Современные методы фармацевтического анализа
Министерство образования РФ
ФГБОУ ВПО «Орловский государственный
университет»
Медицинский институт
Кафедра фармакологии, клинической
фармакологии и фармации
КУРСОВАЯ РАБОТА
«СОВРЕМЕННЫЕ МЕТОДЫ ФАРМАЦЕВТИЧЕСКОГО
АНАЛИЗА»
Мурадян Алина Вартанесовна, группа №15, курс 3 специальность
«Фармация»
Орел 2014
Список сокращения
ГФ - государственная фармакопея
ФСП - фармакопейная статья предприятия
ФС - фармакопейная статья
ОФС - общие фармакопейные статьи
ГОСТ - государственный стандарт
ТУ - технические условия
ЛС - лекарственное средство
ЛВ - лекарственное вещество
ВЭЖХ - высокоэффективная жидкостная хроматография
ДЭС - двойной электрический слой
ИТФ - изотахофорез
ИФЭ - изоэлектрическая фокусировка
КГЭ - капиллярный гель-электрофорез
КЗЭ - капиллярный зонный электрофорез
КИТФ - капиллярный изотахофорез
КИЭФ - капиллярное изоэлектрическое фокусирование
ККМ - критической концентрации мицеллообразования
Оглавление
Введение
Глава 1.
1.1 Критерии
фармацевтического анализа
1.2 Химические методы
установления подлинности
Глава 2.
2.1 Идентификация
элементорганических лекарственных веществ
2.2 Идентификация
органических лекарственных веществ
2.3 Испытание на
специфические примеси
Глава 3.
3.1 Осадительное
титрование
3.2 Кислотно-основное титрование
(метод нейтрализации)
3.3 Титрование в смешанных
растворителях
3.4
Окислительно-восстановительное титрование
3.5 Нитритометрия
3.6 Элементный анализ
3.7 Метод сжигания в колбе
с кислородом
3.8 Физические и
физико-химические методы анализа
3.9 Оптические методы
3.10 Методы, основанные на
поглощении электромагнитного излучения
3.11 Методы, основанные на
испускании излучения
3.12 Методы, основанные на
использовании магнитного поля
3.13 Электрохимические
методы
3.14 Термические методы
анализа
3.15 Методы разделения
Глава 4.
4.1 Валидация методов
анализа
Заключение
Список используемой
литературы
Введение
Фармацевтический анализ - это
наука о химической характеристике и измерении биологически активных веществ на
всех этапах производства: от контроля сырья до оценки качества полученного ЛВ,
изучения его стабильности, установления сроков годности и стандартизации ЛФ.
Фармацевтический анализ имеет свои специфические особенности, отличающие его от
других видов анализа. Эти особенности заключаются в том, что анализу подвергают
вещества различной химической природы: неорганические, элементорганические,
радиоактивные, органические соединения от простых алифатических до сложных
природных биологически активных веществ. Чрезвычайно широк диапазон
концентраций анализируемых веществ. Объектами фармацевтического анализа
являются не только индивидуальные ЛВ (субстанции), но и смеси, содержащие
различное число компонентов [2] <#"818075.files/image001.jpg">
Ионы
калия осаждают винной кислотой:
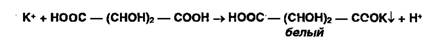
Ионы
натрия осаждают цинкуранилацетатом:
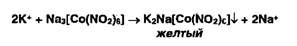
Ионы
калия можно осадить раствором гексанитрокобальтата (III) натрия:
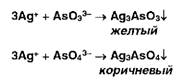
Некоторые
реакции осаждения можно использовать для обнаружения и катионов, и анионов.
Ионы
магния образуют в присутствии фосфат- и аммоний-ионов осадок фосфата
магния-аммония:
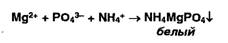
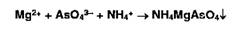
Фосфат-ионы
образуют с раствором молибдата аммония желтый осадок фосфор-молибдата аммония:
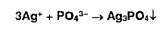

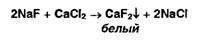
Ионы
бария образуют белый осадок с сульфат-ионами: Аналогичную реакцию дают
сульфиты:
Сульфит
бария, в отличие от сульфата бария, растворим в хлороводородной кислоте. Ионы
фтора открывают реакцией осаждения из раствора хлорида кальция:
Ионы
серебра образуют осадки с хлоридами, бромидами, йодидами:
Образующиеся
галогениды различаются по растворимости в растворе аммиака. Желтый осадок
образуют ионы серебра с фосфатами:
Образует
осадки ион серебра также с арсенит- и арсенат-ионами:
Ионы
магния с растворами карбонатов образуют белый осадок основного карбоната
магния:
Ионы
железа (III) в растворе приобретают красное окрашивание в присутствии
роданид-ионов, образуя малодиссоциирующее соединение:
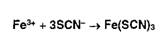
Ряд
реактивов образуют белые или окрашенные осадки с несколькими катионами. Ионы
ртути, цинка, висмута, мышьяка взаимодействуют с сульфидами:

Ионы
железа (III) и цинка осаждаются растворами гексацианоферрата (II) калия:
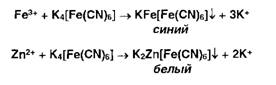
Ионы
железа (II) дают аналогичные результаты с гексацианоферратом (III) калия:
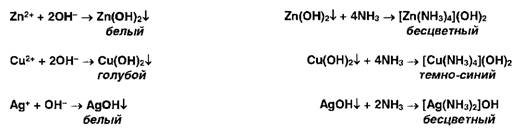
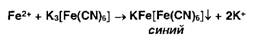
Ионы
цинка, меди и серебра осаждаются гидроксидом аммония с образованием осадков,
растворимых в избытке реактива:
Ионы
ртути (II) и висмута (III) осаждаются йодидами, осадки растворяются в избытке
реактивов:

Окислительно-восстановительные
реакции, используемые для испытаний подлинности, сопровождаются изменением
окраски образующихся продуктов взаимодействия.
Бромид-
и йодид-ионы окисляют хлором (хлорамином, другими окислителями):
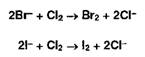
Выделившийся
бром окрашивает слой хлороформа в оранжевый цвет, а йод - в фиолетовый. Йод
обнаруживают также по синему окрашиванию крахмального клейстера.
Фторид-ионы
обесцвечивают красную окраску раствора роданида железа: 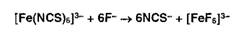 Ионы меди, серебра восстанавливаются из оксидов и
солей до свободных металлов:
Ионы меди, серебра восстанавливаются из оксидов и
солей до свободных металлов:
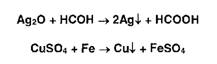
Нитрат-
и нитрит-ионы обнаруживают путем окисления дифениламина до дифенилбензидина, а
затем до дифенилдифенохинондимина гидросульфата (синее окрашивание) в
присутствии концентрированной серной кислоты:
Нитрит-ионы
(в отличие от нитратов) обесцвечивают раствор перманганата калия, подкисленный
серной кислотой:
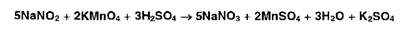
Взаимодействуя
с антипирином (феназоном), нитриты образуют продукт замещения -
нитрозоантипирин (зеленое окрашивание):
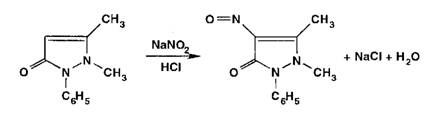
Реакции
разложения сопровождаются образованием газообразных продуктов, которые
обнаруживают органолептически (запах, окраска).
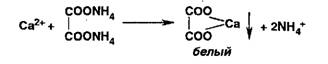
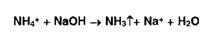
Ионы
аммония разлагаются под действием растворов гидроксидов (запах аммиака или
изменение окраски красной лакмусовой бумаги):
Карбонат-ионы
под действием насыщенного раствора сульфата магния образуют белый осадок, а
гидрокарбонат образует осадок только при кипячении смеси (см. реакцию на
магний).
Карбонат-
и гидрокарбонат-ионы образуют газ - диоксид углерода под действием минеральных
кислот:
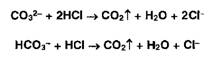
Сульфит-ионы
в тех же условиях образуют диоксид серы (резкий запах):
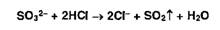
Нитрит-ионы,
в отличие от нитрат-ионов, под действием кислот выделяют оксиды азота (диоксид
азота имеет красно-бурую окраску):
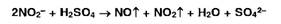
Превращения,
происходящие при нагревании и прокаливании некоторых ЛВ. Йод кристаллический,
соединения мышьяка, ртути возгоняются (испытания выполнять под тягой!). Цинка
оксид при прокаливании желтеет (после охлаждения окраска исчезает). Висмута
нитрат основной разлагается с образованием оксида висмута (желтое окрашивание)
и диоксида азота (желто-бурые пары). Соли алюминия при прокаливании с нитратом
кобальта образуют сплав синего цвета, представляющий собой алюминат кобальта
(тенарова синь). Соли цинка в этих условиях образуют сплав зеленого цвета
(зелень Ринмана).
Установить
наличие ряда элементов в неорганических и элементорганических ЛВ можно по
изменению окраски бесцветного пламени горелки. Так, соль натрия, внесенная в
пламя, окрашивает его в желтый цвет, калия - в фиолетовый, кальция - в
кирпично-красный, лития - в карминово-красный. Соли бора, смоченные этанолом,
окрашивают кайму пламени в зеленый цвет.
Глава 2.
.1 Идентификация элементорганических лекарственных веществ
Поскольку
атомы у большинства элементорганических соединений связаны ковалентно,
необходимым условием испытания их подлинности является предварительная
минерализация. При этом происходит частичное или полное разрушение органической
части молекулы до оксида углерода (IV) и воды. Другие элементы образуют
соответствующие ионы. Последние идентифицируют с помощью рассмотренных выше или
иных реакций[1] <#"818075.files/image031.jpg">
Образовавшийся
при восстановлении органически связанной серы сульфид натрия идентифицируют
цветной реакции с нитропруссидом натрия (красно-фиолетовое окрашивание),
осаждением раствором соли свинца (черное) или по сероводорода:
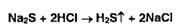
Счисление
органически связанной серы осуществляют действием концентрированной азотной
кислоты или сплавлением со смесью нитрата и карбоната калия. Образовавшийся
сульфат-ион открывают реакцией с солями бария.
Фосфорсодержащие
соединения минерализуют смесью концентрированных серной и азотной кислот до
фосфат-ионов, которые обнаруживают реакциями образования фосфата магния-аммония
или фосфор-молибдата аммония (см. реакции на фосфат-ион).
