Исследование системы гомеостаза железа и развития окислительного стресса методами математического моделирования
Оглавление
Введение
. Обзор литературы
.1 Роль железа в развитии окислительного стресса
.2. Система гомеостаза железа
.2.1. Клеточный гомеостаз железа
.2.2. Системный гомеостаз железа
. Материалы и методы
.1 Материалы
.2 Объект исследования
.3 Выделение клеток
.4 Определение концентрации трансферрина в плазме крови и в
асцитной жидкости опухоленосителя
.5 Определение параметров окислительного стресса в крови и асците
.6 Построение математической модели.
. Результаты и обсуждение
.1 Математическая модель клеточного гомеостаза железа
.2 Математическая модель регуляции системного гомеостаза железа
.3 Изучение динамики развития асцитной гепатомы Зайделя.
.4 Идентификация генов, участвующих в гомеостазе железа.
Выводы
Список литературы
Введение
В постгеномную эру основной задачей системной
биологии является исследование принципов организации и функционирования сложных
молекулярно-генетических сетей, обеспечивающих формирование фенотипических
(молекулярных, биохимических, физиологических, морфологических, поведенческих и
др.) характеристик организмов. Развитие высокоэффективных методов молекулярной
биологии привело к накоплению огромного числа экспериментальных данных по
структурно-функциональной организации генных сетей и их
молекулярно-генетических компонент. Анализ этих экспериментальных данных
принципиально невозможен без использования современных информационных
технологий и эффективных математических методов анализа данных и моделирования
биологических систем и процессов. Это необходимо для понимания принципов их
организации, молекулярных механизмов функционирования, закономерностей
эволюции, оценки влияния мутаций на функцию генных сетей. Таким образом,
теоретическое исследование динамики генных сетей методами математического
моделирования приобретает в настоящее время фундаментальное и первоочередное
значение.
Целью данной работы было исследование системы,
контролирующей гомеостаз железа и развитие окислительного стресса у
млекопитающих. Для достижения этой цели были поставлены следующие задачи:
. Создание формального описания и математической
модели системы клеточного гомеостаза железа.
. Создание формального описания и математической
модели системы, контролирующей гомеостаз железа организма в целом в норме и при
развитии окислительного стресса.
. Изучение с помощью численных методов динамики
системы, контролирующей гомеостаз железа и развитие окислительного стресса.
. Экспериментальное изучение динамики
параметров, связанных с развитием окислительного стресса и метаболизмом железа,
при развитии асцитной гепатомы Зайделя.
Математическая модель, созданная в ходе данной
работы, будет полезна для исследования системы гомеостаза железа у
млекопитающих в норме и при патологии, а также процесса доставки лекарств, в
частности, в ходе химиотерапии.
1.
Обзор литературы
1.1
Роль железа в развитии окислительного стресса
Уникальная способность железа изменять своё
состояние окисления и окислительно-восстановительный потенциал в ответ на смену
состава окружающих лигандов делает этот металл необходимым почти всем живым
существам. Железо легко вступает в одноэлектронные
окислительно-восстановительные реакции, переходя при этом между Fe2+
и Fe3+
состояниями. Железосодержащие белки являются ключевыми компонентами многих
биологических процессов, таких как энергетический обмен, транспорт кислорода,
репликация и репарация ДНК, нейтрализация активных форм кислорода и многих
других, катализируемых ферментами оксигеназами, пероксигеназами и.т.п. Однако
те же самые химические свойства железа делают его опасным для организма. Даже малое
количество “свободного” железа может катализировать образование высокотоксичных
радикалов посредством цикла реакций Фентон/Хабер-Вейса. В присутствии перекиси
водорода железо катализирует образование гидроксил радикалов (уравнение 1) -
реакция Фентона. Далее окисленный металл может быть восстановлен супероксид
радикалом (уравнение 2). Суммарная реакция называется реакцией Хабер-Вейса
(уравнение 3). Она может происходить и в отсутствии переходных металлов, но
присутствие железа значительно увеличивает скорость реакции [1]. Взаимодействие
гидроксил радикала с компонентами клетки может приводить к окислению белков,
липидов, липопротеинов, нуклеиновых кислот, углеводов.
Fe2+
+
H2O2
à Fe3++
-OH + *OH
(уравнение 1),
Fe3+ + O2*-
à Fe2+ + O2
(уравнение
2),2O2 + O2*- à
-OH + *OH
+ O2 (уравнение
3).
Так как “свободное” железо опасно, но в тоже
время жизненно необходимо для организма, существует система, контролирующая его
уровень. В нормальном состоянии доля железа, доступного для продукции свободных
радикалов, составляет 3-5% от общего количества железа клетки. Эта часть также
называется хелатируемым железом (chelatable
iron) или лабильным
пулом железа(labile
iron pool).
Лабильный пул железа содержит обе ионные формы железа (Fe2+
и Fe3+),
связанные с низкой аффинностью с лигандами разной природы: цитратом, фосфатом,
углеводами и карбоксилатами, нуклеотидами, нуклеозидами, полипептидами и
фосфолипидами [2,3].
1.2
Система гомеостаза железа
Систему гомеостаза железа в организме можно
разделить на два основных уровня организации: клеточный гомеостаз, включающий в
себя процессы, происходящие на уровне клетки, и системный, охватывающий
процессы, происходящие в организме в целом. Оба уровня характеризуются
определенным набором белков, веществ и констант реакций, описывающих кинетику
их взаимодействий.
1.2.1
Клеточный гомеостаз железа
Клеточный гомеостаз железа опосредуется
процессами транспорта железа в клетку, его запасания, внутриклеточного
перераспределения и экспорта из клетки в интерстиций и циркуляторное русло. На Рис.
1 представлена схема поддержания клеточного гомеостаза железа.
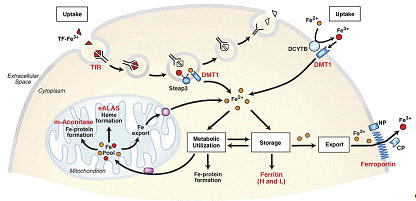
Рисунок 1.
Схема поддержания клеточного гомеостаза железа. Комплекс трансферрина с Fe3+
связывается с TfR1 и TfR2
на поверхности клеточной мембраны. Эти комплексы подвергаются эндоцитозу.
Кислая среда эндосом приводит к диссоциации Fe3+
от трансферрина. Fe3+
восстанавливается редуктазой Steap3
перед тем как транспортироваться из эндосом посредством DMT1(divalent
metal transporter 1). Рецепторы и их комплексы с трансферрином возвращаются на
клеточную мембрану, где могут вступать в новый цикл эндоцитоза. DMT1
также находится на апикальной мембране энтероцитов, где транспортирует в клетку
Fe2+,
восстановленный редуктазой DCYTB.
Железо, поступившее в клетку входит во внутриклеточный лабильный пул железа.
Белки IRP реагируют
на изменение количества железа в этом пуле и регулируют трансляцию мРНК,
содержащих IRE в 5’-UTR
(H- и L-ферритин,
eALAS, m-aconitase,
ferroportin), и стабильность
мРНК, содержащих IRE
в
3’-UTR (TfR1
и DMT1) (взято из [16]).
Потребление железа клетками
Клетки млекопитающих способны получать железо из
плазмы крови, где, в нормальном состоянии, большая часть железа находится в
комплексе с трансферрином. Большинство клеток способно получать железо из этого
комплекса посредством рецептор-индуцируемого эндоцитоза (рецепторы TfR1
и TfR2). Однако, для
некоторых типов клеток, таких как гепатоциты, клетки меланомы, характерен
дополнительный, трансферрин-независимый путь потребления железа. Гепатоциты
способны получать железо из комплекса с ферритином, гем-гемопексином,
гемоглобин-гаптоглобином также посредством рецептор-индуцируемого эндоцитоза
[4, 5]. Альтернативная система потребления железа, имеющая место в
эпителиальных клетках, представлена белком липокалином и его рецептором [6].
Транспорт железа в клетку посредством
трансферринового рецептора 1 (TfR1)
изучен лучше, чем процессы, связанные с TfR2.
Трансферрин - гликопротеид, содержащийся в большом количестве в плазме крови и
с высокой аффиностью связывающий две молекулы Fe3+.
При внеклеточном pH~7.4
холотрансферрин (трансферрин, связанный с двумя молекулами железа) способен
быстро связываться с TfR1.
Более высокое сродство холотрансферрина к TfR1
приводит к вытеснению апотрансферрина (не связанного с железом трансферрина) из
комплекса с рецептором [7]. TfR1
рецептор-индуцируемый эндоцитоз происходит в окаймленных ямках - специальных
структурных образованиях на плазматической мембране. Следует заметить, что TfR1
конститутивно концентрируется в окаймленных ямках и подвергается эндоцитозу
даже в отсутствие лиганда [8]. Вследствие инвагинации окаймленной ямки внутрь
клетки формируется окаймленный пузырек, который быстро теряет клатриновую
оболочку и превращается в эндоцитозный пузырек. Он, в свою очередь, сливается с
ранними эндосомами, где поддерживается низкий pH
за счет работы протонных насосов [9]. Кислая среда вызывает конформационные
изменения как в молекуле TfR1,
так и в трансферрине, что приводит к диссоциации трехвалентного железа от
комплекса. При этом трансферрин остается связанным c
рецептором. Далее редуктаза Steap3
восстанавливает Fe3+
до Fe2+
[10], которое транспортируется из эндосомы в цитоплазму с помощью белка DMT-1.
TfR1 в комплексе с
апотрансферрином возвращается на плазматическую мембрану, где может вступать в
новый цикл эндоцитоза.
Потребление клетками железа из комплекса с
трансферрином может также осуществляться посредством рецептора второго типа - TfR2.
В то время как известно, что TfR1-зависимый
транспорт железа является основным путем доставки его в клетку, роль TfR2
до сих пор изучена слабо. Несмотря на большую гомологию этих рецепторов, TfR2
значительно отличается по своим свойствам и функциям от TfR1.
В то время как TfR1
экспрессируется почти во всех тканях организма, экспрессия TfR2
тканеспецифична и происходит в основном в печени и в меньшей степени в
селезенке, легких, мышцах и предстательной железе. В отличие от TfR1,
рецептор второго типа локализуется на поверхности мембраны не в ямках,
окаймленных клатрином, а в кавеолах, образуемых белком кавеолином. Что,
по-видимому, и приводит к различиям во внутриклеточной локализации этих
рецепторов [8]. TfR2
характеризуется меньшим сродством и меньшей специфичностью по отношению к
различным формам трансферрина [11]. Также было показано, что этот путь
транспорта не насышается трансферрином, по крайней мере, вплоть до концентрации
трансферрина в 2 µM [12].
