Хімічна термодинаміка
Вступ
Хімічна термодинаміка
дозволяє передбачити принципову можливість чи неможливість самочинного перебігу
реакції, а також розрахувати рівноважні концентрації реагуючих речовин. Однак
цього недостатньо для визначення швидкості і механізму реакції та керування
процесом. Тривалість реакції найчастіше не пов’язана із значенням її енергії
Гіббса. Наприклад, термодинамічна імовірність реакції
Н2 + ½ О2 à Н2О
(р), D G0298
= -237,2 кДж/моль
значно вища за
імовірність реакції нейтралізації
Н+ + ОН-
à Н2О
(р), D G0298
= -79,9 кДж/моль.
Але перша реакція за
звичайних умов без каталізатора майже не протікає, а друга реакція відбувається
практично миттєво. Такі якісні та кількісні змінення процесів, що протікають
протягом деякого часу, пояснює хімічна кінетика.
1. Загальні поняття
Хімічна кінетика - це розділ хімії, який вивчає швидкість та механізм перебігу
реакцій.
Отже, хімічна кінетика
вирішує дві конкретні задачі:
визначення механізму
реакції, тобто встановлення елементарних стадій процесу і послідовності їх
протікання;
кількісний опис хімічної
реакції, а саме: встановлення сурових співвідношень, що дають можливість
обчислювати змінення кількості вихідних реагентів і продуктів протягом перебігу
реакції.
Як правило, реакція
протікає через декілька проміжних стадій, при складанні яких одержують сумарне
рівняння реакції, тому кінетичні рівняння, які з урахуванням механізму реакції
описують залежність швидкості від концентрації речовин, можна одержати лише
експериментально. Встановлення механізму спирається на класифікацію реакції за
молекулярністю, що визначається числом молекул, які беруть участь у так
званому елементарному акті - одиничному акті взаємодії або перетворення
частинок, внаслідок чого утворюються нові частинки продуктів реакції чи
проміжних сполук.
Мономолекулярними називаються реакції, в яких елементарним актом є перетворення
однієї молекули. Наприклад:
І2 à 2І.
Бімолекулярні - це такі реакції, елементарний акт у яких здійснюється при зміненні
двох молекул:
Н· + Cl2 à HCl + Cl.
У тримолекулярних реакціях
елементарний акт здійснюється при одночасовому зіткненні трьох молекул:
2NO + H2 à N2O + H2.
Доведено, що одночасове
зіткнення більш, ніж трьох молекул практично неможливе. Наявність у рівнянні
хімічної реакції великих стехіометричних коефіцієнтів (коли їх сума перебільшує
3) однозначно вказує на складний механізм реакції, що містить певну кількість
елементарних актів[1].
2. Швидкість хімічної
реакції
Швидкість хімічної
реакції характеризує інтенсивність хімічного процесу, тобто кількість елементарних
актів взаємодії або розкладання частинок протягом певного часу.
Розглядаючи питання
хімічної кінетики, необхідно розрізнювати гомогенні реакції, які протікають в
одній фазі і відбуваються одночасно в усьому об’ємі системи, і гетерогенні
реакції, перебіг яких можливий лише на поверхні поділу фаз.
Швидкістю гомогенної
реакції називається кількість
речовини, що вступає в реакцію чи утворюється внаслідок реакції за одиницю часу
в одиниці реакційного об’єму.
Оскільки відношення
кількості речовини до одиниці об’єму є концентрація С, то швидкість гомогенної
реакції дорівнює зміненню концентрації вихідних сполук чи продуктів реакції
протягом часу. Причому, завдяки стехіометричному співвідношенню речовин у
хімічній реакції, контроль за зміненням концентрації здійснюється для однієї
сполуки, яку вибирають з практичних міркувань. Розрізняють середню J і миттєву (або істинну) швидкості реакції. Середня швидкість
реакції визначається різницею концентрацій DС речовини протягом певного часу Dt:
J = (C2-C1)/(t2 - t1) = ± DC/ Dt,
де С2 і С1
- концентрації речовини у кінцевий t2 і початковий t1 моменти часу. Знак
± у рівнянні (20) має
такий зміст. Оскільки швидкість реакції завжди додатна, то при використанні DС для вихідної речовини, концентрація якої протягом часу зменшується
(С2,вих < С1,вих, С2,вих - С1,вих <
0), беруть знак мінус. Якщо швидкість визначають за змінюванням концентрацій
одного з продуктів реакції, для якого С2,прод > C1,прод
і С2,прод - С1,прод > 0, то відношення DС/Dt треба
брати із знаком плюс [2].
