Макромолекули лінійних аморфних полімерів
Зміст
Вступ
І. Хімічний зв’язок та будова макромолекул
.1 Хімічний зв’язок, будова молекул
.2 Виникнення і розвиток поняття макромолекула
.3 Макромолекула як об’єкт запису і перетворення
інформації
ІІ. Об’єкти та методи досліджень
.1 Лінійні аморфні полімери та неорганічні
наповнювачі. Отримання зразків
.2 Використання методів розсіювання світла для
визначення макромолекулярних констант
ІІІ. Макромолекулярні константи лінійних аморфних
полімерів та їх систем
.1 Основні геометричні константи макромолекул лінійних
аморфних полімерів
.2 Частотний спектр структуроутворень гетерогенних
полімерних систем
.3 Макромолекулярні константи і дефект модуля зсуву в
гетерогенних полімерних системах
Висновки
Література
ВСТУП
Актуальність роботи. Для прогнозування фізико-механічних та
хімічних властивостей гетерогенних полімерних систем, встановлення
функціональних закономірностей між структурою і властивостями використовуються
різні модельні підходи. Це перш за все зумовлено тим, що багато явищ, які
відбуваються в таких системах, зумовлені індивідуальними взаємодіями. Тому
макромолекули полімерів, навіть якщо вони мають однаковий склад і число ланок,
є індивідуальні і на них записана деяка комбінаторна інформація. Відповідно до
цього система набуває здатності до самоорганізації. При цьому найчастіше
розділяють два різних поняття інформації: структурна інформація (загальна для
значної групи молекул) і конфігураційна (дискретна) - індивідуальна інформація
кожної макромолекули, яка отримується за рахунок комбінації на певних ділянках
ланцюга деяких конкретних первинних положень атомів і груп атомів ланки.
З цієї точки зору особливої ваги набуває розгляд поведінки
структурних елементів макромолекул лінійних аморфних полімерів в силових і
температурних полях, де доводиться враховувати не тільки гармонічні ефекти, але
й ангармонізм їх коливань.
Визначити таку поведінку можливо лише за умови знання
макромолекулярних характеристик ланцюгових молекул. Такі характеристики
макромолекул можна визначити на основі моделювання та експериментальних
досліджень.
Зв’язок роботи з науковими програмами, планами, темами.
Виконання роботи проводилося у межах комплексних досліджень
наукової лабораторії фізики високомолекулярних сполук при кафедрі фізики
Рівненського державного гуманітарного університету відповідно до тем
„Дослідження шляхів напрямленої модифікації властивостей гетерогенних
полімерних та дисперсних систем” і „Шляхи напрямленого регулювання
релаксаційних і термічних властивостей гетерогенних полімерних систем на основі
гнучколанцюгових полімерів” та згідно з держбюджетними науково-дослідними
роботами Міністерства науки і освіти України, зокрема «Вплив нелінійних ефектів
на кібернетику структури і енергообмінні процеси в гетерогенних полімерних
системах» (01.2010-01.2012), №реєстр.0109U000176.
Мета і завдання дослідження.
Для встановлення поведінки макромолекул, їх структурних
елементів у полях різної природи потрібно враховувати як внутрішні так і
міжмолекулярні взаємодії та їх вплив на макромолекулярні параметри. Відповідно
до цієї мети формулюються і завдання дослідження.
. Охарактеризувати макромолекулу як об’єкт запису і
передачі інформації.
. Показати можливості експериментальних методів для
визначення макромолекулярних констант.
. На основі моделювання ланцюгових макромолекул визначити
їх макромолекулярні константи.
Наукова новизна.
. На основі ентропійного підходу та кібернетичних
уявлень макромолекула лінійних аморфних полімерів розглядається як структурний
об’єкт запису і перетворення, передачі польової інформації.
. Методами моделювання макромолекул лінійних аморфних
полімерів введені і визначені їх геометричні константи.
