Химия полимеров
Лекция №1-3. Химические
основы термических и термокаталитических превращений углеводородов нефти
Первичная перегонка нефти позволяет
выделить из нефти в виде отдельных фракций только те вещества, которые в ней
изначально присутствуют. Качество, количество и ассортимент получаемых
продуктов целиком лимитируется химическим составом исходной нефти.
Для увеличения выхода светлых
нефтепродуктов - бензина, керосиновых и дизельных фракций, улучшения качества
полученных при первичной перегонке продуктов широко используются процессы,
которые называют вторичными. Применение вторичных процессов позволяет
дополнительно получить до 30-35% светлых в расчете на нефть, повысить
антидетонационные свойства бензина, улучшить термическую стабильность
реактивного топлива, снизить содержание серы в дизельном топливе и т.д.
С помощью вторичных процессов также
вырабатывается сырье для нефтехимических производств - газообразные и жидкие
алкены, арены, нормальные алканы и др.
Вторичные процессы переработки нефти
подразделяют на термические и термокаталитические.
Вторичные процессы - процессы, в
основе которых способность органических соединений нефти под влиянием высоких
температур распадаться, химически видоизменяться, вступать в различные
вторичные реакции между собой. В нефтеперерабатывающей промышленности
применяются следующие термические процессы: термический крекинг, коксование,
пиролиз.
Термический крекинг осуществляется при давлении до 5 МПа и температурах от 420 до
550°С. Промышленное освоение процесса началось в ХХ в., когда в связи с
развитием автомобильного транспорта значительно вырос спрос на бензин.
Благодаря термическому крекингу дополнительно к прямогонному стали получать
бензин из средних дистиллятов (лигроина, керосино-газойлевых фракций) и мазута.
Выход бензина достигал 30% на исходное сырье, а октановое число полученного
продукта в большинстве случаев было выше, чем у прямогонного бензина.
В 1950-х с повышением спроса на
средние дистилляты произошло изменение сырьевой базы установок: на термический
крекинг стали направлять только тяжелые фракции - остаток перегонки нефти.
Целевым продуктом стал крекинг-остаток, который используется как котельное
топливо. Была разработана особая разновидность термического крекинга - висбрекинг.
Висбрекинг (легкий крекинг) проводится при давлении не выше 2 МПа и температуре
400-480°С и предназначен для превращения гудрона в котельное топливо с низкими
вязкостью и температурой застывания.
Термический крекинг используется
также в специальных целях:
для получения
высокоароматизированного сырья, используемого в производстве технического
углерода (сажи);
для производства б-олефинов,
применяемых затем при синтезе моющих веществ.
Коксование осуществляется при давлении 0,1-0,4 МПа и температуре от 450 до
540°С. Основное назначение процесса - получение электродного кокса, из которого
изготавливаются электроды и анодная масса. В качестве сырья используются
крекинг-остатки, тяжелые пиролизные смолы, гудрон.
Простейшие установки коксования -
коксовые кубы начали строиться еще в 1920-х гг. В настоящее время для получения
кокса в основном применяется процесс коксования в необогреваемых камерах
(«замедленное коксование»). Существует также процесс коксования в
кипящем слое, но он для получения электродного кокса не применяется.
Пиролиз осуществляется при давлении близком к атмосферному и температуре
от 750 до 900°С и является наиболее «старым» из термических процессов
переработки нефти. В настоящее время пиролиз является одним из основных
процессов получения нефтехимического сырья. На пиролизных установках вырабатываются
газы, богатые непредельными углеводородами - этиленом и пропиленом.
Переработкой жидких фракций пиролиза получают широкую гамму ценных продуктов -
бутилен-бутадиеновую фракцию, ароматические углеводороды, сырье для
производства технического углерода, нафталина и др. Пиролизу подвергают
предельные углеводородные газы и бензиновые фракции. Особую разновидность
пиролиза представляет пиролиз метана, который проводится при температурах до
1200°С и предназначается для получения ацетилена, водорода и технического
углерода.
3. Термические превращения нефтяных фракций - весьма сложный
химический процесс. Изучение термических превращений отдельных углеводородов
позволяет делать выводы о характерных для данного классауглеводородов типах
реакций.
Превращения алканов. Основной вид термических превращений для углеводородов этого
класса - реакции распада по связи С - С с образованием алкана и алкена:
CnH2n+2 → CmH2m + CqH2q+2
Полученные предельные осколки вновь
распадаются на алкен и алкан. Распад алканов может происходить по всем связям С
- С, Место разрыва зависит от температуры и давления. Чем выше тампература и
ниже давление, тем место разрыва углеродной цепи все больше смещается к ее
концу и тем больше выход газообразных продуктов. При температуре около 450°С
разрыв происходит посредине цепи. Повышение давления также сдвигает место
разрыва к центру молекулы. Поэтому крекинг под давлением позволяет получать
больше жидких продуктов и меньше газа.
В ряду алканов наиболее термически
устойчив метан, так как в нем отсутствуют связи С - С. Разложение метана на
углерод и водород возможно только при очень высоких температурах (примерно
1500°С). Распад этана и пропана происходит по двум направлениям - по связи С -
С и по связи С - Н (реакция дегидрирования):
CnH2n+2 → CnH2n + H2
Этан наиболее склонен к реакции
дегидрирования, а для пропана уже при 600°С более характерна реакция распада на
этинен и метан.
Превращения алкенов. В сырье крекинга алкены отсутствуют, но их роль в химии крекинга
очень велика, так как они всегда образуются при распаде углеводородов других
классов. Для алкенов характерно большое разнообразие химических превращений.
Низкие температуры и высокие
давления стимулируют реакции уплотнения низкомолекулярных углеводородов:
nCnH2n → (CnH2n)n
Чем выше давление, тем глубже идет
полимеризация, Однако при повышении температуры термодинамическая вероятность
полимеризации резко падает и равновесие смещается в обратную сторону. Чем
больше время пребывания сырья в зоне высоких температур, тем глубже идет распад
продуктов уплотнения. В обычных условиях термического крекинга (около 500°С, 4
МПа) алкены, образовавшиеся при распаде алканов или в результате уплотнения
низкомолекулярных алкенов, в основном претерпевают распад.
Анализ энергетических особенностей молекулы
алкена показывает, что наименьшей энергией диссоциации обладает связь С-С,
находящаяся в в-положении по отношению к двойной связи:
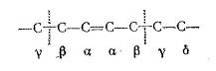
Это так называемое правило
в-связи определяет наиболее вероятное место распада в углеродной цепи.
Превращения
циклоалканов. Для циклоалканов
характерны следующие типы превращений при высоких температурах:
деалкилирование или укорочение
боковых алкильных цепей;
дегидрирование кольца с образованием
циклоалкенов и аренов;
частичная или полная дециклизация
полициклических циклоалканов после деалкилирования;
распад моноциклических циклоалканов.
Деалкилирование - реакция, аналогичная распаду алканов. Термическая устойчивость
боковых алкильных цепей значительно ниже устойчивости кольца. Поэтому
расщепление цепей является преимущественным направлением первичного распада
алкилциклоалканов. Повышение давления препятствует деалкилированию. Укороченная
боковая цепь, так же как и отщепленный осколок, могут быть либо насыщенными,
либо ненасыщенными. При исчерпывающем деалкилировании циклический радикал
насыщается водородом, всегда присутствующим в продуктах распада.
Дегидрирование - более высокотемпературная реакция, ведущая к накоплению в
продуктах крекинга и пиролиза циклоалкенов и аренов. Реакции благоприятствует
пониженное давление:
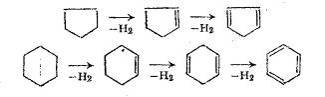
Бициклические циклоалканы при этой
реакции могут дать начало углеводородам рядов тетралина и нафталина. При
пиролизе дегидрирование шестичленных циклоалканов наряду с диеновым синтезом
является наиболее вероятным путем глубокой ароматизации сырья.
Дециклизация полициклических циклоалканов приводит к последовательному
упрощению молекул и сопровождается деалкилированием. Схематично эти превращения
можно представить в следующем виде:

Распад моноциклических циклоалканов (циклопентана, циклогексана)
происходит при 550-600°С с образованием двух алкенов:
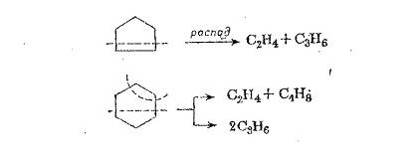
При 700-800°С циклогексан
распадается иначе - с образованием алкена и алкадиена:
С6Н12 →
С2Н6 + С4Н6
Реакции термического распада
циклоалканов протекают не по радикально-цепному, а по молекулярному механизму.
Термодинамически и кинетически реакция распада конкурирует с реакцией
дегидрирования. Поэтому в продуктах крекинга можно обнаружить в сравнимых
количествах и непредельные, и ароматические углеводороды.
Превращения аренов. Арены наиболее термически устойчивы. Поэтому они накапливаются в
жидких продуктах крекинга тем в больших количествах, чем выше температура
процесса.
Арены с длинными боковыми цепями
способны деалкилироваться. Преимущественное место отрыва боковой цепи находится
между первым и вторым атомами углерода в цепи, т.е. в в-положении от углерода
кольца. Поэтому при деалкилировании получаются главным образом
монометилированные ароматические углеводороды.
