют электрохимический параметр
как известную функцию концентрации раствора и по показанию соответствующего
измерительного прибора находят содержание определяемого вещества в растворе.
б) Косвенные методы- это методы титрования, в
которых окончание титрования фиксируют на основании измерения электрических
параметров системы.
В соответствии с данной классификацией
различают, например, прямую кондуктометрию и кондуктометрическое титрование,
прямую потенциометрию и потенциометрическое титрование и т.д.
2. Потенциометрический анализ (патенциометрия)
.1 Принцип метода
Потенциометрический анализ основан на измерении
ЭДС и электродных потенциалов как функции концентрации анализируемого раствора.
Если в электрохимической системе - в
гальваническом элементе - на электродах протекает реакция
aА +
bВ
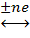 dD
+ eE
dD
+ eE
с переносом n
электронов, то уравнение Нернста для ЭДС E
этой реакции имеет вид:
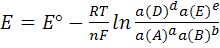 (1)
(1)
где, как обычно,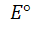 -стандартная
ЭДС реакции (разность стандартных электродных потенциалов), R-
газовая постоянная, T- абсолютная
температура, при которой протекает реакция, F-
число Фарадея; а(А),а(В),а(D)
и а(E)- активности
реагентов- участков реакции. Уравнение (1) справедливо для ЭДС обратимо
работающего гальванического элемента.
-стандартная
ЭДС реакции (разность стандартных электродных потенциалов), R-
газовая постоянная, T- абсолютная
температура, при которой протекает реакция, F-
число Фарадея; а(А),а(В),а(D)
и а(E)- активности
реагентов- участков реакции. Уравнение (1) справедливо для ЭДС обратимо
работающего гальванического элемента.
Для комнатной температуры уравнение (1) можно
представить в форме
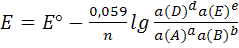 (2)
(2)
В условиях, когда активности реагентов
приблизительно равны их концентрации, уравнение (1) переходит в уравнение (3)
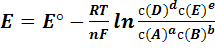 (3)
(3)
где с(А), с(В), с(E), с(D)-
концентрации реагентов. Для комнатной температуры это уравнение можно
представить в виде (4)
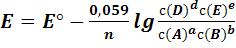 (4)
(4)
При потенциометрических измерениях в
электрохимической ячейке используют два электрода: индикаторный электрод,
потенциал которого зависит от концентрации определяемого
(потенциалопределяющего) вещества в анализируемом растворе, и электрод
сравнения, потенциал которого в условиях проведения анализа остается
постоянным. Поэтому величину ЭДС, определяемую уравнениями (1)-(4), можно
рассчитать как разность реальных потенциалов этих двух электродов.
В потенциометрии используют электроды следующих
типов: электроды первого, второго рода, окислительно-восстановительные,
мембранные электроды.
Электроды первого рода- это электроды, обратимые
по катиону, общему с материалом электрода.
а) Металл М, погруженный в раствор соли того же
металла. На поверхности таких электродов протекает обратимая реакция
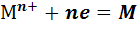
Реальный потенциал такого электрода первого рода
зависит от активности а(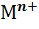 ) катионов металла
и описывается уравнениями (5)-(8).
) катионов металла
и описывается уравнениями (5)-(8).
В общем случае для любой температуры
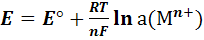 (5)
(5)
Для комнатной температуры
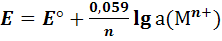 (6)
(6)
При малых концентрациях с(),
когда активность а() катионов металла
приблизительно равна их концентрации
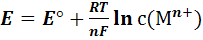 (7)
(7)
Для комнатной температуры
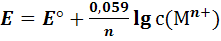 (8)
(8)
б) Газовые электроды, например, водородный
электрод, в том числе и стандартный водородный электрод. Потенциал обратимо
работающего газового водородного электрода определяется активностью ионов
водорода, т.е. величиной pH
раствора, и при комнатной температуре равен
E=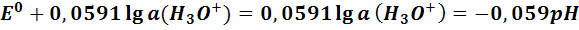 ,
,
поскольку для водородного электрода стандартный
потенциал принимается равным нулю (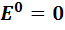 ),
а в соответствии с электродной реакцией
),
а в соответствии с электродной реакцией
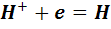
число электронов, участвующих в этой реакции,
равно единице: n=1.
в). Амальгамные электроды, представляющие собой
амальгаму металла, погруженную в раствор, содержащий катионы того же металла.
Потенциал таких электродов первого рода зависит от активности а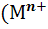 )
катионов металла в растворе и активности а(М) металла в амальгаме
)
катионов металла в растворе и активности а(М) металла в амальгаме
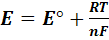 ln
ln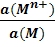
Амальгамные электроды обладают высокой
обратимостью.
Электроды второго рода обратимы по аниону.
Различают следующие виды электродов второго рода.
а) Металл, поверхность которого покрыта
малорастворимой солью этого же металла, погруженный в раствор, содержащий
анионы, входящие в состав этой малорастворимой соли. Примером могут служить
хлорсеребряный электрод 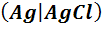 , KCl
или каломельный электрод
, KCl
или каломельный электрод 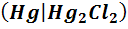 , KCl.
, KCl.
Хлорсеребряный электрод состоит из серебряной
проволоки, покрытой малорастворимой в воде солью AgCl,
погруженной в водный раствор хлорида калия. На хлорсеребряном электроде
протекает обратимая реакция
AgCl+ e
=Ag+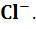
Каломельный электрод состоит из металлической
ртути, покрытой пастой малорастворимого хлорида ртути(I)
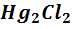 -
каломели, контактирующей с водным раствором хлорида калия. На каломельном
электроде протекает обратимая реакция
-
каломели, контактирующей с водным раствором хлорида калия. На каломельном
электроде протекает обратимая реакция
+2e=
2Hg+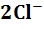 .
.
Реальный потенциал электродов второго рода
зависит от активности анионов и для обратимо работающего электрода, на котором
протекает реакция
MA + ne
= M + 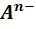
Описывается уравнениями Нернста (9)-(12).
В общем случае при любой приемлимой температуре T
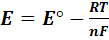 ln
ln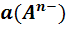 (9)
(9)
Для комнатной температуры
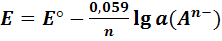 (10)
(10)
Для условий, в которых активность анионов
приблизительно равна их концентрации 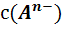
ln (11)\
Для комнатной температуры
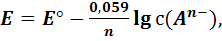 (12)
(12)
Так, например, реальные потенциалы 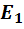 и
и
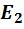 соответственно
хлорсеребряного и каломельного электродов при комнатной температуре можно
представить в виде
соответственно
хлорсеребряного и каломельного электродов при комнатной температуре можно
представить в виде
= 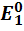 -
0,0591 lg a(
-
0,0591 lg a(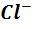 ),
),
=  -
0,0591 lg a().
-
0,0591 lg a().
В последнем случае в электродной реакции
участвуют 2 электрона (n=2)
и образуются также 2 хлорид - иона, поэтому множитель при логарифме равен также
0,059.
Электроды второго рода рассмотренного вида
обладают высокой обратимостью и стабильны в работе, поэтому их часто используют
в качестве электродов сравнения, способных устойчиво поддерживать постоянное
значение потенциала.
б) Газовые электроды второго рода, например,
хлоридный электрод Pt,
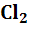 l
KCl. Газов электроды
второго рода в количественном потенциометрическом анализе применяются редко.
l
KCl. Газов электроды
второго рода в количественном потенциометрическом анализе применяются редко.
Окислительно - восстановительные электроды
состоят из инертного материала (платина, золото, вольфрам, титан и др.),
погруженного в раствор, содержащий окисленную Ox
и восстановленную Red
формы данного вещества. Существуют две разновидности окислительно -
восстановительных электродов:
а) электроды, потенциал которых не зависит от
активности ионов водорода, например, Pt
/Fe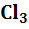 ,
Fe
,
Fe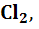 Pt/
Pt/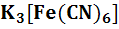 ,
,
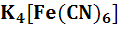 и
т.д.;
и
т.д.;
б) электроды, потенциал которых зависит от
активности ионов водорода, например, хингидронный электрод.
На окислительно - восстановительном электроде,
потенциал которого не зависит от активности ионов водорода, протекает обратимая
реакция
Ox + ne=
Red
Реальный потенциал такого окислительно -
восстановительного электрода зависит от активности окисленной и восстановленной
форм данного вещества и для обратимо работающего электрода описывается, в
зависимости от условий (по аналогии с вышерассмотренными потенциалами),
уравнениями Нернста (13)-(16):
ln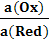 , (13)
, (13)
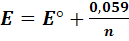 lg, (14)
lg, (14)
ln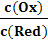 , (15)
, (15)
lg, (16)
где все обозначения - традиционные.
Если в электродной реакции участвуют ионы
водорода, то их активность (концентрацию) учитывают в соответствующих
уравнениях Нернста для каждого конкретного случая.
Мембранные, или ион - селективные, электроды -
электроды, обратимые по тем или иным ионам (катионам или анионам), сорбируемым
твердой или жидкой мембраной. Реальный потенциал таких электродов зависит от
активности тех ионов в растворе, которые сорбируются мембраной.
Мембранные электроды с твердой мембраной
содержат очень тонкую мембрану, по обе стороны которой находятся разные
растворы, содержащие одни и те же определяемые ионы, но с неодинаковой
концентрацией: раствор (стандартный) с точно известной концентрацией определяемых
ионов и анализируемый раствор с неизвестной концентрацией определяемых ионов.
Вследствие различной концентрации ионов в обоих растворах ионы на разных
сторонах мембраны сорбируются в неодинаковых количествах, неодинаков и
возникающий при сорбции ионов электрический разряд на разных сторонах мембраны.
Как результат возникает мембранная разность потенциалов.
Теория мембранных ион - селективных электродов
разработана довольно подробно.
Определение ионов с применением мембранных ион -
селективных электродов называют ионометрией.
Как уже говорилось выше, при потенциометрических
измерениях электрохимическая ячейка включает два электрода - индикаторный
электрод и электрод сравнения. Величина ЭДС, генерируемой в ячейке, равна
разности потенциалов этих двух электродов. Поскольку потенциал электрода
сравнения в условиях проведения потенциометрического определения остается
постоянным, то ЭДС зависит только от потенциала индикаторного электрода, т.е.
от активной (концентраций) тех или иных ионов в растворе. На этом и основана
потенциометрическая определение концентрации данного вещества в анализируемом
растворе.
Для потенциометрического определения
концентраций вещества в растворе применяют как прямую потенциометрию, так
потенциометрическое титрование, хотя второй способ используется намного чаще
первого.
.2 Прямая потенциометрия
Определение концентрации вещества в прямой
потенциометрии проводят обычно методом градуировочного графика или методом
добавок стандарта.
а). Метод градуировочного графика. Готовят серию
из 5 - 7 эталонных растворов с известным содержанием определяемого вещества.
Концентрация определяемого вещества и ионная сила в эталонных растворах не
должны сильно отличаться от концентрации и ионной силы анализируемого раствора:
в этих условиях уменьшаются ошибки определения. Ионную силу всех растворов
поддерживают постоянной введением индифферентного электролита. Эталонные
растворы последовательно вносят в электрохимическую (потенциометрическую)
ячейку. Обычно эта ячейка представляет собой стеклянный химический стакан, в
который помещают индикаторный электрод и электрод сравнения.
Измеряют ЭДС эталонных растворов, тщательно
промывая дистиллированной водой электроды и стакан перед заполнением ячейки
каждым эталонным раствором. По полученным данным строят градуировочный график в
координатах ЭДС - lg
c - концентрация
определяемого вещества в эталонном растворе. Обычно такой график представляет
собой прямую линию.
