1.2
Биологическая активность (со)полимеров N-винилпирролидона
(Со)полимерам N-винилпирролидона (ВП) присуще
сочетание высокой гидрофильности, способность к комплексообразованию,
отсутствие токсичности, хорошие адгезионный свойства, что позволяет
использовать их в различных целях. Несмотря на то, что активные исследования в
этой области проводятся не менее 70 лет, находятся новые области применения
полимеров на основе ВП. Кроме того, продолжается изучение механизма
взаимодействия этих полимеров с белковыми соединениями, низко- и
высокомолекулярными соединениями, способными к образованию комплексов со
звеньями ВП. Данные таких публикаций позволяют судить о новых перспективах
использования и большой практической ценности данных (со)полимеров.
Благодаря своим уникальным свойствам
поливинилпирролидон (ПВП) широко применяется в медицинской практике.
Информация, относящаяся к применению ПВП в медицине до 1997 г., изложена в
монографиях и обзорах, например [1-4]. ПВП входит в число биологически
активных полимеров с неспецифической активностью, образующих комплексы с
широким кругом низко- и высокомолекулярных соединений за счет водородных
связей, гидрофобных и других нековалентных взаимодействий. Этот полимер
нетоксичен, доза LD50 при пероральном и внутривенном применениях
составляет ~ 100 и 10-15 г/кг соответственно [3]. Он не подвергается
метаболизму в живом организме и его выведение зависит от молекулярной массы
(ММ) и способа введения. Стойкость к биодеструкции обуславливает жесткие
требования к ММР ПВП для внутривенного введения, поскольку макромолекулы ПВП с
ММ ≤ 25 тыс. и 25-110 тыс. выводятся в течение нескольких дней и
нескольких месяцев соответственно, а с ММ ≥ 110 тыс. задерживаются на
годы и могут индуцировать патологические процессы [2].
Сополимеры на основе N-винилпирролидона широко
используются в медицине и представляют большой интерес в качестве
физиологически активных веществ лекарственного назначения. При этом важно как
получение образцов с определенным значением молекулярной массы, так и
стабильный состав, а также контроль композиционной однородности.
В Институте фармацевтических технологий (Россия,
Москва) ведутся работы по синтезу сополимеров N-винилпирролидона с
2-метил-5-винилпиридином (МВПр) и 2-метил-5-винилтетразолом (МВТАз) в широком
диапазоне соотношений исходных мономеров и изучению их биологической активности
[34]. Показано, что эти сополимеры обладают выраженной физиологической
активностью, увеличивающейся с повышением доли звеньев МВПр или МВТАз, тогда
как с повышением доли звеньев ВП улучшается растворимость сополимера в воде.
Результаты исследования показали [35-37], что оба сополимера не
токсичны: за 7 суток наблюдения не погибло ни одно животное, которому ввели 0,4
мл раствора, содержащего 10 мас.% сополимера.
Сополимер ВП с МВПр Сополимер
ВП с МВТАз
Оптимальными свойствами обладает сополимер, содержащий
64 мол.% ВП и 36 мол.% МВПр, с Мw=46.6 кДа, получивший торговое
название СовидонTM [34-39]. Показано [39], что введение раствора
сополимера в виде глазных капель способствуют уменьшению митотического индекса
клеток эпителия роговицы мышей и крыс, поврежденной радиационным воздействием,
и уменьшению частоты образования аберрантных митозов. Наилучший эффект
достигается при использовании 10 %-ного раствора Совидона, вводимого сразу
после облучения.
Увеличение доли звеньев МВТАз в его сополимерах с ВП
способствует появлению и усилению иммунодепрессантных (или иммуностимулирующих)
свойств сополимера [36, 37], а также противолучевой эффективности [37].
Найдено, что синтезированные сополимеры с содержанием звеньев МВТАз до 70±5
мол.% растворимы в воде и могут быть использованы в качестве радиопротекторов [37].
Более того, указанные сополимеры обладают свойствами
активаторов фагоцитоза [36, 38]. Оценку фагоцитарной активности
перитонеальных макрофагов проводили по интенсивности поглощения ими красителя -
нейтрофильного красного или в присутствии суспензии коллоидной туши при
введении внутримышечно сополимеров в дозах 5 и 50 мг/кг в объеме 250 мкл.
Обнаружено, что это статистически достоверно увеличивало фагоцитарную
активность на 15-45% по сравнению с контролем в исследованиях с красителем
Нейтрофильным красным, а также повышало индекс завершенности фагоцитоза на
85-138% в исследованиях с частицами коллоидной туши. В последнем случае
отмечалось, что введение сополимеров, практически не изменяя числа
фагоцитирующих клеток, в 1,5-2 раза увеличивает переваривающую способность макрофагов.
(Индекс завершенности фагоцитоза - отношение среднего числа частиц (ФК),
поглощенное одной фагоцитирующей клеткой за 1 ч, к ФК за 2,5 ч, отражает
переваривающую способность фагоцитов.)
Кроме того, исследование противораковой активности
сополимеров ВП с МВТ и с МВП со средневязкостной молекулярной массой (Мµ) 30-50
и 15-28 кДа соответственно [36] показало, что предлагаемые сополимеры в
1.5-2.4 раза увеличивают торможение роста опухоли по сравнению с продигиозаном
и наиболее активны против саркомы М-1 (76 и 52%), саркомы 45 (81 и 50%),
карциносаркомы Уокера (78 и 33%) и аденокарциномы толстого кишечника (АКАТОЛ)
(65% - сополимер, по продигиозану нет данных). Активность ВП с МВП с Мµ 15-28
кДа оказалась в среднем в ~ 1.5 раза выше, чем с Мµ ~ 40 кДа ([40].
При индуцировании подострого поражения введением
подопытным белым крысам дозы нитрата плутония-239 (407 кБк/кг) найдено [36],
что пероральное введение указанных сополимеров способствует улучшению состояния
животных по показателям общего анализа крови. Так, через 430 суток у группы,
получавшей раствор сополимеров, по сравнению с контрольной существенно менее
выражена лейкопения: 11 и 23 тыс./мм3 лейкоцитов (9 - контроль),
снижено содержание эритроцитов: 4.8 и 5.1 млн./мм3 (4.7- контроль),
повышен уровень гемоглобина: 173 и 159 (181- контроль). В результате
проведенных исследований констатируется [36], что введение сополимеров
по предлагаемому изобретению оказывает статистически значимое подавляющее
действие на развитие злокачественных опухолей в печени и почках подопытных
животных, а также улучшает общие показатели анализа крови.
Сотрудниками Института фармацевтических технологий
также установлено [34, 36], что предложенные сополимеры в концентрациях
от 0,01 до 10 мкг/мл крови достоверно повышают продуцирование обеих форм
интерлейкина-1 (ИЛ-1), но при этом не оказывают существенного влияния на
продуцирование интерлейкина-2 и фактора некроза опухоли-альфа. Таким образом,
можно предположить, что они опосредованно препятствуют пролиферации раковых
клеток, задействуя, в частности, механизм фагоцитоза. Кроме того, сополимеры,
как показано авторами [36], могут быть полезны в комбинированной терапии
в сочетании с другими соединениями, повышающими продуцирование ИЛ-1, а также с
лекарственными формами самого ИЛ-1.
Установлено [41], что сополимеры ВП с
диметиламинометилметакрилатом, а также ВП с диметиламинометилметакрилатом и
винилбутиловым эфиром проявляют выраженную детоксицирующую активность по
отношению к эпихлоргидрину, что выражается в увеличении выживаемости животных
(белые крысы-самцы и мыши).
1.3 Использование (со)полимеров
N-винилпирролидона в качестве лекарственных средств пролонгированного действия
Способность ПВП и сополимеров, содержащих звенья ВП, к
комплексообразованию использована для пролонгирования действия физиологически
активных веществ. В этом случае можно выделить также несколько основных
направлений получения лекарственных композиций на основе ПВП и сополимеров:
комплексы ПВП с лекарственными средствами в водных
растворах;
комплексы ПВП с лекарственными веществами в виде
твердых растворов (таблетирование);
комплексы сополимеров N-винилпирролидона с
лекарственными (или другими физиологически активными) веществами, образующиеся
за счет звена сомономера;
сополимеры N-винилпирролидона с мономерами,
являющимися производными лекарственных (или других физиологически активных)
веществ, проявляющие собственную физиологическую активность.
В медицинской практике широко используются комплексы
иода с полимерами, в частности с ПВП («Иодпирон», «Повидон» и др.). В состав
такого комплекса входят ионы I3-, которые не имеют
общетоксического действия и не раздражают кожу, как I2, но обладают
антисептическими, противовирусными, фунгицидными свойствами [1,2]. Этот
комплекс был впервые получен Шеланским более 50 лет назад [1], однако до
сих пор широко используется в мировой медицинской практике в виде растворов,
мазей и аэрозолей [4].
В концентрированных растворах ПВП наблюдается
повышение растворимости ряда лекарственных средств, например, производных
тетрациклина [4], для чего рекомендовано применять ПВП с ММ 10-17 тыс.
Для приготовления инъекционного препарата левомицетина (25-30 % в растворе),
применяемого в ветеринарии, используется ПВП с ММ 40 тыс. [4 4].
Интересно, что предложено также использование ПВП с очень низкой ММ (2.0-3.5
тыс.) в составе композиций с очень высоким содержанием, от 40 до 70 %,
сульфаметоксазола и триметоприма для создания противомикробных средств в виде
как водных растворов, так и мазей. Таким образом, способ применения полимера
связан с величиной его молекулярной массы.
Кроме того, ПВП образует комплексы с пенициллином,
новокаином, гексобарбиталом, сульфаниламидными производными, инсулином,
андекалином, вазопрессином и др., а инъекционные препараты, содержащие ПВП с ММ
от 20 до 60 тыс., проявляют пролонгированное действие [4]. Более того,
эффект побочного вредного воздействия на организм исходной лекарственной формы
в комплексе с ПВП может быть уменьшен или устранен. Это явление было изучено,
например, для комплексов ПВП с иодом [1-4], а также с морфином [42]
(препарат «Морфилонг»). В этом случае возникает в основном анальгезирующий
эффект без наркотического действия, характерного для чистого морфина [3],
причем за счет применения ПВП с ММ 30-40 тыс. достигается также эффект полного
и быстрого рассасывания инфильтрата [42]. Поскольку токсичность
антибиотиков при их иммобилизации на ПВП снижается, и его низкомолекулярные
образцы быстро выводятся из организма человека (за 6 ч при ММ 10-12 тыс.), то
низкомолекулярный ПВП можно использовать как антидот при передозировке
антибиотиками [43 14].
Способность макромолекул ПВП образовывать комплексы с
разнообразными соединениями дает возможность получать твердые дисперсии с
равномерным распределением лекарственного вещества в таблетированных формах,
где обычно применяют ПВП с ММ 20-30 тыс. [3].
К противомикробным средствам относится также Повиаргол
- новый фармацевтический препарат, прошедший клиническое изучение и включающий
в свой состав 7,5 - 8,5% высокодисперсного металлического серебра и ПВП с мол. массами
от 104 до 16.3 105 Да [44, 45 15, 16]. При
изучении защитного эффекта актопротектора биметила и антисептика Повиаргола от
повреждающего действия активных форм кислорода на активность таких ключевых
ферментов метаболизма клеток, как цитозольный фермент креатинкиназа и
мембраносвязанный фермент Na+, K+-АТФаза [46 17] найдено, что Повиаргол
наравне с биметилом может выступать как антиоксидант, повышающий резистентность
организма к повреждающим факторам.
