Метод молекулярної динаміки
Вступ
Багато систем, таких як гази, рідини
і тверді тіла складаються з великого числа взаємодіючих один з одним частинок.
Розглянемо для прикладу дві чашки кави, що зварені в однакових умовах. В кожній
чашці кави утримується приблизно 1023-1025 молекул, рух
яких підкорюється законам класичної фізики. Незважаючи на те, що міжмолекулярні
сили породжують складні рухи молекул, для двох однакових чашок властивості кави
в кожній чашці нерозрізнені і їх легко описати. Відомо, наприклад, що
температура кави, якщо її залишити у чашці, досягає кімнатної і з часом не
змінюється. Як пов’язана температура кави з траєкторіями окремих молекул? Чому
вона не залежить від часу, навіть якщо траєкторії окремих молекул безупинно
змінюються?
Цей приклад з чашкою кави ставить
нас перед проблемою: як можна, виходячи з відомих міжмолекулярних взаємодій,
зрозуміти поведінку складної системи багатьох частинок? Можна вирішити цю
задачу, моделюючи на комп’ютері задачу багатьох частинок. Такий підхід
називають методом молекулярної динаміки. Його застосовують до «невеликих»
систем багатьох частинок (від декількох сотень до декількох тисяч). Він уже
багато дав для розуміння властивостей газів, рідин і твердих тіл, що
спостерігаються. Однак детальне знання траєкторії 104 або 1025
частинок нічого не дасть, якщо ми не знаємо, які питання потребують з’ясування.
Які основні властивості та закономірності проявляють системи багатьох частинок?
Які параметри необхідно використовувати для опису таких систем? До подібних
питань звертається статистична механіка. Єдине, що нам знадобиться для роботи,
це вміння чисельно розв’язувати рівняння Ньютона та деяке знайомство з
кінетичною енергією.
1. Фізична постановка задачі
Молекулярна динаміка ґрунтується на
законах:
) Молекули рухаються за законами
класичної фізики. Їхні рухи описуються рівняннями Ньютона.
) Молекули вважаємо твердими кулями.
) Припустимо, що сила взаємодії
будь-яких двох молекул залежить тільки від відстані між ними.
) Молекула має три ступені свободи в
просторі.
) Якщо немає тертя, то існує тільки
потенціальна енергія взаємодії. Для двох молекул потенціальна енергія V(r12).
У цьому випадку повна потенціальна енергія U визначається сумою двочастинних
взаємодій:
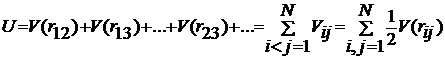 (1)
(1)
де V(rij)
залежить тільки від абсолютної величини відстані rij між
частинками i та j.
Парна взаємодія вигляду (1)
відповідає «простим» рідинам, наприклад, рідкому аргону.
) Розглядаємо прості рідини.
Тобто ми звели потенційну
енергію усієї системи до потенційної енергії двох молекул.
Форму V(r) для
електрично нейтральних атомів можна побудувати шляхом детального розрахунку,
який базується на основних законах квантової механіки. Такий розрахунок дуже
складний і, окрім того, звичайно буває цілком достатньо в якості V(r)
обрати просту феноменологічну формулу. Важливими особливостями V(r) для
простих рідин є сильне відштовхування для малих r і слабке притягання на
великих відстанях. Відштовхування при малих обумовлене правилом заборони.
Інакше кажучи, якщо електронні хмари двох атомів накладаються, деякі електрони
повинні збільшити свою кінетичну енергію, щоб знаходитися в різних квантових
станах. Сумарний ефект виражається в відштовхуванні між електронами, названому
відштовхуванням кора. Слабке притягання при великих r обумовлене
головним чином взаємною поляризацією кожного атома; результуюча сила тяжіння
називається силою Ван-дер-Вальса.
) Міжмолекулярна взаємодія
описується силами Ван-дер-Вальса.
Найбільш поширеною є
феноменологічна формула для V(r), названа потенціалом Ленарду-Джонсу
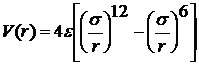 , (2)
, (2)
де σ -
(має розмірність довжини) характерний розмір молекули; ε -
характерна енергія взаємодії.
