Химические свойства аренов
Контрольная работа
Химические свойства аренов
1. Реакции
электрофильного замещения
Ранее нами были рассмотрены реакции
непредельных соединений с электрофильными и нуклеофильными реагентами,
приводящие в большинстве случаев к присоединению, т.е. разрушению p-связи с
образованием более прочных s-связей. Как было сказано выше, ароматические
соединения обладают повышенной устойчивостью, которая определяется энергией
p-электронной делокализации. Поэтому для них более характерны те превращения, в
результате которых сопряженная циклическая p-система сохраняется, т.е. реакции
замещения. Склонность тех или иных аренов к реакциям с электрофилами либо
нуклеофилами определяется, в первую очередь, p-электронной плотностью в
молекуле. Для производных бензола это подразумевает наличие электронодонорных
либо электроноакцепторных заместителей соответственно. Соединения первого типа,
например, фенол, анилин, называют p-избыточными, а второго типа -
p-дефицитными. К ним относятся нитробензол, ацетофенон и др. p-Избыточные арены
более активны, чем бензол, в реакциях с электрофилами и меньше подвержены
нуклеофильной атаке, p-дефицитные - наоборот неохотно воспринимают атаку
электрофилов и более легко реагируют с нуклеофилами.
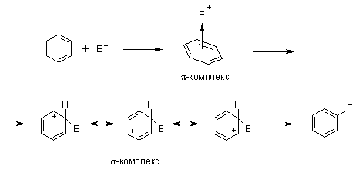
2. Механизм реакций
ароматического электрофильного замещения
Характерными превращениями
производных бензола являются реакции, в ходе которых происходит замещение атома
водорода при углеродном атоме кольца под действием электрофила (для краткости
обозначается - SE2). Менее распространены реакции электрофильного замещения
функциональных групп - так называемое ипсо-замещение.
Электрофилом называют частицу,
имеющую вакантную орбиталь, на которую может быть принята пара электронов для
образования s-связи. По современным представлениям, реакция электрофильного
замещения включает в себя два этапа. Сначала образуется p-комплекс электрофила
с ароматическим кольцом, которое выступает в роли p-донора. p-Комплекс затем
перерождается в катионный s-комплекс - частицу, содержащую один sp3-гибридный атом в кольце. При этом одна пара p-электронов
превращается в связывающую пару новой экзоциклической s-связи. На этой стадии
происходит разрушение ароматичности ядра, для чего требуется большая затрата
энергии. Поэтому активационный барьер данной стадии чаще всего является
максимальным на всей координате реакции, а сама стадия является лимитирующей. В
отличие от s-комплекса, возникающего в реакциях ионного присоединения к
алкенам, аренониевый у-комплекс стабилизирован посредством делокализации
положительного заряда, который оказывается сосредоточенным в пара- и двух
орто-положениях по отношению к реагирующему атому. Второй этап представляет
собой обратный процесс - отрыв протона от s-комплекса с образованием конечного
продукта. Это - стадия ароматизации, т.е. возрождение циклической p-электронной
системы, она протекает при участии противоиона электрофила или частиц среды,
которые отрывают и акцептируют протон.
Изменения энергетики
частиц в процессе замещения в кольце бензола можно представить на графике
энергетического профиля реакции SE2.

3. Правила
ориентации электрофильного замещения под влиянием заместителей в ароматическом
кольце
Замещение в бензоле
равновероятно происходит при любом из шести атомов углерода, т.к. полная и
равномерная делокализация электронной плотности делает все атомы кольца
эквивалентными. Другими словами, при монозамещении в бензоле возможно
образование лишь одного продукта. Напротив, проведение реакции SE2 в бензольном кольце, уже имеющем
заместитель, может с разной степенью вероятности привести к образованию четырех
продуктов, а именно - к орто-, мета-, пара-дизамещенным
бензолам и продукту ипсо-замещения. Практика показывает, что в
большинстве случаев соотношение изомеров отличается от статистического 2:2:1:1.