Для повышения эффективности терапии гнойно-септических
осложнений ран и ожогов разработана [44 15] также водорастворимая
серебросодержащая бактерицидная композиция на основе высокодисперсного
поверхностно-окисленного серебра со степенью окисления 32,60 - 34, 45% и ПВП с
ММ (8÷35) 103 Да, в соотношении (7,5÷8,5) : (91,5÷92,5) мас. %. Такие композиции в водных
растворах сохраняют высокую антимикробную активность в течение 3 лет и являются
в 10-100 раз более сильными бактерицидными средствами, чем высокодисперсные
частицы металлического серебра, стабилизированные более высокомолекулярным ПВП
в Повиарголе, при сравнимом содержании серебра в растворах.
Предложено также использовать комплексы ПВП с
пероксидом водорода, например, при окраске волос [47 18] или для
отбеливания зубов [48 19].
Использование в качестве матрицы сополимеров
N-винилпирролидона с мономерами, имеющими в составе молекулы дополнительные
функциональные группы, представляет большие возможности для создания новых
препаратов. Сополимеры ВП с диацеталем акролеина («Совиаль»), кротоновой
кислотой, эпоксипропилметакрилатом, малеиновым ангидридом и винилфталимидом
можно рассматривать как стандартный набор карбоцепных полимеров-носителей,
содержащих в свободном или скрытом виде наиболее часто применяемые для
связывания физиологически активных веществ функциональные группы (-СНО, -СООН,
-NH2 и др.) [2].
Так, в качестве полимеров-модификаторов свойств
антисептика катамина АБ были использованы сополимеры ВП с метакриловой,
акриловой и кротоновой кислотами [49 20 5 ]. Проведенные авторами
исследования показали, что включение катиона катамина АБ в комплекс с
сополимером ВП и кротоновой кислоты привело к снижению острой токсичности
антисептика (величина LD50 для комплекса в 4 раза больше, чем
исходного катамина АБ), тогда как лечебный эффект при использовании комплексного
лекарственного препарата «Катапол» повышался: в 3.4 раза снижался индекс
поражения и в 2.4 раза уменьшались сроки заживления раны [49 20].
В качестве матрицы-носителей лекарственных средств
может служить также сополимер ВП с ММА и бутилметакрилатом [45 21 50].
Он нерастворим в воде, обладает как гидрофобными, так и гидрофильными
свойствами, рассасывается при имплантации в ткани с программированной
скоростью, не вызывает в местах имплантации деструктивной тканевой реакции,
биологически инертен. Сополимер, медленно рассасываясь, одновременно
обеспечивает такую же медленную десорбцию лекарственных средств.
Описаны [51 30 236] также однородные,
образующие прозрачные водные растворы сополимеры ВП с винилацетатом (ВА),
обогащенные ВП, для использования в фармацевтической и косметологической
продукции. Сополимеры ВП с N-изопропилакриламидом образуют комплексы с ионами
металлов, в частности Сu2+ [52 31 237], они чувствительны к
изменениям температуры, рН среды, составу растворителя [52, 53 31, 32 237,
238] и могут быть пригодны в качестве инъекционного имплантатного материала [52
31 237].
Сополимер ВП с диметилмалеиновым ангидридом
избирательно накапливается в почках, в отличие от ПВП и лекарства
(супероксиддисмутазы) и является перспективным носителем лекарственных средств
для их доставки в почечную систему [54 34240]. Сшитые сополимеры ВП с
N-гидрокси алкилмалеимидами [19 224] изучены как гидрогели для
контролируемого выделения препаратов, в частности теофиллина.
Проводится синтез функциолизированных сополимеров ВП
за счет введения групп, обладающих биологической активностью. На основе
сополимеров ВП с 2-гидроксиэтилметакрилатом синтезированы полимерные эфиры
антибиотика оксациллина [55 35 241]. Модификацией звена МА в терполимере
ВП с МА и винилбутиловым эфиром 4-амино-2,2,6,6-тетраметилпиперидином и
4-амино-2,2,6,6-тетраметилпиперидин-1-оксилом получены полимерные продукты с
устойчивыми нитроксидными радикалами в боковой цепи [56 36 242],
потенциальная биологическая активность которых обусловлена антиопухолевыми
свойствами свободных нитроксильных радикалов [57 33 239]. Изучена
реакция модификации сополимера ВП с глицидилметакрилатом 6-метокси-α-метил-2-нафтилуксусной кислотой
(напроксен) [58 37 243]. С целью получения новых полимерных
иммуностимуляторов синтезированы водорастворимые сополимеры ВП с метакрилоил
или кротоноил амидоглюкозой [59 38 244], содержащие в боковой цепи
фрагменты глюкозы. Сополимеры, включающие звенья ВП и ВА (40:60, ММ 60 тыс.), с
привитыми полиуретановыми цепями [60 45 ] образуют прочные эластичные
пленки и способны к иммобилизации лекарственных препаратов за счет реакции с
участием имеющихся в составе сополимеров свободных NCO группы. Показано [61
46 ], что иммобилизация на них противоспалительного препарата амизона
делает возможным регулируемый вывод лекарственного вещества.
Отдельное направление представляют собой работы,
посвященные получению и всестороннему изучению конъюгатов белков с указанными
(со)полимерами, причем в значительном количестве работ в качестве матрицы
выбран сополимер ВП с МА [62-66 39-43]. На процесс гидролиза, т.е.
транспорта лекарств оказывают влияние такие факторы, как химическая структура
олигопептидных последовательностей [62 39 8], основной цепи [62 39 8],
степень замещения кислотных групп (прививки) [63 40 9], вклад
гидрофобных взаимодействий [64 41 10], рН среды [65, 66 42, 43 11,
12]. При этом в качестве носителя рассмотрен сополимер ВП:МА 1:1 с ММ ~ 20 тыс.
[62-65 39-42 8-11], а также 29 и 58 тыс. [66 43 12].
В последнее время активно развивается поиск новых
полимерных матриц на основе ВП и проводится всестороннее исследование свойств
полученных материалов. Например, показано, что при введении в ПВП боковой
группы β-аланина существенно повышается
уровень его дезинтоксикационного действия в составе кровезаменителей, а также
чувствительность и специфичность диагностической тест-системы на плазминоген в
крови за счет понижения уровня неспецифической адсорбции белков плазмы крови [67
44 47]. Обнаружено, что аминокислотные производные эпоксидсодержащих полимеров
ВП и металлокомплексы на их основе обладают низкой токсичностью и проявляют
высокую иммуностимулирующую, антивирусную, гемостимулирующую активность, а
также усиливают продуцирование интерферона, подавление репликации
микроорганизмов под действием антибиотиков, причем существует взаимосвязь между
биологической активностью полимеров и их химическим строением [68 4748].
Полученные результаты указывают на перспективность использования
синтезированных полимеров и их металлокомплексов в качестве основы лекарственных
препаратов. Разработана рецептура гипоаллергенного, газопроницаемого материала
для закрытия ран различной этимологии на основе смеси хитозана с ПВП и
сополимеров хитозана с акриламидом [69 4849].
Для повышения гемосовместимых свойств поверхности
изделий из полиэтилена, применяемых в качестве имплантантов, предложены [70
49] новые гидрофильные покрытия на основе полиэлектролитных комплексов
биоспецифического модифицированного сополимера ВП и малеиновой кислоты с
хитозаном, амфифильным хитозаном или альбумином. Указанный сополимер содержал
аффинные лиганды к плазминогену - остатки α-аминосвязанного лизина и придавал
модифицируемой поверхности тромборезистентные свойства. Предложенные би- и
многослойные покрытия на основе полимерных комплексов, нанесенные на
имплантанты, могут быть применены для снижения степени тромбогенности
контактирующих с кровью изделий, используемых в медицинской практике.
Из этих примеров видно, что опубликованные в последнее
время работы посвящены в основном получению функционализированных сополимеров
со специальными свойствами, полезных для применения в различных процессах
генной и биоинженерии, систем доставки лекарств, и биомакромолекулярных
конъюгатов.
1.4 Гидрогели на основе (со)полимеров
N-винилпирролидона
В последнее время гидрофильные полимерные гели
(гидрогели) привлекают большое внимание исследователей благодаря своим
уникальным свойствам, которые приближают их к живым человеческим органам. Это
делает возможным и перспективным применение таких гидрогелей в области медицины,
например, в качестве раневых перевязок, контактных линз и систем доставки
лекарственных препаратов [71 1P].
Гидрогели представляют собой нерастворимую сеть
гидрофильных полимеров, способную поглощать воду и биологические жидкости.
Основой для создания гидрогелей может служить целый ряд водорастворимых
высокомолекулярных веществ, как синтетического, так и природного происхождения,
включая белковые молекулы. Включение в структуру гидрогелей азотсодержащих
гетероциклических фрагментов приводит к повышению абсорбционной способности к
водным растворам электролитов [72 2]. Поэтому поли(N-винилпирролидон)
(ПВП) и его сополимеры, которые проявляют высокие степени набухания в водном
растворе благодаря их гидрофильньным функциональным группам, являются одними из
самых популярных полимеров, используемых как биологически совместимые материалы
в качестве основы гидрогелей для медицины [71-75 1-3, 9, 10].
Для получения этих гидрогелей могут быть использованы
два метода: обычная радикальная сополимеризация в водном растворе и
радиационная техника. В первом случае гидрогель получают полимеризацией
гидрофильньных мономеров в присутствии би- или многофункциональных сшивающих
агентов, а также сшивание водорастворимых полимеров [76 11]. К числу
мономеров, используемых для получения гидрогелей, относится N-винилпирролидон
(ВП), который растворим в воде и многих других растворителях. В качестве
сшивающих веществ часто используются такие, как N,N'-метиленбисакриламид,
дивинилбензол и аллилметакрилат. В гидрогелях, содержащих только ВП, необходима
высокая концентрация сшивающего агента (5-20 %) [76 11].
Большое количество исследований, например [77 12],
посвящено получению гидрогелей на основе поли(винилового спирта) и
поли(винилпирролидона) (ПВС/ПВП), которые образуются в водных средах по общей
схеме:
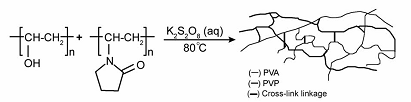
Рисунок 1.5. Упрощенная модель синтеза и
структуры сшитых гидрогелей ПВП/ПВС [77 12 9P].
В качестве исходных полимеров авторы [77 12 9P] использовали ПВС со степенью
полимеризации 300, 1600, 2000 и молекулярной массой 15000, 72000, 100000, и
49000, соответственно, и степенью гидроксилирования 86-89 % мол. (Fluka) и ПВП
со средней молекулярной массой 25000 (Merck). Водные растворы обоих полимеров
смешивали, добавляли персульфат калия и проводили реакцию сшивки при 80ºC при энергичном механическом
перемешивании в атмосфере азота. Более детально процесс образования гидрогеля
может быть представлен схемой [77 12 9P]:
После 5 ч реакционную смесь выливали в пластмассовую
форму и высушивали в течение ночи при 50ºC в вакуумном шкафу. Изучение доли
геля и степени набухания в воде показали [77 12], что с увеличением
концентрации персульфата калия доля геля возрастает, а степень набухания
снижается. Набухание гелей происходило с относительно высокой скоростью в
начале процесса и количество поглощенной воды при погружении достигало около
1400 %, при этом степень набухания возрастала с уменьшением концентрации
персульфата и увеличением доли ПВП. Так, например, для соотношения ПВП/ПВС
50:50 мас.% степень набухания изменяется от 800 до 392% при [K2S2O8]
от 1.1·10-5 до 6.6·10-5 моль/л, а если [K2S2O8]
= 1.1·10-5 моль/л, то эта величина составляет от 800 до 1400 % при
доле ПВП от 50 до 80 мас.%. Влияние молекулярной массы ПВС оказалось
незначительным, т.к. при ее увеличении от 15 до 100 тыс. степень набухания
гидрогеля изменялась в пределах от 800 до 600 % (ПВП/ПВС 50:50 мас.%, [K2S2O8]
= 1.1·10-5 моль/л) [77 12 9P]. Вследствие нейтральности
гидрогелей набухание достигало максимума при pH = 7 и 25ºC, и при более высоких температурах оно
уменьшалось.