Графік потенціалу
Ленарду-Джонсу показаний на мал.1. Хоча залежність r-6 у
формулі (2) отримана теоретично, залежність r-12 обрана
тільки з міркувань зручності. Потенціал Ленарду-Джонсу параметризується
«довжиною» σ та
«енергією» ε.
Помітимо, що при r= σ V(r)=0.
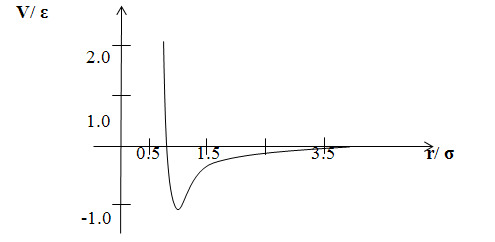
Мал.1
Тут r вимірюється в одиницях
і V(r) - в одиницях.
Параметр ε
являє собою глибину потенціальної ями в точці мінімуму V(r), що
розташований на відстані r=21/6. Помітимо, що даний потенціал
є «координуючим» V(r) для r>2.5 σ
дорівнює нулю.
Будемо виражати довжини,
енергію і масу в одиницях σ, ε і m,
де m - маса частинок. Якщо 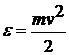 , то вимірюємо швидкість в одиницях
, то вимірюємо швидкість в одиницях 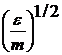 : тобто
: тобто 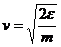 . Час
вимірюємо в одиницях
. Час
вимірюємо в одиницях 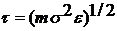 . Для
рідкого аргону параметри ε і σ
потенціалу Ленарду-Джонсу складають
. Для
рідкого аргону параметри ε і σ
потенціалу Ленарду-Джонсу складають 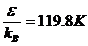 і σ=3.405
і σ=3.405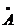 , kB=1/38•10-23
Дж/К. Маса атому аргону дорівнює m=6/69•10-23г, і звідси τ=1.82•10-12.
Таким чином швидкість молекули аргону
, kB=1/38•10-23
Дж/К. Маса атому аргону дорівнює m=6/69•10-23г, і звідси τ=1.82•10-12.
Таким чином швидкість молекули аргону 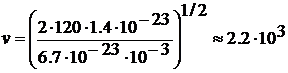 м/с, а час
взаємодії
м/с, а час
взаємодії
S=v*t →
σ= vt
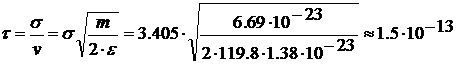 .
.
У новій системі одиниць σ=1, ε=1:
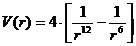 . (3)
. (3)
Усі взаємодії зводяться до
парних взаємодій між i-ю та j-ю молекулами мал. 2:
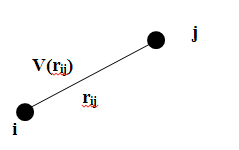
Мал.2
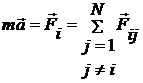 (4)
(4)
2.
Математична модель
За другим законом Ньютона
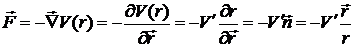 . (5)
. (5)
Тут 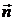 - одиничний
вектор, який має той же напрямок що і
- одиничний
вектор, який має той же напрямок що і 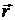 .
.
Сила взаємодії між i-ю
та j-ю молекулами
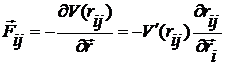 , (6)
, (6)
де похідна від потенціалу
Ленарду-Джонсу
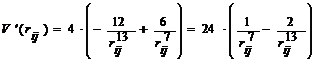 . (7)
. (7)
Якщо відстань між молекулами 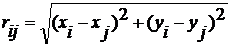 , тоді
, тоді
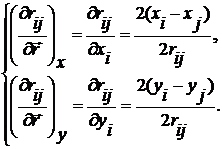 (8)
(8)
х-а та
у-а компоненти діляться на довжину вектора. Це одиничний вектор із точки
і у точку j.