Недавно было обнаружено, что холотрансферрин стабилизирует TfR2,
предотвращая его попадание в лизосомы, где он деградирует. Это привело к
формированию гипотезы о том, что рецептор второго типа может играть роль
сенсора железа в плазме крови [13, 14, 15]. Более подробно этот процесс будет
рассмотрен в разделе 2.2. "Системный гомеостаз железа".
Хотя трансферрин-зависимое потребление железа
превалирует в нормальном состоянии, при патологиях, сопровождающихся
увеличением концентрации железа, не связанного с трансферрином, возможен
другой, трансферрин-независимый путь. Как уже упоминалось выше, железо, не
связанное с трансферрином, в плазме крови связано с низкой аффиностью с
лигандами различной природы [2,3]. Некоторые клетки, к примеру, гепатоциты,
способны получать железо из этих комплексов. Вне зависимости от формы железа,
не связанного с трансферрином, на первом этапе происходит связывание с
поверхностью клетки, где железо диссоциирует из комплекса с лигандом. Далее
может происходить восстановление железа посредством неизвестной на данный
момент гипотетической редуктазы. Считается, что доставка восстановленного
железа в клетку осуществляется посредством транспортера, природа которого на
данный момент не известна, но наиболее вероятными кандидатами являются белки DMT1,
ZIP14 и кальциевые
каналы [5].
Хранение железа в клетках
Основным местом запасания железа в организме
являются клетки печени - гепатоциты. Макрофаги печени, селезенки и костного
мозга также депонируют некоторую, но гораздо меньшую часть железа, полученного
из фагоцитируемых ими эритроцитов. Большая часть железа печени находится в
комплексе с белком ферритином или гемосидерином (примерно 80%), 5%
ассоциировано с трансферрином, 2% - с гемом, а оставшаяся часть находится в
лабильном пуле железа.
Железо, поступившее в клетку, попадает во
внутриклеточный лабильный пул железа, откуда может образовывать комплекс с
ферритином, гемом, другими железосодержащими белками или экспортироваться из
клетки.
железо стресс окислительный гомеостаз
Ферритин - гидрофильная молекула, состоящая из
24 субъединиц, формирующих полую сферу. Внутри этой сферы может разместиться до
4500 атомов железа. Субъединицы ферритина могут быть двух типов: тяжелые и
легкие. Соотношение тяжелых и легких субъединиц варьируется в зависимости от
типа клеток и физиологического статуса. Тяжелая субъединица ферритина обладает
ферроредуктазной активностью. Синтез ферритина индуцируется при высоком уровне
внутриклеточного железа и подавляется при нехватке железа. Эта регуляция
опосредуется IRE-IRP
системой, которая будет описана ниже. Также, синтез ферритина регулируется на
уровне транскрипции цитокинами, такими как интерферон-γ,
интерлейкин
1 и интерлейкин 6. Деградация ферритина и, следовательно, высвобождение железа,
помогает мобилизовать железо для нужд клетки. О деградации ферритина известно
немного, но в этот процесс вовлечены как протеосомный, так и лизосомный аппарат
клетки.
Гемосидерин - нерастворимая в воде молекула,
присутствующая в лизосомах, и, по-видимому, являющаяся промежуточным продуктом
деградации ферритина. Железо, хранящееся в комплексе с гемосидерином, гораздо
менее доступно и менее эффективно в продукции свободных радикалов, чем в
комплексе с ферритином.
Экспорт железа из клетки
Некоторые клетки млекопитающих, такие как
энтероциты двенадцатиперстной кишки, макрофаги, гепатоциты и клетки центральной
нервной системы обладают системой контроля экспорта железа. На данный момент в
литературе известен только один экспортер железа - белок ферропортин. Этот
транспортер переносит железо через клеточную мембрану в восстановленной форме.
В плазме крови железо окисляется различными феррооксидазами прежде чем свяжется
с трансферрином. Чаще всего в роли оксидазы выступает церулоплазмин, в случае
энтероцитов - гефестин, а в случае астроцитов - гликозилфосфатидилинозитольная
форма церулоплазмина, непосредственно взаимодействующая с ферропортином.
Регуляция клеточного гомеостаза
железа
Система белков, участвующих в клеточном
гомеостазе железа, регулируется на нескольких уровнях: транскрипции генов,
стабильности мРНК, трансляции мРНК и посттрансляционной модификации белков.
Посттранскрипционный уровень регуляции наиболее
хорошо изучен. Многие мРНК, кодирующие белки, вовлеченные в поддержание
гомеостаза железа, содержат в нетранслируемой части последовательности IRE
(iron-responsive
element).
IRE -
консервативные последовательности, образующие шпильки, с которыми
взаимодействуют специальные регуляторные белки IRPs
(iron regulatory
proteins). IRE
последовательности располагаются в 5’-нетранслируемых регионах (5’-UTR)
мРНК таких белков как тяжелая и лёгкая субъединицы ферритина, ферропортин (FPN),
ALAS-E
(5-aminolevulinic acid synthase) и митохондриальная аконитаза. Образование IRE/IRP
комплекса на 5’-UTR
приводит к ингибированию ранних этапов трансляции. IRE
также встречаются в 3’-нетранслируемой области (3’-UTR)
мРНК таких белков как рецептор трансферрина первого типа (TfR1)
и DMT1. Образование IRE/IRP
комплекса на 3’-UTR
приводит стабилизации мРНК [16, 17].
Всего на данный момент описаны два вида IRP
белков: IRP1 и IRP2,
которые являются ключевыми сенсорами цитоплазматического железа. При недостатке
железа в клетке IRP
связываются с IRE, ингибируя
трансляцию с участием мРНК, содержащих IRE
в 5’-UTR, и
стабилизируя мРНК, содержащие IRE
в 3’-UTR. Регуляция
активности связывания IRP
c IRE
различна для IRP1 и IRP2.
При недостатке железа в клетке IRP1
связывается с IRE с высокой
аффиностью, а при повышении уровня железа в IRP1
собирается кластер 4Fe-4S,
что приводит к утрате высокой аффинности и приобретению аконитазной активности.
Повышение уровня активных форм кислорода приводит к разборке 4Fe-4S
кластера, а, следовательно, к включению IRE-связывающей
активности IRP1.
Фосфорилирование IRP1
регулирует его активность, что является железо-независимым путем контроля [18,
16].
Несмотря на высокую гомологию IRP
белков, IRP2 не
проявляет аконитазной активности и не способен связывать 4Fe-4S
кластер. Активность IRP2
регулируется на посттрансляционном уровне. В отличие от IRP1,
IRP2 содержит
определенную N-концевую
последовательность, являющуюся сенсором железа. Три из пяти цистеинов этой
последовательности координируют ион железа, а один из них способен окисляться,
формируя дигидроцистеин, что приводит к убиквитинированию и последующей
деградации с участием протеосом. N-концевая
последовательность может связываться с гемом, индуцируя кислород-зависимое
окисление и последующую деградацию IRP2
[19]. Активность IRP2
также регулируется фосфорилированием, что приводит к повышению аффинности
связывания с IRE [18, 16].
Важной составляющей регуляции системы,
поддерживающей гомеостаз железа, является контроль на уровне транскрипции.
Экспрессия генов таких белков, как тяжелая субъединица ферритина, рецептор
трансферрина первого типа (TfR1),
гепсидин и ферропортин, регулируется цитокинами интерфероном-γ,
интерлейкином
1 и 6, а также фактором некроза опухоли α
(TNFα). Гипоксия
также является важным фактором, который способен вносить коррективы в
реализацию информации генов TfR1,
CP (церулоплазмин), FPN
(ферропортин), DCYTB
(дуоденальный цитохром В), HAMP
(гепсидин), по-видимому, посредством активации индуцируемого гипоксией
транскрипционного фактора HIF-1
(hypoxia-inducible
factor 1) [17, 5].
Помимо этого, описано несколько
посттрансляционных механизмов регуляции белков этой системы. Ферропортин
посттрансляционно регулируется гепсидином, который способен индуцировать его
вхождение в цитоплазму и деградацию [20]. Рецептор трансферрина второго типа
регулируется степенью насыщения трансферрина железом: холотрансферрин
ингибирует деградацию TfR2
и, таким образом, повышает стабильность белковой молекулы [21, 22]. Высокий
уровень железа резко снижает представленность белка-транспортера железа DMT1
в апикальной мембране энтероцитов и индуцирует его вхождение в цитозольные
везикулы [5]. Фосфорилирование рецептора трансферрина первого типа протеин
киназой С уменьшает его количество на клеточной мембране [5].
1.2.2
Системный гомеостаз железа
В среднем человек содержит 4 грамма железа. В
нормальном состоянии 1-2 мг железа каждый день потребляется из пищи
дуоденальными энтероцитами. Железо, абсорбированное энтероцитами, по мере
необходимости поступает в плазму крови, где связывается с переносчиком -
трансферрином. Если такой необходимости нет, то железо остается в энтероцитах и
позже выводится из организма черех ЖКТ в составе клеток, отторгнутых по мере
износа. Примерно 3 мг железа циркулирует в комплексе с трансферрином. Это
железо потребляется многими клетками, в основном посредством TfR1-зависимого
рецептор-индуцируемого эндоцитоза. Большая часть этого железа предназначена для
развивающихся эритроцитов костного мозга, где оно потребляется со скоростью
примерно 22 мг в день и используется для синтеза гемоглобина. Большая часть
железа организма (65-70%) находится в циркулирующих эритроцитах. Старые или
поврежденные красные кровяные клетки удаляются из циркуляции макрофагами
ретикуло-эндотелиальной системы, где железо высвобождается из гемоглобина и
экспортируется обратно в плазму или запасается в комплексе с ферритином. Клетки
ретикуло-эндотелиальной системы высвобождают в плазму примерно 22 мг железа в
день, что компенсирует потребление клетками костного мозга. Другие клетки потребляют
железо из плазмы в небольшом количестве для синтеза железосодержащих белков,
таких как гем-содержащие цитохромы и содержащие железо-серный кластер белки.
Примерно 10-15% железа организма содержится в этих белках. Оставшиеся 20%
содержатся в “запасниках”, преимущественно в макрофагах и гепатоцитах. Потеря
железа организмом происходит за счет слущивания клеток кожи и слизистой, а
также при кровопотере [23].
Системные регуляторы
Транспорт железа между описанными выше
компартментами - строго контролируемый процесс, который может регулироваться в
соответствии с потребностями организма. Отдельные клетки поддерживают должный
внутриклеточный уровень железа, изменяя активность соответствующих белков на
различных уровнях (см. 2.1.5. "Регуляция клеточного гомеостаза
железа"). Помимо такой “внутренней” регуляции, клеточный транспорт
железа может контролироваться извне системными регуляторами, что наиболее
характерно для клеток ретикуло-эндотелиальной системы, клеток печени и
энтероцитов тонкой кишки. Открытие новых регуляторов метаболизма железа, таких
как белок гемохроматоза (HFE),
гепсидин, гемоювелин (HJV)
и TfR2 выявило сложность
системы поддержания гомеостаза железа, но, в тоже время, помогло улучшить общее
понимание работы этой системы. Как эти регуляторы взаимодействуют с системой
транспорта железа в местах его абсорбции, использования и запасания, чтобы
поддерживать баланс железа в организме будет описано ниже.