У ході реакції
змінюються концентрації реагуючих речовин і відповідно змінюється швидкість
реакції. Чим менший проміжок часу Dt, тим менше змінення концентрацій DС і тим ближче відношення DС/Dt до
істинної (або миттєвої) швидкості реакції. Однак концентрації речовин у
хімічному процесі змінюються безперервно (рис. 1), тому правильніше говорити не
про середню, а про істинну швидкість реакції, яка є похідною від концентрації
за часом:
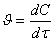 (1)
(1)
Істинна швидкість
реакції графічно визначається тангенсом кута нахилу дотичної до кривої
залежності концентрацій від часу (рис. 1):
J = tg a.
Із визначення
швидкості реакції і аналізу рівняння (1) випливає, що швидкість реакції у
системі СІ вимірюється у моль × м-3 × с-1, однак використовуються й інші одиниці вимірювання
[моль×л-1×с-1], [моль×см-3×с-1] [моль×см-3×хв-1].
Протягом реакції
змінюються концентрації всіх вихідних речовин і продуктів реакції. Якщо вихідні
реагенти мають однакові стехіометричні коефіцієнти, то змінення їх концентрацій
(за модулем) теж однакові. Наприклад, для реакції
СО + Н2О
(г) à CO2 + H2
можна записати
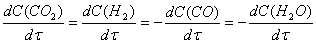
Для реакцій з
різними стехіометричними коефіцієнтами швидкості змінювання концентрацій
реагентів теж будуть різними. Для реакції загального вигляду
аА + bB à lL + mM
Швидкість реакції
дорівнює:
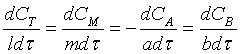
Наприклад, для
реакції
СН4 +
2Н2О (г) à CO2 + 4H2

Як видно,
концентрація Н2О змінюється у 2 рази, а концентрація Н2 -
у 4 рази швидше, ніж концентрації СН4 і СО2, оскільки на
1 моль СН4 витрачається 2 моль Н2О і утворюються 1 моль
СО2 і 4 моль Н2. Тому в рівнянні швидкості реакції
зазначають конкретний реагент (продукт чи вихідну речовину).
Швидкість реакції
залежить від природи реагуючих речовин: деякі реакції протікають миттєво
(вибух), інші можуть продовжуватися роками (корозія). На швидкість реакції
впливає ще багато чинників: концентрація вихідних реагентів, площина поверхні
дотику фаз (для гетерогенних процесів), температура, каталізатор, зовнішні
чинники (наприклад, опромінювання).
3.
Залежність швидкості реакції від концентрації реагентів
Будь-яка реакція
може здійснюватися тільки за умов зіткнення молекул реагуючих речовин, тому
швидкість реакції насамперед залежить від числа зіткнень, яке пропорційне
концентрації реагентів. Ця закономірність була встановлена Гульдбергом і Вааге
(1867 р.) і одержала назву закону діючих мас: швидкість хімічної реакції
пропорційна добутку концентрацій реагуючих речовин у ступенях, які дорівнюють
стехіометричним коефіцієнтам, що стоять перед формулами відповідних речовин у
рівнянні реакції.
Математичний
вираз закону діючих мас для реакції
аА + bB à lL + mM
має вигляд:
J = k CaA
× CbB = k [A]a × [B]b,
де квадратні
дужки позначають концентрацію, а k - константа швидкості, яка не залежить від
концентрації реагентів, але залежить від їх природи і температури. Із вище
наведенного рівняння випливає, що при концентраціях СА = СВ
=1 моль/л, константа швидкості чисельно дорівнює швидкості реакції. Отже, при
постійній температурі константа швидкості має сталу величину і характеризує
природу реагуючих речовин.
Приклад.
При 5090 константа швидкості реакції
Н2 + І2
= 2НІ дорівнює 0,16, а вихідні концентрації (моль/л): [Н2]вих
= 0,04;
[І2]вих
= 0,05. Обчислити початкову швидкість реакції. Як зміниться швидкість реакції,
коли концентрація водню зменшиться до
,03 моль/л?
Розв¢язок. Відповідно до закону діючих мас
початкова швидкість реакції дорівнює:
J = k[Н2]вих
× [І2]вих = 0,16 × 0,04 × 0,05 = 3,2 × 10-4.
До певного часу
прореагувало водню: 0,04 - 0,03 =
=0,01 (моль/л).
Співставляючи коефіцієнти у рівнянні реакції, робимо висновок, що і йоду
прореагувала така ж кількість, тому його концентрація набула значення: [І2]
= 0,05 - 0,01 = 0,04 (моль/л). При цьому швикдість реакції
J = k[Н2]
× [І2] = 0,16 ×
0,03 × 0,04 = 1,92 × 10-4.