Затем в электрохимическую ячейку вносят (после
промывания ячейки дистиллированной водой) анализируемый раствор и измеряют ЭДС
ячейки. По градуировочному графику находят lg
c(X),
где c(X)
- концентрация определяемого вещества в анализируемом растворе.
б) Метод добавок стандарта. В электрохимическую
ячейку вносят известный объем V(X)
анализируемого раствора с концентрацией c(X)
и измеряют ЭДС ячейки.
Рассчитывают концентрацию c(X)
определяемого вещества в анализируемом растворе по формуле (17)
c(X)
= c(ст) 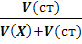 [
[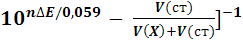 , (17)
, (17)
где 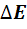 -
разность двух измеренных значений ЭДС, n
- число электронов, участвующих в электродной реакции.
-
разность двух измеренных значений ЭДС, n
- число электронов, участвующих в электродной реакции.
Применение прямой потенциометрии. Метод
применяется для определения концентрации ионов водорода (pH
растворов), анионов, ионов металлов (ионометрия).
Большую роль при использовании прямой
потенциометрии играют выбор подходящего индикаторного электрода и точнее
измерение равновесного потенциала.
При определении pH
растворов в качестве индикаторных используют электроды, потенциал которых
зависит от концентрации ионов водорода: стеклянный, водородный, хингидронный и
некоторые другие. Чаще применяют мембранный стеклянный электрод, обратимый по
ионам водорода. Потенциал такого стеклянного электрода определяется
концентрацией ионов водорода, поэтому ЭДС цепи, включающей стеклянный электрод
в качестве индикаторного, описывается при комнатной температуре уравнением
E = K
+ 0,059 pH
где постоянная K
зависит от материала мембраны, природы электрода сравнения. Стеклянный электрод
позволяет определять pH
в интервале pH = 0 - 10
(чаще - в диапазоне pH
= 2-10) и обладает высокой обратимостью и стабильностью в работе.
Хингидронный электрод, часто применявшийся
ранее, - это окислительно - восстановительный электрод, потенциал которого
зависит от концентрации ионов водорода. Он представляет собой платиновую
проволоку, погруженную в раствор кислоты (обычно HCl),
насыщенный хингидронном - эквимолекулярным соединением хинона с гидрохиноном
состава 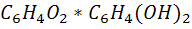 (темно
- зеленый порошок, малорастворимый в воде). Схематическое обозначение
хингидронного электрода: Pt
l хингидрон, HCl.
(темно
- зеленый порошок, малорастворимый в воде). Схематическое обозначение
хингидронного электрода: Pt
l хингидрон, HCl.
На хингидронном электроде протекает окислительно
- восстановительная реакция
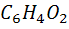 + 2
+ 2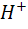 +2e
=
+2e
= 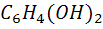
Потенциал хингидринного электрода при комнатной
температуре описывается формулой
E = 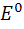 -
0,059 pH/
-
0,059 pH/
Хингидронный электрод позволяет измерять pH
растворов в интервале pH=0-8,5.
При pH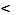 0
хингидрон гидролитически расщепляется: при pH
0
хингидрон гидролитически расщепляется: при pH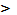 8,5
гидрохинон, являющийся слабой кислотой, вступает в реакцию нейтрализации.
8,5
гидрохинон, являющийся слабой кислотой, вступает в реакцию нейтрализации.
Хингидронный электрод нельзя применять в
присутствии сильных окислителей и восстановителей.
Мембранные ион - селективные электроды
используют, как уже отмечалось выше, в ионометрии в качестве индикаторных для
определения различных катионов (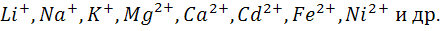 ) и анионов (
) и анионов (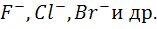 ).
).
К достоинствам прямой потенциометрии относятся
простота и быстрота проведения изверений, для измерений требуются небольшие
объемы растворов.
.3 Потенциометрическое титрование
Потенциометрическое титрование - способ
определения объема титранта, затраченного на титрование определяемого вещества
в анализируемом растворе, путем измерения ЭДС (в процессе титрования) с помощью
гальванической цепи, составленной из индикаторного электрода и электрода
сравнения. При потенциометрическом титровании анализируемый раствор,
находящийся в электрохимической ячейке, титруют подходящим титрантом, фиксируя
конец титрования по резкому изменению ЭДС измеряемой цепи - потенциала
индикаторного электрода, который зависит от концентрации соответствующих ионов
и резко изменяется в точке эквивалентности.
Измеряют изменение потенциала индикаторного
электрода в процессе титрования в зависимости от объема прибавленного титранта.
По полученным данным строят кривую потенциометрического титрования и по этой
кривой определяют объем израсходованного титранта а ТЭ.
При потенциометрическом титровании не требуется
использование индикаторов, изменяющих окраску вблизи ТЭ.
Электродную пару (электрод сравнения и
индикаторный электрод) составляют так, чтобы потенциал индикаторного электрода
зависел от концентрации ионов, участвующих или образующихся в реакции,
протекающей при титровании. Потенциал электрода сравнения во время титрования
должен оставаться постоянным. Оба электрода устанавливают непосредственно в
электрохимической ячейке или же помещают в отдельные сосуды с токопроводящими
растворами (индикаторный электрод - в анализируемый раствор), которые соединяют
электролитическим мостиком, заполненным индифферентным электролитом.
Титрант прибавляют равными порциями, каждый раз
измеряя разность потенциалов. В конце титрования (вблизи ТЭ) титрант прибавляют
по каплям, также измеряя разность потенциалов после прибавления очередной
порции титранта.
Разность потенциалов между электродами измеряют,
используя высокоомные потенциометры. Кривые потенциометрического титрования.
Кривая потенциометрического титрования -
графическое изображение изменения ЭДС электрохимической ячейки в зависимости от
объема прибавленного титранта.
Кривые потенциометрического титрирования строят
в различных координатах:
кривые титрования в координатах E
- V(T)
(иногда такие кривые называют интегральными кривыми титрования);
дифференциальные кривые титрования - в
координатах dE/dV
- V(T)
и 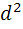 E/d
E/d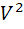 - V(T);
- V(T);
кривые титрования по методу Грана - в
координатах 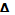 V/E
- V(T),
где E - ЭДС
потенциометрической ячейки, V
и V(T)
- объем прибавленного титранта, E
- изменение потенциала, соответствующее прибавлению V
титранта.
V/E
- V(T),
где E - ЭДС
потенциометрической ячейки, V
и V(T)
- объем прибавленного титранта, E
- изменение потенциала, соответствующее прибавлению V
титранта.
Объем титранта V(ТЭ),
прибавленного в ТЭ, можно определить не только графически, но и расчетным путем
по форме (18):
V(ТЭ) +
(
+
(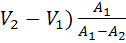 (18)
(18)
где 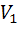 -
объем прибавленного титранта, соответствующий последнему измерению до ТЭ,
-
объем прибавленного титранта, соответствующий последнему измерению до ТЭ, 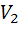 -
объем прибавленного титранта, соответствующий первому измерению после ТЭ
-
объем прибавленного титранта, соответствующий первому измерению после ТЭ
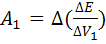 ,
, 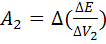
В табл. 1 в качестве примера (фармакопейного)
приведены результаты определений и расчетов при потенциометрическом титровании.
Таблица 1. Пример обработки результатов
потенциометрического титрования
|
V, мл
|
∆V,мл
|
Е,
мВ
|
∆Е,
мВ
|
∆Е/∆V
|
∆(∆E/∆V)=A
|
|
5,00
|
|
250
|
|
|
|
|
0,10
|
|
13
|
130
|
|
|
5,10
|
|
263
|
|
|
+150
|
|
0,10
|
|
28
|
280
|
|
|
5,20
|
|
291
|
|
|
+720
|
|
0,10
|
|
100
|
1000
|
|
|
5,30
|
|
391
|
|
|
-450
|
|
0,10
|
|
55
|
550
|
|
|
5,40
|
|
446
|
|
|
-330
|
|
0,10
|
|
22
|
220
|
|
|
5,50
|
|
468
|
|
|
-120
|
|
0.10
|
|
10
|
100
|
|
|
5,60
|
|
478
|
|
|
|
Рассчитаем по формуле (18) величину V(ТЭ)
с использованием данных табл.1. Очевидно, что максимальное значение 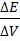 =1000.
Следовательно
=1000.
Следовательно
=5,20 и =5,30;
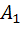 =720,
=720,
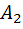 =-450
=-450
Отсюда:
V(ТЭ)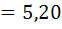 +
(
+
(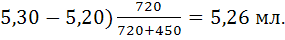
Применение потенциометрического титрования.
Метод универсальный, его можно применять для индикации конца титрования во всех
типах титрования: кислотно-основном, окислительно-восстановительном,
комплексиметрическом, осадительном, при титровании в неводных средах. В
качестве индикаторных используют стеклянный, ртутный, ионселективные,
платиновый, серебряный электроды, а в качестве электродов сравнения -
каломельный, хлорсеребряный, стеклянный.
Метод обладает высокой точностью, большой
чувствительностью: позволяет проводить титрование в мутных, окрашенных,
неводных средах, раздельно определять компоненты смеси в одном анализируемом
растворе, например, раздельно определять хлорид- и иодид-ионы при
аргентометрическом титровании.
Методами потенциометрического титрования
анализируют многие лекарственные вещества, например, аскорбиновую кислоту,
сульфамидные препараты, барбитураты, алкалоиды и др.
3. Кондуктометрический анализ (кондуктометрия)
Основателем кондуктометрического анализа
считается немецкий физик и физико-химик Ф.В.Г. Кольрауш (1840-1910), который
впервые в 1885 г. предложил уравнение, устанавливающее связь между
электропроводностью растворов сильных электролитов и их концентрацией. В
середине 40-х гг. ХХ в. был разработан метод высокочастотного кондуктометрического
титрования. С начала 60-х гг. стали использовать кондуктометрические детекторы
в жидкостной хроматографии.
.1. Принцип метода. Основные понятия
Кондуктометрический анализ (кондуктометрия)
основан на использовании зависимости между электропроводностью (электрической
проводимостью) растворов электролитов и их концентрацией.
Об электропроводности растворов электролитов-
проводников второго рода - судят на основании измерения их электрического
сопротивления в электрохимической ячейке , которая представляет собой
стеклянный сосуд (стакан) с двумя впаянными в него электродами, между которыми
и находится испытуемый раствор электролита. Через ячейку пропускают переменный
электрический ток. Электроды чаще всего изготовляют из металлической платины, которую
для увеличения поверхности электродов покрывают слоем губчатой платины путем
электрохимического осаждения из растворов платиновых соединений (электроды из
платинированной платины).
Во избежание осложнений, связанных с процессами
электролиза и поляризации электродов, кондуктометрические измерения проводят в
переменном электрическом поле.
Электрическое сопротивление R
слоя раствора электролита между электродами, как и электрическое сопротивление
проводников первого рода, прямо пропорционально длине (толщине) l
этого слоя и обратно пропорционально площади S
поверхности электродов:
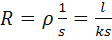 (19)
(19)
где коэффициент пропорциональности ρ
называют
удельным электрическим сопротивлением, а обратную величину к=1/ρ
- удельной
электропроводностью (удельной электрической проводимостью).
Так как электрическое сопротивление R
измеряют в омах, а толщину l
слоя раствора электролита - в см, площадь S
поверхности электродов - в см2, то удельную электропроводность к измеряют в
единицах Ом-1 · см-1, или, поскольку Ом-1 - это сименс (См), то - в единицах См
· см-1 .