Ключевым моментом в понимании регуляции
системного гомеостаза железа было открытие регуляторного белка гепсидина. Этот
небольшой пептид синтезируется гепатоцитами и секретируется в плазму крови, где
ингибирует экспорт железа из различных типов клеток в кровь. Продукция
гепсидина понижается в ответ на стимулы, усиливающие экспорт железа из клеток (недостаток
железа, высокий уровень эритропоэза) и повышается в состоянии, когда необходимо
снизить экспорт железа в системное русло (избыток железа в крови, воспаление).
Гепсидин способен непосредственно взаимодействовать с ферропортином на
поверхности клеточной мембраны и индуцировать его вхождение в цитоплазму и
последующую деградацию, понижая, таким образом, скорость экспорта железа из
клетки [24]. На данный момент известно четыре регуляторных пути, контролирующих
продукцию гепсидина: (1) регуляция в ответ на изменение общего уровня железа,
(2) регуляция в ответ на изменение интенсивности эритропоэза, (3) регуляция в
ответ на воспаление и (4) “обязательный” сигнальный путь (mandatory
signaling
pathway).
Как принято считать на данный момент,
индикатором общего уровня железа в организме является степень насыщения
трансферрина железом в плазме крови. Путь передачи этого сигнала на гепсидин
ясен не до конца, хотя недавние работы по изучению взаимодействий трансферрина
и HFE c
TfR1 и TfR2
привели к гипотетической модели, в которой циркулирующее железо, связанное с
трансферрином, влияет на формирование комплекса HFE
с TfR2 на поверхности
гепатоцитов. Этот комплекс способен увеличивать продукцию гепсидина,
посредством пока неизвестного внутриклеточного сигнального пути [25, 26].
Рецептор трансферрина второго типа стабилизируется холотрансферрином, что
приводит к увеличению количества TfR2
на клеточной мембране при повышении уровня этой формы белка [21, 22]. HFE
и трансферрин связываются с рецептором трансферрина первого типа на частично
перекрывающихся сайтах и конкурируют за связывание. Увеличение уровня
холотрансферрина приводит к вытеснению HFE
из комплекса с трансферрином, и, как следствие, к перемещению HFE
из эндосом, содержащих TfR1,
на плазматическую мембрану. Высвобождение TfR1
из комплекса с HFE
приводит к усилению транспорта железа в клетку. HFE,
вытесненный из комплекса с TfR1,
связывается с рецептором трансферрина второго типа, формируя комплекс,
предположительно передающий сигнал на гепсидин [27, 25, 26] (cм.
Рис. 2).
Рисунок 2. Механизм
регуляции гепсидина при повышении насыщения трансферрина железом в плазме
крови. В норме TfR1
и HFE присутствуют в
виде комплекса на клеточной мембране (A’).
Холотрансферрин и HFE
конкурируют за связывание с TfR1.
В результате повышения насыщения трансферрина HFE
диссоциирует из комплекса c
TFR1 (Б’). Теперь HFE
связывается с TfR2, образуя
комплекс, передающий сигнал на гепсидин (В’) (взято из [25]).
В случае гипоксии или анемии низкое давление
кислорода индуцирует стабилизацию фактора гипоксии HIF-1α,
что приводит к продукции эритропоэтина почкой. Эритропоэтин усиливает
интенсивность эритропоэза и, таким образом, потребность железа костным мозгом.
Это приводит к мобилизации железа из запасников и усилению абсорбции
энтероцитами посредством понижения уровня гепсидина, несмотря на уровень железа
в плазме. Это наводит на мысль, что регуляция в ответ на изменение
интенсивности эритропоэза осуществляется посредством фактора, передающего сигнала
из костного мозга в гепатоциты на гепсидин. Возможными кандитатами на эту роль
могут быть растворимая форма рецептора трансферрина первого типа и GDF-15
(Growth Differentiation
Factor 15) [26].
Третий путь регуляции гепсидина контролируется
воспалением. Этот путь индуцируется преимущественно интерлейкином 6, вследствие
чего активируется Jak/Stat
путь передачи сигнала в ядро и осуществляется регуляция экспрессии гена
гепсидина. Взаимодействие между воспалением и регуляции посредством HJV/BMP
через Stat3 и Smad
белки в результате воздействия фактора роста TGF-β
иллюстрирует координированную работу сигнальных каскадов, участвующих в
регуляции экспрессии гепсидина [26, 28].
Недавние исследования показали, что регуляция
гепсидина при изменении общего уровня железа и интенсивности эритропоэза
зависит от активности дополнительного пути, который контролируется белком HJV(гемоювелином).
Предположительно, гемоювелин поддерживает передачу сигнала посредством BMP/Smad
пути сигнальной трансдукции. Мутации HJV
приводят к нарушению обоих путей сигнализации [26].
При исследование промоторной области гена
гепсидина были найдены сайты связывания различных транскрипционных факторов,
таких как С/EBPα, HNF4α,
USF, p53,
роль которых в регуляции экспрессии гепсидина не до конца понятна [26].
2.
Материалы и методы
2.1
Материалы
Люминол, HEPES,
TRIS∙HCl
(“Агат-Мед”, Россия); гепарин (“Спофа”, Чехия); иммуноферментные наборы для
определения трансферрина (“APTEC”,
Германия); декстран, трипановый синий (“Sigma”,
США); верографин (“Leiras”,
Germany).
2.2
Объект исследования
В качестве объекта исследования была выбрана
модель развития асцитной гепатомы Зайделя. В экспериментах in vivo
использовали крыс-самцов линии Вистар весом 200-220 г. Клетки гепатомы Зайделя
трансплантировали в брюшную полость животного путем введения 1 мл
ресуспендированых в солевом буфере клеток в концентрации 1∙107
кл/мл. Продолжительность жизни животного с трансплантированной опухолью
составляла 12-14 суток. При исследовании соотношения параметров про- и антиоксидантных
систем плазмы крови и асцита ежесуточно производили декапитацию животных с
целью забора образцов асцита и крови.
2.3
Выделение клеток
Полиморфноядерные лейкоциты (ПМЯЛ) из
циркулирующей крови крыс выделяли в градиенте плотности фикол/верографин [29].
Для этого согласно протоколу кровь, обработанная гепарином (из расчета 200 мкл
раствора гепарина с NaCl, 5000 ед. на 10 мл крови) и смешанная с
физиологическим раствором в отношении 2:1, была наслоена на смесь 6% раствора
декстрана Т-500 и 33,9% раствора верографина в отношении 1:2. Спустя 30 мин,
после оседания агрегированных эритроцитов, отбирали плазму (верхний слой)
богатую лейкоцитами и разделяли её далее на двойном градиенте фикол-верографин:
первый градиент (верхний слой) - верографин (10 частей 33,9%-го раствора) -
фикол Т-400 (24 части, 9%-го раствора), плотность 1,070 г/мл; второй градиент
(нижний слой) - верографин (10 частей 50%-го раствора) - фикол Т-400 (20
частей, 9%-го раствора), плотность 1,119 г/мл. Заполненные пробирки центрифугировали
при 800 g в течение 15 мин. После центрифугирования наблюдались три хорошо
выраженные клеточные фракции, разделенные друг от друга слоями градиента. Отбор
ПМЯЛ осуществляли из средней фракции, находящейся на разделе фаз первого и
второго градиентов. ПМЯЛ составили около 92% этой фракции (данные получены с
использованием камеры Горяева). Выделенные ПМЯЛ дважды отмывали 10-кратным
физиологическим раствором с использованием центрифуги Eppendorf
5415 при 1550 об/мин. О жизнеспособности клеток судили по окраске на стекле
трипановым синим, который является индикатором погибших клеток.
2.4
Определение концентрации трансферрина в плазме крови и в асцитной жидкости
опухоленосителя
Бесклеточную асцитную жидкость и плазму крови
получали путем дифференциального центрифугирования асцитной жидкости и крови
при 500 об/мин в течение 10 мин и затем при 14000 об/мин в течение 10 мин на Eppendorf
5415. Далее образцы замораживали при -20°С для последующего анализа.
Концентрацию трансферрина определяли с помощью
иммуноферментного анализа с использованием стандартных наборов (“APTEC”,
Германия). Измерение концентрации трансферрина проводилось в 96-луночном
планшете. Калибровочная кривая строилась по контрольным образцам стандартной
сыворотки (“APTEC”, Германия), содержащей трансферрин в концентрациях 7.38,
0.738, 0.0738 и 0.00738 мг/мл. Измерение оптической плотности проводилось на
планшетном спектрофотометре TECAN Infinite® 200 (Швейцария) при длине волны 570
нм после добавления к 5 мкл образца 250 мкл реагента 1 (100 мМ Tрис-буфер, pH
7,5; NaCl, 180 мМ; полиэтиленгликоль (PEG); детергенты, стабилизаторы) и после
добавления 50 мкл реагента 2 (100 мМ Tрис-буфер, pH 8,0; 180 мМ NaCl; антитела
козы к человеческому трансферрину со стабилизаторами).
2.5
Определение параметров окислительного стресса в крови и асците
Скорость наработки активных форм кислорода (АФК)
в асцитной жидкости определяли хемилюминесцентным методом. Для этого в кювету
объемом 0.5 мл с буфером для регистрации помещали 5 мкл свежеотобранного асцита
и 10 мкл 2 мМ люминола и регистрировали уровень хемилюминесцентного сигнала в
течение 10 минут. АФК-генерирующую активность ПМЯЛ крови определяли
хемилюминесцентным методом. Для этого в кювету с объемом 0.5 мл с буфером для
регистрации помещали 10 мкл свежевыделенных ПМЯЛ с известной концентрацией
клеток и 10 мкл 2 мМ люминола и регистрировали уровень хемилюминесцентного
сигнала в течение 10 минут. Изменение концентрации оценивалось в относительных
единицах.
2.6
Построение математической модели
Для построения модели использовался метод,
основанный на непрерывном описании динамики систем. Формализация осуществляется
на основе блочного принципа, согласно которому моделируемая система
расчленяется на элементарные подсистемы, каждая из которых описывается
изолированно от других. Описание элементарных подсистем проводится в терминах
элементарных процессов. Элементарные процессы описываются на основе формальных
блоков. Формальные блоки однозначно характеризуются упорядоченным списком
формальных динамических переменных X, упорядоченным списком формальных
параметров P и законом преобразования информации F. Для описания биохимических
реакций, активного и пассивного переноса веществ используется принцип
химической кинетики. В условиях полного перемешивания эти процессы можно описывать
на основе закона действующих масс, что в общем случае приводит к системам
обыкновенных дифференциальных уравнений. В этом случае динамическими
переменными являются концентрации соответствующих веществ, параметрами
константы соответствующих процессов, а законом преобразования информации
является соответствующее дифференциальное уравнение кинетики. Для описания
регуляции экспрессии генов применяется подход с использованием функций Хилла.