Галогенсодержащие
соединения под действием цинковой пыли в кислой или щелочной среде образуют
галогениды:
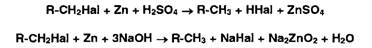
Затем
обнаруживают образовавшиеся галогенид-ионы с помощью рассмотренных выше реакций.
Проба Бейльштейна основана на образовании окрашенных в зеленый цвет галогенидов
меди при внесении в бесцветное пламя медной проволоки с галогенсодержащим
соединением.
Фтор
и хлор открывают аналитическими реакциями на соответствующие ионы после разрушения
органической части молекулы расплавленным металлическим натрием:
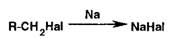
Йод
обнаруживают либо нагреванием йодпроизводного в пробирке на пламени горелки,
либо действуя концентрированной серной кислотой:
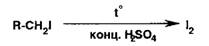
Наблюдают
выделение фиолетовых паров йода или фиолетовую окраску хлороформного
извлечения. Можно также применить спекание со смесью нитрата калия и карбоната
натрия:
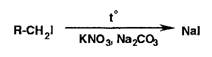
Затем
обнаруживают йодид-ионы.
Метод
спекания можно использовать при наличии в одном соединении хлора и серы с
последующим обнаружением образовавшихся хлорид- и сульфат-ионов.
2.2 Идентификация органических лекарственных веществ
Общие химические реакции
В
фармацевтическом анализе используются различные химические реакции органических
соединений, которые дают определенный аналитический эффект (выпадение осадка,
выделение газа, образование окрашенного раствора и т.д.) [7]
<#"818075.files/image037.jpg">
Под
действием гидроксидов калия (натрия) продукты нитрования образуют окрашенные
соли:
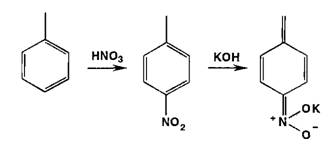
Реакции
нитрозироваиия приводят к образованию окрашенных, флюоресцирующих или имеющих
стабильную температуру плавления нитрозосоединений:
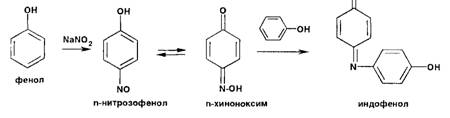
Фенолы
образуют нитрозосоединения, бесцветные или окрашенные в сине-зеленый (фенол),
сине-фиолетовый (резорцин) цвет. При нитрозировании фенолов с последующим
окислением образуются индофенолы (интенсивно-синее окрашивание):
Реакции
диазотирования и азосочетания используют для идентификации производных
первичных ароматических аминов и фенолов. Азосоединения - окрашенные (в
красный, коричневый и оранжевый цвет) продукты, получаемые в две стадии:
Диазотирование
(получение соли диазония):
Азосочетание (взаимодействие соли диазония с фенолом или ароматическим
амином). Сочетание происходит в орто- или пара-положеннях по отношению к
гидроксильной или аминогруппе, но идет легче в пара-положении:
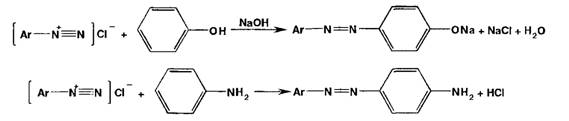
с фенолами (нафтолами) происходит в слабощелочной (рН 9,0-10,0), а с
аминами - в слабокислой среде.
Реакции галогенирования (бромирования и йодирования) по типу реакции
электрофильного замещения используют для производных фенолов и первичных
ароматических аминов. Наличие в их молекулах заместителей первого рода
обусловливает происходящий процесс образования трибромфенола или триброманилина
(белый осадок):
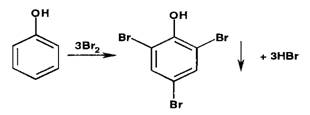
Аналогично происходит процесс образования трийодпроизводных. При наличии
в молекулах фенола и анилина радикалов в пара- или орто-положениях образуются
моно- или дигалогенпроизводные.
Реакции дегалогенирования можно выполнять без предварительной
минерализации (если галогены связаны с углеродом непрочной ковалентной связью).
Отщепление галогена при этом происходит под действием раствора нитрата серебра:

Дегалогенируют также, используя щелочное отщепление, путем нагревания
галогенпроизводного в присутствии цинковой пыли (бромкамфора) или в спиртовом растворе
гидроксида натрия:
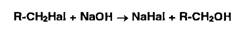
Затем обнаруживают галогенид-ион.
Реакции конденсации альдегидов и кетонов с первичными аминами,
гидроксиламином, гидразинами используются для идентификации всех указанных
групп органических соединений по общей схеме:
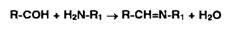
Альдегиды, конденсируясь с первичными аминами, образуют окрашенные в
желтый, красный или оранжевый цвет соли оснований Шиффа:
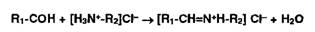
Эта реакция лежит в основе лигниновой пробы на первичные ароматические
амины, которые взаимодействуют с лигнинами, содержащимися в бумаге.
Кетопроизводные образуют гидразоны:
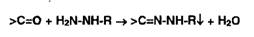
и кетоксимы:
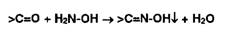
Гидразоны и кетоксимы - белые или окрашенные нерастворимые в воде
соединения со стабильной температурой плавления. По этим признакам можно
идентифицировать исходные для их получения соединения.
Окислительная конденсация с участием альдегидов лежит в основе таких
широко применяемых в фармацевтическом анализе реакций, как образование
ауринового красителя, нингидриновая реакция, мурексидная проба, проба Ле Розена
и др.
Нингидриновая реакция является общей для а-аминокислот, имминокислот,
полипептидов. Нингидрин (1,2,3-трикето-гидринденгидрат) образует с аммиаком,
выделившимся из этих соединений, продукт конденсации - ион
дикетогидриндилидендикетогидрамина, имеющий сине-фиолетовое окрашивание:
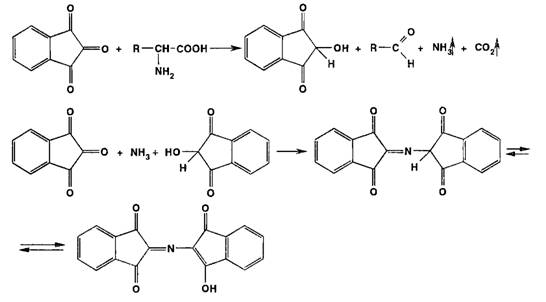
Реакции этерификации, ацилирования и гидролиза. Для подтверждения
подлинности спиртов и карбоновых кислот широко используют реакцию этерификации,
а подлинность сложных эфиров подтверждают с помощью обратного процесса -
гидролиза:
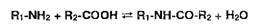
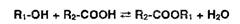
Этерификацию проводят в присутствии дегидратирующих веществ
(концентрированная серная кислота), а гидролиз - в кислой или щелочной среде.
Сходен с этерификацией процесс ацилирования (особенно ацетилирования)
аминопроизводных:
а также обратный процесс - гидролиз ацильных производных.
Образовавшиеся в результате этерификации, ацилирования, гидролиза
продукты идентифицируют по аналитическому эффекту (цвету, запаху, образованию
газа или осадка, температуре плавления осадка и др.).
Очень широко используют, например, реакцию образования этилацетата,
имеющего своеобразный фруктовый запах. Этилацетат образуют органические
соединения, выделяющие при гидролизе этанол или уксусную кислоту:
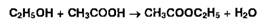
Общим способом испытаний ЛВ, содержащих в молекуле сложноэфирную, лактонную,
лактамную, амидную, имидную группы, является реакция, основанная на образовании
гидроксамовых кислот (гидроксамовая проба):
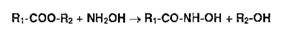
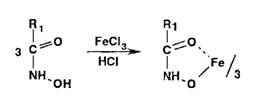
Гидроксамовые кислоты, взаимодействуя с ионами железа (III) или меди
(II), образуют окрашенные соли:
Реакции разложения амидов происходят при нагревании в растворах едких
щелочей с образованием аммиака или алкиламинов. имеющих характерный запах:
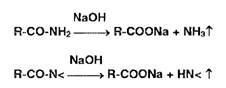
Первичные, вторичные и третичные амины в тех же условиях образуют
соответственно метиламин, диметиламин, триметиламин, например:
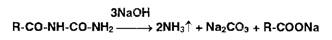
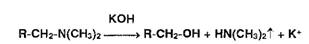
Указанные химические реакции используют для испытания подлинности солей
первичных аммониевых оснований, амидов ароматических и гетероциклических
кислот, производных уретанов.
Ациклические и циклические уреиды, алкилуреиды сульфокислот, производные
гуанидина и семикарбазона, имеющие в молекуле уреидную группу, гидролизуются в
щелочной среде с образованием аммиака. Например, уреиды:
К этой группе реакций можно отнести используемый в фармацевтическом
анализе пиролиз (термическое разложение в сухой пробирке). Используют пиролиз
для идентификации сульфаниламидов, производных бензодиазепина, пиридина и
других ЛВ, которые образуют плавы с различной окраской и выделяют газообразные
продукты с характерным запахом.
Реакции окисления-восстановления.
Процесс гидрирования осуществляют, как правило, водородом в момент
выделения (при взаимодействии металлического цинка с хлороводородной кислотой).
Эту реакцию используют для идентификации непредельных соединений, превращая их
в предельные, или для восстановления нитросоединений до аминопроизводных:
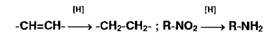
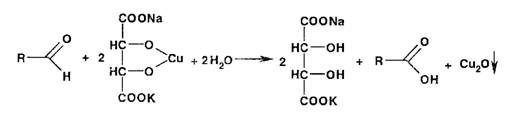
Широко используются в фармацевтическом анализе реакции окисления.
Первичные спирты идентифицируют, последовательно окисляя до альдегидов и
кислот, которые затем обнаруживают с помощью характерных реакций:

Так, например, восстановительные свойства альдегидов устанавливают с
помощью реакции образования «серебряного зеркала»:
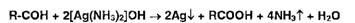
Этот же процесс лежит в основе взаимодействия реактива Несслера с альдегидами:
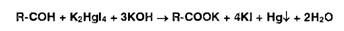
Реакция окисления альдегидов лежит в основе использования реактива
Фелинга, представляющего собой смесь отдельно приготавливаемых растворов
сульфата меди и калий-натриевой соли винной кислоты. В щелочной среде при
нагревании в присутствии альдегидов образуется красный осадок оксида меди (I).
Общая схема этой реакции:
2.3 Испытание на специфические примеси
К числу специфических примесей относят присущие только определенному ЛВ
исходные или промежуточные продукты синтеза, продукты разложения, сопутствующие
биологически активные вещества (алкалоиды, гормоны, белки, полисахариды и др.).