Голоядерные углеводороды (бензол,
нафталин, антрацен и др.), так же как и алкилированные углеводороды с короткими
боковыми цепями, практически не подвергаются распаду. Единственным направлением
их превращений является конденсация с выделением водорода. В результате
происходит накопление полициклических углеводородов. Эта реакция имеет
радикально-цепной механизм, что подтверждено результатами экспериментальных
исследований. Первоначально в результате взаимодействия бензола (нафталина,
антрацена и т, п.) с атомом водорода образуются ароматические радикалы
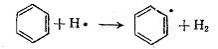
которые в дальнейшем рекомбинируют,
что и приводит к образованию конденсированных молекул. В результате развития реакций
конденсации разнообразных циклических углеводородов образуются карбоиды (кокс).
Постепенное увеличение молекулярной массы, повышение содержания углерода и
потерю водорода в результате конденсации ароматических структур можно
изобразить следующей схемой (на примере нафталина):
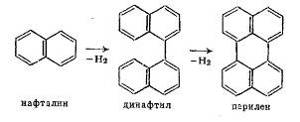
Эта и аналогичные ей реакции по мере
углубления приводят к образованию твердых карбоидов, содержащих минимальное
количество водорода. Следовательно, нефтяной кокс не является модификацией
углерода, получающейся при распаде углеводородов на элементы, а имеет
углеводородное строение.
Превращения
серосодержащих соединений. Серосодержащие
соединения, присутствующие в сырье, либо разлагаются с выделением сероводорода,
меркаптанов и углеводородных осколков, либо, благодаря своей термической
устойчивости (тиофены и им подобные), накапливаются в более высокомолекулярных
продуктах.
Подводя итог рассмотрению всех
химических превращений углеводородов различного строения, следует сделать
вывод, что при термической переработке нефтяного сырья осуществляются следующие
основное реакции: распад, деалкилирование, дегидрирование, полимеризация,
циклизация алкенов, дециклизация циклоалканов, деструктивная конденсация
алкенов, конденсация алканов в алкадиены, конденсация аренов, реакции глубокого
уплотнения до кокса.
Большинство химических превращений
углеводородов нефти, имеющих практическое значение, осуществляется в
присутствии катализаторов. Катализаторы позволяют снижать энергию активации
химических реакций и тем самым значительно повышать их скорость. В самом общем
виде в этом и заключается сущность и значение катализа. Проведение реакции в
присутствии катализаторов позволяет также резко снижать температуру процесса.
Любой катализатор активно
взаимодействует с исходными реагентами, но его участие в процессе
ограничивается только начальными стадиями превращений. В последующих стадиях он
полностью регенерируется и может вновь взаимодействовать с молекулами
реагирующих веществ. Этим и объясняется, что небольшого количества катализатора
достаточно для получения очень больших количеств конечного продукта реакции.
Факт снижения энергии активации химической реакции за счет образования
промежуточных систем с участием катализатора является несомненным. Однако
характер самого взаимодействия с катализатором может быть самым разнообразным.
Различают гомогенный и гетерогенный
катализ. При гомогенном катализе катализатор и реагирующие вещества
образуют однородную систему, например газовые смеси или жидкие растворы. При гетерогенном
катализе катализатор чаще всего находится в твердой фазе, а
реагирующие вещества - в газообразном или парообразном состоянии, т.е. в другой
фазе. Возможны случаи, когда реагенты и катализатор - жидкости, но не
смешивающиеся между собой. В нефтеперерабатывающей промышленности наиболее
распространены каталитические процессы получения топлив - каталитический
крекинг, изомеризация, риформинг, алкилирование, гидрокрекинг.
Крекинг нефтяного сырья в
присутствии катализаторов, или, коротко, каталитический крекинг - в
настоящее время один из основных, методов производства базовых
компонентов автомобильных бензинов. Применение катализатора в крекинг-процессе
вносит значительные изменения как в механизм протекающих превращений
углеводородов, так и в состав получаемых продуктов. Преимущества
каталитического крекинга заключаются, во-первых, в том, что в результате общего
ускорения процесса удается несколько снизить температуру крекинга и проводить
процесс при низком давлении; во-вторых, и это главное, селективное действие
катализатора ускоряет такие реакции, которые приводят к накоплению в
крекинг-бензине аренов, изоалканов и изоалкенов, обладающих большими октановыми
числами.
Процесс проводят в паровой фазе при
450-525°С под давлением не выше 0,15 МПа в присутствии алюмосиликатного
катализатора.
Алканы. Так же как и при термическом крекинге, алканы распадаются на
алкен и алкан меньшей молекулярной массы. Распад происходит в нескольких местах
углеродной цепи, но не на самом ее конце. Выходы метана, этана и этилена незначительны.
В газе накапливаются углеводороды С3-С4. Скорость распада
в десятки раз больше, чем при термическом крекинге.
Алканы не попадают в
хемосорбированный слой. Но и за пределами этого слоя проявляется поверхностная
энергия катализатора. В результате в непосредственной близости от поверхности
катализатора создается зона повышенной концентрации углеводородных молекул с
ослабленными связями С-С. Это и снижает энергию активации реакций распада. При
каталитическом крекинге отмечена также возможность дегидрирования углеводородов
с числом углеродных атомов в молекуле более четырех. Образующиеся при этом
высшие алкены в свою очередь распадаются.
Алкены. Скорость распада алкенов при каталитическом крекинге в тысячи раз
больше, чем при термическом крекинге. Помимо распада алкены вступают в реакции
полимеризации - деполимеризации, перераспределения водорода, изомеризации,
циклизации.
Особенно разнообразны реакции
изомеризации. Здесь имеют место структурная перегруппировка, перемещение
двойной связи вдоль цепи, возможна и цис-, транс-изомерия.
Циклоалканы. Реакции, характерные для крекинга циклоалканов - деалкилирование,
дегидрирование, распад кольца, - ускоряются в присутствии катализатора в
500-4000 раз. В отличие от термического крекинга, кроме того, заметно развиты
перечисленные ниже реакции:
распад кольца с образованием
изоалкенов;
изомеризация циклов;
перераспределение боковых цепей
(миграция заместителей).
Арены. Скорость и направление превращений аренов при каталитическом
крекинге в большой мере зависит от строения и молекулярной массы крекируемого
углеводорода.
Гомологи бензола преимущественно
полностью теряют боковые цепи, что приводит к накоплению бензола. Труднее всего
крекируется толуол. По мере увеличения длины боковой цепи и ее
разветвления глубина деалкилирования резко возрастает.
Полиметилированные гомологи бензола
легко изомеризуются с перераспределением метильных групп.
Вторичные процессы - процессы, в основе которых способность органических соединений
нефти под влиянием высоких температур распадаться, химически видоизменяться,
вступать в различные вторичные реакции между собой.
Деалкилирование - реакция, аналогичная
распаду алканов. (термическая устойчивость боковых алкильных цепей значительно
ниже устойчивости кольца).
Дегидрирование - более высокотемпературная реакция, ведущая к накоплению в
продуктах крекинга и пиролиза циклоалкенов и аренов.
Дециклизация полициклических
циклоалканов приводит к последовательному упрощению молекул и сопровождается
деалкилированием.
При гомогенном катализе катализатор
и реагирующие вещества образуют однородную систему, например газовые смеси или
жидкие растворы. При гетерогенном катализе катализатор чаще всего
находится в твердой фазе, а реагирующие вещества - в газообразном или
парообразном состоянии, т.е. в другой фазе.
Лекция №4. Твердые
горючие ископаемые, происхождение, стадии углеобразования, классификация
Уголь - вид ископаемого топлива,
образовавшийся из частей древних растений под землей без доступа кислорода.
Уголь был первым из используемых человеком видов ископаемого топлива. Он
позволил совершить промышленную революцию, которая в свою очередь
способствовала развитию угольной промышленности, обеспечив её более современной
технологией.
В среднем, сжигание одного
килограмма этого вида топлива приводит к выделению 2,93 кг CO2 и
позволяет получить 6,67 кВт·ч энергии или, при КПД 30% - 2,0 КВ·ч
электричества. В 1960 году уголь давал около половины мирового производства
энергии, к 1970 году его доля упала до одной трети. Использование угля увеличивается
в периоды высоких цен на нефть и другие энергоносители.
По составу основного компонента -
органического вещества - угли подразделяются на 3 генетические группы: гумолиты
(гумусовые угли), сапропелиты и сапрогумолиты. Преобладают гумолиты, исходным
материалом которых явились остатки высших наземных растений. Отложение их
происходило преимущественно в болотах, занимавших низменные побережья морей,
заливов, лагун, пресноводных бассейнов (озёр и рек) - автохтонное накопление;
более ограниченным было отложение при сносе с прилегающих участков суши в
застойные водные бассейны растительного материала и продуктов его
преобразования - аллохтонное накопление. Накапливавшийся растительный материал
в результате биохимического разложения перерабатывался в торф; при этом
значительное влияние оказывали обводнённость и химический состав водной среды.