При малом количестве пероксидисульфата калия прочность
пленок гидрогеля повышается, так как с возрастанием концентрации инициатора
сополимер становится более хрупким и имеет более низкую прочность на растяжение
вследствие дополнительных сшивок. Так, прочность на разрыв изменяется от 90.8
до 10.0 кг/см2 при концентрации K2S2O8
от 1,1×10-5 до 6,7×10-5 моль/л, причем при минимальном
содержании персульфата было достигнуто относительное удлинение при разрыве ~
60%. Найдено, что под нагрузкой деформация образца происходит равномерно по
всей длине, до достижения текучести, что по мнению авторов [77 12 9P], отражает существенно однородную
молекулярную ориентацию полимерной цепи.
Авторами [78 13 10P] изучено получение гидрогелей из
смеси ПВП (Mw = 44000, BDH) и ПВС (Mw = 125000, степень
гидролиза 88 %) при использовании в качестве сшивающего агента глутарового
альдегида (5 % мас.) и молочной кислоты как катализатора. После смешивания
смесь нагревали при 60°C в течение 1 ч, поливали в полистирольных чашках Петри,
с последующей сушкой при 60°C. Далее сшивание было проведено тепловой
обработкой пленок при 120°C и 150°C после 30 минут высушивания при 100°C.
Показано, что при увеличении температуры тепловой обработки от 100 до 120 и
150°C равновесное содержание воды в геле и экстрактируемая масса (полимера?)
уменьшаются от 89.2 до 86.7 и 76.2 % и от 51.2 до 41.8 и 27.6 %. Одновременно с
этим пленки становятся более прочными и жесткими, что выражается в увеличении
предела прочности при растяжении от 0.10 да 0.23 и 0.26 МПа и уменьшении
деформации при разрыве от 70 до 46 и 21 %.
Исследовано [79 14 11P] также получение гидрогеля методом
матричной полимеризации N-винилпирролидона (11.2 г) и метакриловой кислоты (8.6
г) в присутствии полиэтиленоксида (1 г) и различных количеств сшивающего агента
- этиленгликоль-диметакрилата (0.05 и 0.1 г), при использовании АИБН как
инициатора (0.005 г), ТГФ как растворителя (10 мл) при температуре 50ºC. Из свойств гидрогеля изучено
набухание (сорбция им воды) при 20-40°С, которое составило 50-70 % для дисков 3×11.5 мм. Методом электронной микроскопии
показано, что структуры образцов различаются по размеру частиц и распределению
числа полостей в них, которые меньше при большем количестве сшивающего агента,
средний размер пор ~ 1100 нм.
Для получения полимерного гидрогеля биомедицинского
назначения проводили также процесс сополимеризации N-винилпиролидона (45.0 г) с
метилакрилатом и метакриловой кислотой (2.5 и 0.5 г) [80, 81 15, 16 -
укр. Пат.]. Мономеры с добавками 0.2 г этиленгликольдиметакрилата, 0.15 г
персульфата аммония и 0.15 г тетраметилэтилендиамина растворяли в 100 мл
апирогенной воды при 25°С, полученный раствор отфильтровывали, продували азотом
и разливали в плоско-параллельные пресс-формы для получения пластин с толщиной
0,7 мм. Полимеризацию проводили, выдерживая пресс-формы при температуре 25°С на
протяжении двух часов. Полученные гидрогелевые покрытия отмывали в воде при
75°С (соотношение гидрогеля и воды 1:3) на протяжении 7 суток, высушивали при
40°С, упаковывали в полимерную пленку и стерилизовали.
Весьма активно развивается направление радиационного
синтеза гидрогелей на основе водорастворимых мономеров или полимеров, которое
можно разделить на применение излучения с высокой энергией (γ-радиация, электронный луч) и
фотосшивание в водных растворах под воздействием УФ излучения (ртутные лампы).
Достоинством такого способа авторы [82 17 7P] считают отсутствие инициаторов или
сшивающих агентов, которые являются главным образом нежелательными добавками,
подлежащими отмывке.и сотр. [83, 84 18, 19 12, 13P] представили успешную методологию
производства гидрогеля для перевязочных материалов, основанную на воздействии
высокоэнергетической радиации (5-50 кГр) на водные растворы ПВП, ПВС или ПЭГ,
агара. В результате они получили прозрачные листы толщиной в несколько
миллиметров, содержащие более 90 % воды. Доля геля в таких материалах зависит
от дозы облучения, концентрации ПВП и соотношения (ПЭГ). Так, при концентрации
ПВП 4 и 10 мас.% и изменении соотношения ПВС:ПВП от 40:60 до 90:10
водопоглощение полученных гидрогелей изменялось с 45 до 250 % и с 94 до 160 % [84
19 13P].
Материалы на основе полученных гидрогелей выпускаются как перевязочные
материалы для ран, под торговой маркой HDR® и AQUAGEL®.
Получение гидрогелей для раневых перевязок проводили [85
20 14P] также
из смеси Aloe vera, ПВС и ПВП (ПВА:ПВП = 6:4, Aloe vera 0.4-1.2 % от массы
сухой композиции, вода 85%) под воздействием циклов таяние-замораживание и γ-радиации дозой 25, 35 и 50 кГр, или в
двухступенчатом процессе при действии γ-радиации и таяния-замораживания.
В работах бразильских исследователей [86-88 21-23
15-17P] развивается направление получения
гидрогелей на основе ПВП путем фотосшивки в водном растворе с использованием
ртутной лампы низкого давления (λem=254 нм). Доказано [86 21 15P], что полученный таким образом
продукт имеет микро- и макроскопические свойства, подобные гидрогелям,
произведенными высокоэнергетической радиацией, поэтому данный метод является
успешной альтернативой применению высокоэнергетической радиации. Для ускорения
процесса сшивки было предложено введение в раствор пероксида водорода [87 22
16P] и использование системы Н2О2
- Fe2+ [88 23 17P].
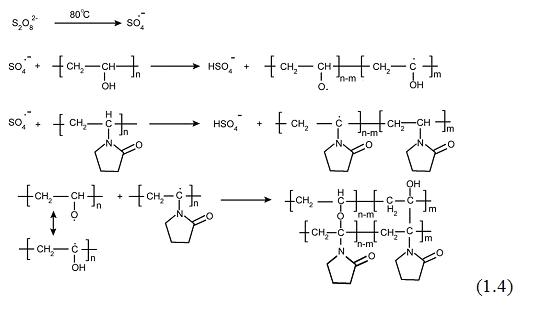
Поскольку гидроксильный радикал - самый активный
радикал среди активных кислородных частиц, авторы [86, 87 21,22 15, 16P] считали, что его реакция с ПВП
приведет к образованию макрорадикалов, центрированных по трем возможным
положениям, принимая во внимание лабильность водородных атомов, присутствующих
в структуре этого полимера (см. схему 1.5).
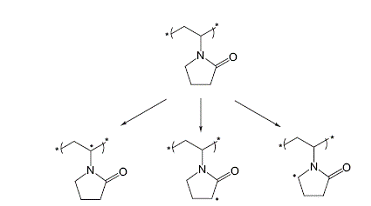 (1.5)
(1.5)
Основные термодинамические расчеты показывают [87 22
16P], что связи C-H в α-положении к гетероатому или карбонилу
ниже по энергии, главным образом из-за стабилизации радикального продукта.
Реакции рекомбинации, следующие за образованием
радикала, приводят к формированию поперечных сшивок. Действительно, было
замечено, что, когда два раствора ПВП, один из которых содержит H2O2,
а другой - FeCl2, смешивают, та практически мгновенно, в течение
5-10 с, образуется густой гидрогель. Несколько попыток исследовать начальную
кинетику этого процесса потерпели неудачу из-за малого времени этого процесса и
высокой вязкости раствора.
УФ-инициированной свободно-радикальной
фотополимеризацией были получены гели сополимеров ВП с метакрилатами (MетA):
метилметакрилатом, н-бутил-метакрилатом, н-октил-метакрилатом и
н-додецил-метакрилатом при исходном соотношении ВП и н-MетA 90:10, с
использованием 0.8 % мас. Irgacure 651 (2,2-диметокси-2-фенилацетофенона) как
фотоинициатора и 0.2 % мас. BIS (N,N-метилен бисакриламида) как сшивающего
агента [73 3 16аP].
Оказалось, что эти гели первоначально набухают быстро
за короткое время (0-100 минут), достигая полностью набухшего состояния в
период 120 - 600 мин в зависимости от системы [73 3 16аP]. При увеличении числа алкильных
атомов углерода в н-MетA гели ВП/MетA имели меньшую равновесную объемную
степень набухания, большую плотность сшивки, меньшую начальную скорость набухания,
более короткое время достижения полностью набухшего состояния и более высокую
механическую жесткость (G). Значения G возрастают линейно от 75 до 760 кПа с
увеличением числа атомов углерода в алкильном заместителе от 1 до 12 вследствие
того, что гидрогели с более длинными алкильными группами поглощают меньше воды
из-за увеличения областей агрегации между гидрофобными алкильными группами. Эти
значения G больше, чем для гидрогелей ВП/ГЕМА (19 кПа), поэтому включение
гидрофобных последовательностей нежелательно для использования в имплантантах.
По содержанию воды и эластичности гидрогели схожи с
биологическими тканями, что дает возможность их широкого биомедицинского
применения, однако основным направлением являются использование их в качестве
материала для контактных линз и перевязочных материалов для ран. Оснóвой большей части выпускаемых в
настоящее время контактных линз является поли(2-гидроксиэтил-метакрилат)
(полиГЭMA), однако есть также большое число различных составов гидрогелей,
которые содержат ПВП, ПВС, полиМАК, хитозан и силикон. В ряде случае
используются сополимеры ГЭMA с ВП (hefilcon A), которые за счет включения
звеньев ВП обладают улучшенной кислородной проницаемостью [70 13 7P8]. При величине минимальной
кислородной проницаемости, требующейся эпителием роговой оболочки, равной 3.5
мкл (STP)/(cм2·ч), для твердых контактных линз из ПММА этот
показатель составляет 0.27, тогда как для мягких контактных линз из поли-ГЭMA
он равен 13.0, а после сополимеризации ГЭMA с ВП возрастает до 25.7.