Функция Хилла 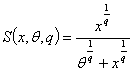 является сигмовидной
функцией, где x -
регулирующая переменная, θ
- константа,
равная величине сигнала x, при котором S достигает
половины от максимального значения, q -
константа, характеризующая крутизну сигмоида. В этом случае динамическими
переменными являются количества продуктов соответствующих генов, параметрами
являются константы θ,
характеризующие
эффективность влияния регулятора и q -
константы, характеризующие степень нелинейности влияния регулятора, а законом
преобразования информации является дифференциальное уравнение, правой частью
которого является функция Хилла.
является сигмовидной
функцией, где x -
регулирующая переменная, θ
- константа,
равная величине сигнала x, при котором S достигает
половины от максимального значения, q -
константа, характеризующая крутизну сигмоида. В этом случае динамическими
переменными являются количества продуктов соответствующих генов, параметрами
являются константы θ,
характеризующие
эффективность влияния регулятора и q -
константы, характеризующие степень нелинейности влияния регулятора, а законом
преобразования информации является дифференциальное уравнение, правой частью
которого является функция Хилла.
Для построения модели и численных расчетов
использовался пакет программ BioUML
(#"587956.files/image004.gif">
Рисунок 3. Схема
модели, описывающей клеточный гомеостаз железа. Железо поступает в клетку
посредством рецептор-индуцируемого эндоцитоза (рецепторы TfR1
и TfR2) и попадает в
лабильный пул железа (LIP).
Активность IRP1 и IRP2
регулируется количеством железа в LIP.
Белки IRP регулируют
экспрессию TfR1, DMT1,
Ferritin, Ferroportin.
Железо запасается в комплексе с белком Ferritin
и экспортируется из клетки посредством белка Ferroportin.
Рецептор-опосредованный эндоцитоз различных форм
трансферрина включает в себя восемь стадий: (1) связывание и диссоциация
рецептора от лиганда на поверхности клеточной мембраны; (2) интернализация
рецептора и рецептор-лигандных комплексов; (3) диссоциация железа от комплекса с
трансферрином при низком pH
эндосомы; (4) диссоциация и связывание различных форм трансферрина с рецептором
в эндосоме; (5) сортировка рецепторов и рецептор-лигандных комплексов в
эндосомах; (6) деградация рецепторов и трансферрина в лизосомах; (7) синтез
новых рецепторов и их транспорт на клеточную мембрану; (8) рециклизация
рецепторов и рецептор-лигандных комплексов на поверхность мембраны.
Модель эндоцитоза различных форм трансферрина
построена в терминах моно- и бимолекулярных реакций. Основная идея модели
эндоцитоза заключается в том, что попавшие в клетку рецептор-лигандные
комплексы могут селективно направляться в лизосомы, где деградируют, либо
возвращаться на поверхность клеточной мембраны, и этот процесс регулируется
насыщением рецептора лигандом. Для описания этих процессов модель эндоцитоза
включает в себя четыре компартмента: эндосомальный везикулярный компартмент,
эндосомальный трубчатый компартмент, рециркуляционный компартмент и компартмент
деградации (Рис. 4). Рецепторы, лиганды и рецептор-лигандные комплексы,
попавшие в эндосому, распределяются между везикулярным компартментом, откуда
попадают в лизосомы, и трубчатым компартментом, откуда попадают в
рециркуляционный компартмент. Также лиганды могут попадать в эндосомы
неспецифически, захватываясь из внеклеточной среды при формировании
эндоцитозного пузырька.
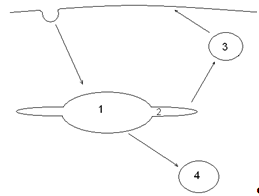
Рисунок 4. Схема
четырех компартментов модели эндоцитоза. (1) эндосомальный везикулярный
компартмент, (2) эндосомальный трубчатый компартмент, (3) рециркуляционный
компартмент, (4) компартмент деградации
В ходе проработки модели на основании данных из
источников [30, 31] мной были получены системы уравнений, описывающие систему
эндоцитоза рецепторов трансферрина. Уравнения, описывающие динамику количества
рецепторов и рецептор-лигандных комплексов, находящихся на плазматической
мембране, имеют вид:
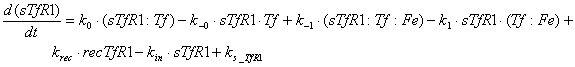
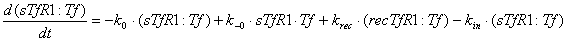
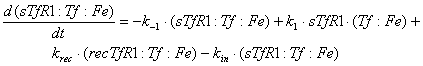
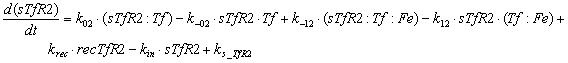
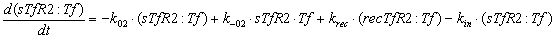
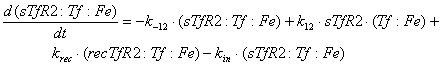
Ниже представлены уравнения для
рецепторов и рецептор-лигандных комплексов, находящихся в эндосомальном
везикулярном компартменте:
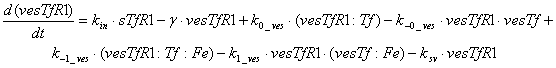
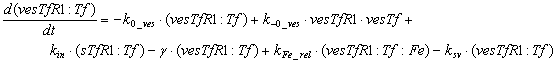
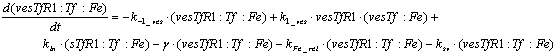
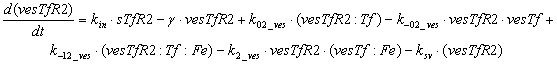
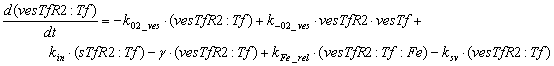
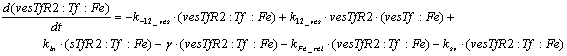
Уравнения для рецепторов и
рецептор-лигандных комплексов, находящихся в эндосомальном трубчатом
компартменте:
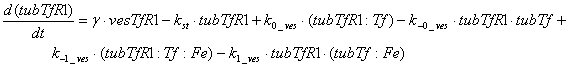
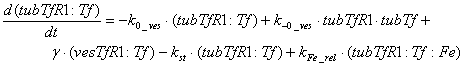
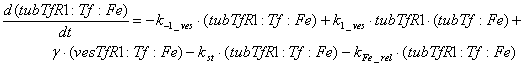
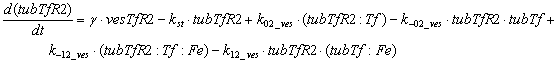
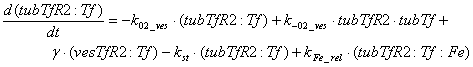

Следующий набор уравнений описывает
димнамику лигандов, находящихся в эндосоме. Размер лиганда влияет на его
распределение между везикулярным и трубчатым компартментами эндосомы. Для
описания этого явления в [30] вводится понятие коэффициента распределения k,
показывающего во сколько раз коцентрация лиганда в трубчатом компартменте
больше, чем в везикулярном. Этот коэффициент отражает эффект исключения объёма
в трубчатом компартменте вследствие размера лиганда, и определяется как k=(1-λ)2,
где λ
- отношение
диаметра лиганда к диаметру трубки. Для молекулы трансферрина k=0.81
[31].
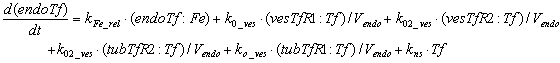
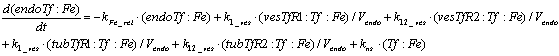
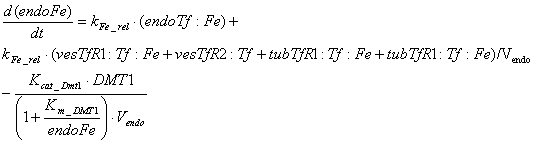
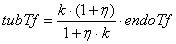
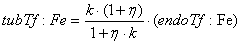
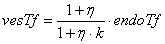
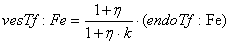
Уравнения для рецепторов, лигадов и
рецептор-лигандных комплексов находящихся в рециркуляционном компартменте имеют
вид:
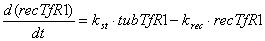
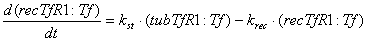
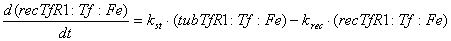
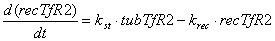
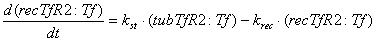

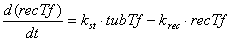
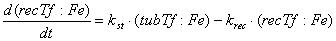
Уравнения для рецепторов, лигадов и
рецептор-лигандных комплексов находящихся в лизосомах:
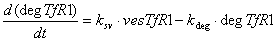
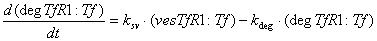
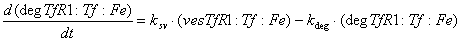
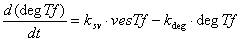
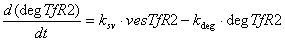
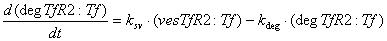
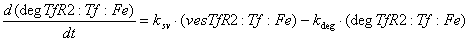
Железо в восстановленной форме
транспортируется из эндосомы в цитоплазму посредством белка DMT1. Кинетика
этого процесса описывается уравнением Михаелиса-Ментена. Железо, поступившее в
цитоплазму, попадает во внутриклеточный лабильный пул железа, откуда может
попадать в комплекс с ферритином, использоваться для нужд клетки или
экспортироваться из клетки посредством белка ферропортина. Высвобождение железа
из комплекса с ферритином происходит при деградации этого комплекса. Кинетика
экспорта железа посредством ферропортина описывается уравнением
Михаелиса-Ментена. Процесс транзита железа через клетку из эндосомы описывается
набором уравнений, приведенным ниже:
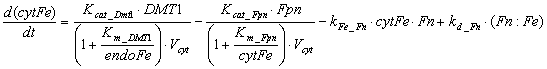
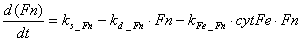
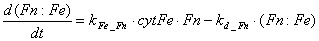
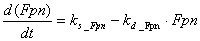
Следует отметить, что экспрессия
многих белков системы поддержания гомеостаза железа контролируется на
посттранскрипционном уровне системой IRP белков.