Анаэробные (в водной среде) условия приводили к гелификации органического
материала - основы образования блестящих - витринитовых, или гелинитовых,
углей; аэробные условия и окислительная среда способствовали фюзенизации тканей
- образованию волокнистых и сажистых фюзинитовых углей. Элювиация - вымывание
проточными водами продуктов окисления лигнино-целлюлозных тканей -
сопровождалась обогащением органической массы остатками наиболее устойчивых
частей растений (оболочками спор, кутикулой, смоляными тельцами, пробковой
тканью коры и т.п.), характерных для матовых лейптинитовых углей. Угли,
сложенные почти полностью стойкими форменными элементами (растительными
остатками, сохранившими своё строение и очертания), выделяются в особую группу
- липтобиолиты.
Сапропелиты (сапропелевые угли) -
продукт преобразования низших растений и микроорганизмов планктона,
накапливавшихся в органогенном иле озёр и морских лагун. На равных стадиях
преобразования органического вещества сапропелиты отличаются от гумолитов более
высоким выходом летучих веществ (60-80%) и содержанием водорода (8 - 12%).
Сапрогумолиты - переходная разность
углей, продукт преобразования высших, а также низших растений. Сапропелиты и
сапрогумолиты обычно залегают в виде прослоев и линз среди гумусовых углей.
Высокозольные разности сапропелитов называют горючими сланцами; они нередко
образуют самостоятельные бассейны (например, Прибалтийский сланцевый бассейн) и
месторождения.
Минеральные примеси находятся либо в
тонкодисперсном состоянии в органической массе, либо в виде тончайших прослоек
и линз, а также кристаллов и конкреций. Источником минеральных примесей в углей
могут быть: неорганические составные части растений-углеобразователей;
терригенный материал, приносимый в области торфообразования водой и ветром, а
также минеральные новообразования, выпадающие из растворов вод, циркулирующих в
торфяниках. Состав минеральных примесей - кварц, глинистые минералы (главным
образом каолиниты), полевые шпаты, пирит, марказит, карбонаты и др. соединения,
содержащие Si, Al, Fe, Ca, Mg, К, Na, Ti, редкие и рассеянные элементы (U, Ge,
Ga, V и др.). Содержание минеральных примесей изменяется в широких пределах;
большая часть из них при сжигании угля превращается в золу.
Различия в исходном материале,
степени обводнённости торфяников, химическом составе среды и фациальных
обстановках осадко- и торфонакопления, обусловливающие направленность и
интенсивность протекания окислительных и восстановительных микробиологических
процессов, создали в торфяной стадии основу для образования различных
генетических типов углей. Торфообразование и торфонакопление завершались
перекрытием торфяника осадками, образующими породы кровли. Происходившие при
относительно невысоких температурах и давлении диагенетические (уплотнение,
дегидратация осадков, газовыделение) и биохимические процессы
восстановительного характера приводили к превращению торфа в бурый уголь. Угли,
включающие слабо разложившиеся древесные остатки, сцементированные землистым
углём, называемые лигнитами.
Бурые угли - одна из разновидностей
углей - имеют широкое распространение. Доля запасов бурых углей и лигнитов в
мировых запасах угля - 42%. Неглубокое залегание и большая мощность угольных
пластов позволяют широко применять открытый способ разработки, экономические и
технические преимущества которого во многом компенсируют относительно низкое
качество сырья.
В результате длительного воздействия
повышенных температур и давления бурые угли преобразуются в каменные угли, а
последние - в антрациты. Необратимый процесс постепенного изменения химического
состава (прежде всего в направлении обуглероживания), физических и
технологических свойств органического вещества в преобразованиях от торфа до
антрацита называются углефикацией. Углефикация на стадиях превращения бурых
углей в каменные и последних в антрациты, обусловленная происходящими в земной
коре процессами, носит название метаморфизма углей. Выделяют 3 основных вида
метаморфизма углей: региональный, вызванный воздействием внутренней теплоты
Земли и давления перекрывающей толщи пород при погружении угля в глубь земной
коры; термальный - под влиянием тепла, выделяемого магматическими телами,
перекрывшими или внедрившимися в угленосную толщу, либо в подстилающие её
отложения; контактовый - под воздействием тепла изверженных пород, внедрившихся
в угольные пласты или пересекших их непосредственно; проблематично признаётся
возможным метаморфизм углей за счёт повышения температур в областях проявления тектонических
сжимающих и скалывающих) усилий - динамометаморфизма.
Структурно-молекулярная перестройка
органического вещества при метаморфизме углей сопровождается последовательным
повышением в них относительного содержания углерода, снижением содержания кислорода,
выхода летучих веществ; в определённых закономерностях с экстремальными
значениями на средних стадиях углефикации изменяются содержание водорода,
теплота сгорания, твёрдость, плотность, хрупкость, оптические, электрические и
другие физические свойства углей.
Каменные угли на средних стадиях
метаморфизма приобретают спекающие свойства - способность гелифицированных и
липоидных компонентов органического вещества переходить при нагревании в
определённых условиях в пластического состояние и образовывать пористый монолит
- кокс. Относительное количество запасов углей с высокой спекающейся
способностью составляет 10-15% от общих запасов каменных углей, что связано с
более высокой интенсивностью преобразования органических вещества на средних
стадиях метаморфизма.
Лекция №5-6. Структура и
классификация полимеров
Основную массу полимеров составляют органические
полимеры, однако известно большое число неорганических и элементорганических
полимеров.
Молекула полимера состоит из молекул
его низкомолекулярных аналогов, соединенных друг с другом п раз
химическими связями, где п - так называемая степень полимеризации
- может принимать очень большие значения (десятки и сотни тысяч).
Соединение большого числа малых
молекул в результате химической реакции в длинную цепную молекулу полимера
приводит к возникновению у последнего целого комплекса новых
физико-механических свойств - упругости, эластичности, способности к пленко- и
волокнообразованию.
Наличие длинных цепных молекул,
имеющих химические, т.е. прочные, связи вдоль цепи, и физические, т.е. слабые,
связи между цепями, является наиболее характерным признаком полимеров. Большая
молекула полимера обладает определенной гибкостью.
Цепная молекула полимера называется макромолекулой.
Составляющие ее низкомолекулярные повторяющиеся структурные единицы, или
звенья, образованы низкомолекулярными веществами, способными к многократному
соединению друг с другом в результате химической реакции синтеза. Эти вещества
называют мономерами, а их соединение в макромолекулу полимера происходит
в результате химических реакций, протекающих по законам цепных или ступенчатых
процессов. Очевидно, что степень полимеризации, т.е. число мономерных звеньев в
одной макромолекуле, определяет молекулярную массу полимера, которая составляет
десятки, сотни тысяч, а иногда и миллионы углеродных единиц и равна
молекулярной массе исходного мономера, умноженной на степень полимеризации.
Совершенно ясно, что в процессе
синтеза полимера, когда степень полимеризации п велика, практически
невозможно получить совершенно одинаковые по размеру макромолекулы.
Молекулярная масса полимеров
является величиной усредненной по отношению к молекулярным массам отдельных
макромолекул.
В этом одно из принципиальных
отличий полимера от низкомолекулярного вещества, так как последнее
характеризуется совершенно определенным значением молекулярной массы. Это
относится и к природным, и к синтетическим полимерам.
Тем не менее, средняя величина
молекулярной массы полимера является его характеристикой, поскольку одинаковые
по химической природе полимеры различной средней молекулярной массы очень
существенно различаются по физическим и механическим свойствам.
Большие размеры макромолекул
полимеров обусловили и еще одну важную особенность их в сравнении с
низкомолекулярными веществами той же химической природы. Как известно, уже у
бутана могут быть два структурных изомера - нормальный и изо-бутан. Огромная
макромолекула полимера может быть линейной и разветвленной, т.е.
иметь боковые ответвления от основной цепи. Если при этом молекулярная масса
линейной и разветвленной молекул одинакова, то они являются изомерами.
Физические и механические свойства полимеров, состоящих из линейных
макромолекул, сильно отличаются от свойств полимеров, состоящих из
разветвленных макромолекул (например, полиэтилен высокой плотности и полиэтилен
низкой плотности).
Разветвленность макромолекул -
важнейший показатель их структуры. Разветвленность макромолекул характеризуют
разными способами, один из которых - по числу разветвлений макромолекул на 1000
атомов С. Так, если полиэтилен содержит всего 20 - 40 разветвлений на 1000
атомов С, то это уже сильно нарушает его регулярность, затрудняет
кристаллизацию, снижает жесткость. Это полиэтилен низкой плотности (ПЭНП). Если
же в молекуле полиэтилена только 5-15 разветвлений на 1000 атомов С, то больше
его склонность к кристаллизации, а также больше жесткость полимера. Это уже
полиэтилен высокой плотности (ПЭВП).
Несколько макромолекул полимера
могут быть соединены между собой химическими связями, что обусловливает еще
большее отличие их свойств. Это так называемые сшитые, или сетчатые, полимеры
(например, резина из каучука). В соответствии с формой и строением молекул
полимеры также называют линейными, разветвленными и сетчатыми (рис. 1).
В случае пространственных (сшитых)
полимеров понятие «молекула» утрачивает смысл, и тогда рассматривают среднюю
молекулярную массу отрезка между химическими связями, соединяющими отдельные
макромолекулы. В принципе весь образец сшитого полимера может представлять
собой одну гигантскую молекулу.

