|
Электродная реакция
|
Е1/2, В
|
|
As3+ + 3 e-
= As
|
-0,7
|
1 моль/л Н2SO4
+ 0,01% желатина
|
|
Cd2+ + 2e-
= Cd
|
-0,60
|
0,1 моль HCl
|
|
Cd2+ + 2e-
= Cd
|
-0,79
|
6 моль/л
HCl
|
|
Co2+ + 2e-
= Co
|
-1,03
|
1
моль/л KSCN
|
|
Cu2+ + 2e-
= Cu
|
0
|
0,5 моль/л
H2SO4 + 0,01% желатина
|
|
Cu2+ + 2e-
= Cu
|
-0,38
|
1 моль/л Na2C4H4O6.
pH=12
|
|
Fe2+ + 2e-
= Fe
|
-1,37
|
1 моль/л HClO4. pH=0-2
|
|
Mn2+ + 2e-
= Mn
|
-1,54
|
0,5 моль/л NH3
+ 0,5 моль/л NH4Cl
|
|
Ni2+ + 2e-
= Ni
|
-1,1
|
HClO4. pH =
0-2, 1 моль/л KCl
|
|
Ni2+ + 2e-
= Ni
|
-1,06
|
1 моль/л NH3
+ 0,2 моль/л NH4Cl + 0,005% желатина
|
|
Zn2+ + 2e-
= Zn
|
-1,02
|
1 моль/л KCl
|
|
Zn2+ + 2e-
= Zn
|
-1,33
|
1 моль/л NH3
+ 0,2 моль/л NH4Cl + 0,005% желатина
|
|
Zn2+ + 2e-
= Zn
|
-1,49
|
1 моль/л NaOH
|
Типичная зависимость силы тока от
приложенного напряжения дана на рисунке:
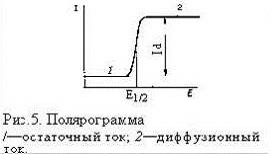
Это полярографическая волна
(полярограмма). Из рисунка видно, что в начале процесса при небольшом
потенциале катода сила тока медленно увеличивается с возрастанием потенциала -
это так называемый остаточный ток, его величина имеет порядок 10-7
А. По достижении потенциала восстановления на катоде начинается разряд ионов и
сила тока резко возрастает, стремясь к предельной величине диффузионного тока.
При I = 1/2Id уравнение (9) переходит в
Е=Еo1/2.
Это соотношение, так же как и (10),
показывает независимость потенциала полуволны от силы тока и, следовательно, от
концентрации восстанавливающегося иона. Потенциал полуволны является, такм
образом, качественной характеристикой иона в растворе данного фонового
электролита и определение потенциала полуволны составляет основу качественного
полярографического анализа.
Однако потенциал полуволны
существенно зависит от среды, природы и концентрации фонового электролита.
Особое значение имеет наличие в
растворе веществ, способных к комплексообразованию с определяемым ионом.
Присутствие в исследуемом растворе лиганда смещает потенциал полуволны в
отрицательную область, что используется для определения сосатва и констант
устойчивости координационных соединений.
Сдвиг потенциала полуволны при
введении в раствор лиганда значительно расширяет возможности полярографического
анализа, позволяя создавать условия для определения нескольких компонентов в
одном растворе без их предварительного разделения. Например, в 1М KCl ионы свинца(II) и таллия (I) имеют потенциалы полуволны,
соответственно, -0,435 и -0,483 В и на этом фоне их раздельное определение
неосуществимо. В 1М NaOH потенциал полуволны свинца становится равным -0,755 В, а у таллия
остается практически без изменений, поэтому в щелочном растворе эти ионы могут
быть определены при совместном присутствии.
Если в растворе находится несколько
веществ, потенциалы полуволны которых различаются на 100 мВ и больше, то на
полярограмме будет не одна волна, а несколько - по числу восстанавливающихся ионов,
а возможно и больше, так как при ступенчатом восстановлении один ион может
давать две волны.
Например, две волны дает ион Сu2+ в присутствии 1М NH3: первую при потенциале
полуволны -0,20 В и вторую при -0,48 В. Можно получить таким образом полярографический
спектр ионов, а затем по этим данным данным и измеренному потенциалу полуволны
идентифицировать неизвестное вещество.
Вполне понятно, что положение
элемента в таком спектре будет зависеть от фонового электролита: его природы и
концентрации.
