Использование жидкостной хроматографии для контроля качества лекарственных средств, применяемых в медицине
Введение
В настоящее время в мире используется большое
количество лекарственных препаратов, в связи с этим, самая важная задача
фармацевтической химии - надежный и точный (качественный, количественный)
анализ веществ, входящих в состав препаратов. Одним из способов установления
качественного и количественного состава лекарственного препарата являются
хроматографические методы анализа, которые позволяют также разделять сложные
смеси, а именно жидкостная хроматография.
Вопросы контроля качества и стандартизации
лекарственных средств усиливают свою актуальность в настоящее время в связи с
общим увеличением числа зарегистрированных лекарственных средств, поступающих,
как правило, от разных производителей.
Цели курсовой работы: проведение анализа
жидкостной хроматографии и установление целесообразности использования для
контроля качества лекарственных средств.
Основные задачи: обобщить и проанализировать
возможность использования жидкостной хроматографии для контроля качества
лекарственных средств, применяемых в медицине.
жидкостный хроматография
лекарственный
1. Жидкостная хроматография (ЖХ)
Жидкостная хроматография - это физико-химический
процесс, суть которого заключается в различии скоростей перемещения компонентов
смеси подвижной и неподвижной фазы в связи с отличающимися силами адсорбции,
сил Ван-дер-ваальса, динамики процессов сорбции и десорбции в системе.
В простейшем виде хроматографическое разделение
смеси осуществляется при прохождении потока жидкости (подвижной фазы),
содержащего анализируемые вещества, через колонку, заполненную сорбентом
(неподвижной фазой). Если молекулы разных компонентов разделяемой смеси
обладают различной адсорбируемостью или растворимостью, то время их пребывания
в неподвижной фазе, а следовательно, и средняя скорость передвижения по колонке
различны. При достаточной длине колонки это различие может привести к полному
разделению смеси на составляющие ее компоненты. После ввода анализируемой смеси
с потоком подвижной фазы в колонку зоны всех веществ расположены в начале хроматографической
колонки. Под действием потока подвижной фазы компоненты смеси начинают
перемещаться вдоль колонки с различными скоростями, величины которых обратно
пропорциональны коэффициентам распределения К (или константам распределения)
хроматографируемых компонентов. Хорошо сорбируемые вещества, значения констант
распределения для которых велики, передвигаются вдоль слоя сорбента по колонке
медленнее, чем плохо сорбируемые. Поэтому быстрее всех из колонки выходит
компонент А, затем компонент Б и последним покидает колонку компонент В
(КА<КБ<КВ). [1,2,18]
2. Классификация жидкостной хроматографии
По агрегатному состоянию фаз хроматографической
системы методы жидкостной хроматографии классифицируют на
жидкостно-адсорбционную, жидкостно-жидкостную и противоточную жидкостную
хроматографию.
По способу перемещения сорбата различают
следующие виды жидкостной хроматографии: вытеснительная, фронтальная,
элюентная, изократическая, градиентная, с программированием температуры,
давления и скорости потока элюента.
По конфигурации разделяющей системы выделяют
планарную (бумажную, тонкослойную), колоночную, микроколоночную,
многоколоночную, циркуляционную, многомерную, перколяционную хроматографию и
мультихроматографию.
По относительной полярности подвижной и неподвижной
фаз различают нормально- и обращенно-фазовую жидкостную хроматографию.
По механизму разделения выделяют адсорбционную,
распределительную, эксклюзионную, афинную, лигандообменную, ионообменную и
другие виды жидкостной хроматографии.
По цели и задачам можно выделить аналитическую,
препаративную и обращенную ситовую хроматографию.
По химическому превращению сорбата выделяют
реакционную и осадочную хроматографию.
По способу детектирования различают
хроматографические методы сочетающие разделение компонентов смеси с прямым
детектированием веществ оптическими детекторами, работающими в
ультрафиолетовой, видимой, инфракрасной области, рефрактометрическими,
эмиссионными, флуориметрическими, хемилюминесцентными, электрохимическими и
другими детекторами.[3,8,25]
3. Определения, относящиеся к области жидкостной
хроматографии
Хроматография - метод разделения смесей веществ
или частиц основанный на различиях в скоростях их перемещения в системе
несмешивающихся и движущихся относительно друг друга фаз.
Колонка - содержит хроматографический сорбент,
выполняет функцию разделения смеси на индивидуальные компоненты.
Элюент - подвижная фаза (растворитель или смесь
растворителей): жидкость или (реже) сверхкритический флюид.
Неподвижная фаза - твердая фаза или жидкость,
связанная на инертном носителе, в адсорбционной хроматографии - сорбент.
Хроматограмма - результат регистрирования
зависимости концентрации компонентов на выходе из колонки от времени.
Детектор - устройство для регистрации
концентрации компонентов смеси на выходе из колонки.
Хроматограф - прибор для проведения
хроматографии.[1,2,22,24]
Критерии пригодности хроматографической системы
- это ряд диапазонов допустимого изменения параметров хроматограммы, соблюдение
которых, по мнению разработчика, необходимо для успешного прохождения процедуры
валидации методики. К параметрам хроматограммы относятся величины,
характеризующие хроматографическое разделение: разрешение R (для пары пиков),
селективность α (для пары пиков),
фактор удерживания k’ (для определенного пика), эффективность N (по
определенному пику с определенным фактором удерживания), коэффициент асимметрии
Ka (для определенного пика). Выполнение критериев пригодности
хроматографической системы - это необходимое условие успешного прохождения
процедуры валидации, но еще не достаточное.