Рис. 1. Схематическое изображение
различных видов макромолекул:
а-линейная; б - разветвленная; в-участок сетчатой структуры
из сшитых линейных макромолекул; г - участок сетчатой структуры, сформированный
из олигомеров с концевыми функциональными группами
По химическому строению основной
цепи полимеры классифицируют следующим образом. Полимер называют органическим,
если цепь его макромолекулы состоит из атомов углерода; элементорганическим,
если цепь составлена атомами кремния, фосфора и другими, к которым присоединены
углеродные атомы или группы; неорганическим, если в цепи и в боковых группах
атомы углерода отсутствуют.
Наиболее широко распространены и
более полно изучены органические полимеры, которые в свою очередь
классифицируются по типу мономера, из которого они получаются. Если основная
цепь макромолекулы полимера содержит только атомы углерода, то полимер называют
карбоцепным. При этом в составе боковых групп могут находиться атомы
водорода, кислорода, азота, серы. Если основная цепь полимера состоит из атомов
углерода и кислорода, углерода и азота, углерода и серы, то такой полимер
называют гетероцепным.
Названия карбо- и гетероцепных
полимеров образуются на основе химических классов и названий мономеров, из
которых состоят эти полимеры. Например, для полимеров, образованных
соответствующими углеводородами, названия образуются с помощью присоединения
слова «поли»: полиэтилен, полипропилен, полиизо-бутилен, полиизопрен,
полибутадиен и др.; названия хлорпроизвод-ных полимеров: поливинилхлорид,
полихлоропрен и др.; названия производных от эфиров: поливинилацетат,
полиметилакрилат и др.
Если функциональные группы с атомами
галогенов, кислорода, азота, серы содержатся в боковых ответвлениях, а не в
основной цепи, то полимер относится к карбоцепным. У гетероцепных полимеров
гетероатом (О, N, S) входит в основную цепь. Это полиэфиры, полиамиды, полисульфиды,
полиэтилентетрафталат, поликапрамид и др.

Если полимер содержит в основной
цепи молекулы двух или более двух разных мономеров, то он является сополимером
и название его образуют обычно из названий этих мономеров (например,
бутадиен-стирольный сополимер). Строение сополимеров более сложное, чем
полимеров, состоящих из одного мономера (гомополимеров). Так, если
звенья двух мономеров соединены в макромолекуле беспорядочно, то такой
сополимер называется статистическим. При правильном чередовании звеньев
мономеров в цепи макромолекулы говорят о чередующемся сополимере. При
достаточно большой протяженности участка, состоящего из одного мономера (он
составляет, как говорят, блок данного мономера), сополимер называют блок-сополимером.
Если блоки одного из мономеров присоединены к основной цепи макромолекулы,
составленной из звеньев другого мономера, в виде больших боковых ответвлений
(т.е. образуется разветвленная макромолекула), то сополимер называется привитым
(рис. 2). Структура сополимера характеризуется химическим составом, длиной
блоков ила привитых цепей, а также числом блоков или прививок в макромолекуле.
Звенья одинаковых или разных
мономеров в молекуле могут соединяться регулярно (конец одного звена - начало
другого звена) или нерегулярно (конец одного звена - конец другого звена,
начало другого звена - начало третьего звена и т.д.), а заместители в боковых
группах могут иметь еще регулярное или нерегулярное пространственное
расположение.
В целом структура макромолекулы
характеризуется ее конфигурацией н конформацией.
Конфигурация - строго определенное
пространственное расположение атомов в молекуле, ие изменяющееся в процессе
теплового движения.
Это понятие справедливо также и для
малых молекул.
Поскольку разные виды конфигураций
стабильны во времени и не изменяются в процессе теплового движения, они
представляют собой стабильные изомеры макромолекул.
Цис - транс-изомеры характеризуются расположением заместителей относительно двойной
связи:
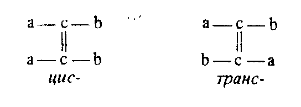
В полимерах цис- или
транс-расположение атомов характерно для каждого повторяющегося звена,
содержащего двойную связь. Это накладывает отпечаток на всю структуру
макромолекулы. Примером могут служить макромолекулы натурального каучука и
гуттаперчи, химически абсолютно идентичные:
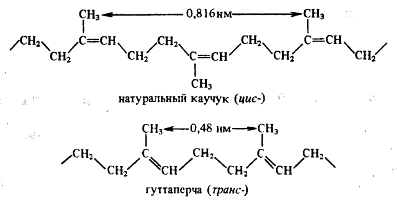
Макромолекула построена из
повторяющихся структурных единиц, что означает наличие химически идентичных
функциональных групп в каждой повторяющейся мономерной единице. Представленная
схема показывает, что с точки зрения конфигурации две соседние мономерные
группы не всегда идентичны: ориентированные в одном и том же направлении
метильные группы в цис-полиизопрене встречаются не в каждой мономерной группе
атомов, а лишь через 0,816 нм, а в гуттаперче через каждые 0,48 нм. Мы говорим,
что у этих двух видов конфигураций разные периоды идентичности. Различие
в конфигурации определяет и различие в свойствах: гуттаперча - пластмасса с
кристаллической структурой, плавящаяся при 50-70°С, а натуральный каучук -
эластомер, сохраняющий эластичность при низких температурах.
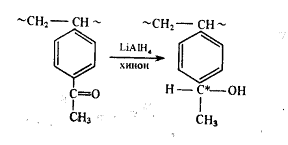
Оптические изомеры характеризуются наличием асимметрического атома углерода в
молекуле:
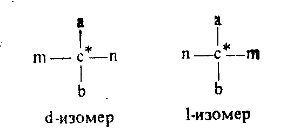
Оптические изомеры способны вращать
плоскость поляризации поляризованного света соответственно вправо или влево,
т.е. имеют разные оптические свойства. В полимерах заместители тип всегда
различны. Это части основной цепи, отличающиеся длиной. Разная длина радикалов
основной цепи (различие m и n) сама по себе не является фактором, достаточным для возникновения
оптической активности. Поэтому для возникновения оптической активности в
полимер следует ввести асимметрический атом специально (например, в
поливинилацетофенон):
Стереорегулярные
полимеры возникают благодаря наличию
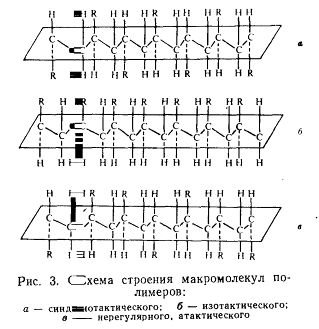
асимметрического атома углерода в макромолекуле полимера. Это - стереоизомеры.
Их строение схематически показано на рис. 3, где зигзагообразная основная
цепь для наглядности помещена в одной плоскости. Легко убедиться, что вращение
вокруг простых связей в основной цепи с учетом валентного угла между связями -
С-С - не приводит к разупорядочиванию относительного расположения заместителей.
Специальные методы синтеза приводят к получению изотактических макромолекул,
когда заместители расположены по одну сторону плоскости, синдиотактических, когда
заместители находятся по разные стороны плоскости, и атактических, когда
заместители ориентированы нерегулярно. Взаимное отталкивание заместителей,
изображенных на рис. 3, приводит к тому, что они смещаются относительно друг
друга в пространстве и поэтому плоскость симметрии оказывается на самом деле
изогнутой в виде спирали. Структура спиралей характерна не только для
макромолекул с углерод-углеродными связями в основной цепи, но и для других
видов макромолекул, в том числе и для биологически активных (например, двойная
спираль ДНК). Различные стереоизомеры имеют и разные механические свойства,
особенно сильно отличающиеся от свойств атактических полимеров того же
химического состава.
Конформацией макромолекулы называют
пространственное расположение атомов в молекуле, которое может меняться под
действнем теплового движения без разрушения химических связей между этими
атомами или группами атомов.
Степень кристалличности, температура
размягчения и плавления, прочность связей и другие характеристики полимеров
определяют их механические свойства. Следовательно, зная строение полимеров и
умея создавать заданную структуру в процессе синтеза, можно широко регулировать
свойства полимеров, а следовательно, и эксплуатационные характеристики изделий
из них.
Генезис, т.е. способ получения
макромолекул из низкомолекулярных молекул мономеров, влияет практически на все
основные свойства полимера. В природе полимеры (за исключением некоторых смол)
образуются, как правило, с высокой степенью химической и пространственной
регулярности, с правильным чередованием звеньев в структуре полимера. Это,
например, молекулы целлюлозы, натурального каучука (1,4 - полиизопрен), белков
и нуклеиновых кислот.
В формировании природных полимеров
принимают участие соответствующие ферменты и катализаторы, которые обеспечивают
направленное протекание реакций. В начальный период развития химии
синтетических полимеров, когда еще не были найдены совершенные катализаторы
синтеза, получали полимеры с нерегулярной структурой, малой молекулярной массой
и вследствие этого с низкими физико-механическими показателями. По мере
развития этой отрасли химической науки и производства были разработаны способы
получения пространственно и химически регулярных полимеров (стереоспецифическая
полимеризация) из промышленнодоступных мономеров (этилен, пропилен, стирол и
др.), что привело к громадному росту производства различных полимеров.
Большинство этих полимеров в природе не существует.