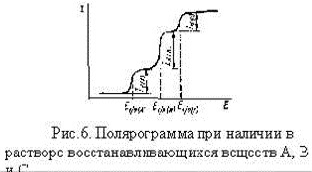
На полярограммах нередко возникают
максимумы различной формы, мешающие определению истинного потенциала полуволны
и силы тока. Различают максимумы I и II рода. Теория связывает их появление с гидродинамическими
явлениями в растворе. Вызываемыми каплями ртути, и адсорбционными процессами.
Для подавления максимумов в полярографируемый раствор обычно вводят
поверхностно-активные вещества: желатин, агар-агар и др. Подавление максимумов
поверхностно-активными веществами лежит в основе нескольких чувствительных (до
10-9 моль/л) аналитических методик определения этих веществ в
растворе.
Связь диффузионного тока Id с концентрацией
иона см и другими величинами передается уравнением Ильковича:
Id = 605zD1/2 m2/3 t1/6 cM (11)
где z - заряд иона; D - коэффициент диффузии; m - масса ртути, вытекающей из
капилляра в 1 с, мг; t - время образования капли (период капания).
Среди величин, входящих в это
уравнение, труднее всего поддается эксперементальному определению коэффициент
диффузии D, а использование соответствующих справочных данных не всегда
возможно. Поэтому коэффициент пропорциональности между концентрацией вещества и
силой диффузионного тока обычно устанавливают с помощью стандартных растворов.
Действительно, при постоянных условиях полярографирования D, m и t постоянны и уравнение переходит в
Id = kcM (12)
В связи с этим в работах по
полярографии всегда указывается так называемая характеристика капилляра,
вычисляемая как m2/3 t1/6. Линейная зависимость
является основой количественного полярографического анализа.
Так как коэффициент диффузии D зависит от температуры, то и
коэффициент пропорциональности К в уравнении Ильковича изменяется при изменении
температуры. Для водных растворов в температурном интервале 20-50 oС коэффициент диффузии
полярографически активных веществ-деполяризаторов увеличивается примерно на 3%
при росте температуры на один градус, что и приводит к повышению среднего
диффузионного тока Id на ~1-2% oС. Поэтому полярографирование проводят при постоянной температуре,
термостатируя полярографическую ячейку обычно при 25 ± 0,5 oС.
Масса ртути m и время каплеобразования t зависят от характеристик ртутного
капающего электрода и высоты столбика ртути в капилляре и в резервуаре,
связанном с капилляром.
Стеклянный капилляр ртутного
капающего микроэлектрода обычно имеет внешний диаметр 3-7 мм, внутренний - от
0,03 до 0,05 мм, длину 6-15 см. высота ртутного столбика от нижнего конца
капилляра до верхнего уровня поверхности ртути в резервуаре составляет 40 - 80
см.
Содержание индифферентного
электролита в анализируемом полярографируемом растворе должно примерно в 100
раз превышать содержание определяемого вещества-деполяризатора, причем ионы
фонового электролита не должны разряжаться в условиях проведения
полярографирования до разряда полярографически активного вещества.
Полярографирование проводят с
использованием в качестве растворителя воды, водно-органических смесей (вода -
этанол, вода - ацетон, вода - диметилформамид и др.) и неводных сред (этанол,
ацетон, диметилформамид, диметилсульфоксид и т.д.).
До начала полярографирования через
анализируемый раствор пропускают ток инертного газа (азота, аргона и др.) для
удаления растворенного кислорода, который также дает полярографическую волну
вследствие восстановления по схеме:
О2 + 2Н+ + 2е-
= Н2О2
Н2О2 + 2Н+
+ 2е- = 2Н2О
Иногда - в случае щелочных растворов
- вместо пропускания тока инертного газа в анализируемый раствор прибавляют
небольшое количество активного восстановителя - сульфита натрия, метола,
которые связывают растворенный кислород, реагируя с ним.
) Схема полярографической установки
Принципиальная схема
полярографической установки представлена на рисунке:
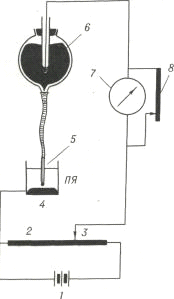
Как уже отмечалось, напряжение,
которое подается на электроды, будет практически целиком определять потенциал
катода (капающего ртутного электрода).