Використовуючи дві компоненти
сили, запишемо рівняння (6):
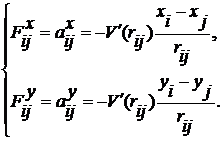 (9)
(9)
Щоб знайти прискорення 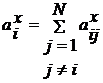
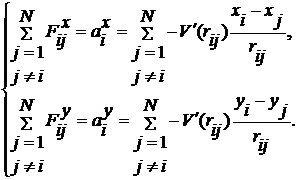 (10)
(10)
3. Чисельний
метод розв’язку
молекулярний динаміка кінетичний макроскопічний
Так як алгоритми Ейлера і
Ейлера-Крамера не можуть забезпечити зберігання енергії на час, що
розглядається при моделюванні молекулярної динаміки, ми використовуємо алгоритм
Верле у швидкісній формі. Він має більш високий порядок по Δt,
ніж Ейлера і Ейлера-Крамера, оскільки нова координата хn+1
обчислюється з використанням не тільки швидкості vn, а також
і прискорення an. Нова координата використовується для
знаходження нового прискорення an+1, що разом з an
використовується для одержання нової швидкості vn+1.
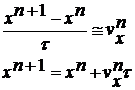
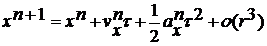 (11)
(11)
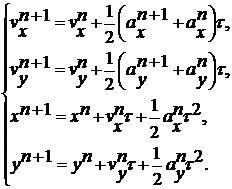 (12)
(12)
4.
Блок-схема алгоритму
. Вхідні
дані
) В замкненому контурі
прямокутної форми знаходяться молекули. Простежити рух однієї молекули.
Початкову швидкість та координати обирати за допомогою генератора випадкових
чисел. Молекула взаємодіє тільки зі стінками. Забезпечити правильний облік
крайових умов.
Крайові умови:
· Стінка тверда
· Протилежні стінки
ототожнюються.
2) Простежити процес
притягнення-відштовхування для двох молекул. Початкові швидкості задати
нульовими. Відстань між молекулами, на якій вони починають притягуватись
дорівнює 1.5σ,
відштовхування має починатись на відстані 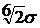 .
.
а) Розділити об’єм на дві
частини. Розташувати в одній половині об’єму рівномірно 12 молекул vx,y[0,1].
Початкові швидкості та координати молекул обирати за допомогою генератора
випадкових чисел.) В деякому місці відкрити частину перегородки. Простежити
заповнення молекулами об’єму в цілому.) Провести розрахунок сумарної кінетичної
енергії для всієї системи:
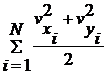
Побудувати графік залежності
середнього значення х від часу:
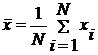 .
.
Теж саме для середнього значення у.)
Розрахувати температуру системи:
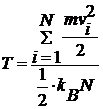
та побудувати графік залежності середнього
значення Т від часу.
е) Одержати тиск. Тиск - це сила, що діє на
одиничну поверхню. Сила - це зміна імпульсу. Вибрати стінку і підрахувати
кількість ударів молекул за одиницю часу об обрану стінку.
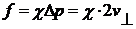 ,
,
χ - кількість ударів. Таким чином тиск
можна розрахувати за формулою
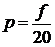 .
.
Побудувати графік залежності Р від t.
6. Аналіз результатів
На першому малюнку ми бачимо рух молекули з
постійною швидкістю. Прискорення частинки дорівнює нулю, так як вона не
взаємодіє з іншими молекулами, тобто за алгоритмом Верле отримуємо що
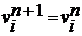 .
.
Також бачимо взаємодію молекули зі стінками, так
як за умовою вони абсолютно тверді.
Тут ми бачимо взаємодію двох молекул, коли їх
початкові швидкості дорівнюють нулю. Спостерігаємо, як дві частинки на відстані
1.5σ (а
так як за нашими припущеннями σ=1, то на
відстані 1.5) починають притягуватись, тобто на частинки вже діє
потенційна енергія. З’являється прискорення. Та на відстані молекули
відштовхуються.
На цьому малюнку ми спостерігаємо 12 молекул,
бачимо як вони взаємодіють одна з одною (притягуються, відштовхуються) та зі
стінками даного об’єму. Бачимо, що температура при зіткненні молекул постійно
змінюється, це пояснюється тим, що коли молекули наближаються одна до одної
більше ніж на відстань 1.5, з’являється прискорення, а, отже, змінюється
швидкість, тому змінюється кінетична енергія, що впливає на температуру.