Лекция №7. Способы
получения полимеров
Синтетические полимеры получают в
результате реакций многократного соединения мономерных структурных единиц
(звеньев) в одну большую макромолекулу. Реакции их получения по своему
характеру подразделяются на цепные и ступенчатые. Последние включают процессы
ступенчатой полимеризации (полиприсоединения) и поликонденсации. Можно
предложить следующую схему классификации процессов синтеза полимеров: они
делятся на две большие группы, каждая из которых характеризуется присущими
только ей закономерностями:
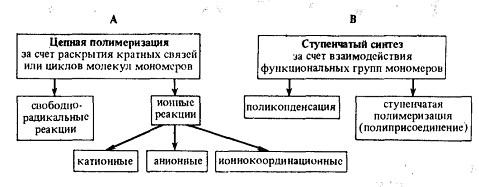
Процессы группы А представляют собой
реакции соединения друг с другом большого числа мономерных молекул в линейные
или разветвленные цепи или сетчатые структуры. Элементный состав исходных
мономеров и образовавшихся полимеров одинаков, т.е. в результате реакций цепной
полимеризации не происходит выделения каких-либо побочных низкомолекулярных
продуктов.
Процессы группы Б представляют собой
реакции соединения друг с другом большого числа мономерных или олигомерных
молекул путем взаимодействия их функциональных групп с образованием линейных,
разветвленных или сетчатых структур. Каждый акт взаимодействия этих
функциональных групп сопровождается выделением низкомолекулярного продукта (поликонденсация)
или в них происходит перестройка атомов и групп атомов в одну устойчивую
молекулярную структуру без выделения такого продукта реакции (ступенчатая
полимеризация).
2. Примерами реакций группы А являются все процессы получения
полимеров из этилена и его производных:
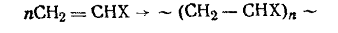
или бутадиена и его производных:
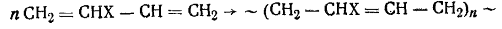
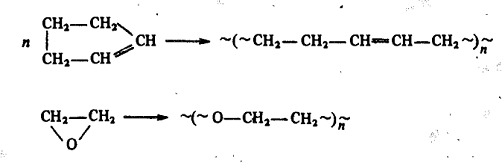
Примерами реакций группы Б является
взаимодействие друг с другом различных би- и полифункциональных соединений с
разными функциональными группами, способными реагировать между собой:
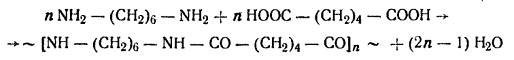
В этой реакции образования линейного
полиамида в каждом акте взаимодействия мономеров выделяется одна молекула
низкомолекулярного вещества (вода). Реакция также идет ступенчато с постепенным
нарастанием молекулярной массы полимера.
Без выделения низкомолекулярных
продуктов образуются, например, полиуретаны из диолов и диизоцианатов, но тоже
по ступенчатому механизму:
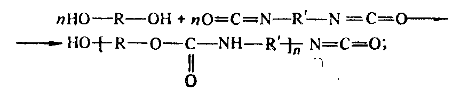
или образование поли-е-капролактама
путем последовательного присоединения циклов капролактама к первичному центру:
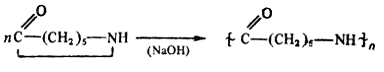
Характерной особенностью всех
полимеров является большая длина их молекулярных цепей, которая значительно
превышает поперечные размеры молекул (степень полимеризации п составляет
десятки, сотни тысяч мономерных единиц, а поперечные размеры эквивалентны
поперечному размеру молекулы мономера).
Лекция №8-10.
Радикальная полимеризация
Радикальная полимеризация всегда
протекает по цепному механизму. Функции активных промежуточных продуктов при
радикальной полимеризации выполняют свободные радикалы. К числу
распространенных мономеров, вступающих в радикальную полимеризацию, относятся
винильные мономеры: этилен, винилхлорид, винилацетат, винилиденхлорид,
тетрафторэтилен, акрилонитрил, метакрилонитрил; метилакрилат, метилметакрилат,
стирол и диеновые мономеры (бутадиен, изопрен, хлоропрен и др.).
Радикальной полимеризации
свойственны все признаки цепных реакций, известных в химии низкомолекулярных
соединений (например, взаимодействие на свету хлора и водорода). Такими
признаками являются: резкое влияние незначительного количества примесей на скорость
процесса, наличие индукционного периода и протекание процесса через
последовательность трех зависящих друг от друга стадий - образование активного
центра (свободного радикала), рост цепи и обрыв цепи. Принципиальное отличие
полимеризации от простых цепных реакций заключается в том, что на стадии роста
кинетическая цепь воплощается в материальную цепь растущего
макрорадикала, и эта цепь растет до образования макромолекулы полимера.
Инициирование радикальной
полимеризации сводится к созданию в реакционной среде свободных радикалов,
способных начать реакционные цепи. Стадия инициирования включает две реакции:
возникновение первичных свободных радикалов инициатора и взаимодействие
свободного радикала с молекулой мономера с образованием радикала.
Свободные радикалы, представляющие
собой частицы с неспаренным электроном, могут образовываться из молекул под
влиянием физического воздействия - теплоты, света, проникающей радиации, когда
в них накапливается энергия, достаточная для разрыва двойной связи. В зависимости
от вида физического воздействия на мономер при инициировании
(образовании первичного радикала) радикальную полимеризацию подразделяют на
термическую, радиационную и фотополимеризацию.
Термическое
инициирование заключается в
самоинициировании при высоких температурах полимеризации чистых мономеров без
введения в реакционную среду специальных инициаторов. В этом случае
образование радикала происходит, как правило, вследствие разложения небольших
количеств перекисных примесей, которые могут возникать при взаимодействии
мономера с кислородом воздуха. На практике таким путем получают так называемый
блочный полистирол. Однако широкого распространения метод термического
инициирования полимеризации не нашел, так как он требует больших затрат
энергии, а скорость полимеризации в большинстве случаев невелика. Ее можно
увеличить, повышая температуру, но при этом снижается молекулярная масса
образующегося полимера.
Фотоинициирование полимеризации происходит при освещении мономера светом ртутной
лампы, при котором молекула мономера поглощает квант света и переходит в
возбужденное энергетическо состояние. Соударяясь с другой молекулой мономера,
она дезактивируется, передавая последней часть своей энергии, при этом обе
молекулы превращаются в свободные радикалы. Скорость фотополимеризации растет с
увеличением интенсивности облучения и, в отличие от термической полимеризации,
не зависит от температуры.
Радиационное
инициирование полимеризации в принципе
аналогично фотохимическому. Радиационное инициирование состоит, в воздействии
на мономеры излучений высокой энергии (г-лучи, быстрые электроны, б-частицы,
нейтроны и др.). Преимуществом фото- и радиационно-химического способов
инициирования является возможность мгновенного «включения и выключения»
излучения, а также проведения полимеризации при низких температурах.
Однако эти способы технологически
сложны и могут сопровождаться протеканием в получаемых полимерах побочных
нежелательных реакций, например, деструкции. Поэтому на практике чаще всего
используют химическое (вещественное) инициирование полимеризации.
Химическое инициирование
осуществляется введением в среду мономера низкомолекулярных
нестойких веществ - инициаторов, легко распадающихся на свободные радикалы под
влиянием теплоты или света. Наиболее распространенными инициаторами радикальной
полимеризации являются перекиси и гидроперекиси (перекись водорода, перекись
бензоила, гидроперекиси трет-бутила и изопропил-бензола и др.), азо- и
диазосоединения (динитрил азобисизомасляной кислоты, диазоаминобензол и др.), персульфаты
калия и аммония. Ниже представлены реакции распада некоторых инициаторов.
Перекись трет-бутила
(алкилперекись):
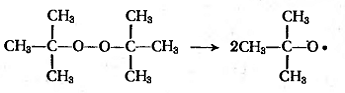
Перекись бензоила (ацилперекись):
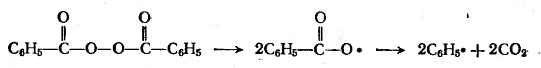
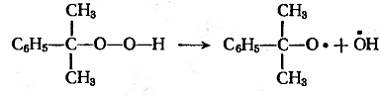
Гидроперекись кумола
(гидроперекись):
трет-бутилпербензоат (перэфир):
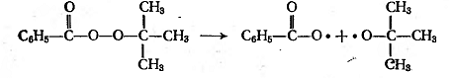
Динитрил азобисизомасляной кислоты,
или 2,2'-азобисизобути-ронитрил:
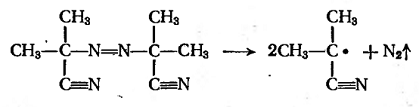
Активность и возможность применения
инициаторов радикальной полимеризации определяется скоростью их разложения,
которая зависит от температуры. Выбор конкретного инициатора обусловливается
той температурой, которая необходима для проведения синтеза полимера. Так,
динитрил азобисизомасляной кислоты применяют при 50-70°С, перекись бензоила -
при 80 - 95°С, а перекись трет-бутила - при 120-140°С.
Эффективными инициаторами,
позволяющими проводить процесс радикальной полимеризации при комнатной и
пониженной температурах, являются окислительно-восстановительные системы. В
качестве окислителей используют обычно перекиси, гидроперекиси, персульфаты и
др. Восстановителями являются соли металлов переменной валентности (Fe, Со, Си) в низшей
степени окисления, сульфиты, амины и др.