В вольамперометрии с успехом
применяют также твердые микроэлектроды, изготовляемые из благородных металлов
(платины, золота и др.) или графита. Основными достоинствами твердых электродов
являются возможность работы в более положительной области потенциалов (до 1,3
В), чем с ртутным электродом (ртутный капающий электрод используется в области
примерно от 0,3 до -2,0 В), и их нетоксичность (пары ртути, как известно,
чрезвычайно ядовиты и работа с ртутным электродом требует строго соблюдения
специальных правил техники безопасности).
Однако использование твердых
электродов также имеет свои трудности, связанные, главным образом, с
обновлением поверхности электродов. Стационарные твердые электроды не нашли
широкого применения в практике из-за медленности установления предельного тока,
невысокой чувствительности и других недостатков.
Значительно более широкое применение
имеют вращающиеся и вибрирующие платиновые микроэлектроды, на которых
устойчивая сила тока устанавливается быстро. При работе таких электродов раствор
непрерывно перемешивается, благодаря чему к поверхности электрода ионы
доставляются не только за счет диффузии, но и за счет механического
перемешивания. Это значительно (в 10-20 раз) увеличивает предельный ток по
сравнению с диффузионным. По точности методы с применением твердых электродов
часто уступают методам, использующим ртутный капающий электрод, однако
применение вращающегося платинового микроэлектрода позволяет существенно
расширить анодную область потенциалов, пригодную для полярографических измерений
до 1,4 В по сравнению с областью, в которой обычно применяется ртутный капающий
электрод (до 0,3 В).
Тем не менее ртутный капающий
электрод сохраняет свое большое практическое значение, так как на твердых
электродах ограничены катодные процессы из-за небольшого перенапряжения
водорода на платине - из кислых растворов на платине он начиняет выделяться при
потенциале около -0,1 В, а на ртути только при -2,0 В. промышленностью
выпускаются полярографы нескольких марок, которые пригодны для выполнения аналитических
работ и проведения научных исследований (ПЭ-312, КАП-225у, ППТ-1 и др.)
) Прямая полярография
Методы прямой полярографии основаны
на непосредственном применении уравнения полярографической волны (9) и
уравнением Ильковича (11) или (12). Потенциал полуволны не зависит от
концентрации и является качественной характеристикой вещества. Обычно потенциал
полуволны определяют графическим методом. Уравнение (9) показывает, что
lg (Id - I)/I
является линейной функцией Е, и,
следовательно, если на график нанести
lg(Id - I)/I
как
функцию Е, то получится прямая, которая пересекает ось абсцисс в точке,
где Е = Е1/2, т.е. когда
lg(Id - I)/I = 0.
Для идентификации неизвестного
вещества можно этим методом определить потенциал полуволны и, пользуясь таблицей
потенциалов полуволны или полярографическим спектром, установить наиболее
вероятный элемент. Однако чаще это свойство используется для выбора фонового
электролита. Зная качественный состав пробы, подбирают по табличным данным
такой фон, на котором полярографическая волна определяемого элемента может быть
получена без каких-либо искажений за счет волны мешающего элемента или иного
электродного процесса.
) Количественный полярографический
анализ
Из изложенного выше следует, что
количественный полярографический анализ основан на измерении диффузионного тока
Id как функции концентрации определяемого полярографически активного
вещества-деполяризатора в полярографируемом растворе.
При анализе получаемых полярограмм
концентрацию определяемого вещества находят методами градуировочного графика,
добавок стандарта, стандартных растворов.
а) Наиболее широко в количественном
полярографическом анализе применяется метод градуировочного графика на основе
уравнения (12). По этому методу готовят серию стандартных растворов, каждый из
которых содержит точно известную концентрацию с определяемого вещества.
Проводят полярографирование каждого раствора (после продувания через него тока
инертного газа) в одинаковых условиях, получают полярограммы и находят значения
Е1/2 (одинаковые для всех растворов) и диффузионного тока Id (разные для всех
растворов). По полученным данным строят градуировочный график в координатах Id - c, представляющий собой обычно прямую
линию в соответствии с уравнением Ильковича.
Затем проводят полярографирование
анализируемого раствора с неизвестной концентрацией сх определяемого
вещества, получают полярограмму, измеряют величину диффузионного тока Id(х) и по
градуировочному графику находят концентрацию сх.
б) Широко распространен в
количественной полярографии метод добавок стандарта.
Пусть при полярографировании
исследуемого раствора сила диффузионного тока равна
Ix = kcx.
Добавим к этому раствору известное
количсетво стандартного раствора сст и снова определим диффузионный ток:
Ix+cт = k(cx
+ ccт)
При почленном делении получаем:
Ix/Ix+cт = cx/cx +cст, откуда сх = сстIx/Ix+ст-Ix.