По физическому смыслу удельная
электропроводность - это электрическая проводимость слоя электролита ,
находящегося между сторонами куба с длиной сторон 1 см, численно равная току,
проходящему через слой раствора электролита с площадью поперечного сечения 1
см2 при градиенте приложенного электрического потенциала 1 В/см.
Удельная электропроводность зависит от природы
электролита и растворителя, от концентрации раствора, от температуры.
С увеличением концентрации раствора электролита
его удельная электропроводность вначале возрастает, затем проходит через
максимум, после чего уменьшается. Такой характер изменения удельной
электропроводности обусловлен следующими причинами. Вначале с увеличением
концентрации электролита возрастает число ионов - токпереносящих частиц - как
для сильных, так и для слабых электролитов. Поэтому электропроводность раствора
(переходящий через него электрический ток) повышается. Затем по мере роста
концентрации раствора увеличиваются его вязкость (понижающая скорости движения
ионов) и электростатические взаимодействия между ионами, что препятствует
возрастанию электрического тока и при достаточно больших концентрациях
способствует его уменьшению.
В растворах слабых электролитов с ростом
концентрации понижается степень диссоциации молекул электролита, что приводит к
уменьшению числа ионов - токпроводящих частиц - и к понижению удельной
электропроводности. В растворах сильных электролитов при высоких концентрациях
возможно образование ионных ассоциатов (ионных двойников, тройников и т.п.),
что также благоприятствует падению электропроводности.
Суммарное действие вышеуказанных фактор и
приводит к описанному изменению характера удельной электропроводности растворов
электролитов.
Удельная электропроводность растворов
электролитов увеличивается с ростом температуры вследствие понижения вязкости
растворов, что приводит к повышению скорости движения ионов, а для слабых
электролитов - также и к увеличению степени их ионизации (диссоциации на ионы).
Поэтому количественные кондуктометрические измерения необходимо проводить при
постоянной температуре, термостатируя кондуктометрическую ячейку.
Кроме удельной электропроводности в
кондуктометрии используют эквивалентную электропроводность λ
и молярную электропроводность µ .
По физическому смыслу эквивалентная
электропроводность λ
- это электрическая проводимость слоя раствора электролита толщиной 1 см,
находящегося между одинаковыми электродами с такой площадью, чтобы объем раствора
электролита, заключенного между ними, содержал 1 г-экв растворенного вещества.
При этом за молярную массу эквивалента принимается молярная масса одинаковых
частиц с единичным зарядовым числом («зарядом»), например, Н+, Br+,
1/2Ca2+, 1/3 Fe3+
и т.д.
Таким образом, если удельная электропроводность
характеризует электрическую проводимость единичного объема раствора электролита
(1 см3) , в котором содержание электролита может быть различным, то
эквивалентная электропроводность характеризует электрическую проводимость
раствора, содержащего один эквивалент электролита, причем объем раствора может
быть различным.
Эквивалентная электропроводность увеличивается с
уменьшением концентрации раствора электролита. Максимальное значение
эквивалентной электропроводности достигается при бесконечном разбавлении
раствора. Эквивалентная электропроводность, как и удельная, возрастает с
повышением температуры.
Эквивалентная электропроводность λ
связана с удельной электропроводностью к соотношением
λ= 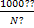 (20)
(20)
где с- молярная концентрация эквивалента, и
изменяется в единицах Ом-1 · см2 (или См· моль-1 · см2, Ом-1 · г-экв-1· см2).
В соответствии с законом независимого движения
ионов Кольрауша эквивалентная электропроводность раствора электролита при
бесконечном разбавлении λ∞
(или λ0) равна
сумме предельных подвижностей катиона λ0+
и аниона λ0- , то есть
их подвижностей при бесконечном разбавлении раствора
λ∞ = λ0+
+ λ0 (21)
Предельная подвижность иона в данном
растворителе при заданной температуре - эктраполированная величина. Она
является константой, характеризующей электрическую подвижность данного иона.
потенциометрия амперометрический титрование
В таблице 2 приведены в качестве примера
предельные подвижности некоторых ионов в водных растворах при 25 °С
|
Катион
|
λ0, См-1 · см2
|
Анион
|
λ0, См· моль-1 ·
см2
|
|
H+
|
349,8
|
OH¯
|
198,3
|
|
Rb+
|
77,5
|
1/4[Fe(CN)6]4-
|
111
|
|
Cs+
|
77,2
|
1/3[Fe(CN)6]3-
|
99,1
|
|
NH4+
|
73,7
|
½CrO42-
|
85
|
|
K+
|
73,5
|
½SO42-
|
80,8
|
|
1/2Rb2+
|
70
|
Br¯
|
78,1
|
|
½Fe3+
|
68
|
I¯
|
76,8
|
|
½Ba2+
|
63,6
|
Cl¯
|
76,4
|
|
Ag+
|
62,2
|
NO3¯
|
71,5
|
|
½Ca2+
|
59,5
|
½CO32-
|
69,3
|
|
½Zn2+
|
54
|
1/3PO43-
|
69
|
|
½Fe2+
|
53,5
|
ClO4-
|
67,3
|
|
½Mg2+
|
53
|
F¯
|
55,4
|
|
Na+
|
50,11
|
HCO3-
|
44,5
|
|
Li+
|
38,68
|
CH3COO-
|
40,9
|
Данные таблицы 3 иллюстрируют влияние
температуры на предельную подвижность ионов в водных растворах, а данные
таблицы 4 - влияние природы растворителя.
Подвижность ионов λ в
растворах с конечной концентрацией не являются постоянными и зависят от
концентрации раствора (таблица 5): с ростом концентрации раствора подвижность
ионов уменьшается.
Молярная электропроводность µ раствора
электролита определяется аналогично его эквивалентной электропроводности
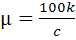 (22)
(22)
где с - молярная концентрация электролита в
растворе. Она измеряется в тех же единицах, что и эквивалентная
электропроводность.
Таблица 3. Значения предельной подвижности λ0
некоторых ионов в водных растворах при разных температурах
|
Ион
|
λ0, См· моль-1 ·
см2 при температуре °С
|
|
0
|
18
|
25
|
55
|
100
|
|
H+
|
225
|
315
|
349,8
|
483,1
|
630
|
|
Li+
|
19,4
|
32,8
|
38,5
|
68,7
|
115
|
|
Na+
|
26,5
|
42,8
|
50,1
|
86,8
|
145
|
|
K+
|
40,7
|
63,9
|
73,5
|
119,2
|
195
|
|
Rb+
|
43,9
|
66,5
|
77,8
|
124,2
|
-
|
|
Cs+
|
44
|
67
|
77,2
|
123,6
|
200
|
|
Ag+
|
33,1
|
53,5
|
62,2
|
-
|
175
|
|
½Ca2+
|
31,2
|
50,7
|
59,5
|
-
|
180
|
|
½Ba2+
|
34
|
54,6
|
63,6
|
-
|
195
|
|
OH¯
|
105
|
198,3
|
-
|
450
|
|
Cl¯
|
41
|
66
|
76,4
|
126,4
|
212
|
|
Br¯
|
42,6
|
68
|
78,1
|
127,8
|
-
|
|
I¯
|
41,4
|
66,5
|
76,8
|
125,4
|
-
|
|
½SO42-
|
41
|
68,4
|
80
|
-
|
260
|
|
CH3COO-
|
20,3
|
34
|
40,9
|
-
|
130
|
Таблица 4. Значения предельной подвижности λ0
некоторых ионов в разных растворителях при 25 °С
|
Ион
|
λ0, См· моль-1 ·
см2
|
|
Вода
|
Метанол
|
Этанол
|
Ацетон
|
Нитробензол
|
|
H+
|
349,8
|
143
|
59,5
|
88
|
23
|
|
Na+
|
50,11
|
45,2
|
18,7
|
80,0
|
17,2
|
|
K+
|
73,5
|
52,4
|
22,0
|
82,0
|
19,2
|
|
Ag+
|
62,2
|
50,3
|
17,5
|
88
|
18,6
|
|
Cl¯
|
76,4
|
52,9
|
24,3
|
111,0
|
17,3
|
|
Br¯
|
78,1
|
55,5
|
25,8
|
113,0
|
19,6
|
Таблица 5. Значения подвижностей ионов λ
при
различной концентрации растворов с
|
c, моль/л
|
λ, См· моль-1 ·
см2
|
|
H+
|
Li+
|
Na+
|
K+
|
OH¯
|
Cl¯
|
F¯
|
|
0,001
|
314,2
|
32,5
|
42,3
|
63,1
|
171
|
65,0
|
45,5
|
|
0,01
|
307,0
|
30,1
|
40,0
|
60,4
|
167
|
61,5
|
43,2
|
|
0,1
|
294,4
|
27,5
|
35,4
|
55,4
|
157
|
55,8
|
38,0
|
Уравнения (20) и (22), связывающие
электропроводность раствора электролита с его концентрацией, лежат в основе
количественного кондуктометрического анализа.
В кондуктометрическом анализе применяют как
прямую кондуктометрию, так и кондуктометрическое титрование.
.2 Прямая кондутометрия
В прямой кондуктометрии концентрацию вещества в
анализируемом растворе определяют по результатам измерений удельной
электропроводности этого раствора. При обработке данных измерений используют
два метода: расчетный метод и метод градуировочного графика.
Расчетный метод. В соответствии с уравнением
(20) молярная концентрация эквивалента с электролита в растворе может быть
рассчитана, если известны удельная электропроводность к и эквивалентная
электропроводность λ:
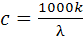 (23)
(23)
Удельную электропроводность определяют
экспериментально на основании измерения электрического сопротивления
термостатированной кондуктометрической ячейки.
Эквивалентная электропроводность раствора λ
равна
сумме подвижностей катиона λ+ и
аниона λ-:
λ = λ+ + λ
Если подвижности катиона и аниона известны, то
концентрацию с можно рассчитать по формуле (24)
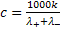
Так поступают при определении методом прямой
кондуктометрии концентрации малорастворимого электролита в его насыщенном
растворе (сульфаты кальция, бария; галогениды серебра и др.)
Рассмотрим, например, определение растворимости
хлорида серебра AgCl
в воде, то есть молярной концентрации насыщенного раствора, методом прямой
кондуктометрии при 25°C.
Согласно формуле (24) имеем
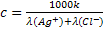
Удельная электропроводность к хлорида серебра в
водном растворе равна разности определяемых экспериментально удельной
электропроводности к1 анализируемого раствора хлорида серебра и удельной
электропроводности к(Н2О) чистой воды:
к=к1-к(Н2О).
Поскольку растворимость хлорида серебра в воде
незначительна, то подвижности катиона серебра и хлорид-иона можно принять
равными их предельным подвижностям, то есть λ(Ag+)=
λ0 (Ag+)=62,2,λ(Cl-)
= = λ0(Cl-)=76,4
(см. табл. 2). Тогда
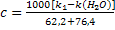 (24)
(24)
Метод градуировочного графика. Готовят серию
эталонных растворов, каждый из которых содержит точно известную концентрацию
определяемого вещества, измеряют их удельную электропроводность при постоянной
температуре в термостатируемой кондуктометрической ячейке. По полученным данным
строят градуировочный график, откладывая по оси абсцисс концентрацию эталонных
растворов, а по оси ординат - значения удельной электропроводности. В
соответствии с уравнением (24) построенный график в относительно небольшом
диапазоне изменения концентраций обычно представляет собой прямую линию.