По "правилам хорошего тона"
разработчик методики должен не только указать критерии пригодности
хроматографической системы, но и объяснить в деталях, как каждый из критериев
рассчитывается.[7,9,21]
. Определения и расчёты общих параметров и
применимых ко всем хроматографическим методам требований для пригодности
системы
Параметры удерживания
Время удерживания и удерживаемый объём
Измерения удерживания в элюентной хроматографии
могут быть представлены временем удерживания (tR),
определённом непосредственно по положению максимума пика на хроматограмме. Из
времени удерживания может быть рассчитан удерживаемый объём (VR).
VR
= tRv
tR - время
удерживания или расстояние, измеренное по базовой линии от точки ввода пробы до
перпендикуляра, опущенного из максимума пика, соответствующего компоненту, v
- объёмная скорость подвижной фазы.
Концентрационный коэффициент распределения.
Концентрационный коэффициент распределения (Dm)
(также известный как коэффициент ёмкости к' или фактор удерживания k)
определяется как:
Dm
= (количество вещества в неподвижной фазе) / (количество вещества в подвижной
фазе)
Kc
= VS
/ VM
Kc
- коэффициент распределения,
Vs
- объём неподвижной фазы,
VM
- объём подвижной фазы.
Значение Dm
компонента может быть определено из хроматограммы при использовании выражения:
Dm = tR-tM
/ tM
tR
- время удерживания (или объём) или расстояние, измеренное по базовой линии от
точки ввода пробы до перпендикуляра, опущенного из максимума пика,
соответствующего компоненту,
tM
- «мёртвое» время (или объём): время (или объём) или расстояние, измеренное по
базовой линии от точки ввода пробы до перпендикуляра, опущенного из максимума
пика, соответствующего неудерживаемому компоненту.
Коэффициент распределения.
Характеристика элюирования в эксклюзионной
хроматографии может быть представлена коэффициентом распределения (K0),
который рассчитывают с помощью выражения:
K
о = (tR
t
о) / (tt
t
о)
tR
- время удерживания (или объём) или расстояние, измеренное по базовой линии от
точки ввода пробы до перпендикуляра, опущенного из максимума пика,
соответствующего компоненту,
t0 - «мёртвое» время
(или объём): время (или объём) или расстояние, измеренное по базовой линии от
точки ввода пробы до перпендикуляра, опущенного из максимума пика, соответствующего
неудерживаемому компоненту,
tt - время
удерживания (или объём) или расстояние, измеренное по базовой линии от точки
ввода пробы до перпендикуляра, опущенного из максимума пика, соответствующего
компоненту, который полностью проникает в поры неподвижной фазы.
Фактор удерживания (подвижности)
Фактор удерживания (подвижности RF
), используемый в плоскостной хроматографии, представляет собой отношение
расстояния от точки нанесения пробы до центра пятна и расстояния, пройденного
фронтом растворителя от точки нанесения пробы.
RF
= b/a
b - расстояние,
пройденное веществом,
a - расстояние,
пройденное фронтом растворителя.
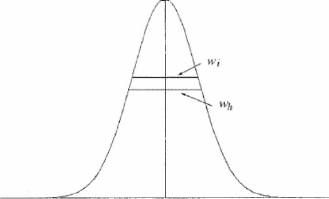
Хроматографические параметры.
Пик может быть охарактеризован площадью пика (A)
или высотой пика (h) и шириной
пика на половине высоты (wh)
или высотой пика (h) и шириной
пика между точками перегиба (w).
Для гауссовских пиков справедливо соотношение:
wh
= 1,18 wt

Фактор асимметрии.
Фактор асимметрии пика (As)
(Рисунок 2.2.46.-2) рассчитывается из выражения:
A
s = w0,05
/ 2d
wo,o5
- ширина пика на одной двадцатой высоты,
d - расстояние между
перпендикуляром, опущенным из максимума пика и передним краем пика на одной
двадцатой высоты.
Если фактор ассиметрии равен 1,0 - это означает
полную (идеальную) симметрию.
Параметры разделения.
Разрешение
Разрешение (Rs)
близких
по высоте пиков двух компонентов может быть рассчитано из выражения:
1>tR2
tRi
и tR2
- времена удерживания или расстояния, измеренное по базовой линии от точки
ввода пробы до перпендикуляров, опущенных из максимумов двух соседних пиков,
wh1
и wh2
- ширина пиков на половине высоты.
Разрешение более 1,5 соответствует разделению
пиков до базовой линии.
Выражение, приведенное выше, не может быть
использовано в случае, когда пики сильно различаются по высоте.
В количественной плоскостной хроматографии
вместо времён удерживания используются пройденные расстояния, и разрешение
может быть рассчитано с использованием выражения:
R = 8a(RPl -RPl)
/ (Wh1+Wh2)
RF1
и RF2
- отношения расстояний от точки нанесения пробы до центров пятен и расстояния,
прошедшего фронтом растворителя от точки нанесения пробы (фактор подвижности),
wh1
и wh2-
ширина пиков на половине высоты,
а - расстояние, прошедшее фронтом растворителя.
Коэффициент разделения пиков.
Коэффициент разделения неполностью разделённых
пиков (p/v-параметр)
может быть использован в качестве требования пригодности хроматографической
системы при выполнении испытания на родственные соединения в случае неполного
отделения примеси от анализируемого вещества (аналита).
p / v
= Hp / Hv
Hp - высота
пика примеси относительно экстраполированной базовой линии,
Относительное удерживание.
Относительное удерживание (г) рассчитывается из
выражения:
Hv -
расстояние между экстраполированной базовой линией и нижней точкой
tR2
*t м1
r = 1 / (tR1
- tM)
tR2
- время удерживания интересующего пика,
tR1
- время удерживания пика сравнения (обычно пик, соответствующий исследуемому
веществу),
tM
- «мёртвое» время (или объём): время (или объём) или расстояние, измеренное по
базовой линии от точки ввода пробы до перпендикуляра, опущенного из максимума
пика, соответствующего неудерживаемому компоненту.