По этому соотношению находим
концентрацию анализируемого раствора. Можно использовать также графический
метод. В этом случае полученные данные наносят на график зависимости Ix+ст от сст. При Ix+ст=0, как показывает
уравнение, cх = -сст, т.е.
при экстраполяции прямая на этом графике при Ix+ст=0 отсекает на оси абсцисс величину, равную концентрации
определяемого вещества. В методе добавок автоматически учитывается влияние фона
и так называемых третьих компонентов, что является важным достоинством метода,
позволяющим применить его при анализе сложных смесей.
Если в анализируемом растворе
присутствует несколько веществ, восстанавливающихся на ртутном катоде, на
полярограмме, как уже отмечалось, появится несколько волн. По величине
потенциала полуволны определяют качественный состав, а по силе диффузионного
тока - концентрацию каждого из компонентов.
в) Метод стандартных растворов. В
одинаковых условиях проводят полярографирование двух растворов: анализируемого
раствора с неизвестной концентрацией сх и стандартного раствора с
точно известной концентрацией сст определяемого вещества. На
полученных полярограммах находят высоты полярографических волн hx и hст, отвечающие диффузионному току при концентрациях соответственно сх
и сст.
hx/hст = сx/cст, cx = hxcст/hст.
Где сх - концентрация
стандартного раствора; hx и hcт - высота волны при
полярографировании соответственно анализируемого и стандартного растворов.
Стандартный раствор готовят так,
чтобы его концентрация была бы как можно ближе к концентрации определяемого
раствора. При этом условии ошибка определения минимизируется. Метод применим
только в условиях строгой стандартизации условий полярографирования.
) Определение цинка в растворе
методом стандарта
В колбу емкостью 50 мл помещают
точно 10 мл раствора ZnSO4 ( Zn = C1 = 0,5 мг/мл), 20 капель раствора
желатина и доводят до метки 1н раствором КСl. Полученный раствор тщательно
перемешивают. Около 20-25 мл этого раствора помещают в сухой (или сполоснутый
этим же раствором) электролизер и пропускают через раствор водород в течение 20
мин. Затем снимают полярограмму (при соответствующем положении шунта), строят
график и определяют высоту волны h1.
К раствору ZnSO4 неизвестной концентрации, помещенному в колбу емкостью 50 мл,
добавляют 20 капель раствора желатина и доводят до метки 1 н раствором КCl. Затем раствор
перемешивают. Подготовленный раствор наливают в электролизер, пропускают через
него водород в течение 20 мин., снимают полярограмму, строят график и
определяют высоту волны hx. Находят концентрации Zn - ионов (Сх) по формуле (в мг/мл):
C1/n1 = Cx/nx
где С1 = 0,5(10/50) = 0,1
мг/мл.
Содержание цинка в миллиграммах в
данном растворе вычисляют, умножая Cx на емкость мерной колбы (50 мл).
полярография комплексообразование
цинк
7) Дифференциальная полярография
Для анализа смесей, содержащих ионы
или вещества с близкими потенциалами полуволны, применяют методы
дифференциальной полярографии, использующие кривые dI/dE - E. Необходимые соотношения можно
получить на основании уравнения (9). Придадим ему вид:
E - E1/2 = RT/nF(ln(Id/I - 1))
И после потенцирования
eE-E1/2 = (Id/I - 1) RT/nF
Решим относительно I:
I = Id/(1+e(E1-E2)nF/RT )
Для удобства дальнейшего
дифференцирования объединим постоянные:
I = Id/ (1+e(E-E1/2)k)
И продифференцируем по Е:
dI/dE = (Idk/(1-e(E-E1/2)k))e(E-E1/2)k
Чтобы найти положение максимума,
продифференцируем уравнение еще раз по Е и приравняем производную нулю. Тогда
получим. Что в точке максимума
(Е)max = Е1/2,
т.е. что потенциал точки,
соответствующей максимуму кривой, является потенциалом полуволны. Величину
ординаты в этой точке находим при подстановке соотношения в уравнение:
(dI/dE)E=E1/2
= Idk/(1+1)2 = Id nF/4RT
Следовательно, ордината в точке
максимума пропорциональна силе диффузионного тока и является, таким образом,
мерой концентрации вещества. Это значение можно использовать, например, при
построении градуировочных графиков.
Получать дифференциальные
полярограммы можно графическим дифференцированием обычных полярограмм или с
помощью специальной электрической схемы, позволяющей посредственно записывать
дифференциальную кривую во время полярографирования.