. Використання підсистемного підходу дало можливість
визначити частотний спектр структурних елементів гетерогенних полімерних систем
і дефект модуля зсуву при поширенні механічних коливань.
Практичне значення отриманих результатів.
. На основі макромолекулярних констант прогнозуються
в’язкопружні властивості гетерогенних полімерних систем.
. Показано, що характер зміни комплексу властивостей
полімерних систем при їх модифікації наповнювачами безпосередньо пов’язаний із
зміною геометричних та енергетичних мікропараметрів макромолекул.
РОЗДІЛ І. ХІМІЧНИЙ ЗВ’ЯЗОК ТА БУДОВА МАКРОМОЛЕКУЛ
1.1 Хімічний зв’язок, будова
молекул
Молекула - найдрібніша частинка речовини, що володіє її
основними хімічними властивостями і складається з атомів, з’єднаних між собою
хімічними зв’язками. Молекули можуть відрізнятися між собою природою або
кількістю атомів, що входять до її складу; напрямком лінії зв’язку між центрами
атомів; віддалю між окремими атомами; енергією зв’язку [1].
У хімії розрізняють два роди зв’язку, що призводять до
утворення молекул: іонний (гетерополярний) і гомополярний. Хімічні властивості
елементів, визначаються в основному електронами зовнішнього шару, який може
містити тільки s- і p-оболонки. Електрони внутрішніх шарів майже не впливають
на хімічні процеси, так як вони набагато сильніше зв’язані з ядром, чим
зовнішні. Тому енергія, що виділяється при хімічних реакціях, значно менша, ніж
енергія зв’язку електронів внутрішніх шарів [2].
Іонний зв’язок реалізується в тих випадках, коли молекулу
можна подати як утворену із позитивних і негативних іонів (наприклад, NaCl).
Знак заряду іона залежить з однієї сторони, від потенціалу іонізації, а з іншої
- від тієї енергії, з якою нейтральний атом може утримувати додатковий електрон
на зовнішньому шарі.
Нехай нейтральний атом з порядковим номером z містить N електронів на
внутрішніх орбітах, тоді на зовнішній орбіті їх буде - 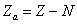 . Кулонівська потенціальна енергія, за рахунок якої
утримуються електрони, дорівнює:
. Кулонівська потенціальна енергія, за рахунок якої
утримуються електрони, дорівнює:
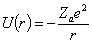 .
(1.1)
.
(1.1)
Для електронів зовнішнього шару електрони внутрішнього шару будуть
екранувати частину заряду ядра, а зовнішні електрони повинні екранувати частину
заряду ядра 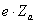 для оболонок, які лежать за межами зовнішнього шару
(тобто оболонок збуджених станів). У самому ж зовнішньому шарі частина заряду
ядра буде зкомпенсована не повністю, і тому ядро за рахунок цієї частини заряду
здатне утримувати в зовнішньому шарі додаткові електрони, що може привести до
утворення негативних іонів.
для оболонок, які лежать за межами зовнішнього шару
(тобто оболонок збуджених станів). У самому ж зовнішньому шарі частина заряду
ядра буде зкомпенсована не повністю, і тому ядро за рахунок цієї частини заряду
здатне утримувати в зовнішньому шарі додаткові електрони, що може привести до
утворення негативних іонів.
Іонний зв’язок може бути пояснений наступним чином: гетерополярна
валентність елементів визначається числом електронів, які треба забрати або
додати, щоб отримати іон, який має електронний шар найближчого інертного газу
[3]. За таких умов основну роль в іонному зв’язку повинні відігравати сили
кулонівського притягання між різнойменно зарядженими іонами. Але одні
електростатичні сили не можуть забезпечити стійку рівновагу такої системи. Тому
для обрахунків потенціальної енергії взаємодії вводяться сили відштовхування у
вигляді 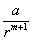 , де a, m - емпіричні константи. Тоді повна
потенціальна енергія двох іонів може бути подана у вигляді:
, де a, m - емпіричні константи. Тоді повна
потенціальна енергія двох іонів може бути подана у вигляді:
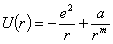 .