В плоскостной хроматографии вместо tr2
и
tr1 используются факторы
подвижности
Отношение сигнал/шум (S/N)
влияет на воспроизводимость (прецизионность) количественного определения и
рассчитывается из уравнения:
S /N
= 2H/h
H - высота пика,
соответствующая рассматриваемому компоненту, на хроматограмме, полученной при
использовании рекомендованного раствора сравнения, измеренная от максимума пика
до экстраполированной базовой линии для сигнала, величина которого эквивалентна
двадцатикратному превышению величины ширины пика на половине высоты,
h - размах для фонового
шума (уровень шума) на хроматограмме, полученной при введении или нанесении
холостой пробы, величина которого эквивалентна двадцатикратному превышению
щирины пика на половине высоты для пика на хроматограмме, полученной при
использовании рекомендованного раствора сравнения и, если возможно,
расположенного на одном и том же расстоянии от места возможного обнаружения
пика.
Сходимость (повторность) отклика выражается в
виде рассчитанного процентного относительного стандартного отклонения (RSD,
%) последовательных серий измерений с участием исследуемой пробы или раствора
сравнения.
у1 - индивидуальное значение площади пика,
высоты пика или отношения площадей методе внутреннего стандарта.
у - среднее индивидуальное значение.
п - число индивидуальных значений.
Пригодность системы
Тесты на определение пригодности системы
являются неотъемлемой частью методики и используются для того, чтобы
удостовериться в адекватном функционировании хроматографической системы. Для
оценки работы колонки обычно используются следующие параметры: эффективность,
концентрационный коэффициент распределения, разрешение, относительное
удерживание и фактор асимметрии.
На хроматографическое поведение могут влиять
такие факторы, как состав, ионная сила, температура и рН подвижной фазы, скорость
потока, длина колонки, температура, давление, а также характеристики
неподвижной фазы: пористость, размер частиц, удельная площадь поверхности, а в
случае обращённой фазы степень химической модификации (блокирование концевых
групп, число атомов углерода и т.д.).
Различные компоненты используемого оборудования
должны быть проверены на соответствие их качества и должны обладать точностью
измерений требуемой для проведения испытания или количественного определения.
Должны быть соблюдены следующие требования при
отсутствии других указаний в частной статье.
· Величина
фактора асимметрии основного пика должна находиться в пределах от 0,8 до 1,5,
если только нет иных указаний в частной статье. Данное требование
распространяется как на тесты, так и на количественные определения, описанные в
частных статьях.
· Максимальное
допустимое относительное стандартное отклонение для повторных измерений для
предписанного раствора сравнения не должно превышать величин, приведенных в
государственной фармакопее. Данное требование распространяется только на
количественное определение вещества и не используется в случае проведения
испытания на родственные соединения.
· Предел
обнаружения пика (соответствующий отношению сигнал/шум равному 3) находится
ниже допустимого содержания примеси (порога, ниже которого присутствие примеси
отрицается) в тесте на родственные соединения.
- Предел количественного определения пика
(соответствующий отношению сигнал/шум равному 10) равен или меньше, чем порог
обнаружения примесей (порог, при котором отрицается присутствие примеси) в
тесте на родственные соединения. Проверка пригодности системы включена для
того, чтобы гарантировать требуемое разделение для удовлетворительного
проведения теста или количественного определения. Неподвижные фазы описаны
только в общем виде, так как существует огромное количество их доступных
коммерческих разновидностей, отличающихся по хроматографическому поведению и
для того, чтобы достигнуть предписанных требований пригодности системы, в ряде
случаев приходится вносить некоторые изменения в хроматографические условия. В
методиках обращено-фазовой хроматографии, в особенности, регулирование
различных параметров не всегда приводит к удовлетворительному разделению. В
этом случае может возникнуть необходимость заменить одну колонку другой,
обеспечивающей желаемое хроматографическое поведение, такого же типа (например,
октадецилсиликагель), но от другого производителя.
Регулирование критических параметров для
гарантии пригодности системы чётко указывается в частной статье. Следует
избегать множественных изменений условий, которые могут оказать совместное
влияние на эффективность системы.
. Аппаратура для жидкостной хроматографии
В современной жидкостной хроматографии
используют приборы различной степени сложности - от наиболее простых систем, до
хроматографов высокого класса, снабженных различными дополнительными
устройствами.[15]
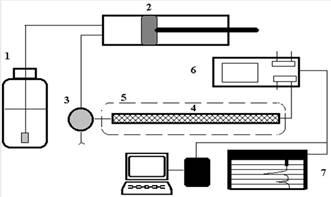
Рис.1 Блок-схема жидкостного хроматографа
- насос предназначен для создания постоянного
потока растворителя. Его конструкция определяется, прежде всего, рабочим
давлением в системе. Для работы в диапазоне 10-500 МПа используются насосы
плунжерного (шприцевого), либо пистонного типов. Недостатком первых является
необходимость периодических остановок для заполнения элюентом, а вторых -
большая сложность конструкции и, как следствие, высокая цена. Для простых
систем с невысокими рабочими давлениями 1-5 МПа с успехом применяют недорогие
перистальтические насосы, но так как при этом трудно добиться постоянства давления
и скорости потока, их использование ограничено препаративными задачами.
- инжектор,обеспечивающий ввод пробы смеси
разделяемых компонентов в колонку с достаточно высокой воспроизводимостью(
используют толстостенные стеклянные колонки).
- термостат обеспечивает постоянноство
температуры.
- детектор(проточная кювета, в которой
происходит непрерывное измерение какого-либо свойства протекающего элюента).