Дифференциальная полярограия имеет
значительно более высокую разрешающую способность, позволяя определить в одном
растворе ионы с близкими потенциалами полуволны. Например, этим методом могут
быть определены свинец и таллий, у которых потенциалы полуволны на фоне 2М КNО3 различаются только на
0,06 В. На интегральной полярограмме оба иона образуют одну общую волну, а на
дифференциальных кривых четко видны два максимума. Кроме того, методы
дифференциальной полярографии более точны, так как фиксировать положение
максимума и измерять его высоту можно с более высокой точностью, чем определять
аналогичные характеристики в методе обычной полярографии.
8) Хроноамперометрия с линейной
разверткой потенциала
Новые возможности в анализе и
изучении электродных процессов открывает хроноамперометрия с линейной
разверткой потенциала. Обычная скорость изменения потенцала в этом методе
составляет до 50 мВ/с вместо 2-3 мВ/с в классичсекой полярографии. Для
измерения силы тока здесь вместо гальванометра используют безынерционный
осциллограф.
При достижении потенциала
восстановления ток резко возрастает и достигает максимума, превышая величину Id классической
полярографии, поскольку происходит электровосстановление практически всех ионов
приэлектродного слоя, и затем падает. Так как приэлектродный слой обедняется
ионами, а скорость диффузии недостаточна, чтобы пополнить дефицит за столь
короткое время. Потенциал максимума Emax на этой кривой является качественной характеристикой иона, а
высота hmax пропорциональна концентрации иона. Хроноамперометрия с линейной
разверткой потенциала имеет более низкий предел обнаружения, чем обычная
вольтамперометрия.
Развитие полярографии в последние
годы привело к появлению новых полярографических методик, таких, как импульсная
полярография, векторная полярографии и др. Во многих из них используется
наложение на обычную медленную постояннотоковую полярографическую развертку
потенциала переменного напряжения. Наложение переменного напряжения понижает
предел обнаружения и улучшает разрешающую способность. Переменнотоковая
вольамперная кривая также является кривой с максимумом.
) Инверсионная вольтамперометрия
Существенное увеличение
чувствительности дат инверсионная вольтамперометрия.
Идея метода инверсионной
полярографии состоит в выделении определяемого элемента из очень разбавленного
раствора на ртутной капле или тонкой пленке ртути на графитовом электроде или
просто на графитовом электроде электролизом с последующим анодным растворением
полученной амальгамы. Процесс накопления происходит при потенциале,
соответствующем предельному току. Зависимость силы тока от напряжения при анодном
растворении имеет вид характерного пика, глубина которого h пропорциональна концентрации
определяемого иона, а потенциал минимума Еmin определяется природой
иона. Предел обнаружения в методике инверсионной вольтамперометрии на 2-3
порядка ниже предела обнаружения в обычных полярографических методиках. Чем
больше продолжительность накопительного электролиза, тем большее каоличество
металла перейдет из раствора в ртутную каплю и тем больше возрастет
чувствительность анализа. Например, при анализе растворов, в которых
концентрация определяемого элементы составляет 10-9 моль/л. Время
электролиза доходит до 1 ч.
) Анализ органических соединений
Объектами вольтамперометрического
анализа являются не только неорганические вещества или ионы, но и многие органические
вещества, способные к электрохимическим превращениям. В электродной реакции
органического вещества обычно принимают участие ионы водорода:
R + nH+ + ne- = RHn
Анализ в связи с этим проводится в
буферных растворах с достаточно высокой буферной емкостью.
К вольамперометрически активным
группировкам относятся, например, =CNO, =С=N, -NO2, - O - O -, - S - S - и многие другие. В условиях вольтамперомтрического анализа
легко восстанавливаются и могут быть определены многие альдегиды, кетоны, азо-
и нитросоединения, органические пероксиды. Органические кислоты (малеиновая,
фумаровая и др.) и эфиры окисляются на платиновом электроде.
) Полярографическое исследование
реакций комплексообразования
Зависимость потенциала полуволны от
свойств фонового электролита эффективно используется в химии и термодинамике
координационных соединений для установления состава и определения констант
устойчивости образующихся комплексов. Чем выше устойчивость образующихся
комплексных соединений и концентрация лиганла, тем больше сдвигается потенциал
полуволны в отрицательную область. При образовании одного соединения эта
зависимость при 25 С выражается уравнением
Где Еo1/2 - сдвиг потенциала полуволны при даннй концентрации лиганда сL по сравнению с
потенциалом полуволны в отсутствие лиганла; бета - константа устойчивости
комплексного соединения; p - координационное число. Величины lgβ и p определяются графически, используя зависимость Еo1/2 от lgcL.