В широком интервале изменения концентраций,
когда подвижности катиона и аниона, входящие в уравнение (24), могут заметно
изменяться, наблюдаются отклонения от линейной зависимости.
Затем строго в тех же условиях измеряют удельную
электропроводность к(Х)определяемого электролита в анализируемом растворе с
неизвестной концентрацией с(Х) и по графику находят искомую величину с(Х).
Так определяют , например, содержание бария в
баритовой воде - насыщенном растворе гидроксида бария.
Применение прямой кондуктометрии.
Методу прямой кондуктометрии присущи простота, высокая чувствительность(до ≈10-4
моль/л), сравнительно малая погрешность определения - до 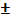 2%. Однако метод малоселективен.
2%. Однако метод малоселективен.
Прямая кондуктометрия имеет
ограниченное применение в анализе. Она используется для определения
растворимости малорастворимых электролитов, для контроля качества
дистиллированной воды и жидких пищевых продуктов (молока, напитков и др.), для
определения общего содержания солей в минеральной, морской, речной воде и в
некоторых других случаях.
3.3 Кондуктометрическое титрование
При кондуктометрическом титровании за ходом
титрования следят по изменению электропроводности анализируемого раствора,
находящегося в кондуктометрической ячейке между двумя инертными
электродами(обычно из платинированной платины). По полученным данным
вычерчивают кривую кондуктометрического титрования, отражающую зависимость
электропроводности титруемого раствора от объема прибавленного титранта.
Конечную точку титрования находят чаще всего экстраполяцией участков кривой
титрования в области изменения ее наклона. При этом не требуется применение
индикаторов, изменяющих окраску вблизи ТЭ.
В кондуктометрическом титровании используют
различные типы реакций: кислотно-основные, окислительно-восстановительные,
осадительные, процессы комплексообразования.
В зависимости от того, какие ионы вступают в
реакцию или образуются при протекании реакции, кривые кондуктометрического
титрования могут быть различными.
NaOH+HCl=NaCl+H2ОX
В ионном виде
(Na+
+ OH-)+(H+
+ Cl-)=(Na+
+ Cl-)+H2O
По мере титрования кислоты электропроводность
титруемого раствора сильно уменьшается, так как в реакции расходуются ионы
водорода Н+ с высокой подвижностью. После ТЭ электропроводность титруемого
раствора также существенно растет, поскольку подвижность гидроксильных групп,
вводимых вместе с титрантом в титруемый раствор, также достаточно высока, хотя
и меньше подвижности ионов водорода.
NaHCO3+HCl=NaCl+H2O+CO2X
В ионном виде
(Na+
+ HCO3-)+(H+
+ Cl-)=(Na+
+ Cl-)+H2O+CO2
При титровании нейтрализуются ионы водорода с
высокой подвижностью, поэтому электропроводность титруемого раствора до ТЭ
резко уменьшается. После ТЭ электропроводность раствора несколько повышается за
счет ионов прибавляемого избыточного титранта.
Примером может служить титрование раствора
хлорида кальция раствором щелочи:
NaOH+CaCl2=Ca(OH)2+2NaCl
В ионном виде
(Na+
+ OH-) + (Ca2+
+2Cl-)=Ca(OH)2
+ 2(Na+ +Cl-)
При титровании до ТЭ электропроводность
титруемого раствора меняется мало, поскольку в реакции расходуются катионы
кальция, но в растворе появляются катионы натрия. После ТЭ электропроводность
раствора увеличивается за счет гидроксид-ионов с высокой подвижностью, вводимых
в титруемый раствор вместе с избыточным титрантом.
Применение кондуктометрического титрования.
Метод кондуктометрического титрования обладает рядом достоинств. Титрование
можно проводить в мутных, окрашенных, непрозрачных средах.
Чувствительность метода довольно низка - до ≈10-4
моль/л; ошибка определения составляет от 0,1 до 2%. Анализ можно
автоматизировать.
К недостаткам метода относится малая
селективность.
Понятие о высокочастотном (радиочастотном)
кондуктометрическом титровании. За ходом титрования следят с помощью
модифицированной переменно-токовой кондуктометрической техники, в которой
частота переменного тока может достигать порядка миллиона колебаний в секунду.
Обычно электроды помещают на внешней стороне
сосуда (кондуктометрической ячейки) для титрования, так что они не
соприкасаются с титрируемым раствором.
По результатам измерений вычерчивают кривую
кондуктометрического титрования. Конечную точку титрования находят
экстраполяцией участков кривой титрования в области изменения ее наклона.
4. Полярографический анализ (полярография,
прямая вольтамперометрия)
.1. Сущность метода
Полярографический анализ (полярография) основан
на использовании следующих зависимостей между электрическими параметрами
электрохимической (в данном случае - полярографической) ячейки, к которой
прилагается внешний потенциал, и свойствами содержащегося в ней анализируемого
раствора.
а) В качественном полярографическом анализе
используют свяп. между величиной приложенного на микроэлектроде внешнего
электрического потенциала, при котором наблюдается восстановление (или
окисление) анализируемого вещества на микроэлектроде в данных условиях, и
природой восстанавливающегося (или окисляющегося) вещества.
б) В количественном полярографическом анализе
используют связь между величиной диффузионного электрического тока,
устанавливающегося в полярографической ячейке после достижения определенного
значения приложенного на микроэлектроде электрического потенциала, и
концентрацией определяемого (восстанавливающегося или окисляющегося) вещества в
анализируемом растворе.
Электрические параметры - величину приложенного
электрического потенциала и величину диффузионного тока - определяют при
анализе получаемых поляризационных, или вольт-амперных, кривых, отражающих
графически зависимость электрического тока в полярографической ячейке от
величины приложенного потенциала микроэлектрода. Поэтому полярографию иногда
называют прямой волыпамперометрией.
Классический полярографический метод анализа с
применением ртутного капающего (капельного) электрода был разработан и
предложен в 1922 г. чешским ученым Ярославом Гейровским (1890-1967), хотя сам
ртутный капающий электрод применялся чешским физиком Б. Кучерой еще в 1903 г. В
1925 г. Я. Гейровский и М. Шиката сконструировали первый полярограф,
позволивший автоматически регистрировать поляризационные кривые. В дальнейшем
были разработаны различные модификации полярографического метода.
Рассмотрим кратко сущность классической
полярографии, основанной на использовании ртутного капающего микроэлектрода.
В сосуде, в который вносится анализируемый
раствор с определяемым веществом, имеются два электрода -- микрокатод и
макроанод, подключенные к внешнему источнику постоянного электрического тока.
На микрокатод прилагается постепенно возрастающий по абсолютной величине
отрицательный электрический потенциал.
Микрокатод, помещаемый в ячейку, представляет
собой стеклянный капилляр, заполненный жидкой ртутью, соединенный шлангом с
резервуаром, содержащим жидкую ртуть. Из капилляра медленно, по каплям вытекает
ртуть (поэтому такой электрод и называют капающим ртутным электродом),
поступающая из резервуара. Макроэлектродом - анодом - в рассматриваемом
варианте служит жидкая ртуть на дне сосуда. Поверх-ность ртутного капающего
микроэлектрода, т.е. ртутной капли, очень мала, тогда как поверхность анода -
большая.
В качестве макроэлектрода - анода на практике
наиболее часто применяют не ртутный, а насыщенный каломельный электрод, по отношению
к которому обычно и измеряют потенциал капающего ртутного микрокатода.
Анализируемый раствор содержит кроме
определяемого вещества также индифферентный - фоновый -- электролит (фон), ионы
которого не разряжаются на электродах в условиях проведения полярографического
анализа, а служат в качестве токопроводящих частиц для поддержания определенной
величины электрического тока в ячейке, когда определяемое вещество еще не
восстанавливается на микрокатоде.
При постепенном повышении приложенного
потенциала вначале электрический ток, обусловленный присутствием ионов фонового
электролита, растворенного кислорода и восстанавливающихся возможных примесей,
возрастает очень медленно - остается почти постоянным. Это - так называемый
остаточный ток. При некотором значении потенциала, называемого потенциалом
выделения, ток в ячейке резко возрастает {фарадеевский ток) и при сравнительно
небольшом дальнейшем повышении потенциала достигает максимального, возможного в
данных условиях значения, после чего снова изменяется мало. Это - так
называемый предельный ток. Разность между предельным и остаточным током
составляет диффузионный ток iD
Ртутная капля по мере ее формирования на конце
капилляра (вытекания из капилляра) остается заряженной отрицательно до тех пор,
пока она не оторвется от капилляра и окруженной раствором.
В поверхностном приэлектродном слое раствора
около ртутной капли находятся катионы Мn+
определяемого вещества, которые разряжаются на ртутной капле по схеме
Mn++ nе
+ Hg = M(Hg)
При достижении величины потенциала выделения,
характерного и специфичного только для данных катионов. После достижения
потенциала выделения эти катионы очень быстро разряжаются на ртутной капле,
поэтому электрический ток в полярографической ячейке резко возрастает.
Концентрация катионов Мn+
в поверхностном приэлектродном слое раствора около ртутной капли столь же резко
понижается (поскольку катионы восстанавливаются до металла) и становится меньше
их концентрации в объеме анализируемого раствора.
Транспорт катионов Мn+
в поверхностный при электродный слой раствора, окружающий ртутную каплю,
поддерживается за счет диффузии катионов Мn+
из объема раствора и зависит от скорости их диффузии. При дальнейшем повышении
приложенного потенциала ртутных капель достигается максимально возможная скорость
диффузии, которая остается практически постоянной, так что при данной
концентрации катионов в растворе ток в полярографической ячейке также достигает
максимального, практически постоянного значения. Устанавливается стационарный
предельный ток.
Если концентрацию катионов Мn+
в растворе увеличить, то увеличится и число стационарно восстанавливающихся
катионов, т.е. возрастает предельный и диффузионный ток. Таким образом,
величина предельного и диффузионного тока в полярографической ячейке зависит от
концентрации определяемого вещества (восстанавливающегося на ртутном капающем
электроде) в анализируемом растворе, тогда как значение потенциала выделения
зависит от природы разряжающихся частиц и не зависит от их концентрации.
Количество катионов, восстанавливающихся на
ртутном капающем электроде, незначительно и практически не сказывается на
изменении концентрации этих катионов в объеме раствора.
Вещество, разряжающееся на микрокатоде, называют
деполяризатором, полярографически активным, электроактивным. Эти названия
условны, поскольку вещество может быть полярографически неактивно при одном
потенциале и полярографически активно при более высоком потенциале.
Вместо потенциала выделения на практике
определяют потенциал полуволны E1/2,
соответствующий половине величины диффузионного тока.
Полученную полярографическую кривую называют,
как отмечалось выше, полярограммой, или полярографической волной. При
использовании капающего ртутного электрода на полярограмме наблюдаются о
цилляции тока (его периодическое небольшое увеличение и уменьшение). Каждая
такая осцилляция соответствует возникновению, росту отрыву ртутной капли от
капилляра микрокатода.
В некоторых современных полярографах
электрический ток измеряется только в конце каплеобразования, что позволяет
устранить осциллции на полярограмме.
На ртутном капающем микрокатоде происходит
постоянное возобновление ртутных капель, на поверхности которых осуществляется
разряд к тионов. Поверхность такого электрода все время обновляется за счет
новы ртутных капель, что исключает изменение его свойств вследствие прокающих
на нем электрохимических процессов и составляет одно из главных достоинств
использования ртутного капающего электрода.
Таким образом, при проведении качественного и
количественного полярографического анализа используют два параметра, получаемые
при рассмотрении полярограмм: потенциал полуволны Е1/2 и величину диффузионного
тока /D (высоту h
полярографической волны).