В настоящее время развитие синтетических перевязок для
ран, которые используются для обработки ожогов, пузырей, трещин, герпеса, и
т.п. представляет большой коммерческий интерес. Идеальный раневый перевязочный
материал должен обладать рядом свойств [70 20 8P13]: быть гибким, достаточно прочным
и не причинять боли при замене перевязки; эффективно поглощать жидкости тела и
предотвращать их потерю, предотвращать загрязнение раны микроорганизмами
снаружи, допускать проникновение кислорода; иметь биологическую совместимость с
кожей и кровью; просто стерилизоваться, быть прозрачным, хорошо придерживаться
на ране, но более сильно на здоровой коже и позволять управлять дозировкой
препаратов; но не быть аллергенным. Гидрогели обладают до известной степени
многими из вышеупомянутых свойств и из-за этого они были исследованы в
различных формах как раневые перевязочные материалы.
Показано [77 12 9P], что гидрогели, полученные на
основе ПВП и ПВС, можно рассматривать как хороший барьер против общих микробов,
включая Sarcina lutea, Escherichia Coli, и Pesudomonase aeruginosa. Тепловой
анализ показал, что эти гели устойчивы до 350ºC. Эти свойства позволяют применять
упомянутые выше гидрогели в качестве раневых повязок.
Коммерчески успешный пример такой повязки известен под
торговой маркой AQUA-GEL, продаваемой в основном в Центральной Европе. Она
производится с помощью радиационных технологий в виде тонких набухших пластин
гидрогеля [82, 83 17, 18 20, 92=12P]. Первым этапом процесса является подготовка водного
раствора компонентов, основными из которых являются ПВП, поли(этиленгликоль) и
агар. После смешивания при повышенной температуре образуется гомогенный
раствор. На втором этапе пресс-формы, которые также могут служить окончательной
упаковкой для перевязочных средств, заполняются раствором. После затвердевания
раствора при охлаждении формы плотно запечатываются в фольгу, которая
непроницаема для воздуха и микроорганизмов. На заключительном этапе этот
полуфабрикат, то есть термообратимый псевдо-гель, укомплектованный в
коммерческие коробки, подвергают обработке ионизирующим излучением. Протекают
два процесса - стерилизация и образование необратимой трехмерной полимерной
сетки. Продукт - полностью стерильной необратимый гидрогель в форме прозрачных
листов, толщиной 3-4 мм, содержащих более 90% воды.
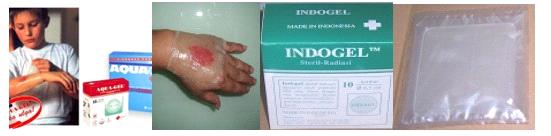
Рисунок 1.5 Товарные гидрогелевые раневые повязки на
основе ПВП, полученные по радиационной технологии и выпускаемые под торговым
названием AQUA-GEL (а), INDOGEL (б), BATAN (в) [70 1P].
В России к настоящему времени также разработаны
гидрогелевые повязки «Апполо», которые рекомендовано применять в качестве
средства первой помощи при ожогах [89 24]. Они изготовлены на
текстильной сетчатой подложке, пропитанной гидрогелем на основе сополимера
акриламида и акриловой кислоты, включающим также йодовидон (комплекс ПВП с
иодом), имеющий широкий спектр антибактериального действия и анилокаин
(анестетик). Кроме того выпускаются ранозаживляющие и протеолитические повязки,
различающиеся по виду дополнительно введенных в их состав лекарственных
препаратов. Размеры повязок - 4 × 5см (ранозаживляющие), 10 × 10см, 20 × 30см (для всех видов).

Рисунок 1.6. Ранозаживляющие гелевые повязки «Апполо»
[89].
При лечении ожогов применяют пленки первичной
обработки ожоговой поверхности из ПВП или смесей ВП с ММА и бутилакрилатом,
содержащие от 8.7 до 16 мкг/л иода, предложена также пленка на основе ПВП,
содержащая фурацилин и анестезин [90 18].
Предложен [91 25 18P] улучшенный биосовместимый
полимерный гелевый материал, который может быть использован для лечения
различных механических, химических, термических повреждений кожи, трофических
язв различной этиологии, а также в комбинации с соответствующими
гемостатическими средствами (например, тромбином) для остановки капиллярного
кровотечения, а также для закрытия донорских участков кожи при дермопластике.
Этот материал представляет собой органо-неорганический гибрид - продукт
объединения кремнийсодержащего продукта и водорастворимого синтетического
органического полимера в целостную структуру, который образуется путем
структурно-химических превращений в водных растворах полимеров при добавлении к
ним либо золей поликремневой кислоты и щелочных агентов (например, гидроксидов
натрия или аммония), либо разнообразных эфиров ортосиликатов. В качестве
органической составляющей может быть использован, один из синтетических
водорастворимых полимеров, например ПВП, поли-М-винилкапролактам, ПВС,
оксипропилцеллюлоза и др. Кремнийсодержащими прекурсорами могут быть различные
эфиры ортосиликатов, например тетраметоксисилан, или золи поликремневой
кислоты, например Сиалит-30. При этом эластические свойства гидрогеля
обеспечивает органическая компонента, а неорганическая - придает прочность.
Гидрогель хорошо самофиксируется к коже вокруг раны, легко и безболезненно
заменяется при перевязках, он достаточно прочен (модуль упругости 0,05÷0,25 МПа), устойчив к щелочным (до
рН=9,5), кислотным средам и кипящей воде, не обладает антигенной активностью,
не образует прочных соединений с белками крови, может поглощать большие
количества раневого экссудата и гноя.
Заявлены также новые гидрогелевые композиции [92 2619P], характеризующиеся специфическим
отношением поливиниллактама к полисахариду, который образует гелеобразную
композицию с водой. Оказалось, что они имеют такую консистенцию, которая
позволяет им эффективно заполнять и оставаться в полостях/отверстиях в теле,
предотвращая попадание микроорганизмов в полости тела или отверстия в теле
млекопитающего. Композиции могут включать агенты, меняющие консистенцию,
агенты, меняющие эксплуатационные свойства, сшивающие агенты и агенты,
улучшающие терапевтические свойства. Кроме того, консистенция этих гидрогелей
позволяет удалить их полностью, когда это необходимо или желательно. После того
как композиции по изобретению образуют гель, его можно разрушить и затем,
неожиданно, через несколько часов, гель снова образуется, т.е. эти гидрогели
полностью обратимы. Авторы полагают, что водородные связи в этих гидрогелях временно
разрушаются, когда такие гидрогели продавливают через небольшие отверстия в
аппликаторах, а через несколько часов они появляются снова.
Таким образом, использование сополимеров ВП,
обладающих биологической совместимостью, в качестве основы гидрогелей весьма
широко распространено в научных исследованиях и медицинской практике. Для
синтеза гидрогелей широко используется термическая полимеризация. Несмотря на
разработку методологии их получения путем радиационного экспонирования g-облучением, электронными лучами и
ультрафиолетом, тепловая полимеризация все еще исследуется благодаря простоте и
низкой стоимости.
2. ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
2.1 Характеристика применяемых
веществ
Таблица 2.1 Характеристика веществ, используемых в
работе
|
Название
|
Mr
|
Плотность, г/см3
|
Показатель преломления, 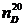
|
Температура, К
|
Формула
|
|
|
d20
|
d40
|
|
Ткип
|
Тпл
|
|
|
N-винилпирролидон
|
111,14
|
1,0483
|
1,0314
|
1,5117
|
338-339 (1,5 мм. рт. ст.)
|
286,9
|
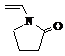
|
|
Малеиновый ангидрид
|
98,06
|
1,48 (тв.)
|
1,314
|
1,6092
|
-
|
333,15
|

|
|
N-метилпирролидон
|
99,13
|
1,0328
|
0,990
|
1,4684
|
355-356 (10 мм. рт. ст.)
|
249
|
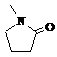
|
|
Янтарный ангидрид
|
100,07
|
1,503 1,23 (тв.)
|
-
|
-
|
-
|
393
|
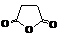
|
|
1,4-Диоксан
|
88,10
|
1,0338
|
0,988
|
1,4224
|
374,32
|
284,8
|
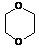
|
|
N,N-диметилформамид
|
73,09
|
0,9445
|
-
|
1,4269
|
426
|
212
|
HCON(CH3)2
|
|
Изопропиловый спирт
|
60,09
|
0,785
|
-
|
1,3776
|
356
|
-
|
(CH3)2CH-OH
|
|
Диэтиловый эфир
|
74,12
|
0,7138
|
-
|
1,4472
|
34,5
|
-116,3
|
С2Н5-О-С2Н5
|
|
АИБН
|
164,21
|
-
|
-
|
-
|
-
|
105-106
|
(CN(CH3)2CN=)2
|
|
Пероксид бензоила
|
242,23
|
1,33 (тв.)
|
-
|
-
|
-
|
380
|
(C6H5CO-O)2
|
|
Персульфат аммония
|
228,18
|
1,98 (тв.)
|
-
|
-
|
-
|
разл. ~ 393
|
(NH4)2S2O8
|
2.2 Очистка мономеров и растворителей
Малеиновый ангидрид очищали
возгонкой.
Янтарный ангидрид (ч.д.а) и
N-винилпирролидон (HPLC, Labscan) использовали без дополнительной очистки.
N,N-диметилформамид (ДМФА) - бесцветная гигроскопичная
жидкость. Может содержать примеси влаги, диметиламина, формиата диметиламмония,
монометилформиата. При высоких температурах ДМФА разлагается, поэтому проводили
вакуумную перегонку над КОН, отбирая наиболее чистую вторую фракцию с tкип
= 50°С /16 мм.рт.ст. [2э 46]
,4-Диоксан (ДО) - бесцветная жидкость с характерным
запахом; огнеопасен, с воздухом образует взрывчатую смесь и может содержать
примеси: СН3СН2СООН, Н2О, этиленацеталь
уксусного ангидрида, перекиси. Предварительно высушенный над гранулами КОН
1,4-диоксан (выдержка в течение двух суток) подвергали фракционной перегонке [2э
46].
Очистку изопропилового спирта и диэтилового эфира
проводили по методикам, описанным в работах [2э, 3э 58, 59].
2.3 Методика проведения эксперимента
2.3.1 Приготовление растворов для УФ и
ЯМР спектроскопии
В мерные колбы на 25 мл помещали навески
низкомолекулярных соединений (N-винилпирролидона, N-метилпирролидона,
малеинового или янтарного ангидрида), которые взвешивали на
аналитических весах с точностью до 0,00005 г, и добавляли небольшое количество
растворителя. Колбы встряхивали и затем растворителем доводили объем до метки,
получая растворы с исходными концентрациями С0 (около ? моль/л). Из
колб пипетками отбирали 0,25 мл раствора и переносили в предварительно
откалиброванные пикнометры, где снова доводили растворителем до метки, получая
растворы с концентрациями С1. Отмеряя пипеткой необходимый объем
раствора с концентрацией С1, переносили их в бюксы и доводили
растворителем до общего объема 4 мл. Полученные растворы использовали для записи
УФ спектров. Навеску полимера около 0,004 г взвешивали на аналитических весах и
переносили в колбу, затем добавляли 2 мл растворителя. Разбавляли раствор до
необходимой концентрации, помещали в кварцевые кюветы толщиной 1 см и
записывали УФ спектры в диапазоне 50000-34000 см-1 (200-294 нм) на
приборе SPECORD UV VIS со скоростью записи 11 минут и растяжением 0,5х, что
соответствует масштабу 2000 см-1=12,5 мм. Оптическую плотность
измеряли в максимуме поглощения, определяя положение максимума по шкале длин
волн и волновых чисел. На основании значений оптической плотности и
концентрации вычисляли соответствующие коэффициенты экстинкции.