Ипсо-электрофильное замещение в бензолах, имеющих свободные положения,
вообще встречается редко, а соотношение орто-, мета- и пара-изомеров
в большой степени зависит от природы заместителя. На этой зависимости
базируются правилами ориентации.
Основные закономерности
состоят в следующем: заместители, способные подавать электронную плотность в
бензольное кольцо, ориентируют вхождение реагента в орто- и пара-положения.
Такие называют заместителями первого рода. Заместители, более
электронодефицитные, чем водород, (заместители второго рода) направляют
реакцию в мета-положение. При этом в первом случае реакция протекает,
как правило, легче, чем в бензоле, во втором - труднее. Исключение составляют
галогенарены, которые реагируют преимущественно своим пара-положением,
но медленнее, чем бензол.
Анализируя
энергетический профиль реакции, можно увидеть два фактора, определяющих
легкость и ориентацию замещения:
а) величина энергии
активации на стадии образования s-комплекса;
б) относительная
стабильность s-комплекса
В связи с этим
существуют два варианта объяснения причин ориентации - кинетический и термодинамический.
В гомологах бензола
симметрия молекулы и электронная плотность на кольцевых атомах углерода
нарушены из-за наличия заместителей. Происходящее перераспределение электронной
плотности приводит к тому, что шесть бензольных углеродных атомов перестают
быть эквивалентными. По этой же причине замещенные бензолы, в отличие от самого
бензола, имеют дипольный момент.
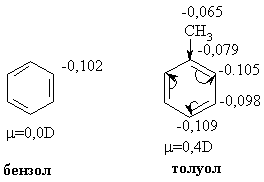
Электрофильное замещение
происходит при том С-атоме, на котором сосредоточен больший отрицательный
заряд, т.к. это связано с большим выигрышем в энергии.
Деформации электронной
системы кольца вызывают не только алкильные группы, но и другие заместители.
Кинетическое объяснение
ориентации требует рассмотреть характер взаимодействия заместителей с
p-электронной системой ароматического кольца. Заместители первого рода в
большинстве случаев обладают более высокой, чем бензольное кольцо, электронной
плотностью, и поэтому проявляют электронодонорный характер. К ним относятся
алкилы (+I-эффект), группы с неподеленным электронными парами: - NH2, - NR2
- OH, - OR, - O - - Cl, - Br, - I и др. (-I, +M-эффект). Мезомерные
доноры обычно ориентируют более эффективно, чем индуктивные. Заместители
второго рода, имея пониженную в сравнении с бензолом электронную плотность,
проявляют электроноакцепторный эффект. К ним относятся группы, имеющие
положительный заряд катионного типа (NR3+),
или такие, в которых положительный заряд обусловлен внутренней поляризацией (-CF3, - NO2,
- SO3H, - CHO, - CN, - COOH и др.). Группы - акцепторы различаются на электромерные (NR3+, SR2+
CF3), обладающие только - I
- эффектом, и мезомерные, способные к сопряжению с p-системой (-I, - M - эффект). К последним
относятся - N2+, - NO2,
- SO3H, - CHO, - CN, - COOH. Влияние заместителей разного рода наглядно представляется
набором резонансных структур для анилина и бензальдегида.

Легко видеть, что
заместитель оказывает влияние на p-заряд одновременно в орто- и пара-положениях.
Это положения называются сопряженными. мета-Положение не сопряжено ни с
донором, ни с акцептором, поэтому влияние заместителя на него минимально.
На второй стадии реакции
SE2 в замещенных бензолах образование
s-комплекса происходит за счет связывания электрофила не с любым углеродным
атомом, а с тем, на котором локализована большая p-электронная
плотность, т.е. отрицательный заряд. В этом случае энергия активации имеет
меньшую величину и лимитирующая стадия реакции идет быстрее. Среди канонических
структур для соединений с донорными заместителями имеются такие, в которых
отрицательный заряд и пара электронов находятся на орто- и пара-углеродных
атомах. Вклад этих структур в резонансный гибрид и определяет ориентацию атаки
электрофила. Электроноакцепторные заместители, напротив, понижают электронную
плотность преимущественно в орто- и пара-положениях и это
обусловливает атаку реагента в мета-положение (положение с наименее
пониженной электронной плотностью). Заметим, что снижение электронной плотности
в кольце вообще затрудняет образование как p-, так и s-комплекса (поскольку
делает кольцо менее восприимчивым к атаке электрофила), т.е. замедляет любую
электрофильную реакцию.