Как указывалось выше, потенциал полуволны Е1/2
характеризует природу восстанавливающегося катиона и не зависит от его
концентрации. Для разных катионов, полярографируемых в одних и тех же условиях,
он неодинаков, что и позволяет открывать различные катионы в растворе.
Потенциал полуволны Е1/2 зависит, кроме природы самого восстанавливающегося вещества,
от природы растворителя, фонового электролита, состава и рН анализируемого
раствора, присутствия веществ комплексообразователей, температуры. Величина
потенциала полуволны открываемого или определяемого катиона должна быть меньше
величины потенциала разряда ионов фонового электролита.
В табл. 10.6 приведены в качестве примера
значения потенциала полуволны для некоторых катионов с указанием состава фона.
Из данных табл. 10.6 следует, что состав фона и рН раствора существенно влияют
на величину потенциала полуволны.
Если в анализируемом растворе присутствуют
несколько восстанавливающихся веществ, причем разность между значениями их
потенциалов полуволны составляет не менее 0,2 В, то на полярограмме наблюдаются
несколько волн, каждая из которых отвечает тому или иному восстанавливающемуся
веществу.
Таблица 10.6. Значения потенциала полуволны Е1/2
некоторых катионов металлов (относительно потенциала насыщенного каломельного
электрода)
|
Электродная
|
£|/2>
в
|
Фоновый
электролит (состав фона)
|
|
реакция
|
|
|
|
As3+ + Ъе =
As
|
-0,7
|
1
моль/л H-.SCX,
+ 0,01 % желатина
|
|
Cd2+ + 2е =
Cd
|
-0,60
|
0,1
моль/л HCI
|
|
Cd2+ + 2е =
Cd
|
-0,79
|
6
моль/л HCI
|
|
Со2+
+ 2е = Со
|
-1,03
|
1
МОЛЬ/Л
|
|
Си2+
+ 2е = Си
|
0
|
0,5
моль/л H2S04
+ 0,01 % желатина
|
|
Си2+
+ 2е = Си
|
-0,38
|
1
моль/л Na2C4H406.
рН = 12
|
|
Fe2+ + 2e = Fe
|
-1,37
|
1
моль/л НСЮ4, рН = 0-2
|
|
Мп2+
+ 2е = Мп
|
-1,54
|
0,5
моль/л NH3 + 0,5
моль/л NH„C1
|
|
Ni2+ + 2e = Ni
|
-1,1
|
НС104,
РН = 0-2, 1 моль/л КС1
|
|
Ni2+ + 2e = Ni
|
-1,06
|
1
моль/л NH3 + 0,2
моль/л NH4CI
+ 0,005% желатина
|
|
Zn2+ + 2е =
Zn
|
-1,02
|
1
моль/л К<~!1
|
|
Zn2+ +
2e
= Zn
|
-1,33
|
1
моль/л NH, + 0,2
моль/л NH4C1
+ 0,005% желатина
|
|
Zn2+ +
2e
= Zn
|
-1,49
|
1
моль/л NaOH
|
Величина среднего диффузионного тока iD
определяется уравнением Ильковича
iD = Kc
где К- коэффициент пропорциональности, с -
концентрация (ммоль/л) полярографически активного вещества-деполяризатора; iD
измеряют в микроамперах как разность между предельным током и остаточным током.
Коэффициент пропорциональности К в уравнении
Ильковича зависит от целого ряда параметров и равен
K = 607nD1/2m2/3
τ 1/6
где n
- число электронов, принимающих участие в электродной
окислительно-восстановительной реакции; D
- коэффициент диффузии восстанавливающегося вещества (см2/с); m-
масса ртути, вытекающей из капилляра в секунду (мг); τ
- время
образования (в секундах) капли ртути при потенциале полуволны (обычно оно
составляет 3-5 с).
Так как коэффициент диффузии D
зависит от температуры, то и коэффициент пропорциональности К в уравнении
Ильковича изменяется при изменении температуры. Для водных растворов в температурном
интервале 20-50 °С коэффициент диффузии полярографически активных
веществ-деполяризаторов увеличивается примерно на 3% при росте температуры на
один градус, что и приводит к повышению среднего диффузионного тока iD
на ~1-2%. Поэтому полярографирование проводят при постоянной температуре,
термостатируя полярографическую ячейку обычно при 25 ± 0,5 °С.
Масса ртути т и время каплеобразования τ
зависят
от характеристик ртутного капающего электрода и высоты столбика ртути в
капилляре и в резервуаре, связанном с капилляром.
Стеклянный капилляр ртутного капающего
микроэлектрода обычно имеет внешний диаметр 3-7 мм, внутренний - от 0,03 до
0,05 мм, длину 6-15 см. Высота ртутного столбика от нижнего конца капилляра до
верхнего уровня поверхности ртути в резервуаре составляет 40-80 cmi
Содержание индифферентного электролита в
анализируемом полирографируемом растворе должно примерно в 100 раз превышать
содержание определяемого вещества-деполяризатора, причем ионы фонового
электролита не должны разряжаться в условиях проведения полярографирования до
разряда полярографически активного вещества.
Полярографирование проводят с использованием в
качестве растио рителя воды, водно-органических смесей (вода - этанол, вода -
ацетон, вода - диметилформамид и др.) и неводных сред (этанол, ацетон, димс
тилформамид, диметилсульфоксид и т.д.).
До начала полярографирования через анализируемый
раствор про пускают ток инертного газа (азота, аргона и др.) для удаления
растворимого кислорода, который также дает полярографическую волну вследствие
восстановления по схеме
О2 + 2Н+ + 2е = Н202
Н202 + 2Н+ + 2е = 2Н20
Иногда - в случае щелочных растворов - вместо
пропускания тока инертного газа в анализируемый раствор прибавляют небольшое
количество активного восстановителя - сульфита натрия, метола, которые
связывают растворенный кислород, реагируя с ним.
.2 Количественный полярографический анализ
Из изложенного выше следует, что количественный
полярографический анализ основан на измерении диффузионного тока iD
как функции концентрации определяемого полярографически активного
деполяризатора в полярографируемом растворе.
При анализе получаемых полярограмм концентрацию
определяемого вещества находят методами градуировочного графика, добавок
стандарта, стандартных растворов.
а) Метод градуировочного графика используют чаще
всего. По этому методу готовят серию стандартных растворов, каждый из которых
содержит точно известную концентрацию с определяемого вещества. Проводят
полярографирование каждого раствора (после продувания через него тока инертного
газа) в одинаковых условиях, получают полярограммы и находят значения Е1/2
(одинаковые для всех растворов) и диффузионного тока iD
(разные для всех растворов). По полученным данным строят градуировочный график
в координатах iD-c,
представляющий собой обычно прямую линию в соответствии с уравнением Ильковича.
Затем проводят полярографирование анализируемого
раствора с неизвестной концентрацией с(Х) определяемого вещества, получают
полярограмму, измеряют величину диффузионного тока iD(X)
и по градуировочному графику находят концентрацию с(Х).
б) Метод добавок стандарта. Получают
полярограмму анализируемого раствора с неизвестной концентрацией с(Х)
определяемого вещества и находят величину диффузионного тока, т.е. высоту h
полярограммы. Затем к анализируемому раствору прибавляют точно известное
количество определяемого вещества, повышающее его концентрацию на величину c(st),
снова проводят полярографирование и находят новое значение диффузионного тока -
высоту полярограммы h
+
Δh, где Δh
- прирост диффузионного тока за счет увеличения концентрации анализируемого
раствора на величину c(st).
В соответствии с уравнением Ильковича можно
написать:
h = Kc(X), h = Kc(st),
откуда
h/Δh=c(X)/c(st)
и
с(X)=(h/Δh)·
c(st)
в) Метод стандартных растворов. В одинаковых
условиях проводят полярографирование двух растворов: анализируемого раствора с
неизвестной концентрацией с(Х) и стандартного раствора с точно известной
концентрацией c(st)
определяемого вещества. На полученных полярограммах находят высоты
полярографических волн h(Х)
и h(st),
отвечающие диффузионному току при концентрациях соответственно с(Х) и c(st).
Согласно уравнению Ильковича имеем:
h(Х) = Кс(Х), h(st)
= Kc(st),
откуда(X)/h(st)=c(X)/c(st),
c(X)=(h(X)/h(st)) · c(st)
Стандартный раствор готовят так, чтобы его
концентрация была 6ы как можно ближе к концентрации определяемого раствора. При
этом условии ошибка определения минимизируется.
.3 Применение полярографии
Условия проведения полярографического анализа.
Из вышеизложенного следует, что при проведении полярографического анализа
требуется соблюдение, по крайней мере, следующих условий.
) Для поддержания необходимой электропроводности
анализируемого раствора в него вводят фоновый электролит, например, хлорид или
нитрат калия, хлорид аммония, соли тетраалкиламмония и др. Ионы фонового
электролита должны разряжаться на ртутном капающем микроэлектроде при более
высоких значениях приложенного потенциала, чем полярографируемое вещество.
Концентрация фонового электролита должна быть
выше концентрации полярографически активного вещества в оптимальном случае не
менее чем в 100 раз. При этом концентрация самого полярографически активного
вещества обычно лежит в пределах от ~102 моль/л до -10-5 моль/л.
) Перед проведением полярографического анализа
из анализируемого раствора должен быть удален растворенный в нем кислород. Это
достигается чаще всего путем пропускания тока инертного газа (например, азота)
через раствор в течение ~15 минут перед началом полярографирования.
) Иногда на полярограмме появляются максимумы,
соответствующие протеканию электрическою тока, превышающего предельный ток.
Появление максимумов обусловлено движением поверхности капли жидкой ртути при
каплеобразовании, что приводит к перемешиванию диффузионного слоя раствора на
поверхности капли, к увеличению числа диффундирующих частиц полярографически
активных веществ и к их разряду на микроэлектроде, следствием чего и является
увеличение электрического тока, протекающего через полярографическую ячейку.
) Необходимо термостатировать полярографическую
ячейку, поддерживая температуру постоянной с точностью ± 0,5 °С.
Применение метода. Полярография используется для
определения малых количеств неорганических и органических веществ. Разработаны
тысячи методик количественного полярографического анализа. Предложены способы
полярографического определения практически всех катионов металлов, ряда анионов
(бромат-, иодат-, нитрат-, перманганат-ионов), органических соединений
различных классов, содержащих диазо-группы, карбонильные, пероксидные,
эпоксидные группы, двойные углерод углеродные связи, а также связи
углерод-галоген, азот-кислород, сера-сера.
Метод - фармакопейный, применяется для
определения салициловой кислоты, норсульфазола, витамина В1, алкалоидов,
фолиевой кислоты, келлина в порошке и в таблетках, никотинамида, пиридоксина
гидрохлорида, препаратов мышьяка, гликозидов сердечного действия, а также
кислорода и различных примесей в фармацевтических препаратах.
Метод обладает высокой чувствительностью (до
10-5-10-6 моль/л); селективностью; сравнительно хорошей воспроизводимостью
результатов (до ~2%); широким диапазоном применения; позволяет анализировать
смеси веществ без их разделения, окрашенные растворы, небольшие объемы
растворов (объем полярографической ячейки может составлять всего 1 мл); вести анализ
в потоке раствора; автоматизировать проведение анализа.
К недостаткам метода относятся токсичность
ртути, ее довольно легкая окисляемость в присутствии веществ-окислителей,
относительная сложность используемой аппаратуры.
Другие варианты полярографического метода.