Порівняно із
початковою швидкість реакції зменшиться у
Jпоч / J = 3,2 × 10-4 / 1,92 × 10-4 = 1,7 разів.
Як довів досвід,
закон Гульдберга-Вааге виявився справедливим тільки для обмеженого кола реакцій
з невеликими стехіометричними коефіцієнтами, сума яких не перевищує 3, а для
складніших процесів розрахунки давали значну похибку. Це пов’язано з тим, що
для більшості взаємодій сумарне рівняння реакції не відображає дійсного
механізму процесу з безліччю проміжних стадій, а є загальним виразом для
вихідних речовин і продуктів. Тому показники ступенів у рівнянні (23) не
повинні дорівнювати стехіометричним коефіцієнтам. Насправді вони мають
формальний характер і визначаються експериментально. При цьому закон діючих мас
набуває математичного вигляду, який має назву кінетичного рівняння:
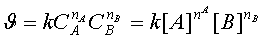
де nA
i nB - частинні порядки реакції за речовинами А і В відповідно, вони
визначаються на практиці для кожної окремої реакції, а їх сума nА +
nВ = n є загальним порядком реакції, який і
характеризує механізм процесу.
Порядок
реакції за реагентом - це експериментально
визначена величина, що дорівнює показнику ступеня, в який необхідно піднести
концентрацію даного реагенту, щоб теоретично розрахована швидкість реакції
дорівнювала встановленій практично.
З урахуванням
цього можна уточнити формулювання закону діючих мас: швидкість реакції
пропорційна добутку концентрацій реагентів у ступенях, що дорівнюють частинним
порядкам реакційза реагентами.
Реакції можуть
мати різні порядки, у тому числі й дробові. Якщо порядок реакції нульовий, швидкість
не залежить від концентрації реагуючих речовин, J
= const.
Швидкість реакції
першого порядку характеризується кінетичним рівнянням
J = kc (2)
Н2 à 2H
N2O5
(г) à 4NO2 (г) + O23OCH3
à CH4 + H2 + CO
Дорівнюючи
рівняння (1) і (2) і розділяючи перемінні, одержимо:
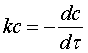
 (3)
(3)
Рішення даного
рівняння за початкових умов Сt=0 = C0 дає вираз:
С = С0
× е-kt. (4)
Підставивши (4) у
(3), знайдемо
= k C0
е-kt.
(5)
Як видно,
концентрація реагентів і швидкість реакції першого порядку зменшується за
експоненціальним законом. Рівняння (5) можна записати у вигляді
(C0/C)
= kt, (6)
звідки константа
швидкості реакції
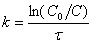 (6)
(6)
Вираз ln(C0/C)
безрозмірний, тому константа швидкості реакції першого порядку має одиницю
вимірювання [с-1]. За графіком змінення концентрації реагенту з
часом (рис. 2а) легко визначити константу швидкості реакції
k = tg a = D lnC/Dt (7)
Швидкість реакції
другого порядку для реагентів А і В підкоряється кінетичному рівнянню
J = k CA
× CB.
Якщо СА
= СВ = С, то
J = k × C2. (8)
Другий порядок
мають, наприклад, такі реакції:
HI = H2
+ I2
NO2 =
2NO + O2.
Із зіставлення
рівнянь (1) і (8) одержуємо:
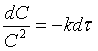 (9)
(9)
Розв¢×язок рівняння (32) для початкової умови Сt=0 = C0 має вигляд
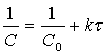 (10)
(10)
Або
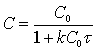 (11)
(11)
звідки константа
реакції
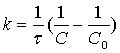 (12)
(12)
Із аналізу
одиниць вимірювання (12) встановлюється розмірність константи швидкості реакції
другого порядку [моль-1×л×с-1]. Константу швидкості можна визначити і графічно за
експериментальною кривою (рис. 2б).
Таким чином,
одиниці вимірювання константи швидкості залежать від порядку реакції.
4.
Енергія активації
Під час хімічної
реакції руйнуються одні молекули та виникають інші, відбувається змінювання
хімічних зв’язків і перерозподіл електронної густини. При цьому реакційна
система проходить через перехідний стан - так званий активований комплекс.
Наприклад,
перебіг реакції
АВ + СD = АС + ВD
В активованому
комплексі старі зв’язки ще не розірвані, але вже послаблені, нові зв’язки
намітилися, але ще не утворилися. Час існування комплексу дуже невеликий (10-13с).