Для записи спектров (со)полимеров навеску образца 0.03
г растворяли в 0.6 мл ДМСО-d6, после чего переносили в специальную
ампулу. Спектры 1Н ЯМР растворов полимеров в регистрировали с
помощью прибора Bruker Avance II (400 МГц) при 298 K, внутренний стандарт -
ТМС. Погрешность измерения химических сдвигов составляет ± 0.0005 м. д.
2.3.2 Методика проведения процесса сополимеризации
Рассчитанный состав смеси мономеров готовили весовым
методом с точностью до 0,00005 г. Приготовленную смесь заливали в ампулы
объемом 5 мл через тонкий капилляр и тщательно продували аргоном для удаления
кислорода. Затем ампулы со смесью запаивали, помещали в термостат,
предварительно нагретый до температуры 40 или 60 °С. По достижению
рассчитанного времени полимеризации ампулу извлекали из термостата и резко
охлаждали. Сополимер высаживали из 1 % раствора в хлороформе диэтиловым эфиром.
Выделенный сополимер сушили в эксикаторе до постоянной массы, в течение 36 ч.
Конверсию полученного полимера рассчитывали по формуле:
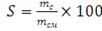 (2.1)
(2.1)
где mс -масса сополимера;см
-масса смеси.
Состав сополимера определяли, исходя из данных
рН-метрии и 1H ЯМР спектроскопии.
2.3.3 Методика определения количества
звеньев малеинового ангидрида методом потенциометрического титрования
Водный раствор 0.1 н NaOH и 0.1 н водный раствор
щавелевой кислоты для проверки его титра готовили из фиксаналов, используя
дистиллированную воду, освобожденную от СО2 кипячением. Титр
раствора NaOH устанавливали по щавелевой кислоте [4э 279]. Титрование
сополимеров ВП с МА проводили в водном растворе, предварительно гидролизовав
звенья ангидрида до кислоты.
Навеску сополимера (0.05 г), взвешенного с точностью
до 0.00005 г, растворяли в 50 мл дистиллированной воды и перемешивали при
нагревании до 50-60ºС в течение 1 часа, при этом его розовая окраска изменялась
на светло-желтую. Титрование осуществляли 0.1 н р-ром NaOH. Измерение рН
проводили с использованием иономера И-160МИ со стеклянным электродом ЭС-10603 и
хлорсеребряным электродом ЭСр-10103 в качестве индикаторного и электрода
сравнения. Количество звеньев МА в сополимере [m2] определяли по
формуле:
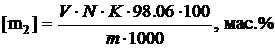 (2.2)
(2.2)
где V - объем NaОН, который соответствует точке
эквивалентности, мл;- нормальность раствора NaОН, моль/л;- поправочный
коэффициент на нормальность;
.06- молекулярная масса звена МА;- навеска сополимера,
г.
Потенциометрическое титрование образцов сополимеров ВП
с МА проводили также в смешанном растворителе «ДМФА - вода», используя
спиртовый раствор КОН.
Для приготовления 0.1 н спиртового раствора KOH 3 г
KOH (ч.д.а.) растирали в ступке и растворяли в 0.5 л ИПС, предварительно
очищенного перегонкой. Титр раствора KOH в ИПС устанавливали по щавелевой
кислоте [4э 279]. 0.05 г сополимера, взвешенного с точностью до 0.00005
г, растворяли в 33 мл очищенного ДМФА, добавляли 5-10 мл дистиллированной воды
и перемешивали при нагревании до 333-343 К в течение 1 ч. Охлажденный раствор
титровали 0.1 н раствором KOH в ИПС, добавляя по 0.1 мл титранта через 1 мин и
определяя величину электродного потенциала (Е) по иономеру И-160МИ со
стеклянным и хлорсеребряным электродами ЭС-10603 и ЭСр-10103. Содержание МА в
сополимере [m2] определяли по формуле (2.14).
Объем титранта (NaOH или КОН), пошедший на титрование,
определяли по кривым титрования в рамках программы Origin: по середине перегиба
(скачка) на исходной кривой «рН-V» («E - V»); по максимуму на дифференциальной
кривой первого порядка или по нулевой точке при переходе из положительной
области в отрицательную на дифференциальной кривой второго порядка.
2.3.4 Определение молекулярной массы
сополимеров по данным вискозиметрии
Вязкость определяли при помощи капиллярного
вискозиметра, измеряя при температуре 30ºС время истечения растворителя (t0)
и раствора полимера (t), концентрацию которого выбирали таким образом, чтобы
получаемые значения относительной вязкости ηотн = t / t0 изменялись в
пределах 1,1÷1,5. По формуле [31 60]
ηуд = t / t0 - 1 (2.3)
находили ведичину удельной вязкости ηуд, затем вычисляли приведенную
вязкость (ηуд/С), строили график зависимость ηуд/С от С и по экстраполяции прямой к
С=0 находили значение характеристической вязкости [η].
2.3.5 Методика получения гидрогелевых
пленок на основе (со)полимеров ВП и определения их степени набухания и
водопоглощения
Для получения сшитой полимерной основы гидрогеля
использовали ПВС с 72000, (АppliChem GmbH, Germany) и ПВП со средней
молекулярной массой 50000 или сополимер ВП с МА (конверсия ~88%, в ДО, АИБН,
333 К), применяя методику, подобную описанной в работе [280]. Предварительно
готовили растворы ПВС и ПВП (или ВП-МА) в воде, сливали, перемешивали,
продували инертным газом, добавляли (NH4)2S2O8
и перемешивали при 353 К 4 ч. Полученный раствор после охлаждения выливали в
чашки Петри и сушили, варьируя температуру и продолжительность сушки.
Полученные образцы пленки взвешивали и погружали в дистиллированную воду при
комнатной температуре. После выдержки набухший образец вынимали, удаляли воду
на его поверхности фильтровальной бумагой и немедленно взвешивали. Для изучения
кинетики набухания его снова погружали в воду, продолжая взвешивание и
погружение в воду через определенные промежутки времени. По окончании набухания
и последнего взвешивания образец высушивали. По результатам взвешивания
находили массовую долю геля (G), степень набухания (DS) и равновесное
содержание воды (водопоглощение WC), пользуясь формулами (2.15), (2.16) и
(2.17) [280-282] соответственно:
= Wс/W0×100 (2.15)= (Wн-Wс)/Wс×100 (2.16)= [(Wн-Wс)/Wн]
× 100 (2.17)
где W0, Wн и Wс -
масса образца перед погружением в воду, набухшей и высушенной пленки
соответственно.
2.4 Техника безопасности
2.4.1 Требования безопасности при
работе со стеклянной посудой
Cобирать стеклянные приборы или отдельные их части
нужно осторожно, применяя, где это необходимо, эластичные соединения и
прокладки. Особенно следует защищать приборы и стеклянные детали в местах
крепления на металлических кольцах штативов или держателях упругими прокладками
(резиной, кожей и т.д.).
В стеклянные ампулы, как правило, разрешается
запаивать сконденсированные вещества, имеющие температуру кипения не ниже 20°C,
заполняя ампулу не более, чем на 50% ее объема. Ампулы перед запаиванием, как и
вскрытием, необходимо охладить ниже температуры кипения помещенного в них
вещества, пользуясь для охлаждения негорючими охлаждающими смесями. Запаянные
ампулы вскрывают, завернув в полотенце, после чего делают надрез ножом или
напильником на капилляре и отломывают капилляр. Все операции с ампулами до их
вскрытия следует проводить под тягой и в защитных очках (защитных масках) и
перчатках. Осколки стекла убираются с помощью щётки и совка, а не руками.
2.4.2 Требования безопасности при
проведении нагревания
Запрещается использование в лаборатории электрических
плиток с открытой спиралью. Для нагрева ЛВЖ и ГЖ необходимо использовать
жидкостные бани с диаметром не менее диаметра нагревательного элемента плитки.
В качестве теплоносителей допускается использовать только чистые жидкости, не
содержащие посторонних примесей и загрязнений.
Запрещается нагревание жидкостей в закрытых колбах или
приборах, не имеющих сообщения с атмосферой. Подобные работы проводятся либо в
лабораторных автоклавах, либо в стеклянных толстостенных ампулах.
2.4.3 Правила безопасной работы с
электрооборудованием и электроприборами
Работы в лаборатории должны проводиться при наличии
исправного электрооборудования. При обнаружении дефектов в изоляции проводов,
неисправности пускателей, рубильников, штепселей, розеток, вилок и другой
арматуры, а также заземления и ограждений следует немедленно сообщать об этом в
энергетический отдел. Запрещается переносить включенные приборы и ремонтировать
оборудование, находящееся под током, работать вблизи открытых токопроводящих
частей и оборудования, а также загромождать подступы к электрическим
устройствам - шкафчикам, ящикам и т.п. - и открывать их. В случае перерыва в
подаче тока все электроприборы, электромоторы и другое электрооборудование
должны быть немедленно выключены. В случае загорания проводов или
электроприборов необходимо их немедленно обесточить и гасить огонь при помощи
сухого углекислотного огнетушителя и покрывала из асбеста.
2.4.4 Требования безопасности при
работе с легковоспламеняющимися жидкостями
Легковоспламеняющиеся и горючие жидкости (за
исключением веществ, имеющих низкую температуру кипения) должны храниться в
лабораторном помещении в толстостенных банках (склянках) с притертыми пробками.
Банки помещают в специальный металлический ящик с плотно закрывающейся крышкой,
стенки и дно которого выложены асбестом. Ящик должен быть установлен на полу
вдали от проходов и от нагревательных приборов, с удобным подходом к нему.
Емкость стеклянной посуды для легковоспламеняющихся жидкостей не должна
превышать 1 л.
Легковоспламеняющиеся и горючие жидкости следует
доставлять в лабораторию в закрытой небьющейся посуде или в стеклянной посуде,
помещенной в футляр. Общий запас одновременно хранящихся в лабораторном
помещении ЛВЖ и ГЖ не должен превышать 3-5 л.(объем емкостей с ЛВЖ не должен
превышать 1 л.). Огнеопасные вещества могут находиться на рабочем месте лишь в
количествах, нужных непосредственно для работы. При работе с диэтиловым эфиром
необходимо соблюдать дополнительные меры предосторожности, связанные с легкой
воспламеняемостью растворителя: его следует хранить изолированно от других
веществ в холодном и темном помещении, в темных склянках, так как под
воздействием солнечного света в нем образуются примеси взрывчатых
пероксосоединений.
Все работы с легковоспламеняющимися веществами и
горючими жидкостями должны проводиться в вытяжном шкафу при работающей
вентиляции.
2.4.5 Техника безопасной работы с
пероксидами
Перекристаллизацию пероксида бензоила ни в коем случае
нельзя проводить при нагревании. Перекристаллизация его из горячего хлороформа
опасна. Для очистки можно брать не более двух грамм пероксида бензоила.
Персульфат аммония (аммоний надсернокислый) негорюч,
пожаро- и взрывобезопасен, но способствует воспламенению других веществ. В огне
выделяет раздражающие, токсичные пары (или газы). Контакт с горючими веществами
недопустим, так вещество является сильным окислителем и реагирует с горючими
материалами и восстановителями.
3. РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
3.1 Изучение основных характеристик
сополимеров ВП с МА
.1.1 Определение содержания звеньев
МА в сополимерах с помощью потенциометрии
Как следует из данных, приведенных в обзоре литературы
(раздел 1.1.2), потенциометрическое титрование широко используется для
определения состава сополимеров МА, в том числе с ВП, однако каждая группа
исследователей проводит его различным образом. Мы также применили свой вариант
методики гидролиза и титрования сополимеров ВП с МА в водном растворе, опираясь
на имеющиеся данные, особенно [8, 25, 66], в то же время предполагая
выполнение анализа в течения рабочего дня, а также выдерживая постоянной
скорость подачи титранта и время выдержки между добавлением раствора NaOH и
записью показаний рН-метра.
Примеры кривых потенциометрического титрования
сополимеров показаны на рис. 3.1 и 3.2. Как видно из формы кривых на рис. 3.1а
и 3.2а, они являются довольно пологими, без четко выраженного перегиба, что
затрудняет определение точки эквивалентности, что согласуется с данными работы
[24]. Поэтому ее определение проводили по дифференциальным кривым
зависимости «ΔpH/ΔV - ΔV» (рис. 3.1б и 3.2б). На них четко виден максимум,
соответствующий точке эквивалентности, хотя имеется также несколько пологих
(рис. 3.1б) или острых (рис. 3.2 б) пиков меньшей интенсивности. Их появление
предположительно связано с разрывом межмолекулярных взаимодействий и изменением
конформации макромолекул при их развертывании и переходе в форму жесткого
стержня.
Форма кривых потенциометрического титрования в виде
зависимости «ΔpH - ΔV» или «ΔpH/ΔV - ΔV» типична для различных образцов.
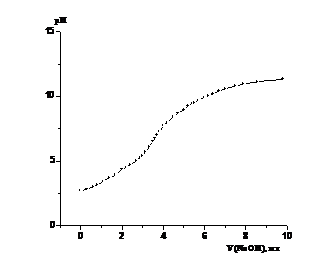
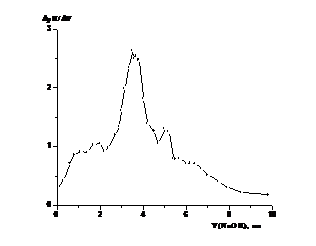
Рисунок 3.1. Кривая потенциометрического титрования
сополимера ВП с МА в водном растворе (а) и ее первая производная (б). Условия
получения сополимера: [ВП]:[МА]=1:1, [ПБ]=3.28·10-2 моль/л, раствор
в ДО Σ[М] = 3 моль/л, 333 К, S~ 88%. (получ.
20.05.09) сополимеров ВП с МА, независимо от условий получения: проведение
синтеза в растворе или в массе, инициатора (ПБ, АИБН, без инициатора),
температуры, степени конверсии. Однако эти факторы опосредованно, через
величину связанной с ними молекулярной массы сополимеров влияют на его
растворимость и скорость титрования.
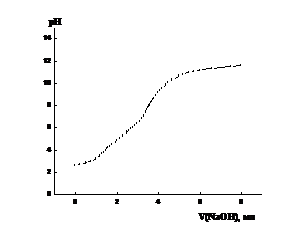

Рисунок 3.2. Кривая потенциометрического титрования
сополимера ВП с МА в водном растворе (а) и ее первая производная (б). Условия
получения сополимера: [ВП]:[МА]=1:1, [ПБ]=3·10-3 моль/л, в массе,
333 К, S~ 16%.
Результаты тирования и данные по составу сополимеров
представлены далее в табл. 3.1 вместе с данными ЯМР спектроскопии.
Чтобы проверить, возможно ли уменьшение влияния
конформационных изменений макромолекулы при замене воды на органический
растворитель, а также для отработки методики функциолизированых сополимеров,
для которых предполагалось ухудшение их растворимости в воде, были проведены
опыты по подбору условий титрования в неводной среде.
В качестве расторителя был первоначально взят ДМФА,
который показал хорошие результаты при титровании сополимера МА и стирола[30].
Однако для изучаемых нами сополимеров оказалось, что определение точки
эквивалентности затруднительно как по кривой титрования, так и по ее первой или
второй производной (рис. 3.3, кривая 2). Условия титрования улучшались при
введении в ДМФА добавок воды. Наилучшие результаты были получены в смешанном
растворителе ДМФА-Н2О при содержании Н2О ~ 17% об. (рис.
3.3, кривая 3). Если при водном титровании сополимеров ВП с МА на кривой имелся
скачок, соответствующий нейтрализации одной карбоксильной группе звена
малеиновой кислоты, то в водно-органической среде титруются обе группы.
Результат, получаемый при титровании первой группы, соответствует данным
водного тирования этого же сополимера (ср. кривые 1 и 3 рис. 3.3).

Рисунок 3.3 - Кривые титрования сополимера ВП с МА в
водном растворе (1), ДМФА (2) и смешанном растворителе ДМФА-Н2О (3).
Условия получения сополимера: [ВП]:[МА]=1:1, [ПБ]=10-3 моль/л, в
массе, 313 К, S~ 10%.
3.1.2 Определение состава сополимеров
ВП с МА методом 1Н ЯМР
Определение состава сополимеров ВП с МА проводили
методом ПМР спектроскопии. Несмотря на то, что ранее его уже применяли для
анализа этих сополимеров [13 222], синтезированных в диоксане, но в
случае получения в массе, использованном в нашей работе, спектры сополимеров
имели специфические особенности (рис. 3.4 - 3.7). Поэтому необходимо было
выбрать диапазон спектра, пригодный для определения их состава.
Согласно опубликованным данным [13 222] по
распределению сигналов протонов групп звена ВП и МА в спектрах бинарных
сополимеров, полученных при 338 К, в ДО, с АИБН, в области 2.33-1.3 м. д.
находятся химические сдвиги 4Н звена ВП, а в диапазоне 4-3 и 4.45-4.1 м. д. -
5Н ВП и 2Н МА соответственно. Однако в спектрах сополимеров, полученных нами в
растворе ДО звено ВП в области 2.5-1.5 м. д. представлено двумя уширенными
сигналами (рис. 3.4), то при получении в массе и инициировании ПБ в спектрах
имеются мультиплетные сигналы (рис. 3.5, 3.6). Кроме того, в спектрах таких
сополимеров и особенно синтезированнных в отсутствии инициатора (рис. 3.7) в
области 1.3-1.0 м. д. обнаруживается большое количество сигналов протонов
метильных групп, а в диапазоне 6.4-5.2 м. д. - групп СН звеньев ВП,
образовавшихся в результате реакций передачи и обрыва цепи. Указанные изменения
положения сигналов звеньев ВП и распределения их интенсивности приводят в тому,
что количество протонов ВП в диапазоне 2.45-1.0 м. д. может изменяться от 4.5
до 6 в зависимости от условий получения.
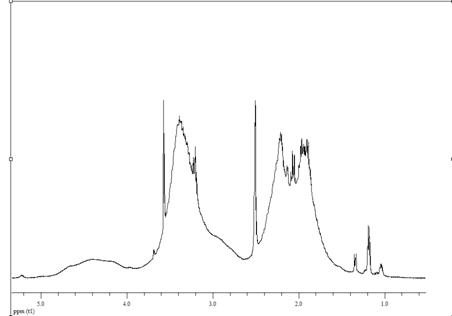
Рисунок 3.4 - 1Н ЯМР спектр сополимера ВП с
МА. Растворитель - ДМСО-d6, стандарт - ТМС, 298 К. Условия получения
сополимера: [ВП]:[МА]=1:1, [АИБН]=3·10-3 моль/л, в растворе ДО Σ[М] = 3 моль/л,, 333 К, S~ 21 %.
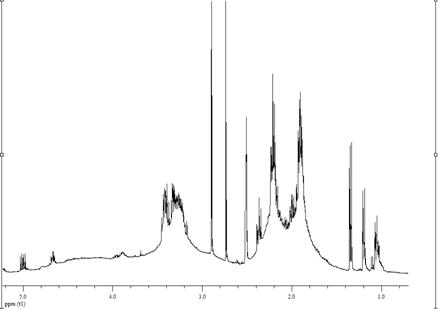
Рисунок 3.5 - 1Н ЯМР спектр сополимера ВП с
МА. Растворитель - ДМСО-d6, стандарт - ТМС, 298 К. Условия получения
сополимера: [ВП]:[МА]=1:1, [ПБ]=3·10-3 моль/л, в массе, 333 К, S~ 16
%.
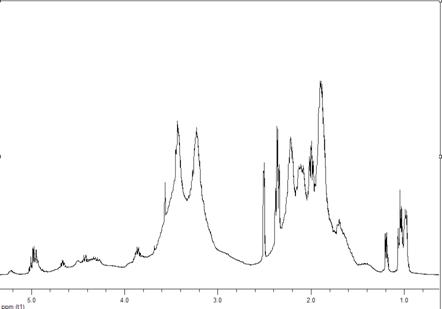
Рисунок 3.6 - 1Н ЯМР спектр сополимера ВП с
МА. Растворитель - ДМСО-d6, стандарт - ТМС, 298 К. Условия получения
сополимера: [ВП]:[МА]=1:1, [ПБ]=10-3 моль/л, в массе, 313 К, S~ 12
%.
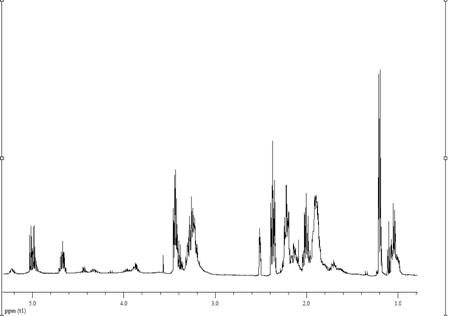
Рисунок 3.7 - 1Н ЯМР спектр сополимера ВП с
МА. Растворитель - ДМСО-d6, стандарт - ТМС, 298 К. Условия получения
сополимера: [ВП]:[МА]=1:1, без инициатора, в массе, 333 К, S~ 27 %.
В связи с выявленными особенностями нами было
проведено апробирование двух методик расчета состава сополимеров. Согласно
первой, интегральную интенсивность в диапазоне 2.5-2.0 м. д. приравнивали 2Н
цикла звена ВП (СО-СН2, вариант 1а) или в диапазоне 2.5-1.7 м. д. -
4Н цикла звена ВП (СО- СН2-СН2, вариант 1б).
Кроме того, выполняли расчет, основанный на величине интенсивности сигнала в
области 4.9-4.0 м. д., которая характерна для протонов звена МА. Поскольку
согласно данным [22] и полученным нами ранее спектрам ПМР сополимеров
стирола с МА химическому сдвигу протонов МА соответствует диапазон 5-3 и
4.3-3.0 м. д. соответственно, мы считаем, что в диапазоне 4.7-4.0 м. д.
находится сигнал 1Н МА. Тогда для определения состава сополимеров ВП-МА
применяли формулы:
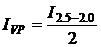 (1а);
(1а); 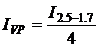 (1б);,
(1б);, 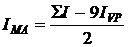 (3.1)
(3.1)
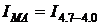 ; (2)
; (2)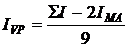 (3.2)
(3.2)
Данные определения состава, представленные в табл.
3.1, показывают, что результаты расчета основанного на величине интенсивности
2.5-2.0 м. д. (1а) и 4.7-4.0 м. д. (2) совпадают между собой, тогда как учет
интенсивности2.5-2.0 м. д. (1б) дает заниженное количество звеньев ВП
(табл.3.1). Отметим, что полученные сополимеры содержат в своем составе от 0.5 до
5 мол. % мономерного МА, видимо, за счет образования им комплекса со звеном ВП.