Помимо описанной выше классической полярографии, использующей капающий ртутный
микроэлектрод с равномерно возрастающим на нем электрическим потенциалом при
постоянном электрическом токе, разработаны другие варианты полярографического
метода - производная, дифференциальная, импульсная, осциллографическая
полярография; переменно-токовая полярография - также в разных вариантах.
5. Амперометрическое титрование
Сущность метода. Амперометрическое титрование
(потенциометрическое поляризационное титрование) - разновидность
вольтамперометрического метода (наряду с полярографией). Оно основано на
измерении величины тока между электродами электрохимической ячейки, к которым
приложено некоторое напряжение, как функции объема прибавленного титранта. В соответствии
с уравнением Ильковича
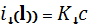
диффузионный ток 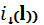 в полярографической ячейке тем
больше, чем выше концентрация с полярографически активного вещества. Если при
прибавлении титранта в анализируемый титруемый раствор, находящийся в полярографической
ячейке, концентрация такого вещества уменьшается или увеличивается, то
соответственно падает или возрастает и диффузионный ток. Точку эквивалентности
фиксируют по резкому изменению падения или роста диффузионного тока, что
отвечает окончанию реакции титруемого вещества с титрантом.
в полярографической ячейке тем
больше, чем выше концентрация с полярографически активного вещества. Если при
прибавлении титранта в анализируемый титруемый раствор, находящийся в полярографической
ячейке, концентрация такого вещества уменьшается или увеличивается, то
соответственно падает или возрастает и диффузионный ток. Точку эквивалентности
фиксируют по резкому изменению падения или роста диффузионного тока, что
отвечает окончанию реакции титруемого вещества с титрантом.
Различают амперометрическое
титрование с одним поляризуемым электродом, называемое также титрованием по
предельному току, полярографическим или поляриметрическим титрованием, и
амперометрическое титрование с двумя одинаковыми поляризуемыми электродами, или
титрование «до полного прекращения тока», биамперометрическое титрование.
Амперометрическое титрование с одним
поляризуемым электродом. Оно основано на измерении тока в полярографической
ячейке в зависимости от количества прибавленного титранта при постоянном
внешнем потенциале на микроэлектроде, несколько превышающем потенциал полуволны
на вольт-амперной кривой титруемого вещества Х или титранта Т. Обычно выбранный
внешний потенциал соответствует области предельного тока на полярограмме Х или
Т. Титрование ведут на установке, состоящей из источника постоянного тока с
регулируемым напряжением, к которому последовательно присоединены гальванометр
и полярографическая ячейка для титрования. Рабочим (индикаторным) электродом
ячейки может служить ртутный капающий электрод, неподвижный иди вращающийся
платиновый либо графитовый электрод. При использовании твердых электродов
необходимо перемешивание раствора во время титрования. В качестве электрода
сравнения применяют хлорсеребряный или каломельный электроды. Фоном служат, в
зависимости от условий, различные полярографически неактивные при данном
потенциале электролиты.
Вначале получают вольт-амперные
кривые (полярограммы) для Х и Т в тех же условиях, в которых предполагается
проведение амперометрического титрования. На основании рассмотрения этих кривых
выбирают значение потенциала, при котором достигается величина предельного тока
полярографически активных Х и Т. Выбранное значение потенциала поддерживают
постоянным в течение всего процесса титрования.
Используемая для амперометрического
титрования концентрация титранта Т должна примерно в 10 раз превышать
концентрацию Х; при этом практически не требуется вводить поправку на
разбавление раствора во время титрования. В остальном соблюдают все те условия,
которые требуются для получения полярограмм. Требования к термостатированию -
менее строгие, чем при прямом полярографировании, поскольку конец титрования
определяется не по абсолютному значению диффузионного тока, а по резкому
изменению его величины.
В полярографическую ячейку вносят
анализируемый раствор, содержащий Х, и прибавляют небольшими порциями титрант
Т, измеряя каждый раз ток і. Величина тока і зависит от концентрации
полярографически активного вещества. В точке эквивалентности величина і резко
изменяется.
По результатам амперометрического
титрования строят кривые титрования. Кривая амперометрического титрования - это
графическое представление изменения величины тока і в зависимости от объема V
прибавленного титранта. Кривая титрования строится в координатах ток і - объем V
прибавленного титранта Т (или степень оттитрованности).
В зависимости от природы титруемого
вещества Х и титранта Т кривые амперометрического титрования могут быть
различного типа. На рис.7 представлены схематически типы кривых
амперометрического титрования.
Объем V(ТЭ)
прибавленного Т, соответствующий точке эквивалентности, определяют по
пересечению прямых на графике амперометрического титрования.
На практике часто наблюдается
искривление (отступление от линейности) прямых - плавный переход - вблизи точки
эквивалентности.
Искривление тем меньше, чем больше
константа равновесия реакции протекающей в полярографической ячейке для
титрования, и чем меньше разбавление раствора. В таких случаях положение точки
эквивалентности определяют по пересечению продолжения линейных участков обеих
ветвей кривой титрования. В начале титрования ток изменяется мало, так как
титруемое вещество Х полярографически неактивно, а прибавляемый
полярографически активный титрант Т связывается веществом Х в продукт реакции и
не восстанавливается на микроэлектроде. После ТЭ начинается восстановление
прибавляемого титранта Т и ток в ячейке возрастает.
Примером может служить титрование
сульфат - ионов 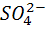 раствором,
содержащим катионы свинца, например, ацетат свинца. При титровании протекает
осадительная реакция
раствором,
содержащим катионы свинца, например, ацетат свинца. При титровании протекает
осадительная реакция
+ 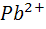 =
PbS
=
PbS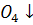
При потенциале микроэлектрода, соответствующем
предельному току катионов свинца (+1,55 В относительно насыщенного каломельного
электрода).
В третьем случае (кривая в) титрование ведут при
потенциале, при котором до ТЭ на микроэлектроде разряжается титруемое вещество
Х, а после ТЭ - титрант Т. В начале титрования ток в ячейке уменьшается за счет
понижения концентрации полярографически активного вещества Х вплоть до ТЭ,
после чего ток снова возрастает за счет восстановления на микроэлектроде
прибавляемого титранта Т, концентрация которого увеличивается.
Примером может служить титрование катионов
свинца раствором,
содержащим хромат - ионы. При этом протекает осадительная реакция
+ 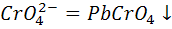
До ТЭ на микроэлектроде происходит
восстановление катионов свинца
+ 2е = Pb
а после ТЭ - восстановление хромат - ионов
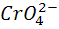 + 3е +
+ 3е +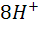 =
= 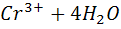
Реже встречаются и другие типы кривых амперометрического
титрования. Так, кривая г на рис. 7 соответствует случаю, когда и титрант, и
титруемое вещество полярографически неактивны, а продукт реакции -
полярографически активен. По мере образования этого продукта реакции в процессе
титрования ток i в полярографической
ячейке возрастает, поскольку увеличивается концентрация полярографически
активного вещества. По достижении ТЭ, когда все титруемое вещество Х
прореагировало и продукт реакции больше не образуется, устанавливается
практически постоянное значение тока, соответствующее постоянной концентрации
образовавшегося продукта реакции. Примером может служить титрование арсенатов
иодидами по схеме
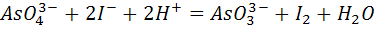
при потенциале, равном -0,05 В по отношению к
потенциалу насыщенного каломельного электрода на фоне 9 моль/л HCl.
В этих условиях арсенат- и иодид-ионы полярографически неактивны, а
образующиеся продукт реакции - иод 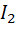 -
полярографически активен и восстанавливается на микроэлектроде.
-
полярографически активен и восстанавливается на микроэлектроде.
Аналогичный тип кривой титрования наблюдается
при титровании селенитов иодидами.
Метод обладает высокой чувствительностью (до 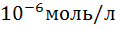 ),
большой точностью, селективность; позволяет проводить титрование мутных и
окрашенных растворов, не требует применения индикаторов, может быть
автоматизирован.
),
большой точностью, селективность; позволяет проводить титрование мутных и
окрашенных растворов, не требует применения индикаторов, может быть
автоматизирован.
Амперометрическое титрование с одним
поляризуемым электродом применяется преимущественно в сочетании с реакциями
осаждения и комплексообразования.
Амперометрическое титрование с двумя поляризуемыми
электродами (биамперометрическое титрование). Этот вариант амперометрического
титрования основан на измерении тока между двумя одинаковыми электродами (из
платины или золота) электрохимической ячейки, на которые налагают небольшую
разность потенциалов. В ячейке протекает ток в том случае, когда в растворе
имеется обратимая окислительно - восстановительная пара (редокс - пара),
например, 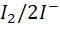 , при таких
концентрациях окислителя и восстановителя, при которых возможно существование
катодного и анодного процессов. При исчезновении в системе одного из
компонентов обратимой окислительно - восстановительной пары или при появлении
обратимой окислительно - восстановительной пары в ток в ТЭ резко прерывается
или мгновенно появляется.
, при таких
концентрациях окислителя и восстановителя, при которых возможно существование
катодного и анодного процессов. При исчезновении в системе одного из
компонентов обратимой окислительно - восстановительной пары или при появлении
обратимой окислительно - восстановительной пары в ток в ТЭ резко прерывается
или мгновенно появляется.
Так, например, методом биамперометрического
титрования можно определять медь(II)
в анализируемом растворе. Для этого к анализируемому раствору, содержащему медь
(II) в сернокислой
среде и помещенному в сосуд для титрования, прибавляют иодид калия KI.
При этом медь (II)
восстанавливается до меди (I)
с образованием эквивалентного количества иода
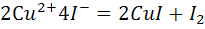
В полученный раствор погружают платиновые
электроды, к которым приложена требуемая разность потенциалов (около 20 мВ).
Поскольку в растворе имеется обратимая редокс - пара, то на платиновом катоде
иод в незначительном количестве восстанавливается до иодид - ионов
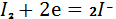
и в электрохимической ячейке фиксируется
электрический ток. Иод, образовавшийся при реакции иодид-ионов с медью (II),
титруют стандартным раствором тиосульфата натрия
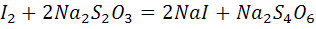
По достижении ТЭ, когда весь иод оттитрован, ток
в ячейке резко падает практически до нуля, так как в растворе уже не содержится
иод - один из компонентов редокс - пары .
Зная количество тиосульфата натрия,
израсходованное на тирование иода, можно рассчитать содержание меди (II)
в исходном анализируемом растворе.
Биамперометрическое титрование ведут при
энергичном перемешивании раствора на упаковке, состоящей из источника
постоянного тока с потенциометром, с которого регулируемая разность потенциалов
(0,05 0,25 В) подается через чувствительный микроамперметр на электроды
электрохимической ячейки. В последнюю перед проведением титрования вносят
титруемый раствор и прибавляют порциями титрант до резкого прекращения или
появления тока, о чем судят по показанию микроамперметра.
Используемые в электрохимической
ячейке платиновые электроды периодически очищают, погружая их на  30 минут в кипящую концентрированную
азотную кислоту, содержащую добавки хлористого железа, с последующим
промыванием электродов водой.
30 минут в кипящую концентрированную
азотную кислоту, содержащую добавки хлористого железа, с последующим
промыванием электродов водой.
Биамперометрическое титрование -
фармакопейный метод; применяется в иодометрии, нитритометрии, акваметрии, при
титровании в неводных средах.