При його розпаданні утворюються або продукти реакції, або знов вихідні
речовини.
Умовою
елементарного акту хімічної взаємодії є зіткнення частинок. Як доводить молекулярно-кінетична
теорія газів і рідин, кількість зіткнень настільки велика, що усі реакції
повинні відбуватися миттєво. Однак цього не спостерігається, оскільки не всі
зіткнення є ефективними, тобто такими, що завершуються хімічною
взаємодією.
Для досягнення
ефективних зіткнень необхідно, щоб кінетична енергія молекул була не тільки
достатньою для послаблення чи розривання старих зв’язків, але і перевищувала
енергію відштовхування (енергетичний бар’єр) між електронними оболонками
реагуючих частинок.
Внаслідок
перерозподілу енергії частина молекул у системі завжди має певний надлишок
енергії порівняно з середньою енергією. Ці молекули здатні подолати
енергетичний бар’єр і вступити в хімічну взаємодію. Такі реакційноздатні
молекули називаються активними. Енергія переходу речовини у стан
активованого комплексу, яка дорівнює різниці між середньою енергією молекул
системи і енергією, необхідною для перебігу хімічної реакції, називається
енергією активації. Отже, під час хімічного процесу перехід системи від вихідних
речовин з енергетичним станом Евих у енергетичний стан продуктів
реакції Епрод здійснюється через енергетичний бар’єр, який
визначається енергією активації реакції Еакт. При цьому різниця
енергій у вихідному і кінцевому станах дорівнює тепловому ефекту реакції:
DН = Епрод
- Евих.
Екзотермічні
реакції потребують з меншої енергії активації, ніж ендотермічні. Велика енергія
активації (тобто високий енергетичний бар’єр) є причиною того, що багато
хімічних реакцій при звичайних температурах не протікають, незважаючи на їх
принципову термодинамічну можливість (DG < 0). Так,
за умов низьких температур самочинно не горять на повітрі нафта, вугілля,
деревина, хоч для реакцій їх окиснення значення DG
від’ємне.
5.
Вплив температури на швидкість реакції
Підвищення
температури зумовлює зростання загальної енергії реакційної системи, а це, в
свою чергу, сприяє підвищенню швидкості руху і збільшенню відносного вмісту
активних молекул. Вплив температури на швидкість реакції оцінюється за
допомогою емпіричного правила Вант-Гоффа: підвищення температури на
кожні 10 градусів збільшує швидкість реакції приблизно у 2-4 рази
2 = V1 × g(T2-T1)/10, (13)
де Т2
- Т1 = DТ - збільшення температури, V1
i V2 - початкова і кінцева швидкість реакції, g
- температурний коефіцієнт швидкості, значення якого для ендотермічних реакцій
вище, ніж для екзотермічних (gенд > gекз). Для більшості реакцій g змінюється у
межах 2-4.
Приклад.
Температурний коефіцієнт реакції дорівнює 3. Як зміниться
швидкість реакції при зниженні температури на 400?
Розв¢язок. Перетворимо рівняння Вант-Гоффа і
підставимо дані, вважаючи, що Т2 - Т1 = - 40 (оскільки
температура знижується).
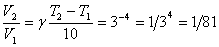
Тобто швидкість
реакції зменшиться у 81 разів.
Рівняння (13)
зручно використовувати лише для приблизних розрахунків, тому що його точність
не дуже висока. Точніше вплив температури на швидкість реакції відображає
рівняння Арреніуса (1889 р.):
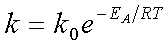 (14)
(14)
де k - константа
швидкості реакції, ЕА - енергія активації (для хімічних реакцій ЕА
= 40-400 кДж/моль), k0 - передекспоненційний множник Арреніуса,
пропорційний числу зіткнень між молекулами.
Інтегрування
рівняння Арреніуса дає вираз для розрахунку енергії активації за відомими
даними про константи швидкості при двох температурах:
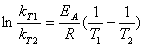 (15)
(15)
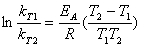 (16)
(16)
Якщо концентрації
реагуючих речовин дорівнюють 1 моль, то рівняння Арреніуса (39) дає змогу
виразити залежність швидкості реакції від температури:
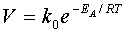
Згідно з
рівнянням Арреніуса константа швидкості зменшується при зростанні енергії
активації. Це рівняння дозволяє обчислювати константи швидкості (і саму
швидкість) реакцій при різних температурах.
6.