- регистрирующая система (дифференциальный
усилитель или самописец).
Желательно также наличие интегратора,
позволяющего рассчитывать относительные площади получаемых пиков. В сложных
хроматографических системах используется блок интерфейса, соединяющий
хроматограф с персональным компьютером, который осуществляет не только сбор и
обработку информации, но и управляет прибором.[15]
. Жидкостную колоночную хроматографию
Жидкостную колоночную хроматографию используют
преимущественно для выделения и очистки простых эфиров и перекисей.
Классическую жидкостную колоночную хроматографию при низких давлениях и неоднородных
сорбентах используют в основном для предварительного разделения. В подавляющем
большинстве случаев применяют жидкостную хроматографию высокой эффективности (
ВЭЖХ) с высокими давлениями, обусловливающими высокие скорости разделения, и
сорбентами высокой степени однородности.
Термин распределительная жидкостная колоночная
хроматография, строго говоря, предполагает наличие двух жидких фаз, неподвижной
и подвижной, при том, что неподвижность одной из них обусловлена ее связью с
твердой матрицей, заполняющей хромато-графическую колонку. Этот вид
хроматографии базируется на явлении растворимости. Движение хроматографических
зон по колонке и элюция пиков подвижной фазой происходят в направлении от %
менее гидрофильных к более гидрофильным компонентам смеси. При изократической
элюции последние лишь понемногу и с трудом диффундируют от неподвижной водной
фазы в подвижную. Для ускорения их элюции можно постепенно (градиентно)
увеличивать полярность подвижной фазы, например уменьшая в ней содержание
органического растворителя в пользу воды.
Основой жидкостной колоночной хроматографии
является разделение веществ, находящихся в растворе (подвижная фаза), на
колонках, заполненных неподвижной фазой. Подвижная фаза перемещается (
фильтруется) вдоль слоя неподвижной фазы со скоростью, зависящей от силы
взаимодействия компонентов с подвижной и неподвижной фазами.[12]
7. Жидкостная бумажная хроматография
Жидкостная бумажная хроматография -
хроматография на бумаге - метод разделения и анализа смесей веществ, основанный
на их распределении между подвижной и неподвижной жидкими фазами; в качестве
носителя неподвижной жидкой фазы используют бумагу. Метод предложен англ.
учёными А. Мартином и Р. Синго в 1941. В Б. х. используют специальные сорта
бумаги, различающиеся по номерам, с возрастанием к-рых плотность бумаги
увеличивается. Бумага удерживает в порах воду, которая и является неподвижной
жидкой фазой. Раствор пробы наносят в виде капель на лист бумаги на некотором
расстоянии от края. После испарения растворителя край листа помещают в
герметичную камеру, содержащую проявитель - подвижную жидкую фазу (напр.,
спирты, кетоны, фенолы, четырёххлористый углерод, хлороформ и др., их смеси, а
также смеси с неорганического растворителями).
При этом происходит передвижение исходного пятна
по току проявителя и разделение смеси на компоненты. Если вещества не окрашены,
то хроматограмму проявляют, напр., опрыскиванием раствором индикатора,
рассматривают в ультрафиолетовых лучах и пр. Отношение расстояния Rf,
пройденного пятном I, к расстоянию, пройденному фронтом проявителя m, при
одинаковых условиях эксперимента является постоянной величиной; Rf для разл.
веществ отличаются по значению и могут быть использованы для идентификации
соединений. Количественные определения различных веществ в пятнах хроматограммы
ведутся обычными аналитическими методами. Различают одномерные, двумерные,
круговые, колоночные и электрофоретические хроматограммы. [11]
8. Жидкостно-адсорбционная хроматография
В основе адсорбционной хроматографии лежит
различная адсорбционная способность компонентов разделяемой смеси на
поверхности выбранного адсорбента. Адсорбенты представляют собой пористые
твердые вещества с сильно развитой поверхностью. Адсорбент должен быть
химически инертным к компонентам разделяемой смеси, избирательным , механически
прочным и термически устойчивым. Наиболее широко применяемыми адсорбентами
являются углекислый кальций, окись магния, окись кальция, окись алюминия,
cиликагель, активированный уголь и цеолиты (алюмосиликаты). Одной из наиболее
распространенных теорий адсорбции является теория Лэнгмюра. Согласно этой
теории на поверхности твердого тела (адсорбента) имеются активные центры, на
которых и происходит адсорбция молекул из раствора или газа.
Адсорбция обуславливается или физическими
ван-дер-ваальсовыми силами межмолекулярного взаимодействия (молекулярная
хроматография), или силами химического сродства, действующими, например, в
процессе реакции при обмене ионов разделяемых компонентов на подвижные ионы
применяемого ионообменного адсорбента (ионообменная хроматография). Адсорбция
зависит от температуры, концентрации раствора, давления газа, от природы
вещества и от природы и структуры адсорбента. [11]
. Эксклюзионная жидкостная хроматография
Эксклюзионная жидкостная хроматография - (ситовая
хроматография), жидкостная хроматография, основанная на различной способности
молекул разного размера проникать в поры неионогенного геля, который служит
неподвижной фазой. Различают гель-проникающую хроматографию (элюент - орг.
растворитель) и гель-фильтрацию (элюент - вода). Ддя эксклюзионной
хроматографии используют макропористые неорганические. или полимерные сорбенты.
Для эксклюзионной хроматографии полярных полимеров неорганические. сорбенты
(силикагели и макропористые стекла) модифицируют кремнийорг. радикалами, а для
эксклюзионной хроматографии гидрофильных полимеров -гидрофильными группами.