(1.2)
.
(1.2)
Поряд з іонними з’єднаннями існують молекули, які утворюються
безпосередньо з нейтральних атомів. Такі молекули отримали назву атомних
утворення атомних молекул пояснити навіть якісно з точки зору класичних уявлень
не можливо. Відповідь на це питання може дати квантова механіка [3]. Для
з’ясування природи гомополярного зв’язку розглянемо молекулу H2. Виражають
потенціальну енергію взаємодії між двома атомами, як функцію відстані, яка
складається з двох частин: із енергії взаємодії ядер 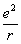 та енергії електронів E, яка залежить від відстані
між ядрами.
та енергії електронів E, яка залежить від відстані
між ядрами.
Таким чином можна записати, що енергія взаємодії дорівнює:
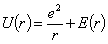 .(1.3)
.(1.3)
Сили хімічного зв’язку володіють такою властивістю як насиченість [4].
Це пояснюється тим, що число електронів у зовнішньому шарі атома обмежено, і
частина з них зв’язана один з одним, пари електронів знаходяться в одному і
тому ж квантовому стані і мають протилежної орієнтації спіни. Такі спарені
електрони не утворюють хімічного зв’язку. Останній виникає тільки завдяки
наявності в зовнішньому шарі неспарених електронів. Кожний атом може утворювати
з іншими атомами стільки ковалентних зв’язків, скільки електронів з вільною
орієнтацією спіну міститься в зовнішньому шарі.
Властивість атомів з’єднуватись один з одним утворюючи молекулу,
описується за допомогою поняття валентності [5]. Кожному атому приписується
певна валентність і при з’єднанні атомів їх валентності повинні взаємно насичуватися,
тобто кожному валентному зв’язку атома повинен відповідати валентний зв’язок
іншого атома. Для кількісної характеристики здатності атомів до взаємного
з’єднання зручно користуватися цілим числом - подвоєних спінів атомів. Це число
співпадає з хімічною валентністю атома. Слід зазначити, що один і той же атом
може мати різну валентність в залежності від того, в якому стані він
знаходиться.
Квантовий підхід до опису утворення молекули дозволяє використати
прийняте в хімії зображення молекул, як сукупності атомів, з’єднаних
локалізованими в просторі валентними зв’язками. Локалізація зв’язків проходить
внаслідок ущільнення електронних хмарин за певними напрямками в інтервалах між
атомами. Електронна хмарина для електронів, які знаходяться в p-стані витягнута
вздовж однієї з осей. Це фактично і зумовлює в просторі напрям ковалентного
зв’язку, так як приєднання інших атомів до даного іде за напрямком найбільшого
перекриття хвильових функцій у валентній парі. (рис.1.1)
Рис.1.1.
У деяких молекулах хімічний зв’язок між парами електронів здійснюється
не однією, а двома чи і трьома парами електронів[6]. Типовим прикладом молекули
з потрійним зв’язком є молекула N2 (рис.1.2)
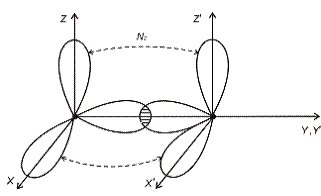
Рис.1.2.
Кожний з атомів азоту має три “пелюстки”. Тому при їх зближенні і
утворенні молекули її вісь відповідає напрямку, вздовж якого перекривається
пара однакових орієнтовних “пелюстків” (наприклад, вісь Y). Зв’язок, який
виникає при цьому найбільш міцний і називається  -зв’язком.
Хвильові функції решти електронів вздовж осей X і Z перекриваються незначно. У
зв’язку з великим взаємним відхиленням цих областей відповідні зв’язки, які
утворюються, значно слабші. Їх називають
-зв’язком.