6. Кулонометрический метод анализа
(кулонометрия)
.1 Принципы метода
Кулонометрический метод анализа (кулонометрия)
основан на использовании зависимости между массой m
вещества, прореагировавшего при электролизе в электрохимической ячейке, и
количеством электричества Q,
прошедшего через электрохимическую ячейку при электролизе только этого
вещества. В соответствии с объединенным законом электролиза М. Фарадея масса m(в
граммах) связана с количеством электричества Q
(в кулонах) соотношением
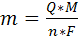 ,
,
где М - молярная масса вещества,
прореагировавшего при электролизе, г/моль; n
- число электронов, участвующих в электродной реакции; F
= 96487 Кл/моль - число Фарадея.
Количество электричества Q
(в Кл), прошедшее при электролизе через электрохимическую ячейку, равно
произведению электрического тока i
(в А) на время электролиза τ (в
с)
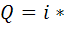 τ.
τ.
Если измерено количество электричества Q,
то согласно (27) можно рассчитать массу m.
Это справедливо в том случае, когда всё количество электричества Q,
прошедшее при электролизе через электрохимическую ячейку, израсходовано только
на электролиз данного вещества; побочные процессы должны быть исключены.
Другими словами, выход (эффективность) по току должен быть равен 100%.
Поскольку в соответствии с объединенным законом
электролиза М.Фарадея (27) для определения массы m
(г) прореагировавшего при электролизе вещества необходимо измерить количество
электричества Q, затраченное на
электрохимическое превращение определяемого вещества, в кулонах, то метод и
назван кулонометрией. Главная задача кулонометрических измерений - как можно
более точно определить количество электричества Q.
Кулонометрический анализ проводят либо в
амперостатическом (гальваностатическом) режиме, т.е. при постоянном
электрическом токе i=const,
либо контролируемом постоянном потенциале рабочего электрода
(потенциостатическая кулонометрия), когда электрический ток изменяется
(уменьшается) в процессе электролиза.
В первом случае для определения количества
электричества Q достаточно как
можно более точно измерить время электролиза τ, постоянный
ток i(A)
и рассчитать величину Q
по формуле (28).
Во втором случае величину Q
определяют расчетным способом, либо с помощью химических кулонометров.
Различают прямую кулонометрию и косвенную
кулонометрию (кулонометрическое титрование).
.2 Прямая кулонометрия
Сущность метода. Прямую кулонометрию при
постоянном токе применяют редко. Чаще используют кулонометрию при
контролируемом постоянном потенциале рабочего электрода или прямую
потенциостатичесую кулонометрию.
В прямой потенциостатической кулонометрии
электролизу подвергают непосредственно определяемое вещество. Измеряют
количество электричества, затраченное на электролиз этого вещества, и по
уравнению (27) рассчитывают массу m
определяемого вещества.
В процессе электролиза потенциал рабочего
электрода поддерживают постоянным, Е = const,
для чего обычно используют приборы - потенциостаты.
Постоянное значение потенциала Е выбирают
предварительно на основании рассмотрения вольт-амперной (поляризационной)
кривой, построенной в координатах ток i
- потенциал Е (как это делают в полярографии), полученной в тех же условиях, в
которых будет проводиться электролиз. Обычно выбирают значение потенциала Е,
соответствующее области предельного тока определяемого вещества и несколько
превышающее его потенциал полуволны Е1/2 (на ~0,05 - 0,2В). При этом значении потенциала,
как и в полярографии, фоновый электролит не должен подвергаться электролизу.
В качестве рабочего электрода чаще всего
применяют платиновый электрод, на котором происходит электрохимическое
восстановление или окисление определяемого вещества. Кроме рабочего электрода
электрохимическая ячейка включает один другой электрод или (чаще) два других
электрода - электрод сравнения, например, хлорсеребряный, и вспомогательный,
например, из стали.
По мере протекания процесса электролиза при
постоянном потенциале электрический ток в ячейке уменьшается, так как
понижается концентрация электроактивного вещества, участвующего в электродной
реакции. При этом электрический ток уменьшается со временем по
экспотенциальному закону от начального значения i0
в момент времени τ=0 до значения i
в момент времени τ
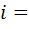 i0e-kτ,
i0e-kτ,
где коэффициент k
зависит от природы реакции, геометрии электрохимической ячейки, площади
рабочего электрода, коэффициента диффузии определяемого вещества, скорости
перемешивания раствора и его объема.
Выход по току будет количественным, когда ток i
уменьшится до нуля, т.е. при бесконечно большом времени τ.
На
практике электролиз определяемого вещества считают количественным, когда ток
достигает очень малой величины, не превышающей ~0,1% от значения i0.
При этом ошибка определения составляет около ~0,1%.
Поскольку количество электричества определяется
как произведение тока на время электролиза, то, очевидно, общее количество
электричества Q, затраченное на
электролиз определяемого вещества, равно
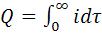 ,
,
т.е. определяется площадью, ограниченной осями
координат и экспонентой.
Для нахождения массы m
прореагировавшего вещества требуется согласно (27) измерить или рассчитать
количество электричества Q.
Способы определения количества электричества,
прошедшего через раствор, в прямой потенциостатической кулонометрии. Величину Q
можно определить расчетными способами либо с помощью химического кулонометра.
а) Расчет величины Q
по площади под кривой зависимости i
от τ.
Измеряют
площадь, ограниченную осями координат и экспонентой (29). если ток i
выражен в амперах, а время τ - в
секундах, то измеренная площадь равна количеству электричества Q
в кулонах.
Для определения Q
без заметной ошибки способ требует практически полного завершения процесса электролиза,
т.е. длительного времени. На практике, как уже отмечалось выше, измеряют
площадь при значении τ, соответствующем
i = 0,001* i0
(0,1% от i0).
б) Расчет величины Q
на основе зависимости ln
i от τ.
В
соответствии с (29) и (30) имеем
= 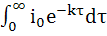 =
i0/k,
=
i0/k,
поскольку
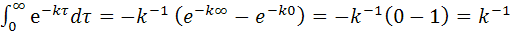 .
.
Таким образом, Q
= i0/k
и для определения величины Q
необходимо
Согласно (29) i
= i0e-kτ.
После логарифмирования этого уравнения получаем линейную зависимость ln
i от τ
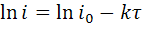
Если измерить несколько значений i
в различные моменты времени τ (например,
воспользовавшись кривой изменения тока i
со временем τ в прямой
потенциометрической кулонометрии или непосредственно опытным путем), то можно
построить график функции, представляющий собой прямую линию.
в) Определение величины Q
с помощью химического кулонометра.
При этом способе в электрическую цепь
кулонометрической установки включают химический кулонометр последовательно с
электрохимической ячейкой, в которой проводят электролиз определяемого
вещества. Количество электричества Q,
проходящее через последовательное соединение кулонометр и электрохимическую
ячейку, одинаково.
Конструкция кулонометра позволяет экспериментально
определить величину Q.
Чаще всего применяют серебряный, медный и
газовые кулонометры, реже - другие. Использование серебряного и медного
кулонометров основано на электрогравиметрическом определении массы серебра или
меди, осаждающейся на платиновом электроде при электролизе. Зная массу металла,
выделившегося на катоде в кулонометре, можно по уравнения (27) рассчитать
количество электричества Q.
Серебряный кулонометр включает анод из
металлического серебра и платиновый катод, погруженные в раствор соли серебра AgNO3.
При электролизе катионы серебра, присутствующие в растворе, разряжаются на
платиновом катоде
Ag+ + e
= Ag
Массу серебра, осажденного на катоде в процессе
электролиза, определяют, взвешивая платиновый катод (после промывания и высушивания)
до и после электролиза. Масса 1,118мг серебра, осажденного на платиновом
катоде, соответствует затрате на электролиз 1Кл электричества.
Медный кулонометр содержит платиновые катод и
анод, погруженные в сернокислый раствор соли меди CuSO4.
При электролизе на катоде выделяется металлическая медь
Cu2+ + 2e
= Cu
Массу которой определяют, взвешивая платиновый
катод (также после промывания и высушивания) до и после электролиза. Масса 0,
3295мг меди, осажденной на платиновом катоде, отвечает затрате на электролиз
1Кл электричества.
Принцип использования газового кулономктра
основан на измерении объема газа, выделившегося во время электролиза. Так, в
водородно-кислородном (водяном) газовом кулонометре во время его работы
происходит электролитическое разложение воды с образованием газообразных
водорода и кислорода. На катоде выделяется водород:
H2O
+ 2e = H2
+ 2OH-
На аноде выделяется кислород:
H2O
- 4e = O2
+ 4H+
При нормальных условиях объем 0,1797 см3
водорода и кислорода соответствует 1 Кл электричества, прошедшего через
кулонометр и через электрохимическую кулонометрическую ячейку.
Кулонометры, особенно - серебряный и медный,
позволяют определить количество электричества Q
с высокой точностью, однако работа с ними довольно трудоемка и продолжительна.
В кулонометрии применяют также электронные
интеграторы, позволяющие регистрировать количество электричества Q,
затраченное на электролиз, по показаниям соответствующего прибора.
Применение прямой кулонометрии. Метод обладает
высокой селективностью, чувствительностью (до 10-8-10-9г или до ~10-5 моль/л),
воспроизводимостью (до ~1-2%), позволяет определить содержание микропримесей. К
недостаткам метода относятся большие трудоемкость и длительность, необходимость
наличия дорогостоящей аппаратуры.
Прямую кулонометрию можно применять для
определения - при катодном восстановлении - ионов металлов, ограниченных нитро-
и галогенпроизводных; при анодном окислении - хлорид-, бромид-, иодид-,
тиоцианат-анионы, ионы металлов в низших степенях окисления при переводе их в более
высокие степени окисления, например
As(III) → As(V), Сr(II)
→ Cr(III), Fe(II) → Fe(III), Tl(II) → Tl(III)
В фармацевтическом анализе прямую кулонометрию
применяют для определения аскорбиновой и пикриновой кислот, новокаина,
оксихинолина и в некоторых других случаях.
Как отмечалось выше, кулонометрия довольно
трудоемка и продолжительна. Кроме того, в ряде случаев начинают заметно
протекать побочные процессы еще до завершения основной электрохимической
реакции, что снижает выход по току и может привести к значительным ошибкам
анализа. Поэтому чаще применяют косвенную кулонометрию - кулонометрическое
титрование.
.3 Кулонометрическое титрование
Сущность метода. При кулонометрическом
титровании определяемое вещество Х, находящееся в растворе в электрохимической
ячейке, реагирует с «титрантом» Т - веществом, непрерывно образующемся
(генерируемом) на генераторном электроде при электроде при электролизе
вспомогательного вещества, также присутствующего в растворе.
Окончание титрования - момент, когда всё определяемое
вещество Х полностью прореагирует с генерируемым «титрантом» Т, фиксируют либо
визуально индикаторным методом, либо с помощью инструментальных методов -
потенциометрически, амперометрически, фотометрически.
Таким образом, при кулонометрическом титровании
титрант не прибавляется из бюретки в титруемый раствор. Роль титранта играет
вещество Т, непрерывно генерируемое при электродной реакции на генераторном
электроде. Очевидно, имеется аналогия между обычным титрованием, когда титрант
вводится извне а титруемый раствор и по мере его прибавления реагирует с
определяемым веществом, и генерацией вещества Т, которое по мере своего
образования также реагирует с определяемым веществом. Поэтому рассматриваемый
метод и получил название «кулонометрическое титрование».
Кулонометричесое титрование проводят в
амперостатическом (гальваностатическом) или в потенциостатическом режиме,
поддерживая электрический ток постоянным в течение всего времени электролиза.
Вместо объема прибавленного титранта в
кулонометрическом титровании измеряют время τ и
ток i электролиза.