Они могут влиять на фармакологический эффект или оказывать токсическое
действие. Несмотря на многообразие химической структуры специфических примесей,
для их обнаружения используют несколько основных способов:
Использование специфической примеси в качестве эталона и фотометрическое
или нефелометрическое определение ее содержания по отношению к этому эталону.
Использование способов, основанных на избирательном взаимодействии
примеси с каким-либо реактивом и последующем ее определении.
Экстрагирование (отделение) примеси с помощью несмешивающихся
растворителей (вода-эфир), отгонка растворителя и гравиметрическое,
титриметрическое или фотометрическое ее определение.
Разделение и исследование примесей с помощью хроматографических методов
(ТСХ, бумажная хроматография).
Испытания на чистоту, основанные на использовании ВЭЖХ, ГЖХ и их
сочетания с другими методами (спектрофотометрия, масс-спектроскопия,
полярография).
Химические методы определения лекарственных веществ
Методики,
основанные на использовании химических методов, включены в ОФС и ФС (ФСП).
Наиболее широко для количественного определения ЛВ применяют титриметрическис
методы анализа. Значительно реже используют гравиметрический метод,
газометрический метод и элементный анализ[8]
<#"818075.files/image064.jpg">
При
прямом аргентометрическом титровании используют индикатор хромат калия (метод
Мора) или адсорбционные индикаторы (метод Фаянса), При обратном титровании
(метод Фольгарда) индикатором служат железоаммониевые квасцы, а избыток нитрата
серебра определяют роданометрическим (тиоцианатометрическим) методом[10]
<#"818075.files/image065.jpg">
Меркуриметрия
основана на образовании малодиссоциированных соединений ртути (II):
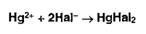
При
титровании хлоридов индикатором служит дифенилкарбазид или дифенилкарбазон:
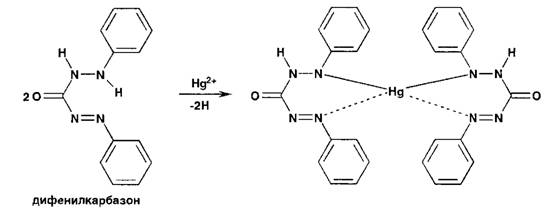
При
титровании йодидов конечную точку титрования устанавливают по выпадению
красного осадка йодида ртути (II) вследствие разрушения образующегося при
титровании тетрайодомеркурат-иона:
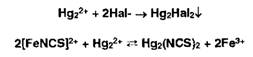
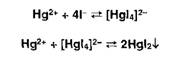
Меркурометрия
- метод определения галогенидов, образующих малорастворимые соединения с
катионами ртути (I). Титрантом служит раствор 0,1М раствор Hg2(NO3)2; индикатор
- тиоцианат железа, который обесцвечивается в точке эквивалентности вследствие
образования тиоцианата ртути (I) [12] <#"818075.files/image071.jpg">
Косвенная (заместительная) нейтрализация основана на реакции осаждения
ионами серебра органических оснований, содержащих в молекуле вторичную
аминогруппу или меркаптогруппу:
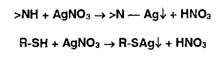
Выделившуюся кислоту титруют алкалиметрическим методом.
Оксимный метод также основан на косвенной нейтрализации эквивалентного
количества хлороводородной кислоты, выделившейся при взаимодействии гидроксиламина
гидрохлорида с кетопроизводными:
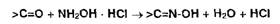
Этерификация в сочетании с алкалиметрией используется при определении
спиртов и фенолов. Их ацетилируют уксусным ангидридом, а его избыток
гидролизуют до уксусной кислоты, которую затем оттитровывают раствором
гидроксида натрия:
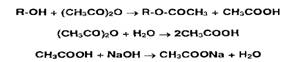
Параллельно выполняют контрольный опыт с тем же количеством уксусного
ангидрида.
Гидролиз сложных эфиров в сочетании с ацидиметрией. Сложные эфиры
гидролизуют титрованным раствором гидроксида натрия, избыток которого титруют
хлороводородной кислотой:
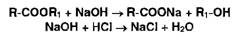
Пиролиз может быть выполнен в кислой среде:
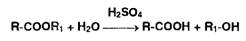
Образовавшуюся при гидролизе органическую кислоту можно извлечь эфиром и
оттитровать алкалиметрическим методом.
3.3 Титрование в смешанных растворителях
Используют в тех случаях, когда ЛВ плохо растворяются в воде или водные
растворы имеют слабо выраженные кислотные (щелочные) свойства. Они усиливаются
в присутствии этанола (ацетона).
Титрование в воде в присутствии несмешивающихся с ней эфира или
хлороформа используют для извлечения органического основания или кислоты из
водной фазы, что исключает их влияние на результаты титрования.
Титрование н среде неводных растворителей (неводное титрование)
Метод
позволяет количественно определить органические вещества, проявляющие в водной
среде очень слабые основные или кислотные свойства. В качестве титрантов
используют растворы сильных кислот или сильных оснований[16]
<#"818075.files/image077.jpg">
Растворение
основания в ледяной СНзСООН:
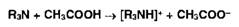
Взаимодействие
ацетоний- и ацетат-ионов:
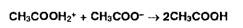
Взаимодействие
протонированного амина с хлорат-ионом:
Очень
слабые органические основания (рК> 12) необходимо титровать хлорной кислотой
в среде уксусного ангидрида ЈУА), т.к. он более активно (чем ледяная уксусная
кислота) усиливает основные свойства аминов.
Взаимодействие
НСlO4 с УА:
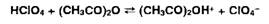
Растворение
амина в УА:
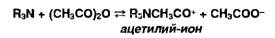
Взаимодействие
кислоты с основанием:
.
Взаимодействие ацетилий-иона с хлорат-ионом:
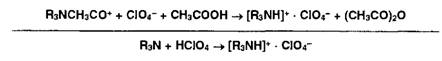
Соли
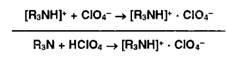
органических оснований с галогеноводородными кислотами (ЯзЫ • НХ) проявляют
кислотные свойства даже в неводной среде. Поэтому их титруют в присутствии
ацетата ртути (II), который нейтрализует галогенпроизводную кислоту.
Малодиссоциированные галогениды ртути (HgX2) и (СНзСОО) не мешают определению.
Образующийся ацетат органического основания оттитровывают хлорной кислотой:
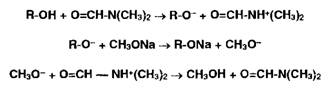
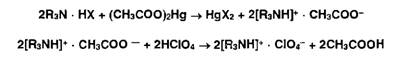
Неводное
титрование галогеноводородов может быть выполнено без добавления ацетата ртути,
если в качестве протогенных растворителей использовать безводную муравьиную
кислоту в присутствии уксусного ангидрида.
Неводное
титрование органических веществ, проявляющих кислотные свойства (фенолы,
барбитураты, карбоновые кислоты, сульфаниламиды и др.), выполняют, используя в
качестве растворителя диметилформамид или его смесь с бензолом. Титрантом
служит раствор гидроксида натрия в смеси метанола и бензола или раствор
метилата натрия. В качестве индикатора используют тимоловый синий.
Суммарно
процесс нейтрализации фенолов (енолов) можно представить так:
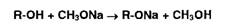
3.4 Окислительно-восстановительное титрование
Йодометрия
- метод, основанный на окислительных свойствах йода и восстановительных
свойствах йодид-ионов[16] <#"818075.files/image087.jpg">
Титрант
- раствор йода (индикатор - крахмал) используют для прямого титрования
неорганических и органических веществ, способных окисляться или образовывать с
йодом продукты присоединения или замещения. Используют также обратное
йодометрическое титрование. При этом избыток йода титруют 0,1 М раствором
тиосульфата натрия:
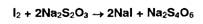
Восстановительные свойства йодида калия используют для количественного
определения веществ, обладающих окислительными свойствами. Выделившееся
эквивалентное количество йода оттитровывают тиосульфатом натрия.
Используют также сочетание реакции замещения (получение нерастворимых в
воде моно-, ди- и трийодпроизводных) и обратной йодометрии. Йодпроизводные
отфильтровывают, а в фильтрате определяют избыток титрованного раствора йода.
Аналогичным образом используют реакцию образования полийодидов и органических
оснований. При выполнении определения необходимо учитывать влияние рН среды.
Йодхлорометрия - отличается от йодометрии использованием в качестве
титранта не раствора йода, а более устойчивого раствора йодмонохлорида.
Аналогично йоду йодмонохлорид образует йодпроизводные органических оснований.
Избыток титранта устанавливают йодометрически:
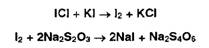
Йодатометрия основана на окислении органических соединений йодатом калия.
Избыток титранта устанавливают йодометрически:
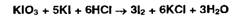
Бромид-броматометрия основана на использовании окислительных свойств или
реакции замещения моно-, ди- или трибромпроизводных) за счет образующегося свободного
брома:
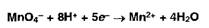
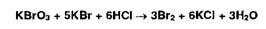
Индикаторами при прямом титровании служат азокрасители, которые
обесцвечиваются бромом. В случае обратного титрования эквивалентную точку
устанавливают йодометрически по избытку титранта (бромата калия).
Перманганатометрия основана на использовании окислительных свойств
титранта - перманганата калия в кислой среде:
Индикатором при прямом титровании служит сам титрант (появляется
фиолетовое окрашивание), а при обратном титровании избыток титранта
устанавливают йодометрическим методом.
Цериметрия основана на использовании окислительных свойств титранта -
соли церия (IV), который в кислой среде восстанавливается до церия (III):
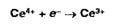
Индикатором служат дифениламин, о-фенантролин, а при обратном титровании
избыток титранта устанавливают йодометрически:
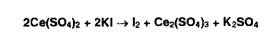
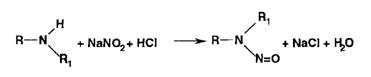
.5 Нитритометрия
Метод количественного определения первичных и вторичных ароматических
аминов, основанный на использовании титранта - раствора нитрита натрия в
присутствии бромида натрия:
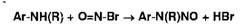
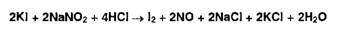

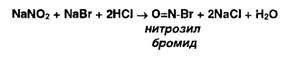
В этих условиях с первичными ароматическими аминами образуются
диазосоединения (в кислой среде):
Вторичные ароматические амины в тех же условиях образуют
Ы-нитрозосоединения:
Эквивалентную точку устанавливают различными путями: потенциометрически,
с помощью указанных в ФС внутренних индикаторов (тропеолин 00, нейтральный
красный), с внешним индикатором (йодкрахмальная бумага). Титрование с
йодкрахмальной бумагой ведут до тех пор, пока капля титруемого раствора, взятая
через 1 мин после прибавления титранта, не вызовет тотчас же посинение бумаги:
На результаты определения влияют температура (смесь охлаждают до 5-15°С),
концентрация хлороводородной кислоты, природа растворителя.