мРНК этих белков содержат последовательности IRE в
нетранслируемой части, с которыми связываются белки IRP. IRE
последовательности располагаются в 5’-UTR мРНК таких
белков как: тяжелая и лёгкая субъединицы ферритина, ферропортин (FPN), и в
3’-нетранслируемой части мРНК белков TfR1 и DMT1.
Образование IRE/IRP комплекса
на 3’-UTR приводит к
стабилизации мРНК, в то время как формирование этого комплекса на 5’-UTR приводит к
ингибированию ранних этапов трансляции [16, 17]. IRP белки
являются сенсорами внутриклеточного лабильного пула железа. При недостатке
железа в клетке IRP1 связывается с IRE с высокой
аффиностью, а при повышении уровня железа, в IRP1 собирается
кластер 4Fe-4S, что
приводит к утрате высокой аффинности и приобретению аконитазной активности.
Экспрессия IRP2
регулируется на уровне деградации. Для моделирования этой системы применялся
подход с использованием функций Хилла, предложенный в [32, 33]. Скорость
изменения концентрации неактивной формы IRP1 можно
описать следующим уравнением:
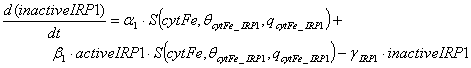 , где
, где
- функция Хилла.
Первый член уравнения утверждает,
что неактивная форма IRP1 образуется с постоянной
скоростью, когда внутриклеточный лабильный пул железа выше порогового значения θcytFe_IRP1. Второй
член отражает скорость, с которой активная форма IRP1
трансформируется в неактивную форму. Эта скорость пропорциональна activeIRP1 и
регулируется внутриклеточным лабильным пулом железа. Последний член уравнения
отражает деградацию IRP1 с постоянной скоростью. Скорость
изменения концентрации активной формы кислорода описывается следующим
уравнением:
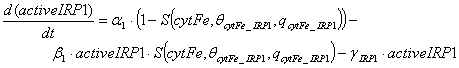 .
.
Первый член уравнения утверждает,
что, когда внутриклеточный лабильный пул железа ниже порогового значения θcytFe_IRP1, скорость
наработки примерно постоянна и равен α1, иначе
скорость наработки близка к нулю. Второй член отражает скорость, с которой
активная форма IRP1 трансформируется в неактивную
форму. Третий член описывает деградацию активной формы IRP1. Скорость
изменения количества IRP2 описывается уравнением:
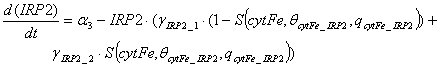 .
.
Первое слагаемое уравнения
утверждает, что IRP2 синтезируется с постоянной
скоростью. Второе слагаемое утверждает, что скорость деградации пропорциональна
количеству IRP2 с
константой скорости деградации, зависящей от внутриклеточного лабильного пула
железа.
Скорость синтеза TfR1
описавается следующим уравнением:
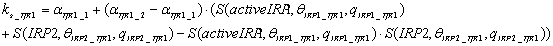 .
.
Предполагается, что TfR1 имеется
базальный уровень синтеза αTfR1_1, и скорость
синтеза увеличивается до αTfR1_2 когда
количество активного IRP1 выше порогового значения θIRP1_TfR1 или
количество IRP2 выше
порогового значения θIRP2_TfR1. Скорость
синтеза ферритина и ферропортина моделируется аналогичным образом.

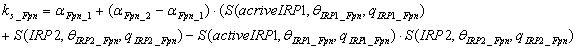
Результаты численных расчетов.
Рецептор трансферрина второго типа
регулируется степенью насыщения трансферрина железом: холотрансферрин
ингибирует деградацию TfR2 и, таким образом, повышает
стабильность белковой молекулы [21, 22]. Нами было предположено, что этот
эффект обусловлен различием в сортировке свободного TfR2, его
комплекса с апотрансферрином и комплекса с холотрансферрином в эндосомах. Для
проверки этой гипотезы были проведены численные расчеты, которые показали, что
этот эффект проявляется за счет увеличения насыщения второго рецептора
трансферрином, так как деградации в эндосомах по большей части подвержены
свободные рецепторы. Этот механизм не проявляется в случае первого рецептора
так как при нормальных условиях он практически полностью насыщен трансферрином.
Сравнение расчетов с экспериментом показано на Рис. 5.
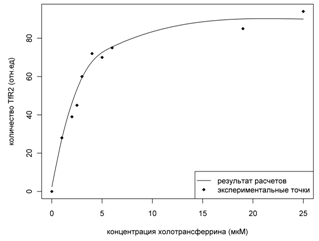
Рисунок 5. Эффект
стабилизации лигандом. На рисунке показаны результаты численных расчетов в
сравнении с экспериментальными точками.
Часто во время развития
окислительного стресса наблюдается повышение степени насыщения трансферрина
железом. Для выяснения динамики системы при изменении степени насыщения
трансферрина железом были проведены численные расчеты модели. Кроме того, для
выяснения роли рецептора второго типа в этом процессе расчеты велись при
наличии и при отсутствии второго типа рецепторов. На Рис.6 представлена
зависимость равновесной скорости транспорта железа от концентрации
холотрансферрина во внеклеточном пространстве. Как видно, при высоком насыщении
трансферрина, клетки, экспрессирующие TfR2, потребляют железо значительно
быстрее клеток, не экспрессирующих данный тип рецептора. Это объясняется тем,
что насыщение TfR1-зависимой системы происходит ещё при малых степенях
насыщения трансферрина.
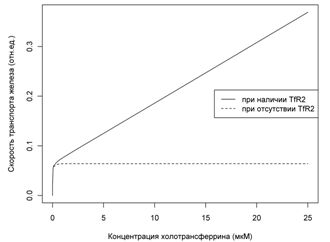
Рисунок 6. Результаты
численных расчетов: зависимость равновесной скорости транспорта железа в клетку
от концентрации холотрансферрина вне клетки. Красная кривая - при наличии TfR2, синяя при
отсутствии.
Кроме того, такая активация TfR2 системы
транспорта при повышении насыщения трансферрина и высокая константа диссоциации
TfR2 трансферринового комплекса приводит к усилению транспорта трансферрина в
клетки, что в литературе известно как явление бифазного захвата трансферрина
клетками, экспрессирующими второй тип рецептора. Этот эффект может
обуславливать падение уровня трансферрина в плазме крови, что иногда
наблюдается во время развития окислительного стресса. На Рис 7.
представлены рассчитанная кривая и экспериментальные точки зависимости
количества трансферрина в клетке от концентрации холотрансферрина вне клетки.
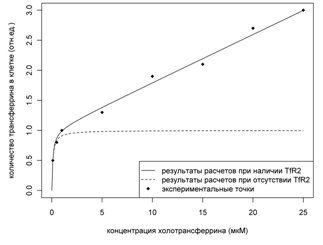
Рисунок 7. Явление
бифазного захвата трансферрина клетками. На рисунке представлены результаты
расчетов при наличии TfR2, при отсутствии TfR2 и экспериментальные точки
Таким образом, математическое моделирование
системы потребления железа клетками выявило механизмы, лежащие за явлениями,
наблюдающимися в экспериментах.
3.2
Математическая модель регуляции системного гомеостаза железа
На основе данных литературы и разработанных
ранее моделей [34-37] мной была создана модель системного гомеостаза железа.
Схема модели представлена на Рис 8.
NTBI
компартмент.
NTBI компартмент
включает в себя железо, экспортированное из внутриклеточного лабильного пула
железа макрофагов, энтероцитов и гепатоцитов, но еще не связавшееся с
трансферрином. Поскольку механизм регуляции экскреции железа организмом неизвестен,
в модели считается, что экскреция происходит с постоянной скоростью. Абсорбция
железа, однако, активно регулируется. Зрелые энтероциты двенадцатиперстной
кишки абсорбируют и экскретируют примерно 1мг железа в день. Эти клетки
экспрессируют транспортер железа ферропортин, посредством которого
осуществляется транспорт из внутриклеточного лабильного пула железа энтероцитов
в плазму крови. Экспрессия ферропортина, и, следовательно, скорость абсорбции
регулируется гепсидином.
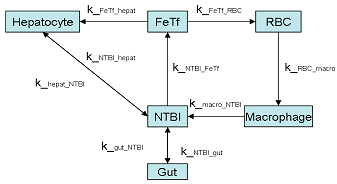
Рисунок 8. Структура
компартментализованной модели системного гомеостаза железа. Каждый компартмент
отражает количество железа, находящегося в определенном состоянии (т.е.
свободное или связанное с трансферрином) или местонахождение (т.е. железо в
макрофагах или гепатоцитах). NTBI
- железо, несвязанное с трансферрином, FeTf
- комплекс железа с трансферрином, RBC
- красные кровяные клетки, Macrofage
- макрофаги, Hepatocyte
- гепатоциты.
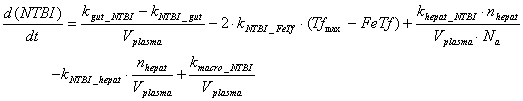
Гепсидин связывается с ферропортином
на клеточной мембране и индуцирует его вхождение в клетку и последующую
деградацию. Согласно современным представлениям продукция гепсидина
регулируется посредством передачи сигнала с рецептора трансферрина второго
типа. Представленные ниже уравнения отражают этот путь передачи сигнала.
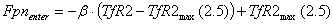
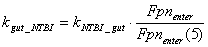
FeTf компартмент
После экспорта железа в плазму крови
посредством ферропортина железо связывается с трансферрином. В таком состоянии
железо потребляется гепатоцитами и красными кровяными клетками.
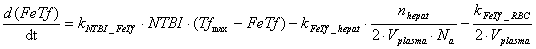
Hepatocyte компартмент
Основным механизмом потребления
железа клетками является TfR1-опосредуемый эндоцитоз
комплекса трансферрина с железом. Гепатоциты также способны получать железо
посредством TfR2 зависимого
эндоцитоза. Было показано, что связывание холотрансферрина с TfR2 в
гепатоцитах увеличивает время полураспада TfR2 c четырех до
16 часов, что позволяет регулировать количество TfR2 в
зависимости от концентрации холотрансферрина в плазме крови. Увеличение
стационарного количества TfR2 при изменении концентрации
холотрансферрина происходит по гиперболическому закону и достигает половины от
максимального значения при концентрации холотрансферрина 2.5 мкМ.
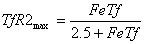
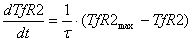
Потребление железа из комплекса с
трансферрином посредством рецептор опосредуемого эндоцитоза опиcывается в
соответствии с моделью клеточеного гомеостаза железа, описанной выше.
Экспорт железа из гепатоцитов
моделируется таким же образом, как и для энтероцитов.