3. Рост цепи осуществляется последовательным присоединением молекул
мономера к радикалам, например:
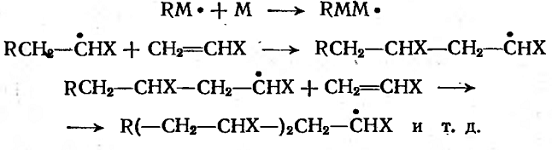
Или в общем виде:
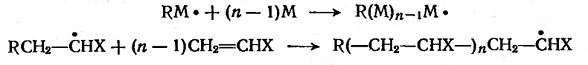
В цепном процессе радикальной
полимеризации рост кинетической цепи происходит практически мгновенно с
образованием материальной цепи макрорадикала и заканчивается ее обрывом.
Обрыв цепи представляет собой
процесс ограничения кинетической и материальной цепей. Он приводит к
исчезновению в системе активных радикалов или к замене их малоактивными
радикалами, не способными присоединять молекулы мономера. На стадии обрыва
образуется макромолекула полимера. Обрыв цепи может происходить по двум
механизмам:
) два растущих макрорадикала,
соударяясь, соединяются друг с другом в единую цепь, т.е. рекомбинируют (3а);
) макрорадикалы, соударяясь,
превращаются в две макромолекулы, причем один из них, отдавая протон,
превращается в макромолекулу с двойной С=С-связью на конце, а другой, принимая
протон, образует макромолекулу с простой концевой С-С-связью; такой механизм
называют диспропорционированием (3б):
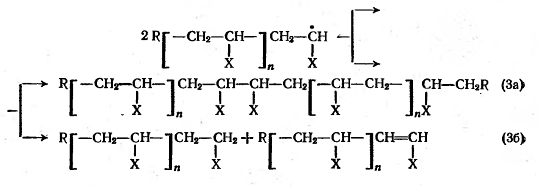
При обрыве цепей рекомбинацией
осколки инициатора находятся на обоих концах макромолекулы; при обрыве цепей
диспропорционированием - на одном конце.
По мере роста цепей макрорадикалов
увеличивается вязкость системы и уменьшается их подвижность. Это приводит к
тому, что обрыв цепей затрудняется, в результате повышается конверсия (скорость
превращения) мономера, т.е. общая скорость полимеризации. Это явление известно
как гель-эффект. Гель-эффект обусловливает повышенную полидисперсность
полимеров, что обычно приводит к ухудшению их механических свойств. Ограничение
материальных цепей при радикальной полимеризации может происходить также путем
присоединения макрорадикала к первичному радикалу (обрыв на инициаторе) и в
результате реакций передачи цепи.
Передача цепи заключается в
отрыве растущим макрорадикалом подвижного атома от молекулы какого-либо
вещества - растворителя, мономера, полимера, примесей. Эти вещества называют передатчиками
цепи. В результате макрорадикал превращается в валентно-насыщенную
макромолекулу и образуется новый радикал, способный к продолжению кинетической
цепи. Таким образом, при реакциях передачи материальная цепь обрывается, а
кинетическая - нет.
Реакцию передачи цепи на
растворитель (например, четыреххлористый углерод) можно представить следующим
образом:
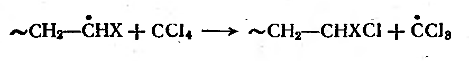
Образующиеся при этом из молекул
растворителя свободные радикалы могут присоединять молекулы мономера, т.е.
продолжать кинетическую цепь:
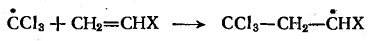
Если их активность отличается от
активности первичных радикалов, то изменяется и скорость полимеризации.
При передаче цепи на полимер
образуются разветвленные макромолекулы:
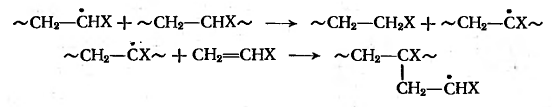
Вероятность передачи цепи на полимер
возрастает при высокой конверсии мономера, когда концентрация макромолекул в
системе велика.
Роль агента передачи цепи в
некоторых случаях может играть сам мономер, если его молекулы содержат
подвижный атом водорода. В таком случае, растущий радикал не присоединяет к
себе новую молекулу мономера по двойной связи, а отрывает у нее подвижный атом
водорода, насыщая свою свободную валентность и одновременно превращая молекулу
мономера в мономерный радикал. Это имеет место при полимеризации винилацетата:
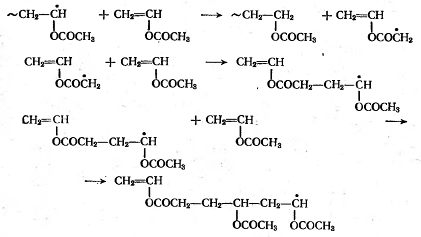
Реакции передачи цепи лежат в основе
получения теломеров. Если полимеризацию какого-либо мономера проводить
при высоких концентрациях растворителя, молекулы которого содержат подвижные
атомы водорода или галогена, то продуктом реакции будут вещества с невысокой
молекулярной массой, состоящие из нескольких мономерных звеньев, содержащих по
концам молекул продукты расщепления растворителя. Эти вещества называют
теломерами, а реакцию их получения - теломеризацией.
Реакции передачи цепи могут быть
использованы для регулирования молекулярной массы полимеров и даже для
предотвращения их образования. Этим широко пользуются на практике, применяя
часто при полимеризации передатчики - регуляторы цепи, а при хранении мономеров
- ингибиторы.
Регуляторы цепи - это вещества, которые обрывая растущие цепи полимера,
практически не влияют при этом на общую скорость процесса. Типичными
регуляторами цепи являются меркаптаны, содержащие подвижный атом водорода в
меркаптогруппе.
Полимеры, синтезированные в
присутствии регуляторов цепи, отличаются оптимальным для переработки значением
средней молекулярной массы и ММР.
Ингибиторы - это вещества, которые обрывают растущие цепи полимера,
превращаясь при этом в соединения, не способные инициировать полимеризацию. В
качестве ингибиторов обычно используют вещества, передача цепи на которые
приводит к образованию неактивных (стабильных) радикалов. На практике для
ингибирования радикальной полимеризации часто применяют гидрохинон, бензохинон,
ароматические амины, нитробензол.
Теоретические и практические
сведения о влиянии различных факторов на радикальную полимеризацию, а именно,
конверсию мономера и соответственно выход полимера, его молекулярные параметры
(молекулярную массу, полидисперсность и ММР) могут быть получены при изучении
закономерностей развития этого процесса во времени, т.е. его кинетики. Из трех
основных элементарных стадий - инициирования, роста и обрыва цепи - самой
медленной и энергоемкой является инициирование. Для ее начала требуется энергия
активации 84-126 кДж/моль, что в 3-4 раза превышает энергию активации стадии
роста цепи и почти в 10 раз энергию активации стадии обрыва.
Общая скорость радикальной
полимеризации V равна скорости расходования мономера М при
взаимодействии его с растущим радикалом.
Исходя из закона действующих масс,
скорость каждой элементарной реакции v процесса полимеризации можно предсгавить следующими уравнениями:
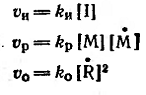
где хи и kи, хр и kр, хо и
kо - скорость и константа скорости реакций инициирования, роста и
обрыва цепи соответственно; [I],
[М], [R], [М] - концентрация инициатора,
первичных радикалов, растущих радикалов и мономера соответственно.
Так как число мономерных молекул,
участвующих при инициировании в реакции с первичным радикалом, очень мало по
сравнению с числом молекул мономера, участвующих в росте цепи (инициатор обычно
вводится в количестве до 1% от массы мономера), концентрацию мономера можно
считать постоянной, и тогда:
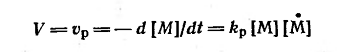
При радикальной полимеризации через
несколько секунд после начала реакции устанавливается стационарный режим
процесса: радикалы возникают при инициировании и исчезают приобрыве с
одинаковой скоростью, т.е.
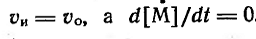
Тогда 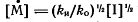 и уравнение общей скорости полимеризации принимает вид:
и уравнение общей скорости полимеризации принимает вид:
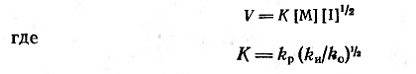
Молекулярная масса полимера так же,
как и степень полимеризации п, определяется длиной кинетической цепи,
которая зависит от соотношения скоростей реакций обрыва и роста цепи:
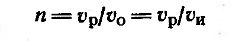
Чем больше хр по
сравнению с хо, тем больше молекул мономера успевает присоединиться
к растущему радикалу до обрыва цепи, тем больше длина цепи.
Учитывая уравнения и условие стационарности
процесса, получают:

т.е. молекулярная масса полимера
пропорциональна концентрации мономера и обратно пропорциональна квадратному
корню из концентрации инициатора.
Молекулярная масса полимера и
скорость радикальной полимеризации находятся в прямой зависимости от
концентрации мономера, повышение которой вызывает ускорение процесса и
увеличение длины цепных молекул. Подобным образом на скорость и молекулярную
массу полимера влияет увеличение давления, так как сжатие сближает реагирующие
молекулы, облегчая процесс полимеризации.
С увеличением концентрации
инициатора в системе растет число радикалов. Эти радикалы реагируют с большим
числом молекул мономера, увеличивая тем самым скорость превращения их в
макрорадикалы, т.е. скорость полимеризации. Но увеличение концентрации
радикалов способствует повышению вероятности их столкновения, т.е. возрастанию
скорости обрыва цепи полимеризации. Это приводит к снижению молекулярной массы
полимера.