Термодинамическое
объяснение ориентации основано на сравнении энергии различных s-комплексов,
образования которых можно ожидать в ходе реакции. Стабильность s-комплекса
непосредственно зависит от степени делокализации в нем положительного заряда.
Для соединений с донорными мезомерными заместителями она является максимальной
в случае присоединении электрофила к орто- или пара-положениям,
т.к. при этом в делокализации положительного заряда наряду с p-электронами
бензольного кольца участвует неподеленная электронная пара самого заместителя.
Резонансные структуры
для s-комплексов, которые могут образоваться в реакции анизола с бромом:
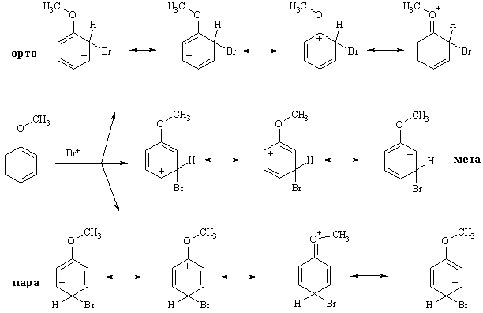
Очевидно, что орто-
и пара - s-комплексы энергетически более выгодны, чем мета-s-комплекс,
т.к. положительный заряд рассредоточен по большему количеству атомов.
Электроноакцепторные
заместители не участвуют в делокализации (+) - заряда в s-комплексе и поэтому
совершенно не способствуют орто- и пара-ориентации электрофила,
но зато в меньшей степени «мешают» атаке электрофила в мета-положение.
Резонансные структуры
для s-комплексов при нитровании бензотрифторида (соединение с электромерным
заместителем второго рода)
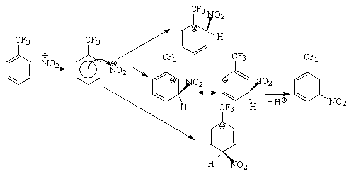
электрофильный
замещение арен ароматический
Атомы галогенов, являясь орто-,
пара-ориентантами, тем не менее затрудняют реакцию. Это объясняют тем, что
их сильный - I-эффект в стационарном состоянии молекулы снижает электронную
плотность в кольце, что замедляет реакцию, но при образовании s-комплекса
вступает в действие +М-эффект, и галоген становится донором электронов.
В тех случаях, когда в кольце
бензола находится два заместителя, ориентацию замещения рекомендуют определять
по следующим правилам:
1. Два заместителя одного
рода ориентируют согласованно, если они находятся в мета-положении
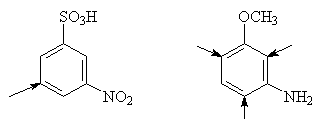
2. Два заместителя разного
родаориентируют согласованно, если они расположены в орто-, пара-положениях
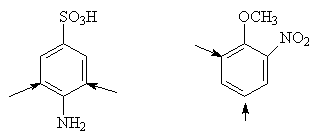
3. Когда отсутствуют условия
согласованной ориентации, замещение может происходить во все свободные
положения, но при этом направление в основном определяется более
электронодонорным заместителем.
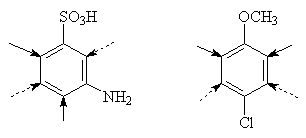
4. Примеры реакций
электрофильного замещения
Гомологи бензола вступают в реакции
электрофильного замещения легче, чем бензол, из-за электронодонорного влияния
алкильных заместителей. В результате монозамещения образуются, как правило, все
три изомера с преобладанием орто- и пара-продуктов.
Дейтерирование
Дейтерирование аренов осуществляют
действием концентрированных дейтерокислот. Так, толуол дейтерируется по
положениям 2, 4 и 6 при комнатной температуре, будучи растворенным в D2SO4.