При
использовании внутренних индикаторов наблюдают изменение их окраски в
эквивалентной точке[5] <#"818075.files/image099.jpg">
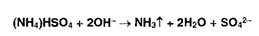
Взаимодействие в приемнике с борной кислотой с образованием
тетрагидроксибората аммония:
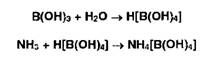
Титрование отгона ОДМ раствором хлороводородной кислоты:

Параллельно выполняют контрольный опыт (без анализируемого вещества) для
повышения точности результатов анализа.
Для
определения веществ, содержащих в молекуле амидную группу, используют
упрощенный вариант метода Кьельдаля, исключающий стадию минерализации. Методика
сводится к гидролизу амида в колбе Кьельдаля 30% раствором гидроксида натрия,
отгонке выделяющегося аммиака или амина в приемник и титровании отгона 0,1 М
хлороводородной кислотой[14] <#"818075.files/image103.jpg">
Для окисления образовавшихся йодидов до йодатов в колбу вносят раствор
ацетата брома до появления желтого окрашивания:
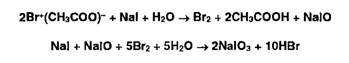
Для удаления избытка брома добавляют концентрированную муравьиную кислоту
до обесцвечивания раствора:
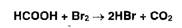
Выдерживают
5 мин в темном месте после добавления йодида калия и раствора серной кислоты, а
затем титруют выделившийся йод, содержание которого эквивалентно его количеству
в испытуемом ЛВ[17] <#"818075.files/image106.jpg">
3.8 Физические и физико-химические методы анализа
Физические и физико-химические методы могут быть классифицированы на
следующие группы: оптические методы, методы, основанные на поглощении
электромагнитного излучения, методы, основанные на испускании излучения,
методы, основанные на использовании магнитного поля, электрохимические методы,
термические методы, методы разделения.
Физико-химические
методы основаны на использовании зависимости физических свойств от химического
состава веществ. В большинстве случаев физико-химические методы отличаются
быстротой выполнения, избирательностью, высокой чувствительностью, возможностью
унификации и автоматизации. Поэтому данная группа методов приобретает все
большее значение для объективной оценки качества ЛС, в т.ч. для испытания на
подлинность, испытания на чистоту и для количественного определения[18]
<#"818075.files/image107.jpg">
где а - измеренный угол вращения, в градусах; / - длина рабочего слоя
кюветы, в дециметрах; С - концентрация раствора вещества (г/100 мл).
Количественно определяют (в %) содержание оптически активного вещества в
растворе по формуле:
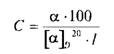
Величину
а измеряют на поляриметрах с точностью до ±0,02°[21]
<#"818075.files/image109.jpg">
где/о - интенсивность излучения, падающего на вещество; /- интенсивность
излучения, прошедшего через вещество; А - величина оптической плотности; х -
показатель поглощения данного вещества; С - концентрация раствора
анализируемого вещества, г; I - длина рабочего слоя кюветы, см.
На основании этого закона содержание вещества в растворе определяют по
формуле:
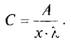
В случае несоответствия закону Бугера-Ламберта-Бера вначале с помощью
стандартного раствора устанавливают зависимость оптической плотности от
концентрации, а затем строят калибровочный график, с помощью которого выполняют
расчеты.
Спектрофотометрия в УФ- и видимой областях - один из наиболее широко
используемых физико-химических методов в фармацевтическом анализе.
Анализируемые ЛВ должны иметь в структуре молекулы хромофорные группы
(сопряженные связи, ароматическое ядро и др.), обусловливающие различные электронные
переходы в молекулах и поглощение электромагнитного излучения.
Кривая зависимости интенсивности светопоглощения от длины волны (нм)
называется спектром поглощения вещества и является его специфической
характеристикой. Измерение спектров поглощения растворов анализируемых веществ
в ультрафиолетовой (190-380 нм) и видимой (380-780 нм) областях производят с
помощью спектрофотометров различных марок (СФ-26, СФ-46 и др.). В качестве
растворителей используют свободные от примесей воду, растворы кислот и щелочей,
этанол, хлороформ и другие органические растворители.
Спектрофотометрической константой является удельный показатель поглощения
( ), который рассчитывают по формуле:
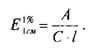
Удельный показатель поглощения представляет собой величину оптической
плотности раствора, содержащего 1,0 г вещества в 100 мл раствора, измеренную в
кювете с рабочей длиной 1 см. Установив по стандартному образцу величину и преобразовав эту формулу, можно
рассчитать концентрацию анализируемого вещества с относительной погрешностью до
±2%.
Идентификацию ЛВ можно провести по Д1^, характеру спектральных
кривых в различных растворителях, положению максимума и минимума
светопоглощения или их отношению (при различных длинах волн). Для
количественного спектрофотометрического анализа важен выбор аналитической
полосы поглощения. Последняя должна быть свободна от наложения полос поглощения
других компонентов смеси и иметь достаточно высокий удельный показатель
поглощения анализируемого вещества.
Фотоколориметрия отличается от спектрофотометрического анализа тем, что
анализируемое вещество с помощью какого-либо реагента переводят (количественно)
в окрашенное соединение. Вначале получают окрашенные растворы, используя
растворы стандартных образцов (ГСО или PCO). Измерение оптической плотности производят на фотоколориметрах. Затем
строят калибровочный график зависимости интенсивности поглощения окрашенных
растворов от концентрации, по которому рассчитывают содержание JTB в испытуемых образцах JIB или ЛФ.
Метод дифференциальной спектрофотометрии и фотоколориметрии основан на
измерении светопоглощения анализируемого раствора относительно раствора
сравнения, содержащего определенное количество стандартного образца испытуемого
вещества или его заменителя. Такой прием приводит к изменению рабочей области
шкалы прибора и снижению относительной погрешности определения до ±0,5-1%, т.е.
сопоставимой с титриметрическими методами.
Производная УФ-спектрофотометрия является одним из вариантов
дифференциальной спектрофотометрии. Если в дифференциальной спектрофотометрии
используют разность оптических плотностей при одной и той же длине волны, то в
производной - при двух длинах волн, разделенных небольшим интервалом. Этот
вариант основан на выделении индивидуальных полос из УФ-спектра, который
представляет собой сумму налагающихся полос поглощения или полос, не имеющих
четко выраженного максимума. При этом на спектральных кривых в координатах
производная-длина волны появляются полосы с отчетливо выраженными максимумами и
минимумами. Благодаря этому можно идентифицировать сходные по химической
структуре вещества, повысить избирательность анализа и выполнять количественное
определение двух-, трехкомпонентных смесей более экономично и эффективно, чем
титриметрическими методами.
Одним из вариантов дифференциальной спектрофотометрии является АЕ-метод.
Он основан на превращении одного из веществ, входящих в состав анализируемой
пробы, в таутомерную (или иную) форму, отличающуюся по характеру и
интенсивности светопоглощения. Затем измеряют светопоглощение раствора одной
таутомерной формы по отношению к другой, т.е. используют в качестве стандарта
раствор анализируемого вещества.
Спектрофотометрии в ИК-области. Природа полос поглощения в ИК области
связана с колебательными переходами и изменением колебательных состояний ядер,
входящих в молекулу поглощающего вещества. Поэтому поглощением в ИК-области
обладают молекулы, дипольные моменты которых изменяются при возбуждении
колебательных движений ядер. Область применения ИК-спектроскопии аналогична, но
более широка, чем у УФ-метода. ПК-спектр однозначно характеризует всю структуру
молекулы, включая незначительные ее изменения. Важные преимущества
ИК-спектроскопии - - высокая специфичность, объективность полученных
результатов, возможность анализа веществ в кристаллическом состоянии. Для
измерения ИК-спектров на однолучевых или двулучевых ИК-спектрофотометрах
используют взвеси веществ в вазелиновом масле или помещают анализируемое
вещество между пластинами из бромида калия. Каждый ИК-спектр представляет собой
серию полос поглощения, максимумы которых определяются волновым числом,
измеряемым в см"1, и
определенной интенсивностью. Для анализа ЛВ обычно используют спектральную
область от 4000 до 400 см-1.
ГФ XI рекомендует два способа установления подлинности по ИК-спектрам.
Один из них основан на сравнении зарегистрированных в идентичных условиях
ИК-спектров испытуемого ЛВ и его стандартного образца. Второй способ
заключается в сравнении ИК-спекгра испытуемого ЛВ с его стандартным спектром,
прилагаемым к ФС и зарегистрированным в соответствии с указанными в ней
требованиями.
Фототурбидиметрия - метод, основанный на измерении интенсивности света,
поглощенного тонкодисперсной суспензией, и фотонефелометрия - метод, основанный
на измерении света, рассеянного взвешенными частицами анализируемого вещества.
Оба метода применяют в фармацевтическом анализе для количественного определения
ЛВ, образующих с различными реактивами тонкие суспензии. Предварительно
устанавливают зависимость между интенсивностью поглощения (рассеяния) света и
концентрацией вещества в анализируемом растворе. Способы расчета аналогичны
фотометрическим методам.
3.11 Методы, основанные на испускании излучения
Атомно-абсорбционная спектрометрия основана на поглощении атомами
излучения с частотой, равной частоте резонансного перехода. Излучение исходит
от лампы с полым катодом, проходит через пламя, в котором распыляется проба,
пропускается через щель монохроматора, и выделенная из спектра резонансная
линия определяемого элемента измеряется фотоэлектрическим способом. Затем
устанавливается зависимость между ослаблением интенсивности излучения источника
света и концентрацией испытуемого вещества.
Флуоресцентные методы основаны на способности веществ флуоресцировать в
УФ-свете, обусловленной либо химической структурой самих органических веществ,
либо продуктов их диссоциации, сольволиза, других превращений. Способностью
флуоресцировать обладают обычно органические соединения с симметричной
структурой молекул, в которых имеются сопряженные связи (нитро-, нитрозо-,
азо-, амидные, карбонильные или карбоксильные группы).
Флуориметрия используется не только для установления подлинности, но и
определения малых количеств веществ, т.к. интенсивность флуоресценции имеет
линейную зависимость от концентрации. Линейная зависимость сохраняется при
постоянстве квантового выхода и интенсивности возбуждающего света для низких
концентраций веществ. При высоких концентрациях эта зависимость нарушается.