) Применение полярографии
Условия проведения
полярографического анализа.
Из вышеизложенного следует, что при
проведении полярографического анализа требуется соблюдение, по карйней мере,
следующих условий:
) Для поддержания необходимой
электропроводности анализируемого раствора в него вводят фоновый электролит,
например, хлорид или нитрат калия, хлорид аммония, соли тетраалкиламмония и др.
Ионы фонового электролита должны разряжаться на ртутном капающем микроэлектроде
при более высоких значениях приложенного потенциала, чем полярографируемое вещество.
Концентрация фонового электролита
должна быть выше концентрации полярографически активного вещества в оптимальном
случае не менее чем в 100 раз. При этом концентрация самого полярографически
активного вещества обычно лежит в пределах от ~102 моль/л до ~10-5
моль/л.
) Перед проведением
полярографического анализа из анализируемого раствора должен быть удален
растворенный в нем кислород. Это достигается чаще всего путем пропускания тока
инертного газа (например. азота) через раствор в течение ~15 минут перед
началом полярографирования.
) Иногда на полярограмме
появляются максимумы, соответствующие протеканию электрического тока,
превышающего предельный ток. Появление максимумов обусловлено движением
поверхности капли жидкой ртути при каплеобразовании, что приводит к
перемешиванию диффузного слоя растворана поверхности капли, к увеличению числа
диффундирующих частиц полярографически активных веществ и к их разряду на
микроэлектроде, следствием чего и является увеличение электрического тока,
протекающего через полярографическую ячейку.
Для подавления максимумов тока в
раствор вводят добавки желатина или других поверхностно-активных веществ
(агар-агар, метиловый красный, фуксин и т.д.). которые изменяют поверхностное
натяжение ртутной капли и препятствуют движению поверхностных слоев ртутной
капли.
) Необходимо термостатировать
полярографическую ячейку, поддерживая температуру постоянной с точностью ±0,5 оС.
13) Применение метода
Полярография используется для
определения малых количеств неорганических и органических веществ. Разработаны
тысячи методик количественного полярографического анализа. Предложены способы
полярографического определения практически всех катионов металлов, ряда анионов
(бромат-, иодат-, нитрат-, перманганат-ионов), органических соединений
различных классов, содержащих диазогруппы. Карбонильные, пероксидные,
эпоксидные группы, двойные углерод-углеродные связи, а также связи
углерод-галоген, азот-кислород, сера-сера.
Метод - фармакопейный, применяется
для определения салициловой кислоты, норсульфазола, витамина В1, алкалоидов,
фолиевой кислоты, келлина в порошке и в таблетках, никотинамида, пиридоксина
гидрохлорида, препаратов мышьяка, гликозидов сердечного действия, а также
кислорода и различных примесей в фармацевтических препаратах.
Метод обладает высокой
чувствительностью (до 10-5 - 10-6 моль/л);
селективностью; сравнительно хорошей воспроизводимостью результатов (до ~2 %);
широким диапазоном применения; позволяет анализировать смеси веществ без их
разделения, окрашенные растворы, небольшие объемы растворов (объем
полярографической ячейки может составлять всего 1 мл); вести анализ в потоке
раствора; автоматизировать проведение анализа.
К недостаткам метода относятся
токсичность ртути, ее довольно легкая окисляемость в присутствии веществ-окислителей,
относительная сложность используемой аппаратуры.
Список литературы
1) Васильев, В.П.
Аналитическая химия. В 2 кн. Кн. 2: Физико-химические методы
анализа. Учебник для студ.вузов, обучающихся на химико-технол.спец.
Издательство «Дрофа», 2002
Стр. 211-230
) Харитонов Ю.Я.
Аналитическая химия (аналитика). В 2 кн. Кн. 2: Количественный
анализ. Физико-химические (инструментальные) методы анализа. Учебник для вузов.
Издательство «Высшая школа», 2001.
Стр. 466-481
) Крешков, А.П.
Основы аналитической химии, ч.2.
Издательство «Химия», Москва, 1976
Стр 412, 443
) Интернет-ресурсы:
<http://eniw.ru/polyarografiya.htm>
<http://www.chem-astu.ru/chair/study/PCMA/r2_4_1.htm>