Среди полимерных сорбентов наиболее
распространены стирол-дивинилбензольные (для эксклюзионной хроматографии
высокополимеров и олигомеров). Для гель-фильтрации биополимеров, прежде всего
белков, используют гидрофильные полимерные сорбенты (сефадексы - декстраны с
поперечными сшивками, а также полиакриламидные гели) или модифицированные
полисахаридами макропористые силикагели. Эксклюзионную хроматографию эффективно
применяют при разработке новых полимеров, технологических процессов их
получения, контроле производства и стандартизации полимеров. Эксклюзионную
хроматографию используют для анализа ММР полимеров, исследования, выделения и
очистки полимеров, в т. ч. биополимеров.[12]
. Реакционная жидкостная хроматография
Реакционная жидкостная хроматография - метод
анализа и физ.-хим. исследования, в котором наряду с хроматографическим
разделением осуществляют химические превращения исследуемых веществ. Термин
"реакционная хроматография" применяют в основном в газовой
хроматографии. Аналогичные разновидности жидкостной хроматографии обычно
называют терминами, например "реакционное
детектирование"-совокупность методов превращения анализируемых соединения
после их выхода из колонки с целью улучшения характеристик последующего
детектирования, "химическая дериватизация" - методы получения
производных анализируемых соединения с целью улучшения характеристик разделения
и детектирования. Иногда ионообменную и лигандообменную (с использованием
хелатообразующих сорбентов) хроматографию рассматривают как частный случай
реакций жидкостной хроматографии. Химические реакции проводят в
хроматографической системе (в спец. микрореакторе или устройстве для ввода
пробы, хроматографич. колонке, детекторе) или вне ее для улучшения разделения
веществ, понижения предела их обнаружения, повышения селективности и т. д. [13]
. Контроль качества лекарственных средств
Контроль качества лекарственных средств можно
осуществить с помощью жидкостной хроматографии. Общие принципы использования
данных методов изложены в общих и частных фармакопейных статьях.
. Кальция глюконат для инъекций
Calcii gluconas at iniectabile
GLUCONATE FOR INJECTION
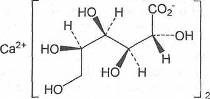
Определение
Кальция глюконат для инъекций содержит не менее
99,0% и не более 101,0% кальция D-глюконата
моногидрата. Белый или почти белый кристаллический или гранулированный порошок.
Умеренно растворим в воде, легкорастворим в кипящей воде.
Применение жидкостной хроматографии для
испытания данного вещества на примеси - оксалаты.
Оксалаты. Не более 0,01 % (100 ррт). Жидкостная
хроматография
Испытуемый раствор. 1,00 г испытуемого образна
растворяют в воде для хроматографии. Р и доводят до объема 100,0 мл этим же
растворителем.
Раствор сравнения. 1,00 г испытуемого .образца
растворяют в воде для хроматографии Р. прибавляют 0,5 мл раствора 0,152 г/л
натрия оксалата Р в воде для хроматографии Р и доводят водой для хроматографии
Р до объема 100,0 мл.
Условия хроматографирования:
- предколонка
длиной 30 мм и внутренним диаметром 4 мм, заполненная подходящей смолой
анионообменной сильноосновной с размером частиц 30-50 мкм;
- колонки
1 и 2 длиной 0,25 м и внутренним диаметром 4 мм, заполненные подходящей смолой
анионообменной сильноосновной с размером частиц 30-50 мкм;
- анион-подавляющая
колонка: расположена между предколонкой и аналитическими колонками, снабжена
микромембраной, которая отделяет подвижную фазу от регенерирующего раствора,
двигающегося противотоком подвижной фазе;
подвижная фаза: 0,212 г натрия карбоната
безводного Р и 63 мг натрия гидрокарбоната Р
- растворяют
в воде для хроматографии Р и доводят до объема 1000,0 мл этим же растворителем;
- скорость
подвижной фазы: 2 мл/мин;
- регенерирующий
раствор: раствор 1,23 г/л кислоты серной Р в воде для хроматографии Р;
- скорость
регенерирующего раствора:
- мл/мин;
- кондуктометрический
детектор;
- объем
вводимой пробы: по 50 мкл каждого раствора трижды.
Пригодность хроматографической системы: раствор
сравнения:
- относительное
стандартное отклонение: не более 2,0% для пика оксалата при пяти повторных
вводах пробы.
Содержание оксалатов в одной части на миллион
частей (ррт) рассчитывают по формуле:
ST
-50
SR
-ST
где: ST
- площадь пика оксалата на хроматограмме испытуемого раствора:
SR
- площадь пика оксалата на хроматограмме раствора сравнения.[1,6]
13.
Бетаметазона
валериат
Betamethasoni valeras
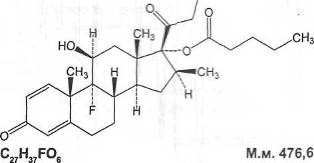
BETAMETHASONE
VALE
диен-17-илпентаноата в пересчете на сухое
вещество.
Описание (свойства)
Белый или почти белый кристаллический порошок.
Практически нерастворим в воде, легкорастворим в
ацетоне и в метилехлориде, растворим в 96% спирте.
Температура плавления: около 192°С (с
разложением).
Применение жидкостной хроматографии для
испытания данного вещества на примеси - сопутствующие примеси.
Раствор А. К 1000 мл подвижной фазь прибавляют 1
мл кислоты уксусной ледяно1 Р и тщательно перемешивают.
Испытуемый раствор. 62,5 мг испытуе мого образца
растворяют в растворе А и до водят до объема 25,0 мл этим же раствори телем.
Раствор сравнения (а). 2 мг ФСО бетаметазона
17-валериата и 2 мг ФСО бетаметазона 21-валериата растворяют в растворе А и
доводят до объема 50,0 мл этим же растворителем.