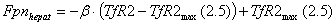
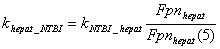
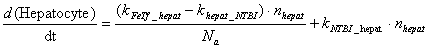
RBC и Macrophage
компартменты
Развивающиеся красные кровяные клетки
костного мозга потребляют железо посредством TfR1-зависимого
рецептор-индуцируемого эндоцитоза. Старые или поврежденные эритроциты
фагоцитируются макрофагами ретикуло-эндотелиальной системы, которые извлекают
железо из комплекса с гемоглобином и экспортируют его в плазму крови. Регуляция
экспорта железа моделируется таким же образом, как и для энтероцитов.
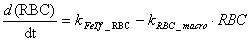
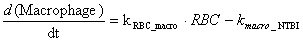
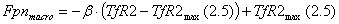
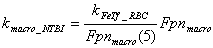
На данный момент проводится поиск и
анализ экспериментальных данных с целью оценки возможных значений параметров,
характеризующих процесс потребления железа гепатоцитами, не связанного с
трансферрином, и верификация созданной модели.
3.3
Изучение динамики развития асцитной гепатомы Зайделя
Для изучения динамики системы гомеостаза железа
в целом при развитии ОС был поставлен эксперимент по измерению ряда параметров
в ходе развития окислительного стресса. Экспериментальная работа была проведена
на базе Института теоретической и экспериментальной биофизики РАН (г. Пущино) в
Секторе функциональной биохимии под руководством канд. биол. наук Ю. Шаталина.
В качестве объекта исследования выбрана асцитная гепатома Зайделя. Развитие
данной опухоли ассоциировано с нарушением окислительно-восстановительного
баланса в плазме крови и асците у крыс (в плазме наблюдается состояние
окислительного стресса, тогда как в опухоли высок уровень
веществ-антиоксидантов), предположительно вызванным нарушением метаболизма
железа. Помимо этого у крыс с данной патологией наблюдается дисбаланс
распределения жидкости и компонентов антиоксидантной системы в организме.
Развитие опухоли в организме, как правило,
ассоциировано с развитием реакции неспецифического иммунного ответа. В
частности следует ожидать изменение численности, активности и локализации
эффекторных клеток. Знание соотношения между опухолевыми и эффекторными
клетками необходимо для оценки интенсивности иммунной реакции на рост опухоли.
Поэтому была исследована динамика количества опухолевых клеток, ПМЯЛ в крови и
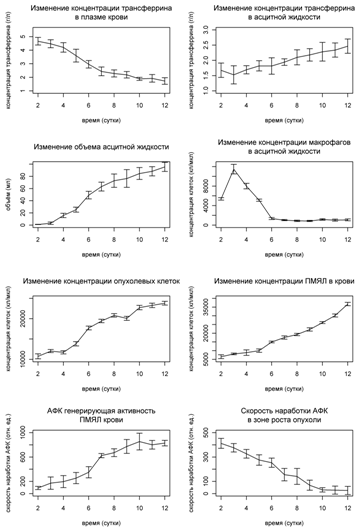
макрофагов в зоне роста опухоли. Соответствующие графики представлены на Рис.9.
При развитии опухоли наблюдается увеличение концентрации опухолевых клеток с
максимальной скоростью роста на 4-7 сутки. Процесс роста числа опухолевых
клеток сопровождается увеличением объема асцитной жидкости, который может
достигать 90-100 мл. Как видно на Рис. 9, развитие опухоли
сопровождается увеличением количества ПМЯЛ примерно в 7 раз, а концентрация
макрофагов в зоне роста опухоли достигает максимума на третьи сутки, после чего
монотонно снижается. Следует отметить, что снижение концентрации макрофагов
вызвано интенсивным увеличением объема асцитной жидкости. Подобное изменение
числа ПМЯЛ в крови и макрофагов в зоне роста опухоли говорит о развитии
иммунной реакции организма.
Для оценки динамики развития окислительного
стресса во время роста асцитной опухоли была измерена АФК генерирующая
активность ПМЯЛ в крови и скорость наработки АФК в асцитной жидкости
хемилюминесцентным методом. На Рис.9 показано, что АФК генерирующая
активность ПМЯЛ к 12-м суткам увеличивается в 7-8 раз, что в совокупности с
увеличением количества ПМЯЛ говорит о развитии окислительного стресса в крови
животных. В то же время, с развитием гепатомы, скорость наработки АФК в зоне
роста опухоли постепенно снижается.
В плазме крови большая часть ионов железа
находится в комплексе с белком трансферрином, защищающим организм от
образования высокотоксичных радикалов по реакции Фентона. Для понимания роли
системы гомеостаза железа в процессе развития асцитной гепатомы Зайделя была
изучена динамика трансферрина в плазме крови и асцитной жидкости иммунноферментным
методом. На Рис.9 показано, что уровень трансферрина в плазме крови
животных в процессе развития опухоли снижается в 2.5-3 раза, тогда как в
асцитной жидкости наблюдается рост концентрации трансферрина.
В совокупности эти данные указывают на противоположность
процессов, происходящих в крови и асците животных. В крови наблюдается усиление
процессов генерации АФК за счет активации и увеличения количества ПМЯЛ и
уменьшение концентрации белка антиоксидантной защиты организма - трансферрина,
в предыдущих работах также было зафиксировано уменьшение концентраций
низкомолекулярных антиоксидантов, таких как α-токоферол
и мочевая кислота. В противоположность этому, в зоне роста опухоли наблюдается
снижение скорости наработки АФК, падение концентрации макрофагов и усиление
антиоксидантной защиты. Такая разнонаправленность отчасти может быть объяснена
перераспределением жидкости и пассивным транспортом веществ из крови в асцит.
Для проверки этой гипотезы была создана
математическая модель пассивного переноса веществ из крови в асцит с током
жидкости. Схема модели представлена на Рис.10. Она состоит из двух
компартментов: плазма крови и асцит, между которыми осуществляется ток жидкости
с тем, чтобы объем асцита соответствовал наблюдаемым значениям. Вместе с током жидкости
из плазмы крови в асцит осуществляется перенос веществ, предсказание
концентраций которых является одной из подзадач данной работы.
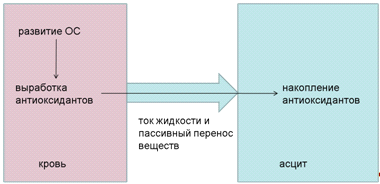
Рисунок 10. Схема
модели пассивного переноса веществ-антиоксидантов из плазмы крови в асцитную
жидкость (ОС - окислительный стресс).
Следующие два дифференциальных уравнения
описывают изменение концентрации веществ в плазме крови и асцитной жидкости,
предполагая механизм пассивного переноса:
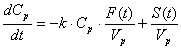 ,
,
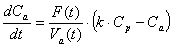 ,
,
где F(t) есть поток
жидкости из плазмы крови в асцит, S(t) - скорость образования вещества в
крови, Vp - объем
плазмы крови, Va(t) - объем
асцитной жидкости. Основная идея этой модели состоит в том, что антиоксиданты плазмы
крови, вырабатывающиеся в большом количестве в ответ на развивающийся
окислительный стресс, с током жидкости переносятся в асцит, и, таким образом,
защищают опухоль. Несмотря на то, что осуществляется пассивный перенос веществ,
и их концентрации в асците, как правило, меньше чем в плазме крови, количество
антиоксидантных веществ в асците значительно превышает соответствующие
показатели плазмы крови (за счет большого объёма асцита). На Рис.11
представлено сравнение предсказанных концентраций трансферрина в плазме крови и
асците с экспериментом. Сплошными линиями показаны рассчитанные концентрации
трансферрина в плазме крови и асците, а точками - результаты экспериментов.
Модель пассивного переноса хорошо описывает динамику трансферрина. Стоит
отметить, что, несмотря на пассивный механизм переноса, заложенный в модели, к
концу развития опухоли количество трансферрина в асците составляет примерно
250мг, в то время как в плазме крови это значение не превышает 100мг.
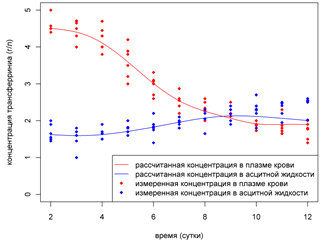
Рисунок 11. Сравнение
предсказанных концентраций трансферрина в плазме крови и асцитной жидкости с
помощью модели пассивного переноса с экспериментальными значениями.
3.4
Идентификация генов, участвующих в гомеостазе железа
С целью расширения модели гомеостаза железа был
произведен поиск генов-кандидатов, участвующих в процессах гомеостаза железа на
основе доступных микрочиповых экспериментов. Для этого из базы данных GEO
(Gene Expression
Omnibus,
http://www.ncbi.nlm.nih.gov/geo/) были отобраны 12 экспериментов, в ходе
которых были проведены измерения экспрессии генов при различных патологических
состояниях, связанных с гомеостазом железа, по сравнению с нормой либо при
диетах, содержащих различное количество железа. Краткое описание экспериментов
приведено в таблице 1.
Анализ микрочиповых данных был проведен с
использованием языка программирования R [38]. Коррекция фона каждого чипа в
отдельности осуществлялась методом normexp
[39, 40]. Затем чипы платформы Affymetrix и одноканальные чипы платформы
Agilent были нормализованы методом quantile [41] в логарифмической шкале.
“Двухцветные” чипы были нормализованы методом loess
[42]. Для идентификации дифференциально экспрессирующихся генов сначала был
применен t-test для каждой пробы (набора проб) предложенным в [43] на основе
аннотации проб в базе данных
Таблица 1. Краткое
описание микрочиповых экспериментов, использованных для поиска генов,
участвующих в гомеостазе железа.
|
Идентифика-тор в базе данных GEO
|
Платформа микрочипа
|
Экспериментальные состояния
|
Ткань/клетки
|
Ссылка
|
Количество микрочипов
|
|
GSE10421
|
Agilent
one-channel
|
диета
сбалансированная, насышенная или дефицитная по железу
|
клетки
печени
|
[44]
|
30
|
|
GSE9726
|
Illumina
BeadChip
|
диета
сбалансированная или насышенная по железу
|
мышечные
клетки сердца и скелетные
|
[45]
|
12
|
|
GSE3573
|
Agilent
two-channel
|
диета
сбалансированная, насышенная или дефицитная по железу
|
caco-2
клетки
|
[46]
|
12
|
|
GSE2269
|
Affymetrix
|
диета
сбалансированная или насышенная по железу
|
клетки
тощей кишки
|
[47]
|
12
|
|
GSE1892
|
Affymetrix
|
диета
сбалансированная или дефицитная по железу
|
клетки
двенадцатиперстной кишки
|
[47, 48]
|
60
|
|
GSE7267
|
Affymetrix
|
+/b
and b/b
|
клетки
тощей и двенадцатиперстной кишки
|
[49]
|
12
|
|
GSE10591
|
Affymetrix
|
клетки
дикого типа и мутантные по гену Tmprss6
|
HepG2
клетки
|
[50]
|
9
|
|
GSE11632
|
Affymetrix
|
клетки
дикого типа и мутантные по гену Tmprss6
|
клетки
печени
|
[51]
|
2
|
Affymetrix
|
клетки
дикого типа и мутантные по гену Tfrc
|
эмбриональные
клетки
|
нет
статьи
|
4
|
|
GSE7357
|
Affymetrix
|
дикий
тип и мутаны по гену HFE
|
клетки
печени и двенадцатиперстной кишки
|
[52]
|
24
|
|
GSE4523
|
Affymetrix
|
дикий
тип и мутаны по гену меланотрансферрина
|
клетки
мозга
|
[53]
|
6
|
|
GSE11777
|
Affymetrix
|
дикий
тип и мутаны по генам stat5a,b
|
клетки
печени
|
нет
статьи
|
6
|
Ensembl
(http://www.ensembl.org/) [54], гены со значением гипергеометрического скора
более 3.0 считались значимо дифференциально экспрессирующимися. Анализ
представленности Gene Ontology (http://www.geneontology.org/) [55] категорий в
списке отобранных генов осуществлялся с помощью гипергеометрического теста.