Аналогичным образом на кинетику
радикальной полимеризации влияет изменение температуры. Обычно скорость
полимеризации возрастает в 2-3 раза при повышении температуры на 10°С.
Повышение температуры облегчает распад инициатора на радикалы, вместе с тем
возрастает подвижность всех частиц системы - молекул и радикалов, -
следовательно, увеличивается вероятность столкновения частиц. Это приводит к
тому, что возрастают скорости реакций роста и обрыва цепи. Таким образом, с
повышением температуры всегда общая скорость полимеризации увеличивается, а
молекулярная масса полимера уменьшается, возрастает доля низкомолекулярных
фракций. Повышение температуры способствует одновременно образованию
разветвленных макромолекул, нарушению химической регулярности построения
полимерной цепи.
На скорость полимеризации и
молекулярную массу полимера существенное влияние оказывают различные примеси и
кислород воздуха, причем кислород в зависимости от природы мономера и условий
полимеризации может ускорять или замедлять полимеризацию. Кислород замедляет
фотополимеризацию винилацетата, но ускоряет фотополимеризацию стирола,
ингибирует инициированную перекисью бензоила полимеризацию винилхлорида,
которая с хорошим выходом полимера и высоким значением молекулярной массы
протекает в атмосфере азота или аргона. Поэтому для получения полимеров используют
мономеры высокой степени чистоты (~99%) и проводят технологический процесс в
атмосфере инертного газа.
Лекция №11-13. Ионная
полимеризация
Полимеры можно получать не только
реакциями цепной радикальной полимеризации, но и цепными реакциями, в которых
растущая цепь является не свободным макрорадикалом, а макроионом. Такой способ
получения полимеров называется ионной полимеризацией, а вещества,
диссоциирующие на ионы и возбуждающие полимеризацию мономеров по ионному
механизму, называются катализаторами.
В зависимости от знака заряда
растущего макроиона различают катионную и анионную полимеризацию.
При катионной полимеризации на атоме углерода конца растущей цепи (карбкатионе)
находится положительный заряд. Заряд возникает на стадии инициирования и исчезает
при обрыве или передаче цепи. При анионной полимеризации заряд растущего
макроиона (карбаниона) отрицателен.
При ионной полимеризации можно
выделить те же элементарные стадии, что и при радикальной: инициирование, рост,
обрыв и передачу цепи. Полимеризация под влиянием ионных катализаторов обычно
происходит с большими, чем при радикальной, скоростями и приводит к получению
полимера большей молекулярной массы. Реакционная система в случае ионной
полимеризации часто является гетерогенной (неорганический или
металлорганический твердый катализатор и жидкий органический мономер).
К ионной относят также
полимеризацию, происходящую путем координации мономера на поверхности твердого
катализатора (координационно-ионная полимеризация). Поверхность
катализатора в этом случае играет особую роль матрицы, которая задает
определенный порядок вхождения мономера в растущую цепь с упорядоченным
пространственным расположением мономерных звеньев. Координационно-ионной
полимеризацией получают все стереорегулярные полимеры.
Катализаторами катионной
полимеризации являются сильные электроноакцепторные соединения. Типичными
катализаторами являются протонные кислоты (H2S04, НС104, Н3Р04
и др.) и апротонные кислоты (BF3, ZnCl2, А1С13, TiCl4 и др.) Последние
проявляют активность в присутствии небольших количеств воды или других веществ
- доноров протонов, называемых сокатализаторами.
В катионную полимеризацию легко
вступают мономеры винилового и дивинилового рядов, содержащие электронодонорные
заместители у двойной связи, например, пропилен, б-метилстирол, эфиры акриловой
и метакриловой кислот и др. В катионной полимеризации активны также некоторые
гетероциклические мономеры: окиси олефинов, лактоны, ряд карбонилсодержащих
соединений, например формальдегид.
Катионная полимеризация начинается с
того, что катализатор, взаимодействуя с сокатализатором, образует комплексное
соединение, которое является сильной кислотой. В реакционной среде происходит
его диссоциация, например:
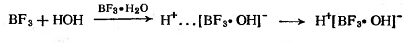
Возникающий протон присоединяется к
молекуле мономера, в результате чего образуется ионная пара, состоящая из иона
карбония и комплексного противоиона:
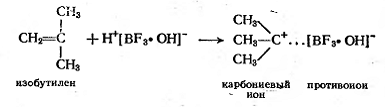
Эти две реакции составляют стадию
инициирования катионной полимеризации.
Рост цепи состоит в последовательном
присоединении молекул мономера к иону карбония, при этом на конце цепи всегда
сохраняется положительный заряд:
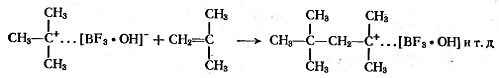
Карбониевый ион поляризует молекулу
мономера, поэтому в цепь оиа входит определенным образом, и образующиеся
макромолекулы всегда имеют регулярную структуру.
Обрыв цепи путем рекомбинации или
диспропорционирования в этом случае невозможен из-за отталкивания одноименно
заряженных ионов. Он происходит путем перестройки ионной пары, при которой
образуется нейтральная молекула полимера с двойной С=С-связью на конце и
генерируется исходный каталитический комплекс:
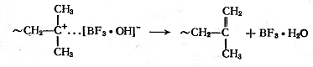
При катионной полимеризации, как и
при радикальной, наблюдается передача цепи на мономер и растворитель:
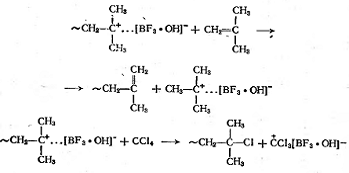
Так как катионная полимеризация
связана с образованием и диссоциацией ионной пары, то на скорость процесса
оказывает влияние диэлектрическая проницаемость среды. Повышение
диэлектрической проницаемости существенно ускоряет процесс, но мало сказывается
на молекулярной массе полимера. В сравнении с радикальной, катионная
полимеризация характеризуется низкой энергией активации (60 кДж/моль), поэтому
она протекает с высокой скоростью, которая снижается с повышением температуры.
Катализаторами анионной
полимеризации служат вещества, которые являются донорами электронов:
щелочные металлы, щелочи, гидриды и амиды щелочных металлов, металлорганические
соединения. В реакциях анионной полимеризации наиболее активны виниловые
мономеры с электроноакцепторными заместителями, например стирол СН2=СН-С6Н5
акрилонитрил СН2=СН-C=N. При анионной полимеризации в качестве активного центра выступает
карбанион - соединение с трехвалентным углеродом, несущим отрицательный заряд,
а сама растущая цепь представляет собой макроанион.
Механизм анионной полимеризации в
присутствии амидов щелочных металлов и металлорганических соединений
описывается одинаковыми схемами. Так, полимеризация стирола в среде жидкого
аммиака, катализируемая амидом натрия, протекает следующим образом.
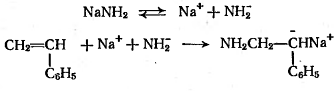
Рост цепи:
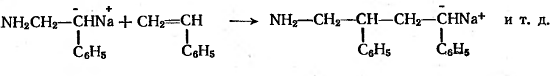
т.е. молекула мономера внедряется
между ионами ионной пары.
Обрыв цепи, как и при катионной
полимеризации, невозможен путем соединения растущих макроанионов из-за наличия
у них одинакового заряда. Он чаще всего происходит в результате реакций
передачи цепи на растворитель или мономер:
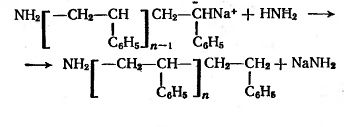
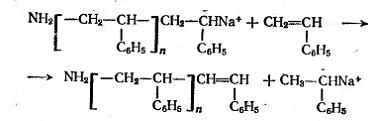
Если катализаторами анионной
полимеризации являются щелочные металлы (Li, Na), то на стадии
инициирования образуются ион-радикалы мономера, которые, соединяясь
превращаются в двухцентровое металлорганическое соединение - бианион. Рост
цепи осуществляется внедрением мономера между ионами ионной пары по обоим
центрам возникшего бианиона, т.е. цепь растет одновременно в двух направлениях.
Таким путем осуществляется полимеризация бутадиена под действием металлического
натрия:
Инициирование
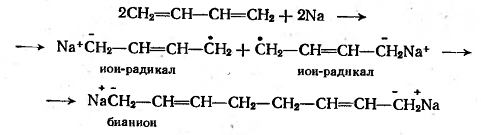
рост цепи (по обоим концам бианиона)
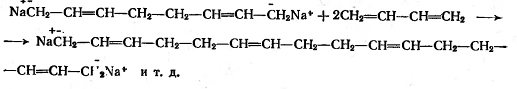
Этот вид полимеризации, связанный с
возникновением ион-радикалов, интересен тем, что дает возможность получать
«живые» полимерные цепи, т.е. растущий макробианион длительное время способен
возбуждать полимеризацию при добавлении новых порций мономера. Обрыв цепи даже
способами передачи на растворитель или мономер исключен полностью.
Полимеризация прекращается только после исчерпания всего мономера. Полимеры,
получаемые этим способом, характеризуются высоким значением молекулярной массы
и малой полидисперсностью.