Содержание звеньев МА в сополимере, определенное
методами ЯМР и потенциометрического титрования, удовлетворительно совпадает в
пределах 2.5-6 мол. %. Снижение температуры сополимеризации с 333 до 313 К,
концентрации ПБ до 10-3 моль/л и проведение процесса в массе
способствуют образованию сополимеров, содержащих ~60 мол.% ВП, т. е.
неэквимолярного состава (табл. 3.1).
Таблица 3.1 - Результаты определения состава
сополимеров ВП с МА
|
Условия получения
сополимера
|
Содержание МА в сополимере,
мол. % (потенциом. титрование
|
ВП : МА в сополимере по
данным ПМР, мол. %
|
Методика расчета1)
|
|
|
Прямой расчет
|
Состав с учетом неотмытого
МА
|
|
|
333 К, в ДО, ПБ 0.03
моль/л, S~ 88%
|
43.1; 42.4
|
46.8:53.2
|
45.9:54.1
|
2
|
|
333 К, в ДО, АИБН 10-2
моль/л, S~ 21 %
|
47.4
|
49.6:50.4
|
49.0:51.0
|
2
|
|
333 К, в массе, ПБ 10-2
моль/л, S~ 25 %
|
49.7
|
57.4:42.6 59.5:40.5
|
55.3:44.7
|
1a 2
|
|
333 К, в массе, ПБ 0.003
моль/л, S~ 16%
|
44.8:55.2
|
40.4:59.5
|
58.9
|
2
|
|
313 К, в массе, ПБ 10-3
моль/л, S~ 25 %
|
32.8
|
64.9:35.1 63.0:37.0
|
64.7:35.3 62.7:37.3
|
1б 2
|
|
1) интегральная интенсивность в диапазоне 2.5-2.0 м.д.
приравнена 2 Н ВП (1а); 2.5-1.7 м.д. - 4 Н ВП (1б); 4.7-4.0 - 1 Н МА (2).
|
3.1.3 Определение характеристической
вязкости и молекулярной массы сополимеров ВП с МА
От величины молекулярной массы (со)полимеров ВП
зависит область их применения в медицине, поэтому определение ММ имеет
первостепенное значение. Для ПВП вискозиметрические измерения проводят обычно в
водных растворах, однако для сополимеров ВП нам необходимы были
сопоставительные измерения и в неводных растворителях.
Первоначально нами была сделана попытка определить
вязкость сополимеров ВП с МА в ДМФА, чтобы избежать нежелательных эффектов при
превращении ангидридных групп в кислотные и их диссоциации в воде. Оказалось,
что приведенная вязкость ([η]уд/С) с уменьшением концентрации (С) не
уменьшается линейно, а возрастает (рис. 3.8а), как характерно для
полиэликтролитов. Поскольку в литературе [5э 50] есть сведения о том,
что молекулярную массу сополимеров на основе ВП можно определять в растворе
ДМФА с добавлением KBr (0,05н), то нежелательное возрастание вязкости
устраняли, вводя добавки этой соли. Однако зависимость [η]уд/С от С в этом случае имела вид
прямой с отрицательным тангенсом угла наклона, характерный для неполного
подавления диссоциации в растворах полиэлектролитов (рис. 3.8б), возможно,
из-за низкой растворимости соли в ДМФА, что не позволяло определить величину [η].
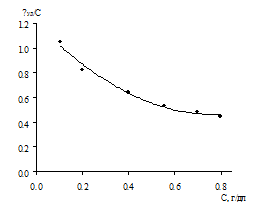
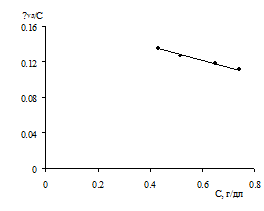
Рисунок 3.8 - График зависимости приведенной вязкости
от концентрации для сополимера ВП с МА в водном растворе (а) и в ДМФА с
добавкой KBr (б). Условия получения сополимера: [ВП]:[МА]=1:1, [АИБН]=3·10-3
моль/л, в растворе ДО Σ[М] = 3 моль/л,, 333 К, S~ 13 %.
Ранее авторами [24, 33] было установлено, что
сополимеры ВП с МА проявляет характер неэлектролита в разбавленном водном
растворе HCl с рН 2.1 ([НСl] = 0.0085 М) при 25°С. Мы также провели определение
характеристической вязкости полученных нами сополимеров ВП с МА в кислой среде
согласно [24]. Как показано на рис. 3.9, в этих условиях они не
проявляют свойства полиэлектролитов и дают прямолинейные зависимости в
координатах «[η]уд/С - С», что позволяет определить значение характеристической вязкости [η], равной 0.197 и 0.174 дл/г. Исходя из
них и используя уравнение (1.?), были найдены величины молекулярной массы
сополимеров, которые для показанных на рис. 3.9 образцов составили 3,08·104
и 2,53·104.
Условия получения сополимеров: 1 - [ВП]:[МА]=1:1,
[ПБ]=3.28·10-2 моль/л, раствор в ДО Σ[М] = 3 моль/л, 333 К, S~ 88%; 2 -
[ВП]:[МА]=1:1, [ПБ]=10-3 моль/л, в массе, 313 К, S~ 25 %.
Рисунок 3.9. Зависимость приведенной вязкости от
концентрации сополимеров ВП с МА для растворов в воде при рН 2.1 ([НСl] =
0.0085 М) и 30°С.
Поскольку титрование сополимеров оказалось возможным в
смешанном растворителе ДМФА-Н2О, то эта смесь, но с добавлением КСl
(0.1 н) была также применена для определения характеристической вязкости. Здесь
нашей целью было показать возможность сопоставительного анализа влияния ДМФА на
размер макромолекул и предварительное определение величин коэффициентов К и α в водно-органическом растворителе.
Оказалось, что в выбранном растворителе ДМФА-Н2О величина [η]уд/С для обоих сополимеров также
линейно уменьшается с концентрацией (рис. 3.10). При этом были получены близкие
к определению в водной среде (рН 2.1) величины [η], равные 0.232 и 0.193 дл/г.

Рисунок 3.10. Зависимость приведенной вязкости от
концентрации сополимеров ВП с МА для растворов в смеси ДМФА-Н2О
(70:30) с содержанием КСl 0.1 моль/л при 30°С. Обозначения прямых аналогично
рисунку 3.9.
Сопоставление значений [η] в смешанных растворителях с
величинами молекулярной массы, рассчитанными по уравнению (1.?) для кислых
водных растворов, приводит к коэффициентам уравнения Куна Хувинка: α = 0,936 и К = 1,46·10-5.
Судя по величине α, смесь ДМФА-Н2О является лучшим
растворителем, хотя для окончательного заключения необходим больший объем
экспериментальных данных.
3.1.4 Изучение электронных спектров
(со)полимеров N-винилпиролидона
Получение данных о полосах поглощения сополимеров ВП с
МА в электронных спектрах интересовало нас, поскольку в дальнейшем
предполагалось проводить исследование взаимодействия этих полимеров с
различными органическими соединениями, для чего практически полезной может быть
УФ спектроскопия. В литературе данные по этому вопросу практически отсутствуют.
При сравнении электронных спектров водных растворов
поливинилпиролидона (рис. 3.11, кр. 3), сополимеров N-винилпирролидона с
малеиновым ангидридом (рис. 3.11, кр. 4, 5) и N-метилпирролидона (рис. 3.11,
кр. 6) обращает на себя внимание их подобие. Все изучаемые соединения поглощают
в области 200-230 нм с максимумом полосы поглощения при 205-212 нм. При этом не
имеет значения способ получения и ММ СПЛ. Раствор янтарного ангидрида, взятого
как низкомолекулярный аналог звена МА в СПЛ, при той же концентрации в области
200-240 нм практически не поглощает (рис. 3.11, кр. 2). Из этого следует, что
поглощающим элементом структуры является пирролидониевое кольцо.
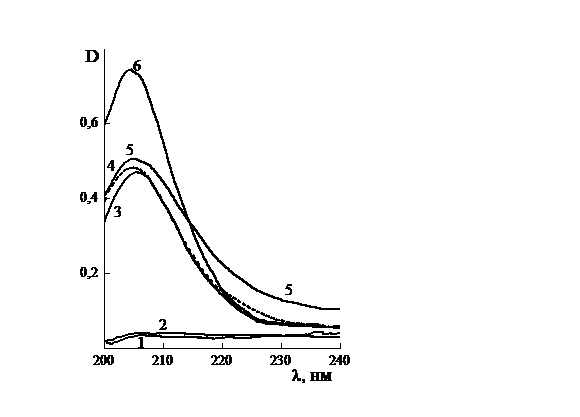
Рис. 3.11. Электронные спектры растворов в воде (1)
поливинилпиролидона (3), сополимеров N-винилпирролидона с малеиновым ангидридом
(4, 5), N-метилпирролидона (6) и янтарного ангидрида (2) с концентрацией 0.02
г/л. Условия получения сополимеров: в растворе ДО (4), в массе (5), инициатор -
ПБ, 333 К.
Максимум полосы поглощения смещается в длинноволновую
сторону с ростом концентрации от 0,03 до 0,12 г/л (ПВП, рис 3.12а); от 0,015 до
0,07 г/л (СПЛ, рис. 3.12б) и от 0,02 до 0,08 г/л (МП, рис. 3.12в). При
пересчете концентрации для полимеров на осново-моль/л звена ВП или моль/л для
МП оказалось, что величина λмакс растворов полимеров линейно зависит
от концентрации (рис. 3.13, кр. 1 и 3). Однако наклон прямой для СПЛ в 2.5 раз
превышает таковой для ПВП, хотя они отсекают практически одинаковый отрезок на
оси ординат, равный ~204 нм. Для МП зависимость «λ - С» (рис. 3.13, кр. 2) имеет вид
кривой, начало которой параллельно кр.1 (СПЛ), а конец - кр. 3 (ПВП). Наличие
таких зависимостей, по-видимому, связано с образованием ассоциатов МП и
конформационными изменениями в цепях (со)полимеров.
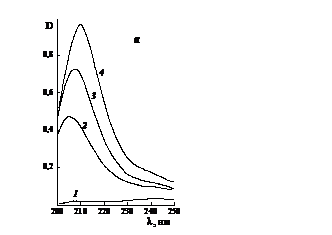

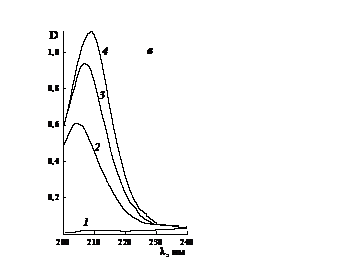
Рис. 3.12. Электронные спектры растворов в воде (1)
поливинилпиролидона (а), сополимеров N-винилпирролидона с малеиновым ангидридом
(б) и N-метилпирролидона (в) с концентрацией, г/л: а) 2 - 0.02, 3 - 0.03, 4 -
0.06; б) 2 - 0.0187, 3 - 0.0374, 4 - 0.075; в) 2 - 0.0163, 3 - 0.0327, 4 -
0.0496. Условия получения сополимера: в массе, ПБ, 333 К.

Рис. 3.13. Изменение длины волны максимума полосы
поглощения (λ) в зависимости от концентрации СПЛ (1), МП (2) и ПВП (3) в
водном растворе.