Категории с p-value меньше чем 0.01 считались значимо перепредставленными.
Десять наиболее представленных категорий приведены в таблице 2.
В список выявленных дифференциально
экспрессирующихся генов входят ранее известные участники гомеостаза железа,
такие как, TfR1, Dmt1,
цитохром b редуктаза,
ферропортин, Hfe, Cp,
Hp, Slc11a2,
Steap3, Slc25a37
(категории "iron
ion transport",
"response
to iron
ion" и " cellular
iron ion
homeostasis").
А также группы генов ранее не замеченных в
регуляции гомеостаза железа, из которых следует отметить группу генов,
участвующих в гомеостазе ионов меди, представленные в таблице 3 и
участники липидного гомеостаза таблица 4.
Таблица 2. Список
категорий Gene
Ontology, наиболее
представленных среди генов с измененной экспрессией.
|
Gene Ontology категория
|
p-value
|
|
cholesterol
metabolic process
|
4,54E-09
|
|
angiogenesis
|
2,73E-05
|
|
lipid
binding
|
2,95E-05
|
|
electron
carrier activity
|
3,60E-05
|
|
lipid
transport
|
6,20E-05
|
|
phospholipid
metabolic process
|
1,30E-03
|
|
cellular
copper ion homeostasis
|
1,80E-03
|
|
cellular
iron ion homeostasis
|
2,40E-03
|
|
responce
to iron ion
|
2,70E-03
|
|
iron
ion transport
|
3,30E-03
|
Таблица 3. Гены,
регулирующие гомеостаз меди, экспрессия которых значимо коррелирует с уровнем
железа в ткани.
|
Ген
|
Название
|
|
Mt1
|
metallothionein
1
|
|
Mt2
|
metallothionein
2
|
|
Atp7a
|
ATPase, Cu++ transporting, alpha
polypeptide
|
|
App
|
amyloid beta (A4) precursor
protein
|
|
Prnp
|
prion
protein
|
Таблица 4. Гены,
регулирующие гомеостаз липидов, экспрессия которых значимо коррелирует с
уровнем железа ткани
|
Ген
|
Название
|
|
Apol7a
|
apolipoprotein
L 7a
|
|
Apoa5
|
apolipoprotein
A-V
|
|
Apoc3
|
|
Apoa1
|
apolipoprotein
A-I
|
|
Fabp4
|
fatty
acid binding protein 4
|
|
Plcg1
|
phospholipase
C, gamma 1
|
|
Acsl1
|
acyl-CoA synthetase long-chain
family member 1
|
|
Srd5a1
|
steroid
5 alpha-reductase 1
|
|
Ppara
|
peroxisome proliferator-activated
receptor alpha
|
|
Cyp39a1
|
cytochrome P450, family 39,
subfamily A, polypeptide 1
|
|
Slc27a2
|
solute carrier family 27 (fatty
acid transporter), member 2
|
|
Pnpla8
|
patatin-like phospholipase domain
containing 8
|
|
Pemt
|
phosphatidylethanolamine
N-methyltransferase
|
|
ALOX15
|
arachidonate
15-lipoxygenase
|
|
GPAM
|
glycerol-3-phosphate
acyltransferase, mitochondrial
|
В дальнейшем, результаты анализа экспрессии
генов, регулирующих метаболизм железа, меди и липидов, будут использованы для
уточнения, расширения и интеграции созданных в ходе выполнения данной дипломной
работы моделей.
Актуальность проведенной работы по исследованию
и моделированию процессов, происходящих в асцитной опухоли у крысы и в плазме
крови при развитии окислительного стресса на фоне дисбаланса метаболизма
железа, видится несомненной. Помимо того, что был сделан очередной шаг к
полному пониманию данной патологии и разработке методов эффективного лечения
пациентов в будущем, также был расширен набор моделей системы метаболизма
железа, что в дальнейшем позволит создать комплексную интеграционную модель для
проведения экспериментов in
silico.
В
настоящее время в мировой науке прослеживается сильная тенденция в
направлении минимизации использования лабораторных животных и экспериментов с
участием добровольцев засчет разработок в области моделирования биологических
процессов, отдельных типов клеток, тканей и органов животных и человека. Эти
тенденции подчеркивают важность и в достаточной степени новизну проведенной
работы.
Выводы
1. Согласно результатам моделирования,
стабилизация рецептора TfR2 холотрансферрином обусловлена различием в
эндосомальной сортировке комплексов данного рецептора с лигандом, помимо этого
наблюдается бифазный режим захвата трансферрина клетками, экспрессирующими
данный рецептор, что хорошо согласуется с экспериментальными данными других
авторов.
. Во время развития асцитной гепатомы Зайделя:
а). в крови крыс наработка АФК увеличивается в
7-8 раз, тогда как в зоне роста опухоли она непрерывно снижается, что
свидетельствует о работе защитных механизмов асцита;
б). в крови крыс происходит непрерывный рост
числа ПМЯЛ, в то время как концентрация макрофагов в асците достигает максимума
на 3 сутки и в дальнейшем падает, что свидетельствует о недостаточности средств
иммунной системы для подавления опухоли;
в). в плазме крови концентрация трансферрина
падает, а в асцитной жидкости растет, при этом к концу развития опухоли большая
часть трансферрина переходит из крови в асцит.
. На основе экспериментальных данных и
результатов моделирования показано, что перераспределение трансферрина и
низкомолекулярных антиоксидантов между кровью и асцитом обусловлено их
пассивным транспортом в опухоль.
. В результате анализа микрочиповых данных
показана значимая корреляция экспрессии ряда генов, регулирующих метаболизм
липидов и меди, с уровнем железа в ткани. В дальнейшем эти гены и их белки
будут учтены при моделировании системы метаболизма железа и ее роли в развитии
окислительного стресса.
Список
литературы
1. Kruszewski, M. Labile iron
pool: the main determinant of cellular response to oxidative stress // Mutation
Research. - 2003. - N 531. - P. 81-92.
. Kakhlon, O., Cabantchik, Z.I.
The labile iron pool: characterization, measurement, and participation in
cellular processes // Free Radic. Biol. Med. - 2002. - N 33. - P. 1037-1046.
. Petrat, F., Groot, H.D.,
Sustmann, R., Rauen, U. The chelatable iron pool in living cells: a
methodically defined quantity // Biol. Chem. - 2002. - N 383. - P. 489-502.
. Delanghe, J.R., Langlois,
M.R. Hemopexin: a review of biological aspects and the role in laboratory
medicine // Clinica Chimica Acta. - 2001. - V. 312. - P. 13-23.
. Chua, C.G., Graham, R.M.,
Trinder, D., Olynyk, J.K. The regulation of cellular iron metabolism //
Critical Reviews in Clinical Laboratory Sciences. - 2007. - V. 44. - P.
413-459.
. Kaplan, J. Mechanisms of
cellular iron acquisition: another iron in the fire // Cell. - 2002. - V. 111.
- P. 603-606.
. Richardson, D.R., Ponka, P.
The molecular mechanisms of the metabolism and transport of iron in normal and
neoplastic cells // Biochimica et Biophysica Acta. - 1997. - V. 1331. - P.
1-40.
. Calzolari, A., Oliviero, I.,
Testa, U. Transferrin receptor 2 is emerging as a major player in the control
of iron metabolism. // CEJB. - 2007. - V. 2. - N 1. - P. 34-55.
. Smythe, E., Warren, G. The
mechanism of receptor-mediated endocytosis // Eur. J. Biochem. - 1991. - V.
202. - P. 689-699.
. Ohgami, R.S., Campagna, D.R.,
Greer, E.L., Antiochos, B., McDonald, A., Chen, J., Sharp, J.J., Fujiwara, Y.,
Barker, J.E., Fleming, M.D. Identification of a ferrireductase required for
efficient transferrin-dependent iron uptake in erythroid cells // Nat. Genet. -
2005. - V. 37. - N 11. - P. 1264-1269.
. West, A.P., Bennett, M.J.,
Sellers, V.M., Andrews, N.C., Enns, C.A., Bjorkman, P.J. Comparison of the
interactions of transferrin receptor and transferrin receptor 2 with
transferrin and the hereditary hemochromatosis protein HFE // J. Biol. Chem. -
2000. - V. 275. - N 49. - P. 38135-38138.
. Robb, A.D., Ericsson, M.,
Wessling-Resnick, M. Transferrin receptor 2 mediates a biphasic pattern of
transferrin uptake associated with ligand delivery to multivesicular bodies //
Am. J. Physiol. Cell Physiol. - 2004. - V. 287. - P. 1769-1775.
. Johnson, M.B., Chen, J.,
Murchison, N., Green, F.A., Enns, C.A. Transferrin receptor 2: evidence for
ligand-induced stabilization and redirection to a recycling pathway //
Molecular Biology of the Cell. - 2007. - V. 18. - P. 743-754.
. Johnson, M.B., Enns, C.A.
Diferric transferrin regulates transferrin receptor 2 protein stability //
Blood. - 2004. - V. 104. - N 13. - P. 4287-4293.
. Ganz, T. Is TfR2 the iron
sensor? // Blood. - 2004. V. 104. - N 13. - P. 3839-3840.
. Wallander, M.L., Leibold,
E.A., Eisenstein, R.S. Molecular control of vertebrate iron homeostasis by iron
regulatory proteins // Biochim. Biophys. Acta. - 2006. - V. 1763. - N 7. - C.
668-689.
. Hentze, M.W., Muckenthaler,
M.U., Andrews, N.C. Balancing acts: molecular control of mammalian iron
metabolism // Cell. - 2004. - V. 117. - P. 285-297.
. Schalinske, K.L., Eisenstein,
R.S. Phosphorylation and activation of both iron regulatory protein 1 (IRP1)
and IRP2 in HL60 cells // J. Biol. Chem. - 1996. - V. 271. - P. 7168-7176.