Середні координати х та у також змінюються, це можна пояснити
тим,що частинки постійно змінюють напрямок своєї швидкості при зіткненні одна з
одною або зі стінками. Тиск змінюється при зіткненні молекул з однією із
стінок, чим більше молекул взаємодіє з цією стінкою, тим більшим стає тиск.
Висновки
Основні цілі цієї роботи були у тому, щоб ввести
деякі поняття кінетичної теорії рідин та ознайомитись із методом молекулярної
динаміки. Було виявлено, що моделювання системи з двадцяти частинок вже
виявляли деякі якісні властивості макроскопічних систем, але для того щоб
отримати кількісні результати нам знадобиться моделювати системи з великою
кількістю частинок. Як правило, більше всього ліченого часу займає формування
рівно масового стану та велика арифметика, яка необхідна для обчислення сили та
енергії. Якщо радіус дії сили достатньо малий, то є багато способів зменшити
час отримання рівно масового стану. Обчислення енергії та сил потребує
сумування по всім параметрам N взаємодіючих частинок. Однак, якщо
взаємодія є короткодіючою, то часу, потрібного для підрахунку цих сум на
кожному кроці по часу, можна зменшити. Тобто, у будь-який даний момент часу
більшість пар частинок розділяє відстань набагато більша, ніж ефективний радіус
rij між часткового потенціалу. Звідси, при обчисленні сили та
енергії потрібно врахувати у суму взаємодії тільки ті пари, які стоять одна від
одної менше ніж на rij. Звичайно, перевірка кожної пари на
виконання цієї умови також займає багато часу. Очевидно, що необхідно зменшити
кількість пар що перевіряються. Але як дізнатися, чи достатньо великий розмір
системи? Все просто - повторюємо моделювання з різними N.
Метод молекулярної динаміки також дозволяє
проводити численні експерименти при постійній температурі та тиску, а не при
постійній енергії та об’ємі. Також багато висновків можна зробити з траєкторій
системи.
Перелік посилань
1. Х.Гульд,
Я.Тобочник. Компьютерное моделирование в физике. Часть первая./ Перевод с англ.
канд. физ.-мат. наук А.Н.Полюдова и В.А.Панченко. - Москва «Мир». 1990 - 350 с.
. Д.В.Хеерман.
Методы комп’ютерного эксперимента в теоретической физике. Перевод с англ.
В.Н.Задкова - Москва «Наука». Главная редакция физико-математической
литературы. 1990 - 174 с.
. К.Жаблон,
Ж.-К.Симон. Применение ЭВМ для численного моделирования в физике./ Перевод с
франц. А.В.Арсентьевой - Москва «Наука». Главная редакция физико-математической
литературы. 1983 - 234 с.
Додаток
#include<iostream.h>
#include<math.h>
#include<graphics.h>
#include<conio.h>
#include<stdlib.h>
#include<stdio.h>
#include<dos.h>metod (double
x[],double y[],double vx[],double vy[],double ax[],double ay[],int N,int
i);generator (int max)
{
return random (max);
}setka (double mx,double my,double
k)
{
char str[4];
int i;
setcolor(11);
for (i=0;i<=30;i+=5)
{
line (347,i*my+5+k,353,i*my+5+k);