Это предположение подтверждает зависимость величины
оптической плотности полосы 205-212 нм от концентрации (со)полимеров (по звену
ВП) или их низкомолекулярного аналога МП (рис. 3.14). Для сополимера ВП с МА
эта зависимость представлена двумя пересекающимися прямыми, первая из которых
выходит из нуля (рис. 3.14, кр. 1), т. е. имеет вид типичной зависимости
Бугера-Ламберта-Бера: D = ε C l. Для соответствующего этой части
кривой диапазона концентраций 0÷0,000126 осново-моль ВП/л, (0÷0,0265 г/л сополимера) коэффициент ε составил 5170±30 моль/(л∙см).
На втором участке кривой точки описываются уравнением:
= (1425±150)×C +(0.48±0.04).

Рисунок 3.14. Изменение оптической плотности (D) в
максимуме полосы поглощения в зависимости от концентрации сополимера ВП с МА
(1), МП (2) и ПВП (3) в водном растворе.
Как видно из этих данных, величины ε снижается в 3.6 раз. Для МП тот же диапазон
массовых концентраций (0.016÷0.09 г/л), что и для сополимера (0.015÷0.075 г/л) соответствует более высоким
величинам концентрации в мольном выражении: 1.6÷9.9×10-4 моль/л по сравнению с 0.7÷3.6×10-4 осново-моль ВП/л в случае
сополимера. На графике зависимости «D - C» для МП (рис. 3.14, кр. 2) видно
отсутствие І участка, однако точки можно разделить также на две прямые, первая
из которых по своему наклону, равному 1510±150 моль/(л∙см), соответствует
ІІ участку кр.1. Поэтому можно считать, что оставшиеся точки образуют участок
ІІІ с наименьшим наклоном 603±130 моль/(л∙см), что в 8.6 и 2.5 раз
меньше, чем для І и ІІ участков соответственно. Малое количество точек для ПВП
не позволяет сделать определенные выводы, хотя можно представить, что в этом
случае также может быть выявлено разделение смеси на 3 участка.
Причиной такого вида зависимости «D - C» может быть
то, что в водном растворе сополимер ВП с МА диссоциирует с образованием из
ангидридных звеньев кислотных, проявляя свойства полиэлектролита. Поэтому в
разбавленных растворах макромолекула имеет вид стержня, окруженного гидратной
оболочкой, которая возрастает с увеличением разбавления, на что указывает
резкий рост приведенной вязкости при концентрации сополимера менее 0.02 г/л
(см. рис. 3.9). В этой же области концентраций мы наблюдаем перелом на кривой 1
(рис. 3.14). При более высоких концентрациях в растворе возможно взаимодействие
между макромолекулами, как и ассоциация МП, что приводит появлению участка ІІ
на кривых 1 и 2 (рис. 3.14). Можно предположить, что участок ІІІ соответствует
дальнейшей агрегации молекул.
Таким образом, ПВП и сополимеры ВП с МА обладают
слабым поглощением с полосой при 205-212 нм, которое обусловлено наличием в них
лактамного цикла. Результаты, полученные методами УФ спектроскопии,
вискозиметрии и рН-метрии согласуются друг с другом и дают общее представление
о поведении макромолекул сополимеров ВП с МА в разбавленных водных растворах.
3.2 Получение пленочных гидрогелевых
материалов на основе (со)полимеров N-винилпиролидона
Пленочные материалы на основе (со)полимеров ВП,
пригодные для применения в медицинских целях, получали по двум вариантам
методик:
) сшивка ПВС (ММ 72000, АppliChem GmbH, Germany) и ПВП
(ММ 50000) или ПВС и сополимера ВП с малеиновым ангидридом - МА (СПЛ 1,
[ВП]:[МА] = 1:1, ММ ~ 35000) в водном растворе в присутствии персульфата
аммония подобно методике [77 5] с последующей сушкой;
) получение пленок на основе ПВП или СПЛ 1 и ПВС,
содержащих лекарственные препараты (фурацилин, новокаин) с их термообработкой.
Полученные на основе ПВП или СПЛ 1 и ПВС материалы
после сушки (табл. 1) представляют собой прозрачные пленки. Для них определяли
такие показатели, как массовая доля геля (G), степень набухания (DS) и
равновесное содержание воды (EWC) или водопоглощение по уравнениям [71, 77,
6э 1, 5, 6]:
= Wс/W0×100 (3.1)= (Wн-Wс)/Wс×100 (3.2)= [(Wн-Wс)/Wн]
× 100 (3.3)
где W0, Wн и Wс -
масса образца перед погружением в воду, набухшего и высушенного соответственно.
Результаты определения показали, что пленка, полученная
путем выдержки смеси ПВП и ПВС в присутствии персульфата с сушкой при 50°С в
течение суток (обр. 1), полностью растворима. Более жесткий режим получения
материалов - выдержка при 150°С (обр. 2) или дополнительное введение в раствор
полимеров персульфата с выдержкой раствора при 50°С в течении еще 5 ч (обр. 3)
дают пленки, содержащие ~70 и 75 % сшитого продукта соответственно (табл. 3.2).
В то же время пленка 2 является более жесткой, на что указывает величина
степени набухания, которая для нее примерно в 2 раза меньше, чем у пленки 3:
338 и 746 %.
Таблица 3.2. Условия получения и свойства гидрогелевых
пленок на основе (со)полимеров ВП
|
Образец
|
Исходные полимеры
|
Время выдержки раствора при
80°С (ч)
|
Режим сушки
|
Доля геля, %
|
Толщина пленки, мм
|
|
1
|
ПВП:ПВС (18:82 мас. %)
|
|
24 ч ~50°С
|
0
|
0.04
|
|
2
|
|
|
+ 5ч ~150°С
|
70.3
|
0.04
|
|
3
|
|
|
24 ч ~50°С
|
75.5
|
0.04
|
|
4/11)
|
СПЛ 1:ПВС (18:82 мас. %)
|
|
7.5 ч~50°С, 15 ч ~80°С
|
80.8
|
0.03
|
|
4/21)
|
|
|
|
95.1
|
0.03
|
|
5
|
|
|
+7 ч ~80°С2)
|
96.5
|
0.03
|
Для того, чтобы ввести в пленочные гидрогелевые материалы
группы, способные к ионизации или дальнейшей функционализации, мы заменили ПВП
на сополимер ВП с МА (СПЛ 1), содержащий звенья сомономеров в соотношении ~1:1
(обр. 4, 5 в табл. 1). Из одного исходного раствора были политы пленки 5-7,
высушенные в относительно мягких условиях с увеличением продолжительности сушки
от 7.5 ч при 50°С и 15 ч при 80°С (обр. 4), а также 7.5 ч при 50°С и 22 ч при
80°С (обр. 5). Найдено, что доля геля, т. е. сшитого полимера при этом возросла
от 80,8 (обр. 4/1) до 96.5 % (обр.5), а степень набухания и водопоглощения -
снизилась от 660 до 200 % и от 87 до 67 % соответственно.
Для получения пленочных материалов с добавками
фурацилина и новокаина вводили эти препараты к прогретому с персульфатом
раствору СПЛ1 и ПВС и проводили высушивание 7,5 ч при 50°С, а потом еще 15 ч
при 70-80°С (обр. 6). При выдержке образца 6 в воде на протяжении 2 ч раствор
приобретал желтое окрашивание и становился вязким, т. е. происходил выход в
жидкую среду лекарственных веществ и части полимерной основы, но образцы
сохраняли целостность (доля геля 56 %), степень их набухания составила 767 %.
Опыты по определению характеристик пленок проводились
в нескольких циклах «набухание-сушка», причем исходные эластичные образцы после
сушки становились жесткими и хрупкими, но снова приобретали гибкость и
эластичность после погружения в воду (рис. 3.15).
Найдено, что способность образцов пленок к поглощению
воды в трех циклах набухания отличается мало. Для нескольких образцов пленок в
третьем цикле набухания проводили периодический контроль за массой набухающих
образцов на протяжении 2 ч. Найденные величины степени набухания (рис. 3.16)
показали, что временем достижения равновесного состояния можно считать 1-1.5 ч,
причем за 15 мин достигается 75-90% от максимальной степени набухания.

Рисунок 3.15. Фотографии пленок образцов 4/2 (а, б) и
6 (в. г) в высушенном состоянии (а, в) и через 5 мин погружения в воду (б. г).
Размеры пленок (мм): а - 32×24; б - 43×27; в - 25×20; г - 37×29.
Ход набухания образцов 4/1 и 6, полученных при
одинаковом режиме сушки, в течение 2 ч практически совпадает (рис. 3.16, кривые
2 и 3). Это позволяет считать, что основным фактором, обусловливающим
показатели полученных сшитых пленочных материалов на основе сополимера ВП с МА
и ПВС, является способ высушивания.
Поверхность и торцевой скол образцов изучены методом
растровой электронной микроскопии с помощью н.с. В.В. Бурховецкого (Донецкий
физико-технический институт им. А.А. Галкина). Применение этого метода
показало, что полученные полимерные материалы имеют глобулярную структуру,
особенно хорошо видно на торцевом сколе (рис. 3.17). Введение лекарственных
препаратов, как видно из сравнения микрофотографий образцов 4/2 и 6 (рис. 3.17,
а и б), сопровождается увеличением размера глобул, что свидетельствует о
связывания лекарственных средств полимерной основой.
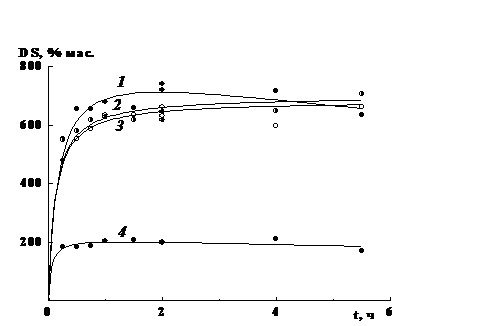
Рис. 3.16. Зависимость величины степени набухания (DS)
от продолжительности выдержки в воде для образцов пленок 3 (1), 4/1 (2), 5 (4)
и 6 (3) на основе ПВС и ПВП (1), ПВС и сополимера ВП с МА (2-4).

Рисунок 3.17. Микрофотографии торцевого скола пленки
на основе СПЛ 1 и ПВС (образец 4/2 - а), а также с добавками фурацилина и
новокаина (образец 6 - б), полученные с помощью растрового электронного
микроскопа JEOL JSM-6490LV при напылении углеродом.
Эти результаты показали, что на основе сополимера ВП с
МА и ПВС возможно получить пленки, способные к многократному обратимому
набуханию в воде (гидрогели) с долей геля в конечном продукте от 80 до 97% и
степенью набухания от 490 до 200%. Это указывает на возможность их применения в
качестве перевязочных материалов для ран, поскольку с их помощью осуществляется
постепенный выход лекарственных средств, выделения (со) полимеров ВП, имеющих
свойство создания антимикробного барьера [1, 6], а также поглощения раневого
экссудата. Полученные предварительные результаты предполагается использовать
при создании лекарственных пленочных материалов.
ВЫВОДЫ
Показана возможность получения пленок, способных к
многократному обратимому набуханию в воде (гидрогелей) на основе сополимера ВП
с МА и ПВС, имеющих от 54 до 97 % геля в своем составе и степень набухания от
490 до 200 %, а также иодсодержащих пленок на основе двух бинарных сополимеров
ВП - с ММА и МА. Полученные данные служат основой методики получения
лекарственных пленочных материалов.
сополимер винилпирролидон малеиновый ангидрид
ССЫЛКИ