. Kang, D.K., Jeong, J., Drake,
S.K., Wehr, N.B., Rouault, T.A., Levine, R.L. Iron regulatory protein 2 as iron
sensor. Iron- dependent oxidative modification of cysteine. // J. Biol. Chem. -
2003. - V. 278. - P. 14857-14864.
. Knutson, M.D., Oukka, M.,
Koss, L.M., Aydemir, F., Wessling-Resnick, M. Iron release from macrophages
after erythrophagocytosis is up-regulated by ferroportin 1 overexpression and
down-regulated by hepcidin // PNAS. - 2005. - V. 102. - N 5. - P. 1324-1328.
. Johnson, M.B., Chen, J.,
Murchison, N., Green, F.A., Enns, C.A. Transferrin receptor 2: evidence for
ligand-induced stabilization and redirection to a recycling pathway //
Molecular Biology of the Cell. -2005 - V. 18. - P. 743-754.
. Johnson, M.B., Enns, C.A. Diferric
transferrin regulates transferrin receptor 2 protein stability // Blood. -
2004. - V. 104. - N 13. - P. 4287-4293.
. Anderson, G.J., Darshan, D.,
Wilkins, S.J., Frazer, D.M. Regulation of systemic iron homeostasis: how the
body responds to changes in iron demand // Biometals. - 2007. - V. 20. - P.
665-674.
. Nemeth, E., Tuttle, M.S.,
Powelson, J., Vaughn, M.B., Donovan, A., Ward, D.M., Ganz, T., Kaplan, J.
Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing
its internalization // Science. - 2004. - V. 306. - P. 2090-2093.
. Goswami, T., Andrews, N.C.
Hereditary hemochromatosis protein, HFE, interaction with transferrin receptor
2 suggests a molecular mechanism for mammalian iron sensing // Journal of
Biological Chemistry. - 2006. - V. 281. - N 39. - P. 28494-28498.
. Erwin, H.J., Kemna, M.,
Tjalsma, H., Willems, H.L., Swinkels, D.W. Hepcidin: from discovery to
differential diagnosis // Haematologica. - 2008. - V. 93. - N 1. - P. 90-97.
. West, A.P., Bennett, M.J.,
Sellers, V.M., Andrews, N.C., Enns, C.A., Bjorkman, P.J. Comparison of the
interactions of transferrin receptor and transferrin receptor 2 with
transferrin and the hereditary hemochromatosis protein HFE // J. Biol. Chem. -
2000. - V. 275. - N 49. - P. 38135-38138.
. Falzacappa, M.V., Spasic,
M.V., Kessler, R., Stolte, J., Hentze, M.W., Muckenthaler, M.U. STAT3 mediates
hepatic hepcidin expression and its inflammatory stimulation // Blood. - 2007.
- V. 109. - N 1. - P. 353-358.
29. Лобашевский, А.Л.
Выделение полиморфно-ядерных лейкоцитов из малых объемов крови после осаждения
декстраном // Лаб. дело. - 1983. - Т. 11. - С. 28-31.
30. French, A.R.,
Lauffenburger, D.A. Controlling receptor/ligand trafficking: effects of
cellular and molecular properties on endosomal sorting // Annals of Biomedical
Engineering. - 1997. - V. 25. - P. 690-707.
. Lao, B.J., Tsai, W.P.,
Mashayekhi, F., Pham, E.A., Mason, A.B., Kamei, D.T. Inhibition of transferrin
iron release increases in vitro drug carrier efficacy // Journal of Controlled Release.
- 2007. - V. 117. - P. 403-412.
. Omholt, S.W., Kefang, X.,
Andersen, O., Plahte, E. Description and analysis of switchlike regulatory
networks exemplied by a model of cellular iron homeostasis // J. Theor. Biol. -
1998. - V. 195. - P. 339-350.
33. Plahte, E., Mestl, T.,
Omholt, S.W. A methodological basis for description and analysis of systems
with complex switch-like interactions // J. Math. Biol. - 1998. - V. 36. - P.
321-348.
. Nathanson, M.H., Mclaren,
G.D. Computer simulation of iron absorption: regulation ofand systemic iron
kinetics in dogs // The Journal of Nutrition. - 1986. - V. 117 - N 6. - P.
1067-1075.
. Lao, B.J., Kamei, D.T. A
compartmental model of iron regulation in the mouse // Journal of Theoretical
Biology. - 2007. - V. 243. - C. 542-554.
. Franzone, P.C., Paganuzzi,
A., Stefanelli, M. A mathematical model of iron metabolism // J. Math. Biology.
- 1982. - V. 15. - C. 173-201.
. R: A language and environment
for statistical computing, Vienna, 2008. - 1667 p.
. Smyth, G.K. Limma: linear
models for microarray data // Bioinformatics. - 2005. - V. 23. - P. 397-420.
. Gautier, L., Cope, L.,
Bolstad, B.M., Irizarry, R.A. Affy - analysis of affymetrix genechip data at
the probe level // Bioinformatics. - 2004. - V. 20. - P. 307-315.
. Bolstad, B.M., Irizarry,
R.A., Astrand, M., Speed, T.P. A comparison of normalization methods for high
density oligonucleotide array data based on bias and variance //
Bioinformatics. - 2003. V. 19. - P. 185-193.
. Yang, Y.H., Dudoit, S., Luu,
P., Lin, D.M., Peng, V., Ngai, J., Speed, T.P. Normalization for cDNA
microarray data: a robust composite method addressing single and multiple slide
systematic variation // Nucleic Acids Research. - 2002. - V. 30. - P. 15-19.
. Kondrakhin, Y.V., Sharipov,
R.N., Kel, A.E., Kolpakov, F.A. Identification of differentially expressed
genes by meta-analysis of microarray data on breast cancer // In Silico
Biology. - 2008. - V. 8. - P. 5-23.
. Kautz, L., Meynard, D.,
Monnier, A., Darnaud, V., Bouvet, R., Wang, R.H., Deng, C., Vaulont, S.,
Mosser, J., Coppin, H., Roth, M.P. Iron regulates phosphorylation of Smad1/5/8
and gene expression of Bmp6, Smad7, Id1, and Atoh8 in the mouse liver // Blood.
- 2008. - V. 112. - P. 1503-1509.
. Rodriguez, A., Hilvo, M.,
Kytomaki, L., Fleming, R.E., Britton, R.S., Bacon, B.R., Parkkila, S. Effects
of iron loading on muscle: genome-wide mRNA expression profiling in the mouse
// BMC Genomics. - 2007. - V. 8. - P. 379.
. Chicault, C., Toutain, B.,
Monnier, A., Aubry, M., Fergelot, P., Treut, A., Galibert, M.D., Mosser, J.
Iron-related transcriptomic variations in CaCo-2 cells, an in vitro model of
intestinal absorptive cells // Physiol. Genomics. - 2006. - V. 26. - P. 55-67.
. Collins, J.F. Gene chip
analyses reveal differential genetic responses to iron deficiency in rat
duodenum and jejunum // Biol. Res. - 2006. - V. 39. - P. 25-37.
. Collins, J.F., Franck, C.A.,
Kowdley, K.V., Ghishan, F.K. Identification of differentially expressed genes
in response to dietary iron deprivation in rat duodenum // Am. J. Physiol.
Gastrointest. Liver. Physiol. - 2005. - V. 288. - P. 964-971.
. Collins, J.F., Hu, Z.,
Ranganathan, P.N., Feng, D., Garrick, L.M., Garrick, M.D., Browne, R.W.
Induction of arachidonate 12-lipoxygenase (Alox15) in intestine of
iron-deficient rats correlates with the production of biologically active lipid
mediators // Am. J. Physiol. Gastrointest. Liver. Physiol. - 2008. - V. 294. -
P. 948-962.
. Du, X., She, E., Gelbart, T.,
Truksa, J., Lee, P., Xia, Y., Khovananth, K., Mudd, S., Mann, N., Moresco,
E.M., Beutler, E., Beutler, B. The serine protease TMPRSS6 is required to sense
iron deficiency // Science. - 2008. - V. 320. - P. 1088-1092.
. Folgueras, A.R., Lara, F.M.,
Pendas, A.M., Garabaya, C., Rodriguez, F., Astudillo, A., Bernal, T.,
Cabanillas, R., Lopez-Otin, C., Velasco, G. Membrane-bound serine protease
matriptase-2 (Tmprss6) is an essential regulator of iron homeostasis // Blood.
- 2008. - V. 112. - P. 2539-2545.
. Coppin, H., Darnaud, V.,
Kautz, L., Meynard, D., Aubry, M., Mosser, J., Martinez, M., Roth, M.P. Gene
expression profiling of Hfe-/- liver and duodenum in mouse strains with
differing susceptibilities to iron loading: identification of transcriptional
regulatory targets of Hfe and potential hemochromatosis modifiers // Genome
Biol. - 2007. - V. 8. - C. 221.
. Dunn, L.L., Sekyere, E.O.,
Rahmanto, Y.S., Richardson, D.R. The function of melanotransferrin: a role in
melanoma cell proliferation and tumorigenesis // Carcinogenesis. - 2006. - V.
27. - P. 2157-2169.
. Hubbard, T.J., Aken, B.L.,
Ayling, S., Ballester, B., Beal, K., Bragin, E., Brent, S., Chen, Y., Clapham,
P., Clarke, L., Coates, G., Fairley, S., Fitzgerald, S., Fernandez-Banet, J., Gordon,
L., Graf, S., Haider, S., Hammond, M., Holland, R., Howe, K., Jenkinson, A.,
Johnson, N., Kahari, A., Keefe, D., Keenan, S., Kinsella, R., Kokocinski, F.,
Kulesha, E., Lawson, D., Longden, I., Megy, K., Meidl, P., Overduin, B.,
Parker, A., Pritchard, B., Rios, D., Schuster, M., Slater, G., Smedley, D.,
Spooner, W., Spudich, G., Trevanion, S., Vilella, A., Vogel, J., White, S.,
Wilder, S., Zadissa, A., Birney, E., Cunningham, F., Curwen, V., Durbin, R.,
Fernandez-Suarez, X.M., Herrero, J., Kasprzyk, A., Proctor, G., Smith, J.,
Searle, S., Flicek, P. Ensembl 2009 // Nucleic Acids Res. - 2009. - V. 37. - P.
690-697.
. Ashburner, M., Ball, C.A.,
Blake, J.A., Botstein, D., Butler, H., Cherry, J.M., Davis, A.P., Dolinski, K.,
Dwight, S.S., Eppig, J.T., Harris, M.A., Hill, D.P., Issel-Tarver, L.,
Kasarskis, A., Lewis, S., Matese, J.C., Richardson, J.E., Ringwald, M., Rubin,
G.M., Sherlock, G. Gene ontology: tool for the unification of biology // Nat.
Genet. - 2000. - V. 25. - P. 25-29.