Анионная полимеризация эффективна
при пониженных температурах в тщательно освобожденных от воздуха
(деаэрированных) и осушенных растворителях основного характера.
Координационно-ионная полимеризация осуществляется под действием комплексных
катализаторов, обладающих высокой избирательностью. Такие катализаторы
представляют собой комплексы, образующиеся при взаимодействии алкилов металлов I-III групп периодической системы
Д.И. Менделеева с галогенидами переходных металлов IV-VIII групп. Типичным
катализатором является комплекс триэтилалюминия и треххлористого титана:
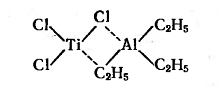
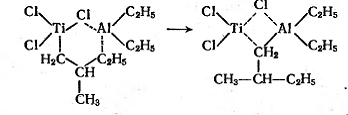
На стадии инициирования атом титана
катализаторного комплекса определенным образом координирует мономер. При такой
координации происходит разрыхление связей мономера и перераспределение связей в
катализаторном комплексе. Возникает р-комплекс между мономером и катализатором.
Так, инициирование стереоспецифической полимеризации пропилена можно
представить таким образом:
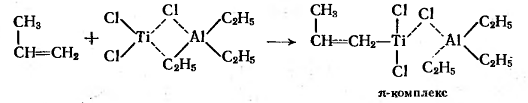
р-комплекс перегруппировывается в
шестичленное кольцо, в структуру которого внедряется мономер:
Далее генерируется катализаторный
комплекс исходной структуры, в поле притяжения которого находится первое
мономерное звено. Внедрение каждого следующего мономерного звена происходит
через стадию образования перегруппированного р-комплекса, и растущая цепь полимера
как бы отодвигается от катализатора:
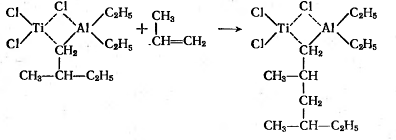
Этого не происходит ни при
радикальной, ни при катионной, ни при анионной полимеризации.
При координационно-ионной
полимеризации для образующихся макромолекул характерно не просто химически
регулярное соединение мономерных звеньев (что вообще присуще ионной
полимеризации), но и строгое чередование в пространстве заместителей при атомах
углерода основной цепи полимера. Стереоспецифичность макромолекул полимеров,
синтезированных при координационно-ионной полимеризации обеспечивается природой
комплексного катализатора. Соединения алюминия и титана аналогичной
структуры, но взятые в отдельности, не являются стереоспецифическими
катализаторами.
Лекция №14-16. Типы и
принципы реакций поликонденсации
химический углеводород
поликонденсация полимер
Поликонденсация наряду с
полимеризацией является одним из основных методов получения полимеров. Поликонденсацией
называется ступенчатый процесс образования полимеров из двух- или
полифункциональных соединений, сопровождающийся в большинстве случаев
выделением низкомолекулярного вещества (воды, спиртов, галогеноводородов и
др.). Необходимым условием поликонденсации является участие в реакции молекул,
каждая из которых содержит две или более функциональные группы, способные
взаимодействовать между собой. В общем виде процесс поликонденсации может быть
представлен следующим образом:
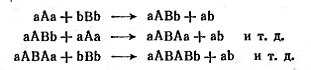
где А и В-остатки реагирующих
молекул; а и b - функциональные группы; ab - низкомолекулярный
продукт.
Приведенная схема показывает
ступенчатость образования полимера при поликонденсации: сначала взаимодействуют
между собой молекулы мономеров с образованием димеров, затем димеры
превращаются в тримеры, тримеры- в тетрамеры и т.д., т.е. в олигомеры. Благодаря
наличию функциональных групп, олигомеры могут взаимодействовать и между собой и
с мономерами. Такое взаимодействие определяет рост полимерной цепи. Если
молекулы исходных мономеров содержат по две функциональные группы,
рост полимерной цепи происходит в одном направлении и образуются линейные
макромолекулы. Наличие в молекулах исходных мономеров более двух функциональных
групп приводит к образованию разветвленных макромолекул или сшитых (трехмерных)
структур. Бифункциональные вещества могут обладать функциональными группами
одинакового или различного строения. В результате каждого акта взаимодействия
образуется продукт с концевыми функциональными группами, способными к
дальнейшему взаимодействию. Например, полиамиды можно получать из диаминов и
дикарбоновых кислот или из аминокислот. На первой стадии реакции образуются
димеры, которые далее превращаются в более высокомолекулярные продукты:
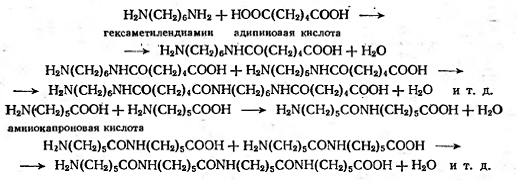
Три- и тетрафункциональные вещества,
а также их смеси с бифункциональными соединениями образуют при поликонденсации
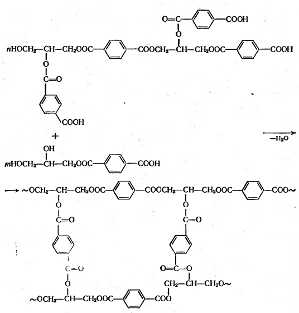
разветвленные или трехмерные продукты. Например, конденсация глицерина с
фталевой кислотой протекает по следующей схеме:
. Образование димера:
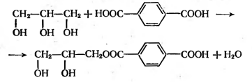
. Образование разветвленных
продуктов:
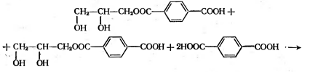
3. Образование трехмерных структур
из разветвленных продуктов:
. Полимеризация - цепной процесс,
идущий по механизму присоединения; поликонденсация - ступенчатый процесс,
идущий по механизму замещения. Промежуточные продукты на отдельных стадиях
процесса поликонденсации могут быть выделены и охарактеризованы.
. Полимеризация не сопровождается
выделением низкомолекулярных продуктов; при поликонденсации это происходит в
большинстве случаев.
. Выделение низкомолекулярного
продукта приводит, в свою очередь, к двум особенностям: во-первых, химическая
структура повторяющегося звена молекулярной цепи полимера, полученного
поликонденсацией, не соответствует составу исходных мономеров; во-вторых,
выделяющийся низкомолекулярный продукт реакции может взаимодействовать с
возникающей полимерной молекулой с образованием при этом исходных веществ. Это
означает нарушение установившегося равновесия реакции. Сместить его в сторону
образования полимера можно, удаляя из сферы реакции низкомолекулярный продукт.
. При полимеризации молекулярная
масса полимера, как правило, не зависит от продолжительности реакции; при
поликонденсации она увеличивается по мере протекания реакции.
В зависимости от природы
функциональных групп исходных веществ поликонденсацию разделяют на гомофункциональную
и гетерофунациональную. Процесс, который происходит в результате
взаимодействия функциональных групп одинаковой химической природы, является гомополиконденсацией.
Гомополиконденсацией получают, например, полиэфиры из гликолей:
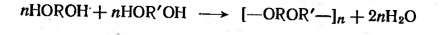
Гетерополиконденсация представляет собой процесс взаимодействия функциональных групп
разной химической природы. Примером гетерополиконденсации может служить
взаимодействие диаминов с дихлорангидридами:
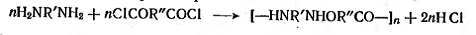
В зависимости от строения исходных
веществ поликонденсация может быть представлена химическими процессами
различных типов: этерификацией, аминированием, амидированием, циклизацией и
т.д. Поликонденсация является основным методом получения гетероцепных
полимеров.
При поликонденсации большое значение
имеет соблюдение стехиометрического соотношения между мономерами, что является
основной предпосылкой получения полимеров высокой молекулярной массы. Если
соотношение мономеров в смеси эквимолекулярно, т.е. функциональные группы обоих
типов мономеров содержатся в равных количествах, процесс поликонденсации
протекает до конца, до полного исчерпания обоих мономеров. Если в реакционной
смеси один из мономеров содержится в избытке, процесс поликонденсации протекает
до тех пор, пока израсходуется мономер, присутствующий в меньшем количестве. В
этом случае в момент окончания реакции в макромолекулах образующегося полимера
на обоих концах будут находиться одинаковые функциональные группы компонента,
имеющегося в избытке в реакционной среде. Это приведет к остановке процесса
поликонденсации и, следовательно, к снижению молекулярной массы полимера.
Аналогичный результат наблюдается, если, например, в исходную эквимолекулярную
смесь двух бифункциональных соединений ввести монофункциональное.
Монофункциональное вещество блокирует функциональные группы другого типа, в
результате чего прекращается процесс поликонденсации. Такой прием используют на
практике, когда при синтезе полиамидов в реакционную смесь из диаминов и
дикарбоновых кислот вводят добавки монокарбоновых кислот.
Стехиометричность соотношения
исходных веществ в течение процесса может нарушаться, если эти вещества
обладают различной летучестью, а также если в ходе реакции происходит изменение
природы функциональных групп.
Повышение температуры (до
определенных пределов) ускоряет реакцию поликонденсации, облегчает удаление
низкомолекулярного продукта, что при равновесной поликонденсации приводит к
смещению равновесия в сторону образования более высокомолекулярных полимеров. В
некоторых случаях повышение температуры изменяет ход реакции и характер
образующегося продукта.