Теорія активних зіткнень
Теорія активних зіткнень
ґрунтована на підрахунку числа зіткнень між реагуючими частками, які
представляються у вигляді твердих сфер. Передбачається, що зіткнення приведе до
реакції, якщо виконуються дві умови: 1) поступальна енергія часток перевищує
енергію активації EA; 2) частки правильно орієнтовані в просторі один відносно
одного. Перша умова вводить у вираження для константи швидкості множник exp (-
EA/RT), який дорівнює долі активних зіткнень в загальному числі зіткнень. руга
умова дає так званий стеричний множник P - константу, характерну для цієї
реакції.
У ТАЗ отримано два
основні вираження для константи швидкості бімолекулярної реакції. Для реакції
між різними молекулами (A + B продукти) константа швидкості рівна
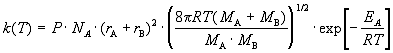 (1)
(1)
Тут NA - постійна
Авогадро, r - радіуси молекул, M - молярні маси речовин. Множник у великих
круглих дужках - це середня швидкість відносного руху часток A і B.
Константа
швидкості бімолекулярної реакції між однаковими молекулами (2A продуктів)
рівна:
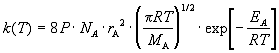 (2)
(2)
З(1) і(2)
витікає, що температурна залежність константи швидкості має вигляд:
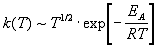 .
.
Згідно ТАЗ,
передекспоненціальний множник слабо залежить від температури. Досвідчена
енергія активації Eоп, визначувана по рівнянню, пов'язана з аррениусовской, або
істинною енергією активації EA співвідношенням:
Мономолекулярні
реакції у рамках ТАС описують за допомогою схеми Линдемана, в якій константу
швидкості активації k1 розраховують по формулах(1) і(2).
7.
Теорія активного комплексу
У теорії
активованого комплексу елементарну реакцію представляють як мономолекулярний
розпад активованого комплексу за схемою:
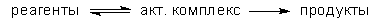
Передбачається,
що між реагентами і активованим комплексом існує квазірівновага. Константу
швидкості мономолекулярного розпаду розраховують методами статистичної
термодинаміки, представляючи розпад як одновимірну поступальну ходу комплексу
по координаті реакції.
Основне рівняння
теорії активованого комплексу має вигляд:
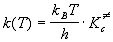 ,
,
де kB = 1.38.10-23 Дж/К
- постійна Больцмана, h = 6.63.10-34 Дж. с - постійна Планка, - константа
рівноваги утворення активованого комплексу, виражена через молярні концентрації
(у моль/л). Залежно від того, як оцінюють константу рівноваги, розрізняє
статистичний і термодинамічний аспекти ТАК.
Найбільш потужним
засобом інтенсифікації хімічних процесів є застосування каталізаторів.
Каталізатор - це речовина, що збільшує швидкістьреакції, кількісно і якісно при
цьому не змінюючись. Явище змінювання швидкості реакції під впливом
каталізатора називається каталізом.
Речовини, які
уповільнюють швидкість хімічних процесів, а самі при цьому не змінюються, називаються
інгібіторами.
Важливою властивістю
каталізаторів є їх селективність (вибірність), тобто здатність
спрямовувати взаємодію одних і тих же самих речовин у різних напрямках для
одержання бажаних продуктів, наприклад:
2O3, ThO2
С2Н5ОН
-àC2H4
+ H2O,, Cu2H5OH -à CH3CHO + H2.
Каталітична активність
багатьох каталізаторів зростає при додаванні невеликих кількостей промоторів
- каталітично неактивних речовин, присутність яких посилює дію
каталізаторів. Наприклад, швидкість окиснення SO3 на каталізаторі V2O5
зростає у сотні разів при додаванні промоторів - сульфатів лужних металів.
У той же час існують
речовини, які погіршують каталітичну активність - каталітичні отрути. Так,
до каталітичних отрут платинових каталізаторів належать сполуки сірки, миш’яку,
ртуті.
Каталізатори не
впливають на термодинамічні показники реакції (DН, DG, DS) і на константу хімічної рівноваги, вони рівною мірою збільшують
швидкість як прямої, так і зворотної реакцій.
Механізм дії
каталізаторів дуже складний і не до кінця вивчений. Однак доведено, що вони
зменшують енергію активації процесу, який у присутності каталізаторів протікає
по іншому шляху, через інші проміжні стани. Активований комплекс з участю
каталізаторів має меншу енергію, ніж комплекс без каталізаторів, тому енергія активації
каталітичної реакції ЕА,к нижча за енергію активації некаталітичної
реакції ЕА (рис. 3.