Идентификацию проводят по цвету излучаемого света, специфичного для флуоресцирующих
веществ. Спектр дает широкие полосы излучения (от 100 до 200 нм). Метод
отличается очень высокой чувствительностью. Количественное определение
выполняют на спектрофлуориметрах. Расчет концентрации производят с помощью
калибровочного графика или шкалы стандартных растворов, аналогично
фотометрическим методам.
3.12 Методы, основанные на использовании магнитного поля
Спектроскопия ядерного магнитного резонанса (ЯМР) - метод, основанный на
регистрации индуцированных радиочастотным полем переходов между ядерными
магнитными энергетическими уровнями молекул вещества, помещенного в магнитное
поле. Метод позволяет изучать магнитные переходы ядер со спиновыми квантовыми
числами больше нуля (ядра 'Н, 13С, 19Р, 31Р).
Совокупность сигналов переходов между энергетическими уровнями ядер молекул
составляет спектр ЯМР. Каждый спектр ЯМР регистрируется для одного типа ядер и
специфичен для каждого вещества. Чаще всего используют спектроскопию на
протонах (ПМР) и ЯМР 13С.
Спектры регистрируют при помощи ЯМР-спектрометров. Каждый спектр является
отражением числа ядер, порядка их связи и геометрии расположения ядер в
молекуле. Спектр представляет собой совокупность пиков с различной шириной,
площадью и интенсивностью сигналов. По характеру протонных сигналов можно
сделать заключение о наличии в молекуле тех или иных групп атомов. Величина
химического сдвига имеет порядок 10~6 или млн~!
(миллионная доля). Она зависит от наличия в молекуле тех или иных групп,
например, химический сдвиг у ароматических протонов находится в интервале 7-9
млн4, альдегидных - 9-10 млн-1 и т.д.
Метод ЯМР- и ПМР-спектроскопии используют для объективной идентификации
органических Л В и для количественного определения относительного содержания
вещества или примеси. Подлинность может быть подтверждена либо пут ем сравнения
со стандартным образцом, либо по наиболее характерным сигналам спектра, либо по
полному набору спектральных параметров.
Масс-спектроскопия - метод, позволяющий определить массу ионов,
ионизированных молекул или фрагментов молекул по отклонению в магнитных и
электрических полях или по кинетической энергии. Ионизация молекул происходит в
результате воздействия пучка электронов. Интенсивность пика в масс-спектре
пропорциональна числу образовавшихся ионов данного вида. Состав и массовые
числа характеристических ионов позволяют установить принадлежность исследуемого
соединения к определенному классу веществ, осуществить его идентификацию.
Масс-спектроскопия отличается большой информативностью и очень высокой
чувствительностью.
3.13 Электрохимические методы
Потенциометрия - метод, основанный на измерении равновесных потенциалов,
возникающих на границе между испытуемым раствором и погруженным в него
электродом. В фармацевтическом анализе наиболее широко используют
потенциометрическое титрование. Оно основано на установлении эквивалентного
объема титранта путем измерения ЭДС, возникающей при титровании за счет
разности потенциалов индикаторного электрода и электрода сравнения, погруженных
в анализируемый раствор. Метод потенциометрии используют для определения рН
(рН-метрия) и установления концентрации отдельных ионов.
Преимущества потенциометрического метода определения по сравнению с
индикаторным состоят в возможности титрования окрашенных, коллоидных, мутных
растворов, смеси нескольких компонентов в водных и неводных средах. Метод
применим в различных видах титриметрии, основанных на реакциях нейтрализации,
осаждения, окисления-восстановления. Электродом сравнения служит каломельный
электрод, индикаторным - стеклянный. Измерение ЭДС между индикаторным
электродом и электродом сравнения производят с помощью высокоомных
потенциометров. Титр ант прибавляют равными объемами, причем вблизи точки
эквивалентности по 0,1-0,05 мл. Около точки эквивалентности изменение ЭДС
происходит наиболее сильно. Результаты титрования представляют либо графически,
обозначая точку эквивалентности на кривой титрования, либо расчетным методом.
Ионометрия основана на использовании зависимости между ЭДС гальванической
цепи с ионселективным электродом и концентрации анализируемого иона в
электродной ячейке цепи. Метод отличается высокой чувствительностью,
экспрессностыо, хорошей воспроизводимостью, несложным оборудованием, доступными
реагентами. Широко применяют для определения ионов натрия, калия, кальция,
галогенидов в многокомпонентных смесях, в т. ч. ЛФ.
Полярография - метод, основанный на измерении силы тока, возникающего на
микроэлектроде, при электровосстановлении анализируемого вещества в растворе.
Растворителем служит вода или органические и смешанные растворители. Электролиз
проводят в полярографической ячейке, состоящей из электролизера и двух
микроэлектродов: ртутного капающего и внешнего насыщенного каломельного. При
соблюдении идентичных условий измерений для идентификации используют величину
потенциала полуволны, а для количественного определения - высоту волны
(измерение предельного диффузного тока). Количественный анализ выполняют
методами калибровочных кривых с использованием стандартных растворов и методом
добавок.
3.14 Термические методы анализа
Термические методы основаны на изменениях, которые вызывает нагревание
вещества в зависимости от их природы, температуры, условий нагревания. При этом
происходят полиморфные превращения, удаление сорбционной и кристаллизационной
воды, сублимация, плавление, кипение, разложение. Разложение веществ
сопровождается такими химическими превращениями, как структурирование,
термическая, окислительная или гидролитическая деструкция. Термическая
деструкция сопровождается поглощением или выделением теплоты, а также образованием
газообразных продуктов. Эти процессы лежат в основе термографии - оценке
термической стабильности по температурам термоэффекта, связанного с деструкцией
вещества.
Термический анализ основан на точной (до 0,1°С) регистрации равновесного
состояния между кристаллической и жид- кон фазами анализируемого вещества при
медленном нагревании или охлаждении. Лучшей воспроизводимостью отличается
дифференциальный термический анализ, основанный на регистрации изменения
энергии в зависимости от температуры. Одной из модификаций этого метода
является дериватография, сущность которой состоит в регистрации изменений
температуры образца (термических характеристик), вызванных дегидратацией,
плавлением, термической деструкцией и другими процессами, происходящими при нагревании.
Особенно широкие возможности создают термические методы при исследовании
стабильности ЛВ.
3.15 Методы разделения
В фармацевтическом анализе для разделения смесей ЛВ используют
экстракцию, хроматографические методы и электрофорез.
Экстракция - метод разделения, основанный на использовании экстрагента,
не смешивающегося с исходной фазой и легко отделяющегося от нее и от
экстрагируемых компонентов. В зависимости от исходной фазы различают экстракцию
из твердого вещества и экстракцию из раствора (жидкостную). По количеству
операций экстракция может быть однократной и многократной. В фармацевтическом
анализе экстракцию широко используют для разделения компонентов, входящих в
состав ЛФ. Кроме того, ее сочетают с фотометрией в экстракционно-фотометрическом
методе, основанном на образовании испытуемым веществом цветных продуктов
реакции, способных экстрагироваться каким-либо органическим растворителем.
Затем в органической фазе выполняют фотометрическое определение
экстрагированного продукта.
Хроматографические методы разделения веществ основаны на их распределении
между двумя фазами: подвижной и неподвижной. Подвижная фаза - жидкость или газ;
неподвижная - твердое вещество или жидкость, адсорбированная на твердом
носителе. Относительная скорость перемещения частиц вдоль пути разделения
зависит от их взаимодействия с неподвижной фазой. Поэтому каждое вещество
проходит на носителе определенный путь. Отношение пути перемещения вещества к
пути перемещения растворителя есть величина постоянная, обозначаемая Rf. Она является константой для данных
условий разделения и используется для идентификации ЛВ.
Хроматография на бумаге. Носителем неподвижной фазы (например, воды)
служит специальная хроматографическая бумага. Распределение происходит между
водой, находящейся на поверхности бумаги, и подвижной фазой, которая
представляет собой систему из нескольких растворителей. Испытание выполняют
согласно требованиям ГФ XI (в. 1, с. 98) или ФС (ФСП). Для подтверждения
подлинности одновременно хроматографируют испытуемое вещество и стандартный
образец. Если они идентичны, то пятна на хроматограммах будут иметь одинаковый
вид и равные значения Rf. Чтобы
исключить влияние на ошибку определения условий хроматографирования, пользуются
более объективной константой Rs, которая
представляет собой отношение величин Rf испытуемого и стандартного образцов. Хроматографию используют при
испытании на чистоту. О наличии примесей судят по появлению дополнительных
пятен на хромато грамме. Анализируемое вещество и примесь обычно имеют разные значения
Rf.
Количественное содержание вещества можно определить непосредственно на
хроматограмме, используя планиметрический, денситометрический, люминесцентный и
другие методы. Используют также способы, основанные на элюировании
анализируемого вещества из вырезанного и измельченного участка хроматограммы с
соответствующим пятном. В элюате содержание испытуемого вещества определяют
фотометрическим или электрохимическим методом.
Хроматография в тонком слое сорбента (ТСХ) отличается от хроматографии на
бумаге тем, что процесс хромато графирования происходит на носителе (сорбенте),
нанесенном тонким слоем на инертную поверхность. Твердый сорбент может быть
закрепленным или незакрепленным на этой поверхности. Сорбентом служит
силикагель или оксид алюминия. Для закрепления добавляют небольшие количества
крахмала или сульфата кальция. Используют также пластинки промышленного
изготовления типа «Силуфол УФ-254», «Сорбфил» и др.
Преимуществами ТСХ является простота приемов и оборудования, более
высокая чувствительность, чем у бумажной хроматографии, устойчивость пластинок
к температурным и химическим воздействиям, значительно большие возможности
процессов разделения, детектирования, элюирования, меньшая продолжительность
выполнения испытания. Все это создает широкие возможности в использовании ТСХ
для выполнения испытаний на подлинность, чистоту, для количественного
определения ЛВ в ЛФ.
Двумерное хроматографирование отличается повторным (после высушивания)
пропусканием той же или иной подвижной фазы, но в перпендикулярном по отношению
к первоначальному направлении. При этом используют квадратные пластины или
листы бумаги.
В фармацевтическом анализе широко применяют сочетание ТСХ с
физико-химическими методами анализа. Такие комбинированные методы, как
хромато-спектрофотометрия, хромато-флуориметрия, хромато-масс-спектроскопия
особенно эффективны в анализе ЛРС и препаратов, содержащих большое число
сопутствующих компонентов.
Газожидкостная хроматография (ГЖХ) основана на распределении компонентов
смеси между газовой и жидкой или твердой фазами. Распределение происходит в
результате многократных актов сорбции и десорбции анализируемых веществ,
которые вводятся в поток газа-носителя, испаряются и в парообразном состоянии
проходят через колонку с сорбентом. Поэтому метод ГЖХ применим для анализа
летучих веществ или веществ, которые могут быть переведены в газообразное
состояние. Разделенные вещества элюируются из колонки потоком газа-носителя,
регистрируются детектором и фиксируются на хроматограмме в виде пиков, по
которым можно идентифицировать или определять содержание каждого компонента
смеси.