}
for (i=0;i<=300;i+=50)
{
itoa (i,str,10);
outtextxy (i*mx+347,147+k,str);
}
setcolor (1);
line (320,140+k,640,140+k);
line (350,0+k,350,160+k);
}metod (double x[],double y[],double
vx[],double vy[],double ax[],double ay[],int N,int i)
{
double
axn[20],ayn[20],r[20],vxn[20],vyn[20],xn[20],yn[20];
double V,t=0.01;
axn[i]=0;
ayn[i]=0;
for (unsigned j=0;j<N;j++)
{
if (i!=j)
{
r
[i]=sqrt(pow((x[i]-x[j]),2)+pow((y[i]-y[j]),2));
V=24*((1/pow(r[i],7))-(2/pow(r[i],13)));
axn[i]+=(-V)*(x[i]-x[j])/r[i];
ayn[i]+=(-V)*(y[i]-y[j])/r[i];
}
}[i]=0.5*(axn[i]+ax[i])*t;[i]=0.5*(ayn[i]+ay[i])*t;[i]+=vx[i]*t+0.5*ax[i]*t*t;[i]+=vy[i]*t+0.5*ay[i]*t*t;[i]=axn[i];[i]=ayn[i];
}main()
{
clrscr();
int driver=DETECT,mode,e;
initgraph(&driver,&mode,"c:\\BorlandC\\BGI");
e=graphresult();
if(e!=grOk)
{
printf("%s",grapherrormsg(e));
}
{
double
vx[20],y[20],x[20],vy[20],ax[20],ay[20],T=0,t=0.01;
int i=0,N=1,max=200;
randomize();
ax[i]=0;
ay[i]=0;
vy[i]=generator(max);
vx[i]=generator(max);
y[i]=generator(max);
x[i]=generator(max);
do
{
line(40,30,600,30);
line(40,30,40,450);
line(40,450,600,450);
line(600,30,600,450);
metod(x,y,vx,vy,ax,ay,N,i);
T+=t;
fillellipse(x[i]+300,-y[i]+300,10,10);
if((x[i]+300>590)||(x[i]+300<50))
vx[i]=-vx[i];
if((-y[i]+300<40)||(-y[i]+300>440))
vy[i]=-vy[i];
delay(10);
cleardevice();
}
while(T<15);=2;=0;[0]=0; vy[0]=0;
vx[1]=0; vy[1]=0; x[0]=0; x[1]=1.5;[0]=60; y[1]=60; ax[0]=0; ax[1]=0; ay[0]=0;
ay[1]=0;
{
for(i=0;i<N;i++)
{
T+=t;
metod(x,y,vx,vy,ax,ay,N,i);
fillellipse(x[i]*20+300,-y[i]+300,9,9);
}
delay(20);
cleardevice();
}(T<7);=12;=0;=0.1;=5;Ek=0,xs=0,ys=0,Tm,v=0,P;Kv=1.38;poly[8]={0,0,0,400,320,400,320,0};k=0,m=10,ydar=0;mx=1.8,my=4.5;(320,160,640,160);(320,320,640,320);(mx,my,k);=160;(mx,my,k);=320;(mx,my,k);(i=0;i<N;i++)
{
ax[i]=0;
ay[i]=0;
vx[i]=generator(max);
vy[i]=generator(max);
}=0;(i<12)
{
k=0;
for(unsigned j=0;j<3;j++)
{
x[i]=5+k;
k+=5;
i++;
}
}=0;(i<12)
{
k=0;
for(unsigned j=0;j<4;j++)
{
y[i]=5+k;
k+=9;
i++;
}
}
{
for(i=0;i<N;i++)
{
metod(x,y,vx,vy,ax,ay,N,i);
setcolor(WHITE);
setfillstyle(SOLID_FILL,WHITE);
fillellipse(x[i]*m,-y[i]*m+400,4,4);
Ek+=(vx[i]*vx[i]+vy[i]*vy[i])/2.0;
if(T<50)
}
else
{(160,0,160,100);(160,300,160,400);(x[i]*m<160)
{
if(x[i]*m<5){vx[i]=-vx[i];ydar++;v+=vx[i];}
if((x[i]*m>155)&&((-y[i]*m+400<105)||(-y[i]*m+400>295)))
vx[i]=-vx[i];
}(x[i]*m>160)
{
if(x[i]*m>315) vx[i]=-vx[i];
if((x[i]*m<165)&&((-y[i]*m+400<105)||(-y[i]*m+400>295)))
vx[i]=-vx[i];
}(((x[i]*m<165)&&(x[i]*m>155))&&((-y[i]*m+400<105)||(-y[i]*m+400>295)))
vy[i]=-vy[i];((-y[i]*m+400<5)||(-y[i]*m+400>395))
vy[i]=-vy[i];
}
line(320,0,320,480);
xs+=x[i]/N;
ys+=y[i]/N;
}
P=ydar*v/10.0;
Tm=(2*Ek)/N*Kv;
T+=t;
putpixel(T*mx+350,-xs*my+150,3);
putpixel(T*mx+350,-ys*my+150,5);
putpixel(T*mx+350,-Tm*my+310,6);
putpixel(T*mx+350,-P*my/15+400,9);
xs=0;ys=0;Ek=0;//ydar=0;v=0;
delay(20);(4,poly);}(T<150);