За своїм агрегатним
станом каталізатори бувають твердими, рідкими і газоподібними, тому каталітичні
процеси поділяються на гомогенні і гетерогенні.
При гомогенному каталізі
всі реагуючі речовини утворюють з каталізатором одну фазу (газоподібну або
рідку). Механізм гомогенного каталізу (Сабат’є, Зелінський) пояснюється на
основі проміжних сполук, які каталізатор утворює з реагентами. Це сприяє
зменшенню енергії активації. Наприклад, реакція
А + В à A…B à AB
у присутності
каталізатора може відбуватися за схемою:
А + К à A…K à AK+ B à A…K…B à AB + K
Прикладами газофазних
каталізаторів у гомогенних реакціях можуть бути:
2O(г)
СO(г) + O2
(г) à 2CO2(г),2(г)3CHO(г)
à CH4(г)
+ CO(г).
Більш поширені гомогенні
каталітичні реакції у рідкій фазі, де роль каталізатора іноді можуть
відігравати розчинники (особливо вода), іони водню або гідроксид-іони. Каталіз
іонами Н+ і ОН - називають кислотно-основним.
Приклади гомогенного рідкофазного каталізу:
I-(p)
або Fe2+(p)
2Н2О2
(р) -à 2H2O(р) + O2,
СН3СООС2Н5(р)+Н2О(р)à CH3COOH(р)
+ C2H5OH(р).
До гомогенних
каталітичних реакцій належать численні природні процеси, що каталізуються
ферментами.
При гетерогенному
каталізі реагенти і каталізатори перебувають у різних фазах і мають межу
поділу. Як правило, гетерогенними є тверді каталізатори, на поверхні яких
реагують газоподібні речовини. Сумарна швидкість перетворення на гетерогенному
каталізаторі залежить від площини його поверхні, тому звичайно використовують
каталізатори з розвиненою поверхнею або наносять їх тонким шаром на пористе
вугілля, силікагель тощо.
Існує декілька теорій
гетерогенного каталізу. Згідно з найбільш вичерпною теорією Баландіна для
здійснення каталізу необхідна геометрична відповідність між параметрами
кристалічної решітки каталізатора і довжинами хімічних зв’язків у молекулах
реагентів та продуктів реакції. У більшості теорій припускається, що реакція
протікає не на всій поверхні каталізатора, а лише на активних центрах -
ділянках, де забезпечуються оптимальні умови процесу. Кількість активних
центрів визначається складом поверхневого шару, способом приготування
каталізатора і обробки його поверхні.
Каталізатори мають
важливе значення, оскільки забезпечують економію енергії та сировини і
допомагають вирішувати, екологічні проблеми (очищення стічних вод, промислових
та автомобільних викидів). Застосування каталізаторів необхідне і при створенні
екологічно чистих маловідходних технологій.
8. Швидкість гетерогенних
реакцій
Гетерогенні реакції
відбуваються на поверхні поділу фаз, яка і вважається реакційним простором.
Швидкістю гетерогенної
реакції називається кількість речовини, що вступає в реакцію чи
утворюється внаслідок неї за одиницю часу на одиниці площини поверхні фаз.
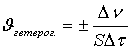
де Dn - різниця між кількістю речовини в кінцевий t2 і початковий t1 моменти часу (Dn
= n2 - n1), S - площина поверхні. Необхідно
зазначити, що площину поверхні твердого тіла не завжди легко виміряти, тому
іноді швидкість гетерогенної реакції відносять не до одиниці поверхні, а до
одиниці маси чи до одиниці об’єму.
Особливістю
кінетики гетерогенних реакцій є вплив площини реакційної поверхні на швидкість
реакції.
Однак, якщо в
реакції безпосередньо бере участь тверда речовина, то в кінетичне рівняння не
входить його концентрація, яка вона постійною. Наприклад, для гетерогенної
реакції
Са (к) + СО2
(г) à CaCO3 (к)
кінетичне
рівняння має вигляд:
= K × C(CO2) = k [CO2].
Більшість
гетерогенних реакцій складається з трьох основних стадій:
підведення однієї
реагуючої речовини до поверхні іншої;
2
хімічна взаємодія на
поверхні;
3
відведення продукту від
поверхні.
Повільніша стадія, яка
визначає швидкість реакції в
цілому, називається
лімітуючою. Якщо енергія активації хімічної реакції невелика, то
лімітуючими стадіями є перенесення речовини. Для підвищення швидкості таких
реакцій посилюють конвекцію найчастіше за допомогою перемішування. Так, горіння
вугілля, хімічна стадія якого потребує невеликої енергії активації,
відбувається тим швидше, чим інтенсивніше подається до вугілля кисень.