Раствор сравнения (Ь). 1,0 мл испытуемого
раствора доводят раствором А до объема 50,0 мл.
Условия хроматографирования:
- колонка
из нержавеющей стали длиной 0,25 м и внутренним диаметром 4,6 мм, заполненная
силикагелем октадецилсилильным для хроматографии Р с размером частиц 5 мкм;
- подвижная
фаза: смешивают 350 мл воды Р с 600 мл ацетонитрила Р, выдерживают до
установления равновесия, доводят объем раствора водой Р до объема 1000 мл и
снова перемешивают;
- скорость
подвижной фазы: 1 мл/мин;
- спектрофотометрический
детектор, длина волны 254 нм;
- объем
вводимой пробы: 20 мкл;
- время
установления равновесия: не менее 45 мин;
- время
хроматографирования: 2,5-крат- ное время удерживания бетаметазона 17-валериата.
Время удерживания пиков (раствор сравнения (а)):
бетаметазона 17-валериат - около
- мин;
бетаметазона 21-валериат - около
- мин.
Пригодность хроматографической системы: раствор
сравнения (а):
разрешение: не менее 5,0 между пиками бетаметазона
17-валериата и бетаметазона 21-валериата; при необходимости изменяют
концентрацию ацетонитрила в подвижной фазе.
Предельное содержание примесей:
любая примесь (не более 1,5%): на хроматограмме
испытуемого раствора площадь любого пика, кроме основного, не должна превышать
0,75 площади основного пика на хроматограмме раствора сравнения (Ь), и площадь
не более чем одного из таких пиков может превышать 0,5 площади основного пика
на хроматограмме раствора сравнения (Ь) (1 %);
сумма примесей (не более 3,0%): на хроматограмме
испытуемого раствора сумма площадей всех пиков, кроме основного, не должна
превышать 1,5-кратную площадь основного пика на хроматограмме раствора
сравнения (Ь).
На хроматограмме испытуемого раствора не
учитывают пики с площадью менее 0,025 площади основного пика на хроматограмме
раствора сравнения (Ь) (0,05%).[6]
14.
Бетаметазона
дипропионат
Betamethasoni dipropionas
DIPROPIONATE
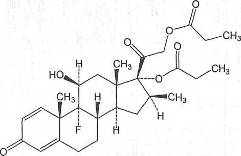
C28H37F07 Mm.
504,6
Определение
Бетаметазона дипропионат содержит не менее 97,0%
и не более 103,0% 9-фтор-11р- гидрокси-16(3-метил-3,20-диоксопрегна-1,4-
диен-17,21-диилдипропионата в пересчете на сухое вещество.
Описание (свойства)
Белый или почти белый кристаллический порошок.
Практически нерастворим в воде, легкорастворим в
ацетоне и в метилехпориде. умеренно растворим в 96% спирте.
Применение жидкостной хроматографии для
испытания данного вещества на примеси - сопутствующие примеси.
Испытуемый раствор. 62,5 мг испытуемого образца
растворяют в подвижной фазе и доводят до объема 25,0 мл этим же растворителем.
Раствор сравнения (а). 2,5 мг ФСО бетаметазона
дипропионата и 2,5 мг ФСО беклометазона дипропионата безводного растворяют в
подвижной фазе и доводят до объема 50,0 мл этим же растворителем.
Раствор сравнения (Ь). 1,0 мл испытуемого
раствора доводят подвижной фазой до объема 50,0 мл.
- колонка
из нержавеющей стали длиной 0,25 м и внутренним диаметром 4.6 мм, заполненная
силикагелем сктасеиилсилильным для хроматографии Р с размером частиц 5 мкм;
- подвижная
фаза: смешивают 350 мл воды Р с 600 мл ацетонитрила Р, выдерживают до
установления равновесия, доводят объем раствора водой Р до объема 1000 мп и
снова перемешивают;
- скорость
подвижной фазы: 1 мп/мин:
- спектрофотометрический
детектор. длина волны 254 нм;
- объем
вводимой пробы: 20 мкл;
- время
установления равновесия: не менее 45 мин;
- время
хроматографирования: 2,5-кратное время удерживания бетаметазона дипропионата.
Время удерживания пиков (раствор сравнения (а)):
бетаметазона дипропионат - около
- мин,
бекпометазона дипропионат - около 10,7 мин.
Пригодность хроматографической системы: раствор
сравнения (а):
- разрешение:
не менее 2,5 между пиками бетаметазона дипропионата и беклометазона
дипропионата. При необходимости изменяют концентрацию ацетонитрила в подвижной
фазе.
Предельное содержание примесей:
- любая
примесь: на хроматограмме испытуемого раствора площадь любого пика, кроме
основного, не должна превышать 0,75 площади основного пика на хроматограмме
раствора сравнения (Ь) (1,5%) и площадь не более одного из таких пиков может
превышать 0,5 площади основного пика на хроматограмме раствора сравнения (Ь) (1
%);
- сумма
примесей (не более 2,5%); на хроматограмме испытуемого раствора сумма площадей
всех пиков, кроме основного, не должна превышать 1,25-кратную площадь основного
пика на хроматограмме раствора сравнения (Ь).
На хроматограмме испытуемого раствора не
учитывают пики с площадью менее 0,025 площади основного пика на хроматограмме
раствора сравнения (Ь) (0,05%).
Потеря в массе при высушивании (2.2.32). Не
более 1,0%. 0,500 г испытуемого образца сушат при температуре 105°С.[6]
. Бисопролола фумарат
Bisoprololi
fumaras
BISOPROLOL
FUMARATE
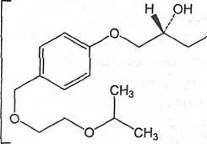
М.м. 767
Определение
Бисопролола фумарат содержит не менее 99,0% и не
более 101,0% (RS)-1-[4-[[2-(1-
метилэтокси)этокси]метил]фенокси]-3-[(1 - метилэтил)амино]пропан-2-ола фумарата
в пересчете на безводное вещество.