Газовый хроматограф включает в себя систему измерения и регулирования
скорости потока газа-носителя, систему ввода пробы испытуемого образца,
газохроматографическую колонку, систему термостатирования и контроля
температуры в различных узлах прибора и систему детектирования, регистрации и
обработки информации, полученной на приборе.
Подлинность JIB методом ГЖХ
можно подтвердить либо с помощью свидетелей, либо методом относительных удерживаний.
В первом случае доказательством идентичности служит совпадение времени
удерживания вещества-свидетеля и одного из компонентов смеси JIB при хроматографировании каждого в
отдельности в одинаковых условиях. Во втором случае вещество-свидетель добавляют
к пробе, затем анализируют по рекомендуемой методике. Рассчитывают по формуле
величину относительного удерживания, которая является постоянной для JTB в конкретных условиях. Количественный
анализ выполняют в тех же условиях, используя для расчетов такие параметры, как
площадь или высота пиков ЛВ, Площадь пиков устанавливают на хроматограмме с
помощью планиметра, интегратора или умножением высоты пика на его полуширину.
Высокоэффективная жидкостная хроматография (ВЭЖХ) отличается от ГЖХ тем,
что подвижной фазой служит не газ, а жидкость, причем она проходит через
колонку, наполненную сорбентом, с большой скоростью за счет значительного
давления. Поэтому ВЭЖХ позволяет разделять многокомпонентные смеси на
индивидуальные вещества высокой степени чистоты. ВЭЖХ отличается высокой
чувствительностью (до Ю-6 г). На разделение 10-15 компонентов
затрачивается 20-30 мин.
Жидкостный хроматограф включает такие узлы, как дозатор, насос высокого
давления, высокоэффективную колонку, детектор с регистрирующим устройством. Колонки
изготавливают из нержавеющей стали, они имеют длину 10-25 см, внутренний
диаметр 0,3-0,8 см и плотно набиваются адсорбентом с размером частиц 5-10 мкм.
В качестве элюента используют различные углеводороды в сочетании с этанолом.
Детектором обычно служит спектрофотометр с переменной длиной волны (190-900
нм), но существуют также флуориметрические, электрохимические и другие
детекторы.
Подлинность испытуемых JIB подтверждают
по времени выхода каждого компонента смеси из колонки, которое будет стабильно
при одинаковых условиях проведения эксперимента. Количественное содержание
рассчитывается по площади пика, которая пропорциональна количеству ЛВ в пробе.
Электрофорез - метод анализа, основанный на способности заряженных частиц
к перемещению в электрическом поле. Скорость перемещения ионов зависит от
напряженности электрического поля, величины заряда, размера частицы, вязкости, pH среды, температуры и других факторов.
Электрофоретическая подвижность - величина, характерная для испытуемого
вещества. Различают абсолютную (измеряемую в сантиметрах в секунду) и
относительную электрофоретическую подвижность (отношение к подвижности
стандартного образца). По технике выполнения и аналитическим возможностям
электрофорез на бумаге и в тонких слоях сорбента сходен с ТСХ. Он позволяет
разделять и идентифицировать компоненты различных смесей.
Глава 4.
.1 Валидация методов анализа
Валидация - это подтверждение обоснованности выбора метода анализа для
установления норм качества Л С по каждому разделу НД. Она проводится при
подготовке проектов НД на новые ЛС или при последующем пересмотре НД. Валидации
подвергаются аналитические методы, используемые для идентификации ЛВ,
установления содержания в нем различных примесей, количественного определения
индивидуальных ЛВ и содержания их в ЛФ, определения вспомогательных веществ и
консервантов.
Валидация метода анализа предполагает оценку его специфичности, линейной
зависимости результатов испытаний, аналитической области методики,
правильности, воспроизводимости результатов, предела обнаружения.
Ревалидация необходима в тех случаях, когда произошли изменения в синтезе
ЛВ, в составе ЛС, в аналитической методике. Параметры аналитического метода,
устанавливаемые при его валидации и ревалидации, рассчитываются в соответствии
с существующими правилами статистической обработки результатов анализа.
Линейная зависимость аналитических сигналов от концентрации ЛВ
устанавливается графически. Оценивается она на основании не менее 5 испытаний,
выполненных с помощью используемой аналитической методики. Параметрами,
подтверждающими линейную зависимость, являются коэффициент регрессии, угол
наклона линии регрессии и остаточная сумма площадей.
Аналитическая область методики охватывает интервал между верхним и нижним
пределами содержания испытуемого вещества, в котором соблюдается линейная
зависимость. При этом данная методика должна обеспечивать определение с
требуемыми воспроизводимостью и точностью. Аналитическая область выражается в
тех же единицах, что и результаты испытаний с помощью данной методики
(проценты, миллионные доли).
Правильность (точность) аналитического метода характеризует близость
результатов, полученных с помощью данной методики, к истинному значению. При
установлении этого параметра для количественного определения субстанций,
примесей могут быть использованы стандартные образцы, другие независимые
методики, метод добавок.
Правильность оценивается не менее чем на трех повторениях определения для
трех аналитических концентраций в пределах аналитической области.
Воспроизводимость аналитического метода отражает степень совпадений
результатов отдельных испытаний при многократном использовании методики. Она
устанавливается при количественном определении не менее 9 аликвот образца и
выражается в результате статистической обработки по величинам стандартного
отклонения, коэффициента вариации и доверительного интервала.
Межлабораторная воспроизводимость аналитического метода показывает
степень воспроизводимости результатов испытаний, выполненных по разработанной
методике в различных лабораториях на соответствующем оборудовании, разными
аналитиками, в разное время.
Предел обнаружения - минимальное содержание анализируемого вещества,
которое можно обнаружить с помощью данной методики (выражается в процентах или
миллионных долях). Устанавливается для химических методов визуально. Для
физико-химических методов устанавливается по минимальной концентрации
испытуемого вещества, которое может быть достоверно обнаружено или
рассчитывается по величине стандартного отклонения и углу наклона калибровочной
кривой.
Предел количественного определения - минимальное содержание (в процентах)
анализируемого вещества, которое может быть определено с достаточной точностью
и воспроизводимостью. Устанавливается для любых методов визуально или расчетным
путем подобно установлению предела обнаружения.
Пригодность системы - интегральная часть аналитических методик,
подтверждающая надежность анализа в заданных условиях его проведения.
лекарственный титрование излучение экстракция
Выводы
Фармацевтическая химия - наука, которая, базируясь на общих законах
химических наук, исследует способы получения, строение, физические и химические
свойства лекарственных веществ, взаимосвязь между их химической структурой и
действием на организм; методы контроля качества лекарств и изменения,
происходящие при их хранении.
Основными методами исследования лекарственных веществ в фармацевтической
химии являются анализ и синтез - диалектически тесно связанные между собой
процессы, взаимно дополняющие друг друга. Анализ и синтез - мощные средства
познания сущности явлений, происходящих в природе.
Задачи, стоящие перед фармацевтической химией, решаются с помощью
классических физических, химических и физико-химических методов, которые
используются как для синтеза, так и для анализа лекарственных веществ.
Чтобы познать фармацевтическую химию, будущий провизор должен иметь
глубокие знания в области общетеоретических химических и медико-биологических
дисциплин, физики, математики. Необходимы также прочные знания в области
философии, ибо фармацевтическая химия, как и другие химические науки,
занимается изучением химической формы движения материи.
Фармацевтическая химия занимает центральное место среди других
специальных фармацевтических дисциплин - фармакогнозии, технологии лекарств,
фармакологии, организации и экономики фармации, токсикологической химии и
является своеобразным связующим звеном между ними.
Вместе с тем фармацевтическая химия занимает промежуточное положение
между комплексом медико-биологических и химических наук. Объектом применения
лекарств является организм больного человека. Исследованием процессов,
происходящих в организме больного человека, и его лечением занимаются
специалисты, работающие в области клинических медицинских наук (терапия, хирургия,
акушерство и гинекология и т.д.), а также теоретических медицинских дисциплин:
анатомии, физиологии и др. Многообразие применяемых в медицине лекарств требует
совместной работы врача и провизора при лечении больного.
Являясь прикладной наукой, фармацевтическая химия базируется на теории и
законах таких химических наук, как неорганическая, органическая, аналитическая,
физическая, коллоидная химия. В тесной связи с неорганической и органической
химией фармацевтическая химия занимается исследованием способов синтеза
лекарственных веществ. Поскольку их действие на организм зависит как от
химической структуры, так и от физико-химических свойств, фармацевтическая
химия использует законы физической химии.
При разработке способов контроля качества лекарственных препаратов и
лекарственных форм в фармацевтической химии применяют методы аналитической
химии. Однако фармацевтический анализ имеет свои специфические особенности и
включает три обязательных этапа: установление подлинности препарата, контроль
его чистоты (установление допустимых пределов примесей) и количественное
определение лекарственного вещества.
Развитие фармацевтической химии невозможно и без широкого использования
законов таких точных наук, как физика и математика, так как без них нельзя
познать физические методы исследования лекарственных веществ и различные
способы расчета, применяемые в фармацевтическом анализе.
В фармацевтическом анализе используются разнообразные методы
исследования: физические, физико-химические, химические, биологические. Применение
физических и физико-химических методов требует соответствующих приборов и
инструментов, поэтому данные методы называют также приборными, или
инструментальными.
Использование физических методов основано на измерении физических
констант, например, прозрачности или степени мутности, цветности, влажности,
температуры плавления, затвердевания и кипения и др.
С помощью физико-химических методов измеряют физические константы
анализируемой системы, которые изменяются в результате химических реакций. К
этой группе методов относятся оптические, электрохимические,
хроматографические.
Химические методы анализа основаны на выполнении химических реакций.
Биологический контроль лекарственных веществ осуществляют на животных,
отдельных изолированных органах, группах клеток, на определенных штаммах
микроорганизмов. Устанавливают силу фармакологического эффекта или токсичность.
Методики, используемые в фармацевтическом анализе, должны быть
чувствительными, специфическими, избирательными, быстрыми и пригодными для
экспресс-анализа в условиях аптеки.
Список используемой литературы
1. Аксенова Э.М. Руководство к лабораторным занятиям по
фармацевтической химии. - М.: «Медицина», 1987г. - 381с.
2. Ануфриева Р.М. «Система капиллярного
электрофореза»/Ануфриева Р.М., Бессчетнова Т.Ю., Каменцев Я.С. и др. - СПб:
Изд-во «Петрополис», 2001. - 768с.