Однак для реакцій з
високою енергією активації лімітуючою є друга стадія, в цьому випадку
переміщування не буде прискорювати взаємодію. Наприклад, ржавіння заліза на
вологому повітрі не посилюється при збільшенні подачі кисню, оскільки енергія
активації цієї реакції досить значна.
Другою особливістю
швидкості гетерогенної реакції є її залежність від швидкості подачі реагенту в
реакційну зону.
Найбільше змінювання
концентрації спостерігається у дифузійному шарі - тонкому шарі реагенту поблизу
реакційної поверхні. Перенесення речовини в ньому здійснюється за рахунок
дифузії. При перемішуванні товщина дифузійного шару зменшується і відповідно
зростає швидкість підведення реагентів. Якщо швидкість дифузії нижча, ніж
швидкість хімічної взаємодії, то лімітуючою стадією є дифузія. В такому випадку
говорять, що має місце дифузійний контроль. Коли швидкість дифузії достатньо
висока, то спостерігається кінетичний контроль, при якому процес лімітується
власне хімічною реакцією. А при змішаному контролі швидкості дифузії і хімічної
реакції порівнянні [5].
Гетерогенні процеси
мають важливе значення в техніці; до них належать корозія металів і сплавів,
горіння твердого палива, випалювання сульфідних руд тощо.
9. Теорія
мономолекулярної адсорбції Ленгмюра
термодинаміка реакція
гетерогенний ленгмюр
Адсо́рбція - вибіркове поглинання речовини з газового чи рідкого середовища
поверхневим шаром твердого тіла (адсорбенту) чи рідини. Компонент що поглинається,
який вміщується в суцільному середовищі (газі, рідині), називають адсорбтивом,
а той що вміщується в адсорбенті - адсорбатом.
Теорія мономолекулярної
адсорбції, яку розробив американський хімік И. Ленгмюр, ґрунтується на
наступних положеннях.
) Адсорбція є
злокалізованою і викликається силами, близькими до хімічних.
) Адсорбція відбувається
не на усій поверхні адсорбенту, а на активних центрах, якими є виступи або
западини на поверхні адсорбенту, що характеризуються наявністю т.з. вільних
валентностей. Активні центри вважаються незалежними (тобто один активний центр
не впливає на адсорбційну здатність інших), і тотожними.
) Кожен активний центр
здатний взаємодіяти тільки з однією молекулою адсорьату; в результаті на
поверхні може утворитися тільки один шар адсорбованих молекул.
) Процес адсорбції є
оборотним і рівноважним - адсорбована молекула утримується активним центром
деякий час, після чого десорбується; т.ч., через деякий час між процесами
адсорбції і десорбції встановлюється динамічна рівновага.
Згідно електронної
теорії гетерогенного каталізу, каталітична активність напівпровідників
пов'язана з об'ємною концентрацією носіїв струму (електронів і дірок).
Адсорбція частки на поверхностисти напівпровідника призводить до освіти
доповнить. (домішкового) енергетичного рівня в забороненій зоні. Перехід
електрона або дірки на цей рівень змінює їх об'ємну концентрацію і властивості
поверхні (напр., роботу виходу електрона), на якій виникають заряджені центри,
що беруть участь в каталитич. перетворенні. Можна представити, напр., що
дегідрування изопропилового спирту відбувається по механізму:
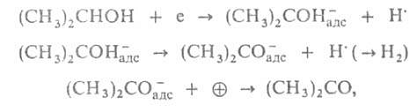
де е-электрон
каталізатора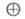 -
вільна дірка. Оскільки об'ємна концентрація носіїв струму залежить від
положення рівня Ферми і змінюється при всякому зрушенні останнього,
передбачалася можливість регулювання каталітичної активності напівпровідника
зміщенням рівня Ферми. Подальші дослідження, проте, не підтвердили існування
електронної рівноваги між поверхнею і об'ємом каталізатора-напівпровідника в
умовах гетерогенного каталізу.
-
вільна дірка. Оскільки об'ємна концентрація носіїв струму залежить від
положення рівня Ферми і змінюється при всякому зрушенні останнього,
передбачалася можливість регулювання каталітичної активності напівпровідника
зміщенням рівня Ферми. Подальші дослідження, проте, не підтвердили існування
електронної рівноваги між поверхнею і об'ємом каталізатора-напівпровідника в
умовах гетерогенного каталізу.
10.
Активні центри гетерогенного каталізатора
Поняття про
активні центрах з’явилося в 1924 р., коли Г. Тейлор висловив припущення про те,
що каталітично активною є не вся поверхня гетерогенного каталізатора, а лише
деякі ділянки її - так звані активні центри (АЦ).