Описание (свойства)
Белый или почти белый порошок. Слегка
гигроскопичен.
Очень легко растворим в воде, легкорастворим в
метаноле.
Обладает полиморфизмом
Применение жидкостной хроматографии для
испытания данного вещества на примеси - сопутствующие примеси.
Смесь растворителей. Ацетонитрил р1 - вода для
хроматографии Р (20:80, об/об).
Испытуемый раствор. 25 мг испытуемого образца
растворяют в смеси растворителей и доводят до объема 25,0 мл этим же
растворителем.
Раствор сравнения (а). 1,0 мл испытуемого
раствора доводят смесью растворителей до объема 100,0 мл. 2,0 мл полученного
раствора доводит см.есью растворителей до объема 10,0 мл.
Раствор сравнения (b).
Содержимое контейнера ФСО бисопролола для проверки пригодности
хроматографической системы метода В (содержит примеси А и G)
растворяют в 1,0 мл смеси растворителей.
Условия хроматографирования:
- колонка
длиной 0,25 м и внутренним диаметром 4,6 мм, заполненная силикагелем
октадецилсилильным для хроматографии Р с размером частиц 5 мкм;
- температура:
30°С;
- подвижная
фаза:
-подвижная фаза А: раствор 10 г/л кислоты
фосфорной Р
подвижная фаза В: раствор 10 г/л кислоты
фосфорной Р в ацетонитриле Р1
- скорость
подвижной фазы: 1,0 мл/мин;
- спектрофотометрический
детектор, длина волны 225 нм;
- объем
вводимой пробы: 10 мкл.
Таблица 1
|
Время
(мин)
|
Подвижная
фаза А (%, об/об\
|
Подвижная
фаза В (%, об/об]
|
|
0-35
|
90
-> 20
|
|
|
35-40
|
20
-* 90
|
|
|
40-50
|
90
|
|
Идентификация пиков примесей: идентифицируют
пики примесей А и G, используя
хроматограмму раствора сравнения (b)
и хроматограмму, прилагаемую к ФСО бисопролола для проверки пригодности
хроматографической системы метода В.
Относительное удерживание (по отношению к
бисопрололу; время удерживания - около 13,4 мин): примесь А - около 0,4;
примесь G - около 1,02;
примесь Е - около 1,2.
Пригодность хроматографической системы: раствор
сравнения (Ь):
- коэффициент
разделения пиков: не менее 2,5 (Нр - высота пика примеси G
относительно базовой линии; Hv
- расстояние между базовой линией и нижней точкой кривой, разделяющей пики
примеси G и бисопролола).
Предельное содержание примесей:
- примесь
G (не более 0,5 %):
на хроматограмме испытуемого раствора площадь пика, соответствующего примеси G,
не должна превышать 2,5-кратную площадь основного пика на хроматограмме
раствора сравнения (а);
- примесь
А (не более 0,3 %): на хроматограмме испытуемого раствора площадь пика,
соответствующего примеси А, не должна превышать 1,5-кратную площадь основного
пика на хроматограмме раствора сравнения (а);
- неспецифицированные
примеси (не более 0,10 %У- на хроматограмме испытуемого раствора площадь любого
пика, кроме основного и пиков примесей А и G,
не должна превышать 0,5 площади основного пика на хроматограмме раствора
сравнения (а);
- -сумма
примесей (не более 0,5%): на хроматограмме испытуемого раствора сумма площадей
всех пиков, кроме основного, не должна превышать 2,5-кратную площадь основного
пика на хроматограмме раствора сравнения.[6]
.Гидроксикарбамид
Hydroxycarbamidum
HYDROXYCARBAMIDЕ
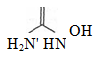
ch4n2o2
Определение
Гидроксикарбамид содержит не менее 97,5% и не
более 102,0% N-гидроксимочевинь.
Описание (свойства)
Белый или почти белый кристаллический порошок.
Гигроскопичен.
Легкорастворим в воде, практически нерастворим в
96% спирте.
Применение жидкостной хроматографии для
испытания данного вещества на примеси - сопутствующие примеси.
Испытуемый раствор (а). 0,100 г испытуемого
образца растворяют в подвижной фазе и доводят до объема 10,0 мл этим же
растворите- л«ем.
Испытуемый раствор (b).
5,0 мл испытуемого раствора (а) доводят подвижной фазой до объема 50,0 мл.
Раствор сравнения (а). Раствор готовят
непосредственно перед использованием. 0,100 г гидроксиламина гидрохлорида Р и 5
мг испытуемого образца растворяют в подвижной фазе и доводят до объема 10,0 мл
этим же растворителем.
Раствор сравнения (b).
0,1 мл испытуемого раствора (а) доводят подвижной фазой до объема 100,0 мл.
Раствор сравнения (с). 0,100 г ФСО
гидроксикарбамида растворяют в подвижной фазе и доводят до объема 10,0 мл этим
же растворителем. 5,0 мл полученного раствора доводят подвижной фазой до объема
50,0 мл.
Условия хроматографирования:
- колонка
длиной 0,25 м и внутренним диаметром 4,6 мм, заполненная силикагелем окта-
децилсилильным для хроматографии Р с размером частиц 5 мкм;
- подвижная
фаза: метанол Р - вода Р (5:95, об/об);
- скорость
подвижной фазы: 0,5 мл/мин;
- спектрофотометрический
детектор, длина волны 214 нм;
- объем
вводимой пробы: по 20 мкл испытуемого раствора (а) и растворов сравнения (а) и
(b);
- время
хроматографирования: 3-кратное время удерживания гидроксикарбамида.