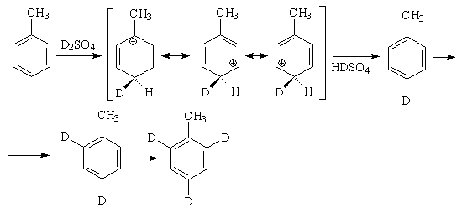
Как видно из рисунка,
правила ориентации соблюдаются.
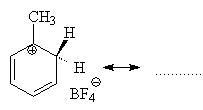
Факты протонирования
аренов зафиксированы не только приборными методами, некоторые s-комплексы
получены и выделены как индивидуальные вещества. Например, ониевая «соль» толуола
с тетрафторбороводородной кислотой устойчива при низких температурах и имеет
Т.пл. -65 оС (толуол проявляет свойства основания). Ее строение
изображается формулой
Галогенирование
Для введения галогена в
ароматическое кольцо в качестве реагентов используют комплексы галогенов с
кислотами Льюиса. Роль последних заключается в поляризации связи
галоген-галоген, в результате чего один из атомов приобретает положительный
заряд, тогда как другой образует связь с кислотой Льюиса за счет ее вакантной d-орбитали.

На практике вместо
безводного бромида алюминия часто применяют железные стружки. При добавлении в
реакционный сосуд галогена (хлора или брома) железо превращается в галогенид
железа (III), который является сильной кислотой Льюиса и тоже катализирует
реакцию.
Нитрование
Бензол и его гомологи
превращаются в нитросоединения действием нитрующей смеси, которая состоит из
концентрированных серной и азотной кислот. Нитрующей частицей (электрофилом)
является катион нитрония NO2+,
существование которого в нитрующей смеси доказано криоскопическим методом:
измерения температур замерзания азотной и серной кислот и их смеси указывает на
присутствие четырех частиц в растворе.

Можно нитровать азотной
кислотой без применения серной, но реакция идет медленнее, т.к. равновесие
смещено влево. Еще медленнее реакция идет, когда азотную кислоту используют в
растворах уксусной кислоты, нитрометана и т.д., поэтому такие условия применяют
для нитрования соединений с повышенной электронной плотностью.
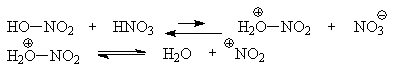
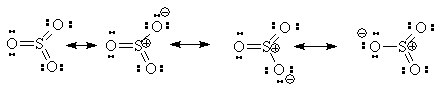
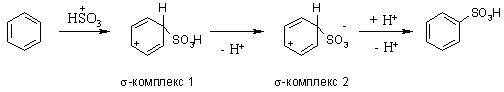
Сульфирование
Реакция сульфирования
аренов, как считают, протекает в олеуме при действии триоксида серы, а в серной
кислоте - с участием катиона HSO3+.
Триоксид серы проявляет электрофильный характер благодаря полярности связей S-O.
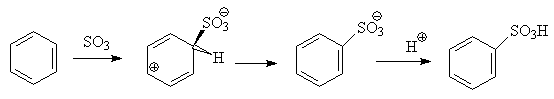
В результате атаки
триоксида серы образуется s-комлекс, который представляет собой биполярную
нейтральную частицу. Вероятно, поэтому, в отличие от других реакций
электрофильного замещения, где у-комплекс положительно заряжен, в этом случае
медленной стадией является не образование s-комплекса, а отщепление протона и
реароматизация.
При сульфировании серной
кислотой катион гидросульфоксония образуется в результате следующей обменной
реакции:
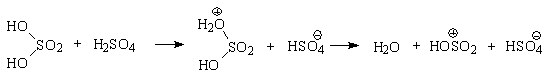
Образующийся при
сульфировании серной кислотой заряженный s-комплекс находится в равновесии с
нейтральным s-комплексом. Это объясняет тот факт, что скорость реакции и в этом
случае определяется скоростью стадии реароматизации.