В середині ХХ
століття було запропоновано кілька приватних теорій дії каталізаторів. В цих
теоріях розглядаються структури АЦ і механізм їх дії.
Відповідно до
теорії проміжних поверхневих сполук (Поляни), на АЦ відбувається активована
адсорбція реагує молекули, що призводить до розриву зв’язків між атомами в ній
і утворення проміжного з’єднання з атомами каталізатора.
У теорії
перехідних станів (Ейрінг і Поляни) передбачається, що АЦ надають деформуючому
(розпушує) дія на зв’язку в адсорбованих молекулах.
Структура АЦ
розглядається в мультиплетной теорії каталізу (А.А. Баландін). АЦ - мультиплет
- складається з невеликого (від двох до шести) кількості атомів, розташованих
певному чином. Важливу роль в каталітичної активності відіграє структурний
відповідність відстаней між атомами в молекулах реагентів параметрам кристалічної
решітки каталізатора. У багатьох випадках виявляється зв’язок каталітичної
активності з міцністю хімічного зв’язку між молекулою реагенту та АЦ.
Відповідно до теорії мультіплетов, дегідрування граничних одноатомних спиртів
відбувається на дублеті, а дегидрирование циклогексану - на секстеті
Таким чином,
теорія мультіплетов дозволила зв’язати каталітичну активність металів з
величиною їх атомного радіусу і типом кристалічної решітки.
АЦ можуть, бути
не тільки частиною кристалічної поверхні (наприклад, металу), а й певною
ділянкою макромолекули в ферментативному каталізі.
У теорії активних
ансамблів (Н.І. Кобозев) передбачається, що при нанесенні каталітично активного
металу на інертний носій каталітичні властивості проявляють окремо розташовані
атоми або сукупності невеликого числа атомів - «активні ансамблі».
11.
Активні центри гомогенних каталізаторів
У гомогенному
каталізі в ідеальному випадку всі молекули розчиненої каталізатора є АЦ і
утворюють з реагентами проміжні реакційноздатні продукти. Характер проміжного
хімічної взаємодії при цьому дуже різноманітний і залежить від типу гомогенного
каталізу.
В
окисно-відновному каталізі, коли утруднений перенос електронів, що віддаються
відновником і прийнятих окислювачем, каталізатор у вигляді іона бере участь в
електронному перенесення і процес протікає по багатостадійному механізму.
В
кислотно-основному гомогенному каталізі АЦ служать кислоти і підстави або в
формі (загальний кислотно-основний каталіз), або іони Н3О + і ОН - (специфічний
кислотно-основний каталіз). Під їх дією зазвичай посилюються електрофільні або
нуклеофільні властивості молекул реагентів.
В
металокомплексної гомогенному каталізі АЦ є перехідні метали (Pd, Pt, Rh, Ti,
Fe, та ін.), що входять до складу координаційних з’єднань і здатні до утворення
комплексів з молекулами реагентів.
Висновок
Можна зробити висновок,
що хімічна кінетика вирішує дві конкретні задачі, це визначення механізму
реакції, тобто встановлення елементарних стадій процесу і послідовності
їх протікання та кількісний опис хімічної реакції, а саме: встановлення сурових
співвідношень, що дають можливість обчислювати змінення кількості вихідних
реагентів і продуктів протягом перебігу реакції. Це дає змогу розраховувати
швидкість перебігу реакцій, а також дозволяє визначити механізми, за допомогою
яких можна з високою точністю прискорити або уповільнити хід реакції.
Найбільш потужним
засобом інтенсифікації хімічних процесів є застосування каталізаторів.
Механізм дії
каталізаторів дуже складний і не до кінця вивчений. Однак доведено, що вони
зменшують енергію активації процесу, який у присутності каталізаторів протікає
по іншому шляху, через інші проміжні стани.
Література
1. Загальна та неорганічна хімія, А.М. Голуб, Київ, 1968. - 328
с.
2. Кнорре Д.Г., Эмануэль Н.М. «Курс химической кинетики».
4-е издание, М.: Высшая школа, 1984. - 463-468 с.
. В.И. Коробова, В.Ф. Очкова «Химическая кинетика: введение
с Mathcad/Maple/MCS» М.: Горячая линия-Телеком, 2009. - 93, 98 с.
. Г.С. Яблонский, В.И. Быков, А.Н. Горбань, «Кинетические модели
каталитических реакций», Новосібірск: Наука (Сиб. отделение), 1983. - 48-52 с.