. Арзамасцев А.П. Фармакопейный анализ. - М.:
«Медицина», 1971г. - 239 с.
. Арианова Е.А. Подходы к определению качества и
безопасности лекарственных средств на основе низкомолекулярных гепаринов / Е.А.
Арианова, М.Н. Богачук, О.И. Передеряев // Вопросы биологической, медицинской и
фармацевтической химии. - 2012. - № 2. - С. 3-8.
. Бендрышева С.Н. Возможности термолинзового
детектирования в капиллярном электрофорезе: Автореф. дис… канд. хим. наук. -
М., 2007. - 26 с.
. Богачук М.Н. Определение водорастворимых витаминов
в поливитаминных препаратах методом капиллярного зонального электрофореза /
М.Н. Богачук, О.И. Передеряев, Г.В. Раменская // Вопросы биологической,
медицинской и фармацевтической химии. - 2011. - № 9. - С. 14-22.
. Буданова Н.Ю. Капиллярное электрофоретическое
разделение энантиомеров при использовании олиго- и полисахаридных хиральных
селекторов: Автореф. дис… канд. хим. наук. - М., 2005. - 26 с.
. Быковский С.Н. Руководство по инструментальным
методам исследований при разработке и экспертизе качества лекарственных
препаратов. - М.: Изд-во «Перо», 2014. - 656 с.
. Глазков И.Н. Определение органических примесей в
фармацевтических препаратах / И.Н. Глазков, Н.Л. Бочкарёва, И.А. Ревельский //
Журн. аналит. химии. - 2005. - №2. - 136 с.
. Духин С.С. «Электрофорез»/ Духин С.С., Дерягин
Б.В.- М: «Наука», 1976 - 332с.
. Каменцев Я.С. Основы метода капиллярного
электрофореза. Аппаратурное оформление в области применения / Я. С. Каменцев,
Н.В. Комарова // Журн. «Аналитика и контроль». - 2002. Т.6. - №1. - 18 с.
. Карцова Л.А. Возможности и ограничения различных
режимов капиллярного электрофореза для количественного определения катехинов и
кофеина в чёрном и зелёном чае / Л. А.Карцова, О. В. Ганжа, А. В. Алексеева //
Журнал аналитической химии. - 2010. - Т. 65, № 2. - С. 212-217.
. Карцова Л.А. Различные варианты on-line
концентрирования при электрофоретическом определении аминов, аминокислот и
стероидных гормонов / Л.А. Карцова, А.А. Сидорова, Е.А. Бессонова // Журнал
аналитической химии. - 2012. - Т. 67, № 7. - С. 715-720.
. Комарова Н.В. Практическое руководство по
использованию систем капиллярного электрофореза «КАПЕЛЬ»/Комарова Н. В.,
Каменцев Я. С. - СПб.: ООО «Веда», 2006. - 212 с.
. Максютина М.П. Методы идентификации лекарственных
препаратов. - Киев «Здоровье», 1978г. - 240с.
. Машковский М.Д. Лекарственные средства. - М.:
Медицина, 1978 - 240с.
. Морзунова Т.Г. Капиллярный электрофорез в
фармацевтическом анализе (обзор) [Текст]/Т.Г.Морзунова//
Химико-фармацевтический журнал. - 2006. - №3. - С. 39-52.
. Старцев М.Ф. Идентификация лекарственных препаратов
- М.: «Медицина», 1981г. - 240с.
. Хомов Ю.А. Капиллярный электрофорез как
высокоэффективный аналитический метод (обзор литературы) / Ю. А. Хомов, А. Н.
Фомин // Современные проблемы науки и образования. - 2012. - № 5;
. Черноглазов В.Н. Развитие капиллярного
электрофореза и его аппаратурного оформления / В. Н. Черноглазов, П. Н.
Нестеренко // Рос. хим. журн. - 1996. - №1. - 110 с.
. Юрьев А.В. Применение метода капиллярного
электрофореза при анализе фармпрепаратов// Актуальные проблемы аналитической
химии: тезисы. докл. Всерос. конф. (Москва, 11-15 марта 2002 г.). - М., 2002. -
108 с.
22. X. Zheng. An online field-amplification sample
stacking method for the determination of diuretics in urine by capillary
electrophoresis-amperometric detection / [et al.] // - 2008. - Vol. 76, № 1. -
P. 15-20.
. F. Tagliaro [et al.] .Capillary
electrophoresis: a new tool in forensic toxicology. Applications and prospects
in hair analysis for illicit drugs // Forensic Science International. - 1995. -
Vol. 70, № 1-3. - P. 93-104.
. Capillary electrophoretic determination of
triamterene, methotrexate, and creatinine in human urine / J. R. Flores [et
al.] // J. Separation Science. - 2005. - Vol.28, №7. - P. 658-664.
. J. Caslavska, W. Thormann. Rapid analysis of
furosemide in human urine by capillary electrophoresis with laser-induced
fluorescence and electrospray ionization-ion trap mass spectrometric detection
// Journal of Chromatography B. - 2002. - Vol. 770, № 1-2. - P. 207-216.
. Chankvetadze B. Simultaneous
enantioseparation of cis-diltiazem hydrochloride and its metabolite
cis-desacetyldiltiazem using high-performance liquid chromatography and
capillary electrophoresis / B. Chankvetadze, I. Kartozia, G. Blaschke //
Journal of Pharmaceutical and Biomedical Analysis. - 2002. - Vol. 27, № 1-2. -
P. 161-166.
. S. Torres-Cartas. Comparison between
micellar liquid chromatography and capillary zone electrophoresis for the
determination of hydrophobic basic drugs in pharmaceutical preparations //
Biomedical Chromatography. - 2007. - Vol. 21, № 1. - P. 21-28.
. R.M. Krisko [et al.]. Determination of
bupivacaine and metabolites in rat urine using capillary electrophoresis with
mass spectrоmetry detection //
Electrophoresis. - 2003. - Vol.24, №14. - P. 2340-2347.
. Z.D. Peterson[et al.]. Determination of
catecholamines and metanephrines in urine by capillary
electrophoresis-electrospray ionization-time-of-flight mass spectrometry//
Journal of Chromatography B. - 2002. - Vol. 776, № 2. - P. 221-229.
. Y.Mrestani [et al.]. Determination of
cephalosporins in urine and bile by capillary zone electrophoresis // Analytica
Chimica Acta. - 1997. - Vol. 349, № 1-3. - P. 207-213.
. J. Song [et al.]. Determination of metformin
in plasma by capillary electrophoresis using field-amplified sample stacking
technique // Journal of Chromatography B: Biomedical Sciences and Applications.
- 1998. - Vol. 708, № 1-2. - P. 277-283.
. X. Zhang [et al.]. Determination of morphine
by capillary electrophoresis immunoassay in thermally reversible
hydrogel-modified buffer and laser-induced fluorescence detection // Journal of
Chromatography A. - 2000. - Vol. 895, № 1-2. - P. 1-7.
. K.P. Edward [et al.]. Determination of
nicotine and its metabolites in urine by solid-phase extraction and sample
stacking capillary electrophoresis-mass spectrometry // Journal of
Chromatography B. - 2003. - Vol. 796, № 2. - P. 303-313.
. F.J. Lara [et al.]. Determination of
phenothiazines in pharmaceutical formulations and human urine using capillary
electrophoresis with chemiluminescence detection // Electrophoresis. - 2006. - Vol.
27, № 12. - P. 2348-2359.
. Development and validation method for
determination of fluoxetine and its main metabolite norfluoxetine by nonaqueous
capillary electrophoresis in human urine / J. R. Flores[et al.] // Talanta. -
2005. - Vol. 65, № 1. - P. 163-171.
. J.R.Flores [et al.]. Development of a
Capillary Zone Electrophoretic method to determine six antidepressants in their
pharmaceutical preparations. Experimental design for evaluating the ruggedness
of method // J. Separation Science. - 2004. - Vol.27, №1-2. - P. 33-40.
. M. Valenzuela [et al.]. Development of a
non-aqueous electrophoresis method for the simultaneous determination of
tricyclic antidepressants in human serum // Electrophoresis. - 2009. - Vol. 30,
№ 6. - P. 1052-1058.
. J.Olsson[et al.]. Enantiomeric separation of
omeprazole and its metabolite 5-hydroxyomeprazole using non-aqueous capillary
electrophoresis // Journal of Chromatography A. - 2006. - Vol. 1129, № 2. - P.
291-295.
. Enantiomeric separation of some common
controlled stimulants by capillary electrophoresis with contactless
conductivity detection / T. Mantim[et al.] // Electrophoresis. 2012. - Vol.33,
№2. - P. 388-394.
. Enantioseparation of atropine by capillary
electrophoresis using sulfated в-cyclodextrin:
application to a plant extract / L.Mateus[et al.] // Journal of Chromatography
A. - 2000. - Vol. 868, № 2. - P. 285-294.
. Fan B. Determination of lamivudine
(didanosine)saquinavir in human serum using capillary zone electrophoresis //
J. Liq. Chromatogr. and Relat. Technol. - 2002. - № 2. - P. 241-249.
. Field-amplified sample stacking in capillary
electrophoresis for the determination of clozapine, clozapine N-oxide, and
desmethylclozapine in schizophrenics’ plasma / Y.Ho[et al.] // Journal of
Chromatography B. - 2004. - Vol. 809, № 1. - P. 111-116.
. Gбspбr A. Application of capillary zone
electrophoresis to the analysis and to a stability study of cephalosporins / A.
Gбspбr, M. Andrбsi, S. Kardos // Journal of Chromatography B. - 2002. - Vol.
775, № 2. - P. 239-246.
. Gonzбlez E. Direct determination of diuretic
drugs in urine by capillary zone electrophoresis using fluorescence detection /
E. Gonzбlez, A. Becerra, J. Laserna // Journal of Chromatography B: Biomedical
Sciences and Applications. - 1996. - Vol. 687, № 1. - P. 145-150.
. Hempel G. Enantioselective determination of
zopiclone and its metabolites in urine by capillary electrophoresis / G.
Hempel, G. Blaschke // Journal of Chromatography B: Biomedical Sciences and
Applications. - 1996. - Vol. 675, № 1. - P. 139-146.
. Horstkцtter C. Stereoselective determination
of ofloxacin and its metabolites in human urine by capillary electrophoresis
using laser-induced fluorescence detection / C. Horstkцtter, G. Blaschke //
Journal of Chromatography B: Biomedical Sciences and Applications. - 2001.
-Vol. 754, № 1. - P. 169-178.
. Investigation of the metabolic fate of the
neuroleptic drug haloperidol by capillary electrophoresis-electrospray
ionization mass spectrometry / A.J. Tomlinson[et al.] // Journal of
Chromatography B: Biomedical Sciences and Applications. - 1993. - Vol. 621, №
2. - P. 239-248.