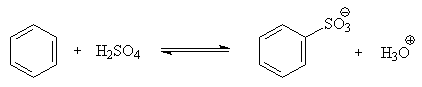
Реакция сульфирования
обратима. Это подтверждается тем, что выход бензолсульфокислоты увеличивается,
если при проведении реакции производится азеотропная отгонка воды избытком
бензола. Кроме того, нагревание аренсульфокислот с 25-50%-ной серной кислотой
приводит к удалению сульфогруппы.
Поэтому сульфирование
зачастую применяют для защиты определенного положения в ядре в многостадийных
синтезах, например:
Алкилирование по
Фриделю-Крафтсу
Одним из способов
получения гомологов бензола является реакция алкилирования. Превращение носит
имена Фриделя и Крафтса, которые его открыли. Алкилирующими агентами в этой
реакции являются галогеналканы, активированные галогенидом алюминия. Считают,
что катализатор - кислота Льюиса - поляризует связь С-галоген, создавая на
атоме углерода дефицит электронной плотности, т.е. механизм аналогичен реакции
галогенирования

Этот углеродный атом
является электрофилом, и атакует молекулу арена. При планировании синтеза
Фриделя-Крафтса очень важно учитывать строение алкильного радикала, который
предполагается ввести в ароматическую молекулу. Природа алкилов, входящих в
комплекс с кислотой Льюиса, близка к катионной. Известно, что устойчивость
карбокатионов зависит от их структуры и падает в ряду третичный > вторичный
> первичный. Поэтому при электрофильном алкилировании может происходить и
часто происходит перегруппировка промежуточных катионов в более устойчивые:
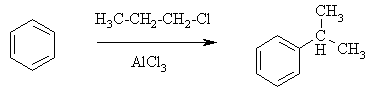
первичных во вторичные и третичные, вторичных - в третичные. Так,
взаимодействие 1-хлорпропана с хлоридом алюминия может быть представлено одной
из схем
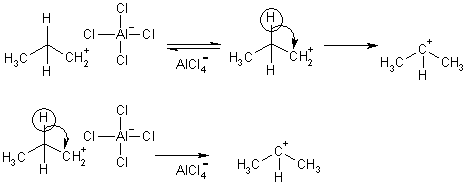
Именно такой
перегруппировкой объясняется то, что при попытке синтеза н-пропилбензола из
бензола и 1-хлорпропана получают изопропилбензол (кумол).
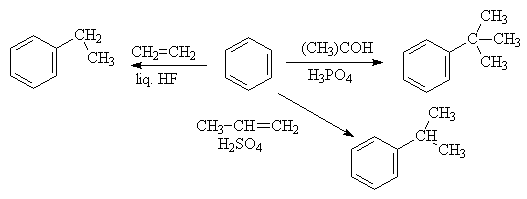
Для алкилирования аренов
не обязательно применять галогеналканы. В реакции могут использоваться алканолы
и алкены, этиленоксид; из них также генерируются карбокатионы при действии
сильных протонных и Льюисовских кислот.
Ацилирование по
Фриделю-Крафтсу
Сходной с реакцией
алкилирования является реакция ацилирования ароматических соединений. В
качестве реагентов применяют ангидриды или галогенангидриды карбоновых кислот,
продуктами являются ароматические кетоны. Механизм этой реакции включает
образование комплексного соединения между ацилирующим реагентом и кислотой
Льюиса. В результате положительный заряд на атоме углерода несравненно
возрастает, что делает его способным к атаке ароматического соединения.

Нужно отметить, что, в
отличие от реакции алкилирования, в данном случае необходимо брать избыток
катализатора по отношению к количеству реагентов, т.к. продукт реакции (кетон)
сам способен к комплексообразованию и связывает кислоту Льюиса.
5. Реакции боковых
цепей аренов
Бензольное кольцо в
обычных условиях устойчиво к действию окислителей (KMnO4,
K2Cr2O7), алкильные заместители, напротив, весьма легко подвергаются
окислению и взаимодействуют с другими реагентами радикального характера.
Наиболее реакционноспособным центром боковой цепи является атом углерода,
непосредственно связанный с ароматическим ядром, так называемый a-атом.
Действие перманганата
калия в воде или бихромата калия в кислой среде окисляет алкильный заместитель.