Пригодность хроматографической системы: раствор
сравнения (а):
разрешение: не менее 1,0 между пиками примеси А
и гидроксикарбамида.
Предельное содержание примесей:
любая примесь (не более 0,1%): на хроматограмме
испытуемого раствора площадь любого пика, кроме основного, не должна превышать
площадь основного пика на хроматограмме раствора сравнения (b);
- сумма
примесей (не более 0,2 %): на хроматограмме испытуемого раствора сумма площадей
всех пиков, кроме основного, не должна превышать 2-кратную площадь основного
пика на хроматограмме раствора сравнения (b).
На хроматограмме испытуемого раствора не
учитывают пики с площадью менее 0,2 площади основного пика раствора сравнения (b)
(0,02%).[6]
Заключение
Начало ХХ века ознаменовалось открытием
хроматографического метода анализа, обогатившего и объединившего различные
области науки, без которых немыслим научный прогресс XXI века. Внедрение
хроматографических методов, и в первую очередь жидкостной хроматографии, в
медицину позволило решить многие жизненно важные проблемы: исследование степени
чистоты и стабильности лекарственных средств, препаративное выделение
индивидуальных гормональных препаратов (например, инсулина, интерферона),
количественное определение в биологических объектах нейромедиаторов:
адреналина, норадреналина. С наличием этих веществ в живом организме связывают
способность к запоминанию, обучению, приобретению каких-либо навыков. Идентификация
методами ВЭЖХ стероидов, аминокислот, аминов и других соединений оказалась
крайне важной при диагностике некоторых наследственных заболеваний: инфаркта
миокарда, диабета, различных заболеваний нервной системы. Одной из актуальных
задач клинической медицины для экспресс-диагностики является проведение так
называемого профильного анализа компонентов биологического объекта,
осуществляемого методами жидкостной хроматографии, что позволяет не проводить
идентификацию каждого пика, а сопоставлять профили хроматограмм для заключения
о норме или патологии.
В моей работе были изучены и проанализированы
возможности использования жидкостной хроматографии для количественного и
качественного анализа различных классов лекарственных средств.
Список литературы
1. Жерносек А.К. Аналитическая
химия для будущих провизоров. Часть 1. Учебное пособие / А.К. Жерносек, Т.Е.
Талуть; Под ред. А.И. Жебентяева. − Витебск: ВГМУ, 2003. - 362 с.
2. Википедия. Свободная
энциклопедия
. Химик. Сайт о химии
. Государственная Фармакопея
Республики Беларусь. Т.1: Общие методы контроля качества лекарственных средств
/ МЗ РБ, УП «Центр экспертиз и испытаний в здравоохранении» // Под общ.ред.
Г.В. Годовальникова - Минск: Минский госуд. ПТК полиграфии. − 2006. - 656
с.
. Эпштейн Н.А. О требованиях
и пригодности хроматографической системы при контроле качества лекартсвенных
субстанций и препаратов методом ВЭЖХ / Н.А. Эпштейн, С.В. Ешманова //
Химико-фармацевтический журнал. - 2008. − № 11. - С. 34-40.
. Государственная Фармакопея
Республики Беларусь. Т.3. Контроль качества фармацевтических субстанций / МЗ
РБ, УП «Центр экспертиз и испытаний в здравоохранении» // Под общ. ред. А.А.
Шерякова - Молодечно: «Победа». - 2009. - 728 с.
. Беликов В.Г.
Фармацевтическая химия. В 2 частях: Ч. 1 Общая фармацевтическая химия; Ч. 2
Специальная фармацевтическая химия: Учеб. пособие / В.Г. Беликов - 2-е изд. -
М.: МЕДпресс-информ, 2008. - 616 с.
. Спутник хроматографиста.
Методы жидкостной хроматографии / О.Б. Рудаков [и др.]; под ред. В.Ф.
Селеменева. - Воронеж: Водолей, 2004. - 528 с.
. Сычев К.И. Оформление
методик жидкостной хроматографии / К.И. Сычев // научно-технологический журнал.
- 2004. - т.44, №7. - С. 12-14.
. Параметры
хромматографической системы
. Колоночная жидкостная
хроматография
. Бумажно-абсорбционная
хроматография
. Эклюзионная хроматография
. Реакционная хроматография
. Хроматографический прибор
16. Фармацевтическая химия: учебное
пособие / под ред. А.П. Арзамасцева. - 3 -е изд., испр. - М.: ГЭОТАР - Медиа,
2006.
17. Фармацевтическая химия /
Глущенко Н.Н., Плетнева Т.В., Попков В.А.
a. - М., 2004.-
382с.
. Адсорбционная
хроматография
19. Лабораторное
пособие под ред. Пол С. Садек. - "Как избежать ошибок в Высокоэффективной
Жидкостной Хроматографии." - М.,2000. - 447с.
. Высокоэффективная
жидкостная хроматография: Основы теории. Методология. Применение в
лекарственной химии / под ред. В.Д. Шатц - М., 2006. - 267с.
. Высокоэффективная
жидкостная хроматография на микроколоночных хроматографах серии «Милихром» /
под ред. С.Н. Сычев - М.,2000. - 345с.
. Сычев
С.Н., Сычев К.С., Гаврилина В.А. "Высокоэффективная жидкостная
хроматография на микроколоночных хроматографах серии "Миллихром" Орел
2002 / С.Н. Сычев - 2002. - 656с.
. Рудаков
О.Б. Методы жидкостной хроматографии / О.Б. Рудаков. - 2007. - С. 234.
. Бражников
В.В. Детекторы для хроматографии / В.В. Бражников. 2004. - С. 241.
. Хроматография