. Lдmmerhofer M. Chiral separations by
capillary electromigration techniques in nonaqueous media: I. Enantioselective
nonaqueous capillary electrophoresis. // Journal of Chromatography A. - 2005. -
Vol. 1068, № 1. - P. 3-30.
. Li J. Simultaneous determination of
psychotropic drugs in human urine by capillary electrophoresis with
electrochemiluminescence detection / J. Li, F.Zhao, H. Ju // Analytica Chimica
Acta. - 2006. -Vol. 575, № 1. - P. 57-61.
. Liu Y.Determination of quinolone antibiotics
in urine by capillary electrophoresis with chemiluminescence detection / Y.Liu,
Y.Jia, W.Tian // J. Separation Science. - 2008. - Vol. 31, № 21. - P.
3765-3771.
. Lombardo-Agьн M. Capillary zone
electrophoresis with diode-array detection for analysis of local anaesthetics
and opium alkaloids in urine samples / M. Lombardo-Agьн, C. Cruces-Blanco, A.
M. Garcнa-Campaсa // Journal of Chromatography B. - 2009. - Vol. 877, № 8-9. -
P. 833-836.
. Martin-Girardeau A. Optimization of a
capillary electrophoresis-electrospray mass spectrometry method for the
quantitation of the 20 natural amino acids in childrens blood / A.
Martin-Girardeau, M. Renou-Gonnord // Journal of Chromatography B: Biomedical
Sciences and Applications. - 2000. - Vol. 742, № 1. - P. 163-171.
. Micelle to solvent stacking of two alkaloids
in nonaqueous capillary electrophoresis / H. Zhu [et al.] // Journal of
Chromatography A. - 2011. - Vol. 1218, № 34. - P. 5867-5871.
. Migration behaviour and separation of
tramadol metabolites and diastereomeric separation of tramadol glucuronides by
capillary electrophoresis/ P.Lehtonen [et al.] // Journal of Chromatography A.
- 2004. - Vol. 1041, № 1-2. - P. 227-234.
. Mrestani Y. Thiamine analysis in biological
media by capillary zone electrophoresis with a high-sensitivity cell / Y.
Mrestani, R. H. Neubert // Journal of Chromatography A. - 2000. - Vol. 871, №
1-2. - P. 351-356.
. Nonaqueous capillary electrophoresis method
for the analysis of tamoxifen, imipramine and their main metabolites in urine /
J.R.Flores [et al.] // Talanta. - 2005. - Vol. 65, № 1. - P. 155-162.
. Nonaqueous capillary electrophoresis with
laser-induced fluorescence detection: A case study of comparison with aqueous
media/ L.Zhou [et al.] // Analytica Chimica Acta. - 2008. - Vol. 611, № 2. - P.
212-219.
. On-line coupling of cyclodextrin mediated
nonaqueous capillary electrophoresis to mass spectrometry for the determination
of salbutamol enantiomers in urine / A. Servais [et al.] // Journal of
Pharmaceutical and Biomedical Analysis. - 2006. - Vol. 40, № 3. - P. 752-757.
. On-line field-amplified sample stacking in
capillary electrophoresis for analysis of amitriptyline and its metabolite
nortriptyline in plasma / C. Chen [et al.] // Analytica Chimica Acta. - 2004. -
Vol. 517, № 1-2. - P. 103-110.
. On the perspectives of capillary
electrophoresis modes for the determination of morphine in human plasma without
sample pretreatment / S. Emara [et al.] // Biomedical Chromatography. - 2004. -
Vol. 18, № 1. - P. 21-27.
. Pajchel G. Adaptation of capillary
electrophoresis to the determination of selected cephalosporins for injection /
G. Pajchel, S. Tyski // Journal of Chromatography A. - 2000. - Vol. 895, № 1-2.
- P. 27-31.
. Pioch M. Capillary electrophoresis/mass
spectrometry relevant to pharmaceutical and biotechnological applications /M.
Pioch, S. Bunz, C. NeusьЯ // Electrophoresis. 2012. - Vol. 33, № 11. - P.
1517-1530.
. Plenis A. Modern chromatographic and
electrophoretic measurements of antidepressants and their metabolites in
biofluids / A. Plenis, T. B№czek // Biomedical Chromatography. - 2011. - Vol.
25, № 1-2. - P. 164-198.
. Potential of capillary electrophoresis,
tandem mass spectrometry and coupled capillary electrophoresis-tandem mass
spectrometry as diagnostic tools / K. Р. Elgstoen [et al.] // Journal of Chromatography A. - 2001. - Vol. 914, №
1-2. - P. 265-275.
. Pucci V. Separation of antipsychotic drugs
(clozapine, loxapine) and their metabolites by capillary zone electrophoresis /
V. Pucci, M. Raggi, E. Kenndler // Journal of Chromatography A. - 1999. - Vol.
853, № 1-2. - P. 461-468.
. Quantitative bioanalysis of enantiomeric
drugs using capillary electrophoresis and electrospray mass spectrometry / E.
K. Kindt[et al.] // Journal of Pharmaceutical and Biomedical Analysis. - 2003.
- Vol. 31, № 5. - P. 893-904.
. Rapid analysis of
3,4-methylenedioxymethamphetamine: a comparison of nonaqueous capillary
electrophoresis/fluorescence detection with GC/MS / C. Fang[et al.] // Forensic
Science International. - 2002. - Vol. 125, № 2-3. - P. 142-148.
. Screening for the presence of drugs in serum
and urine using different separation modes of capillary electrophoresis / C. M.
Boone[et al.] // Forensic Science International. - 2001. - Vol. 121, № 1-2. -
P. 89-96.
. Scriba G. Nonaqueous capillary
electrophoresis-mass spectrometry. // Journal of Chromatography A. - 2007. -
Vol. 1159, № 1-2. - P. 28-41.
. Sensitive determination of d-carnitine as
enantiomeric impurity of levo-carnitine in pharmaceutical formulations by
capillary electrophoresis-tandem mass spectrometry / L. Sбnchez-Hernбndez [et
al.] // Journal of Pharmaceutical and Biomedical Analysis. - 2010. - Vol. 53, №
5. - P. 1217-1223.
. Separation and determination of levodopa and
carbidopa in composite tablets by capillary zone electrophoresis with
amperometric detection / L. Zhang [et al.] // Analytica Chimica Acta. - 2001.
-Vol. 431, № 2. - P. 287-292.
. Simple method for determination of cocaine
and main metabolites in urine by CE coupled to MS / Costa J. [et al.] //
Electrophoresis. - 2009. - Vol.30, №12. - P. 2238-2244.
. Simultaneous analysis of cocaine and its
metabolites in urine by capillary electrophoresis-electrospray mass
spectrometry using a pressurized liquid junction nanoflow interface /
V.Hezinovб[et al.] // Electrophoresis. - 2012. - Vol.33, №4. - P. 653-660.
. Simultaneous determination of
sulfamethoxazole and trimethoprim in human plasma by capillary zone
electrophoresis / D.Teshima [et al.] // Biomedical Chromatography. - 2004. -
Vol. 18, № 1. - P. 51-54.
. Simultaneous determination of ten
amphetamine designer drugs in human whole blood by capillary electrophoresis
with diode array detection / Maria Nieddu [et al.] // Biomedical Chromatography.
- 2005. - Vol. 19, № 10. - P. 737-742.
. Smith F.P. Detection of cocaine metabolite
in blood - stain, perspiration stain and hair / F.P. Smith. R.H. Lui // J.
Forens. Sci. - 1986. - Vol.31, № 4. - P. 1269-1273.
. Song L. Separation and determination of
chiral composition in penicillamine tablets by capillary electrophoresis in a
broad pH range / L. Song, Z. Guo, Y. Chen // Electrophoresis. - 2012. - Vol.
33, №13. - P. 2056-2063.
. A. Mohammadi [et al.]. Stability evaluation
of tramadol enantiomers using a chiral stability-indicating capillary
electrophoresis method and its application to pharmaceutical analysis // J.
Separation Science. - 2011. - Vol. 34, №13. - P. 1613-1620.
. Suntornsuk L. Capillary Electrophoresis in
Pharmaceutical Analysis: A Survey on Recent Applications // J. Chromatogr. Sci.
- 2007. - Vol. 45, № 9. - P. 559-576.
. H. Teng, B. Yuan, T. You Recent Advances in
Application of Nonaqueous Capillary Electrophoresis // Chinese Journal of
Analytical Chemistry. - 2010. - Vol. 38, № 11. - P 1670- 1677.
. J.Senior [et al.]. The analysis of basic and
acidic compounds using non-aqueous CE and non-aqueous CE-MS // Journal of
Pharmaceutical and Biomedical Analysis. - 2000. - Vol. 22, № 3. - P. 413-421.
. The use of CE ECL with ionic liquid for the
determination of drug alkaloids and applications in human urine/ Y. Liu [et
al.] // Electrophoresis. - 2009. - Vol. 30, № 8. - P. 1406-1411.
. M. Tomita, T. Okuyama. Tomita M. Application
of capillary electrophoresis to the simultaneous screening and quantitation of
benzodiazepines // Journal of Chromatography B: Biomedical Sciences and
Applications. - 1996. - Vol. 678, № 2. - P. 331-337.
. N.D. Pietro [et al.]. Use of Capillary
Electrophoresis and Poly(ethylene Oxide) as the Coating Agent for the
Determination of Substances Related to Heroin Addiction and Treatment // J.
Anal. Toxicol. - 2006. - Vol. 30, № 9. - P. 679-682.
. M.S. Anderson [et al.]. Utility of
nonaqueosus capillary electrophoresis for the determination of lidocaine and
its metabolites in human plasma: a comparison of UV and MS detection // Rapid
commun. mass spectrum. - 2004. - №18. - P. 2612-2618.
. S.J. Prasanna [et al.]. Validation of
Capillary Electrophoresis Method for Determination of N-Methylpyrrolidine in
Cefepime for Injection // J. Chromatogr. Sci. - 2010. - Vol. 48, № 10. - P.
830-834.
. A.B. Wey, J. Caslavska, W. Thormann.
Analysis of codeine, dihydrocodeine and their glucuronides in human urine by
electrokinetic capillary immunoassays and capillary electrophoresis-ion trap
mass spectrometry // Journal of Chromatography A. - 2000. -Vol. 895, № 1-2. -P.
133-146.
. A.B. Wey, W. Thormann. Capillary
electrophoresis-electrospray ionization ion trap mass spectrometry for analysis
and confirmation testing of morphine and related compounds in urine // Journal
of Chromatography A. - 2001. - Vol. - 916, № 1-2. - P. 225-238