Окисление происходит таким образом, что вся боковая цепь «отгорает», а
связанный с кольцом атом углерода окисляется до карбоксильной группы, и
образуется аренкарбоновая кислота. Если алкильных заместителей несколько, то
образуются многоосновные кислоты.
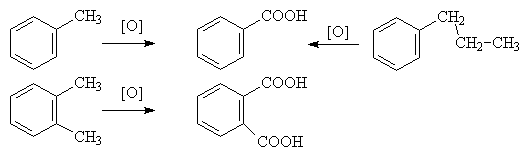
Гомологи бензола легко
реагируют с галогенами на свету. В отличие от реакции с участием кислот Льюиса
(см. выше), в данном случае происходит галогенирование боковой цепи. Как и при
действии окислителей, в реакцию вступает a-углеродный атом. Промежуточно
образующийся бензильный радикал по стабильности близок к аллильному благодаря
тому, что ароматическое кольцо принимает участие в делокализации неспаренного
электрона.
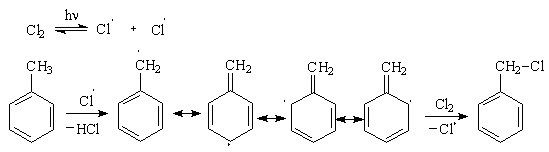
Процесс хлорирования
протекает до полного замещения всех a-водородов галогеном и образования
бензотрихлорида; в то же время, бромирование можно остановить на стадии
замещения одного или двух атомов водорода, и в качестве продукта реакции
получить бромистый бензил или бромистый бензилиден.
6. Реакции
присоединения к ароматическому кольцу
Бензол и его гомологи
присоединяют три моля водорода в присутствии катализатора при повышенной
температуре. Превращение бензольного кольца в циклогексановое сопровождается
выделением тепла (теплоты гидрирования), количество которого служит
сравнительной мерой устойчивости соединения
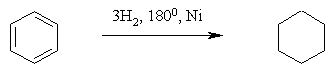
При действии на арены
натрием в жидком аммиаке в присутствии этанола происходит присоединение двух
атомов водорода в пара-положения кольца (восстановление по Берчу).
Реакция протекает с двумя стадиями одноэлектронного переноса от атома натрия к
ароматическому субстрату.
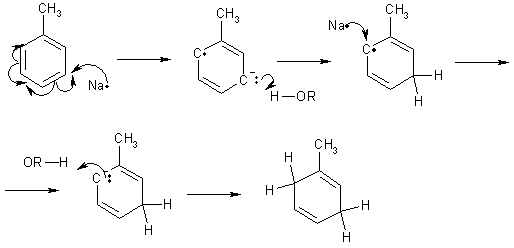
Реакция останавливается
на стадии моноприсоединения по причине того, что полученный циклогексадиен не
имеет циклического сопряжения и не способен к образованию стабильного
анион-радикала.
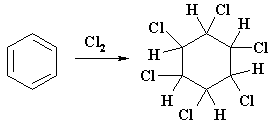
Бензольное кольцо при
облучении ультрафиолетом присоединяет хлор, превращаясь в смесь стереоизомеров
гексахлорциклогексана (гексахлоран). Это вещество ранее широко использовалось в
качестве пестицида, хотя лишь один из стереоизомеров является активным.
Литература
1. Грандберг, И.И.
Органическая химия: Учебник для студ. вузов, обучающихся по агроном. спец.
[Текст]. - Изд. 5-е, стереотип. - М.: Дрофа, 2002. - 672 с. - 10 000 экз. -
ISBN 5-7107-6129-Х.
. Петров, А.А., Бальян,
Х.В., Трощенко, А.Т. Органическая химия: Учебник для вузов [Текст] / Под ред.
Стадничука М.Д. - Изд. 5-е, перераб. и доп. - СПб.: «Иван Федоров», 2002. - 624
с. - 5000 экз. - ISBN 5-81940-067-4.
. Лабораторные работы по
органической химии [Текст] / Под ред. Гинзбурга, А.А. и Петрова, А.А. - М.:
Высшая школа, 2002. - 269 с.