Процесс образования вещества Т в кулонометрической ячейке во время электролиза
называется генерация титранта.
Кулонометрическое титрование при постоянном
токе. При кулонометрическом титровании в амперостатическом режиме (при
постоянном токе) измеряют время τ, в
течение которого проводится электролиз, и количество электричества Q,
израсходованное при электролизе, рассчитывают по формуле (28), после чего
находят массу определяемого вещества Х по соотношению (27).
Так, например, стандартизацию раствора
хлороводородной кислоты HCl
методом кулонометрического титрования проводят путем титрования ионов водорода H3O+
стандартизируемого раствора, содержащего HCl,
электрогенерируемыми на платиновом катоде гидроксид-ионами ОН- при электролизе
воды:
Н2О + 2е = 2ОН- + Н2↑
Образовавшийся титрант - гидроксид-ионы -
реагирует с ионами Н3О+ в растворе
Н3О+ + ОН- = 2Н2О
Титрование ведут в присутствии индикатора
фенолфталеина и прекращают при появлении светло-розовой окраски раствора.
Зная величину постоянного тока i
(в амперах) и время τ (в секундах),
затраченное на титрование, рассчитывают по формуле (28) количество
электричества Q (в кулонах) и по
формуле (27) - массу (в граммах) прореагировавшей НСl,
содержавшуюся в аликвоте стандартизируемого раствора НСl,
внесенного в кулонометрическую ячейку (в генераторный сосуд).
Генераторный платиновый анод и вспомогательный
платиновый катод помещены в генераторный сосуд и вспомогательный сосуд.
Генераторный сосуд заполнен испытуемым раствором, содержащим определяемое
вещество Х, фоновый электролит с вспомогательным электроактивным веществом и
индикатором. Вспомогательное вещество и само может быть фоновым электролитом; в
таких случаях нет необходимости вводить в раствор другой фоновый электролит.
Генераторный и вспомогательный сосуды соединены
солевым мостиком, заполненным сильным индифферентным электролитом для
обеспечения электрического контакта между электродами. Концы трубки солевого
мостика закрыты пробками из фильтровальной бумаги. В генераторном сосуде
имеется магнитный стержень ждя перемешивания раствора посредством магнитной
мешалки.
Электрохимическая ячейка включается в
электрическую цепь установки для кулонометрического титрования, способную
поддерживать ток постоянным и требуемой величины (например, используют
универсальный источник питания типа лабораторного прибора УИП-1 и т.п.
аппаратуру).
До проведения кулонометрического титрования
электроды тщательно промывают дистиллированной водой, в генераторный сосуд
внося раствор с вспомогательным электроактивным (в данных условиях) веществом,
при необходимости - фоновый электролит и индикатор.
Поскольку приготовленный таким путем фоновый
раствор может содержать электровосстанавливающиеся или электроокисляющиеся
примеси, то в начале проводят предэлектролиз фонового раствора с целью
электровосстановления или электроокисления примесей. Для этого замыкают
электрическую цепь установки и ведут электролиз некоторого (обычно -
небольшого) времени до изменения окраски индикатора, после чего цепь размыкают.
После завершения предэлектролиза в генераторный
сосуд вносят точно измеренный объем анализируемого раствора, включают магнитную
мешалку, замыкают электрическую цепь установки, одновременно включая
секундомер, и ведут электролиз при постоянном токе до момента резкого изменения
окраски индикатора (раствора), когда сразу же останавливают секундомер и
размыкают электрическую цепь установки.
Если анализируемый раствор, вводимый в
кулонометрическую ячейку для титрования, содержит примеси электровосстанавливающихся
или электроокисляющихся веществ, на превращение которых при электролизе
затрачивается некоторое количество элеткричества, то после предэлектролиза (до
прибавления в ячейку анализируемого раствора) проводят холостое титрование,
вводя в кулонометрическую ячейку вместо анализируемого раствора точно такой же
объем раствора, который содержит все те же вещества и в тех же количествах, что
и прибавленный анализируемый раствор, за исключение определяемого вещества Х. В
простейшем случае к фоновому раствору прибавляют дистиллированную воду в
объеме, равном объему аликвоты анализируемого раствора с определяемым
веществом.
Время, затраченное на холостое титрование, в
дальнейшем вычитают из времени, затраченного на титрование испытуемого раствора
с определяемым веществом.
Условия проведения кулонометрического
титрования. Из вышеизложенного следует, что условия проведения
кулонометрического титрования должны обеспечить полный выход по току. Для этого
необходимо выполнять следующие требования.
а) вспомогательный реагент, из которого на
рабочем электроде генерируется титрант, должен присутствовать в растворе в
большом избытке по отношению к определяемому веществу (~1000-кратный избыток).
В этих условиях обычно устраняются побочные электрохимические реакции, основная
из которых - это окисление или восстановление фонового электролита, например,
ионов водорода
Н+ + 2е = Н2↑
б) величина постоянного тока i
= const при проведении
электролиза должна быть меньше величины диффузного тока вспомогательного
реагента во избежание протекания реакции с участием ионов фонового электролита.
в) необходимо как можно точнее определять
количество электричества, израсходованное при проведении электролиза, для чего
требуется точно фиксировать начало и конец отсчета времени и величину тока
электролиза.
Индикация конца титрования. Как уже говорилось
выше, при кулонометрическом титровании ТЭ определяют либо визуальными
индикаторными, либо инструментальными методами.
При электрохимической индикации ТЭ в испытуемый
раствор (в генераторный сосуд) помещают еще пару электродов, входящих в
дополнительную индикаторную электрическую цепь. Окончание титрования можно
фиксировать с помощью дополнительной индикаторной электрической цепи
потенциометрически (рН-метрически) или биамперометрически.
При биамперометрической индикации ТЭ строят
кривые титрования в координатах ток i
- время τ,
измеряя
ток i в дополнительной
индикаторной электрической цепи как функцию времени τ
электролиза
в кулонометрической ячейке. При этом получают кривые титрования, основные типы
которых схематически показаны на рисунке.
Кривая а соответствует случаю, когда
электроактивно определяемое вещество Х, а генерируемый титрант Т и продукты
реакции не являются электроактивными в данных условиях. При взаимодействии Х с
генерирумым титрантом Т концентрация Х по мере протекания электролиза
понижается, поэтому уменьшается и ток i
в дополнительной индикаторной электрической цепи вплоть до достижения ТЭ, после
чего ток i в дополнительной
цепи уже не возрастает (или вообще отсутствует). Положение ТЭ находят порезкому
излому на кривой титрования. Если этот излом не резкий, а плавный, то ТЭ
фиксируют по пересечению продолжения линейных ветвей кривой.
Описанная картина наблюдается, например, при
кулонометрическом титровании железа(II)
церием(IV),
генерируемым из церия (III)
на генераторном электроде
Се3+ - е = Се4+ (генерация титранта - Се4+)
Се4+ +Fe2+
= Ce3+ + Fe3+
(реакция в растворе)
По мере генерации титранта - ионов Се4+ и
протекания реакции титрования концентрация ионов Fe2+
в растворе понижается. Ток i,
обусловленный разрядом катионов Fe2+,
уменьшается до тех пор, пока прореагируют все катионы Fe2+,
т.е. при достижении ТЭ, после чего остается постоянным i
= const.
Кривая б отвечает тому случаю, когда
определяемое вещество Х и продукты реакции титрования не являются
электроактивными, а титрант Т - электроактивен. Вначале по мере протекания
реакции вещества Х с генерирумым Т ток i
в дополнительной индикаторной электрической цепи остается постоянным или
отсутствует, поскольку генерируемый электроактивный Т сразу же реагирует с Х.
после того, как все определяемое вещество Х прореагирует с Т, ток i
в дополнительной индикаторной электрической цепи начинает расти, так как
увеличивается концентрация генерируемого электроактивного Т. На криво титрования
наблюдается резкий излом в ТЭ или плавный переход. Подобный ход криво
наблюдается при кулонометрическом титровании катионов цинка Zn2+
ферроцианид-ионами [Fe(CN)6]4-,
генерируемыми в процессе электролиза из имеющихся в растворе феррицианид-ионов
[Fe(CN)6]3-
(в раствор до замыкания электрической цепи вводят феррицианид калия) на
генераторном электроде:
[Fe(CN)6]3-
+ e = [Fe(CN)6]4-
Образовавшиеся ферроцианид-ионы реагируют с
определяемыми катионами цинка по схеме
3Zn2+ + 2K+ + 2[Fe(CN)6]4- = K2Zn3[Fe(CN)6]2↓
После достижения ТЭ, когда все катионы Zn2+
прореагирует с генерируемыми электроактивными ферроцианид-ионами, ток i
в дополнительной индикаторной электрохимической цепи возрастает вследствие
увеличения концентрации электроактивных ферроцианид-ионов.
Кривая в относится к такому случаю
кулонометрического титрования, когда электроактивны и определяемое вещество Х,
и генерируемый титрант Т. Вначале по мере генерации Т ток i
в дополнительной индикаторной цепи уменьшается , так как понижается содержание
в растворе электроактивного вещества Х. после ТЭ ток возрастает, так как в
растворе накапливается электроактивный Т. На кривой титрования в ТЭ наблюдается
резкий излом или плавный переход.
Кулонометрическое титрование при постоянном
потенциале. Потенциостатический режим в кулонометрическом титровании
используется реже.
Кулонометрическое титрование в
потенциостатическом режиме ведут при постоянном значении потенциала,
соответствующем потенциалу разряда вещества на рабочем электроде, например, при
катодном восстановлении катионов металлов на платиновом электроде. По мере
протекания реакции потенциал остается постоянным до тех пор, пока прореагируют
все катионы металла, после чего он резко уменьшается, поскольку в растворе уже
нет потенциалопределяющих катионов металла.
Применение кулонометрического титрования. В
кулонометрическом титровании можно использовать все типы реакций
титриметрического анализа: кислотно-основные, окислительно-восстановительные,
осадительные, реакции комплексообразования.
Так, малые количества кислот можно определять
кулонометрическим кислотно-основным титрованием электрогенерированными
ОН--ионами, образующимися при электролизе воды на катоде:
Н2О + 2е = 2ОН- + Н2↑
Можно титровать и основания ионами водорода Н+,
генерируемыми на аноде при электролизе воды:
Н2О - 4е = 4Н+ +О2↑
При окислительно-восстановительном
бромометрическом кулонометрическом титровании можно определять соединения
мышьяка(III), сурьмы(III),
йодиды, гидразин, фенолы и другие органические вещества. В роли титранта
выступает электрогенерируемый на аноде бром:
Br- - 2e
= Br2
Осадительным кулонометрическим титрованием можно
определять галогенид-ионы и органические серосодержащие соединения
электрогенерированными катионами серебра Ag+,
катионы цинка Zn2+ -
электрогенерированными ферроцинид-ионами и т.д.
Комплексонометрическое кулонометрическое
титрование катионов можно проводить анионами ЭДТА, электрогенерированными на
катоде из комплексоната ртути(II).
Кулонометрическое титрование обладает высокой
точностью, широким диапазоном применения в количественном анализе, позволяет
определять малые количества веществ, малостойкие соединения (поскольку они
вступают в реакции сразу же после их образования), например, меди(I),
серебра(II), олова(II),
титана(III), марганца(III),
хлора, брома и др.К достоинствам метода относится также и то, что не требуется
приготовление, стандартизация и хранение титранта, так как он непрерывно
образуется при электролизе и сразу же расходуется в реакции с определяемым
веществом.
Недостатком метода можно считать необходимость
использования сравнительно сложной и дорогостоящей аппаратуры.