Хвильові функції решти електронів вздовж осей X і Z перекриваються незначно. У
зв’язку з великим взаємним відхиленням цих областей відповідні зв’язки, які
утворюються, значно слабші. Їх називають  -зв’язками.
Зв’язки -типу легко руйнуються і дозволяють отримувати нові
з’єднання. Однак роль цих зв’язків значна, особливо в з’єднаннях карбону.
Прикладом потрійного зв’язку може бути молекула ацетилену C2H2 (рис.1.3)
-зв’язками.
Зв’язки -типу легко руйнуються і дозволяють отримувати нові
з’єднання. Однак роль цих зв’язків значна, особливо в з’єднаннях карбону.
Прикладом потрійного зв’язку може бути молекула ацетилену C2H2 (рис.1.3)
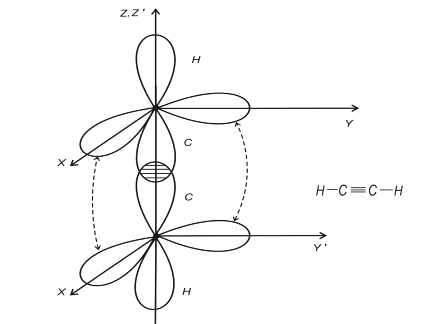
Рис.1.3
Два зв’язки між атома С - це зв’язки -типу,
вони орієнтовані вздовж осей X або Y; решта С-С зв’язки відносяться до зв’язків
- типу. За рахунок таких зв’язків молекула C2H2 -
лінійна. Подвійний зв’язок спостерігається в молекулах етилену C4H2 (рис.1.4)
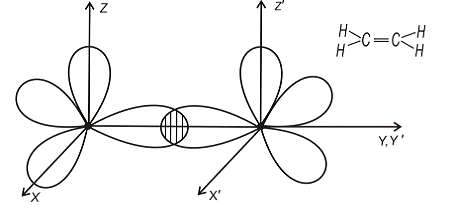
Рис.1.4.
У цьому випадку кожний атом С має три рівноцінних -зв’язки: С-С; С-Н; С-Н. Тому зв’язки групи CH2
розміщені в площині під кутом 1200. Враховуючи, що максимальне перекриття
хвильових функцій в -зв’язках досягається за рахунок паралельності їх
напрямків, отримують плоску структуру молекули етилену.
У розглянутих вище зв’язках приймають участь електрони двох сусідніх
атомів. Утворені таким чином зв’язки називають локалізованими. Зовсім інший
зв’язок спостерігається в молекулах типу бензолу C6H6. У цій молекулі три
електрони кожного атома С приймають участь в утворенні трьох -зв’язків: один з атомом Н і два з сусідніми атомами
С. Оскільки ці зв’язки рівноцінні, вони утворюють плоску структуру з кутом 1200
між зв’язками. Крім того, у кожному атомі С наявний один неспарений електрон,
здатний утворювати зв’язок -типу.
Внаслідок цього молекула C6H6 є плоским правильним шестикутником (рис.1.5)
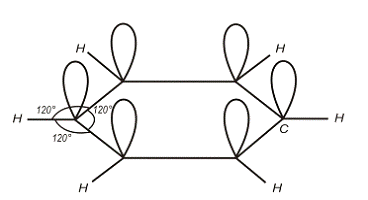
Рис.1.5.
Проте у молекулі бензолу є відмінності від інших кратних зв’язків, а
саме: у молекулі C6H6 на кожний s-зв’язок
припадає по два електрони; для шести p-зв’язків
наявні не 12, а лише 6 електронів. Оскільки молекула бензолу симетрична, то це
означає, що на кожний s-зв’язок між атомами С
припадає половина локалізованого p-зв’язку. З
точки зору квантової механіки, цей факт трактується таким чином: кожний
неспарений електрон у зовнішньому шарі атома карбону бере участь в обміні
одночасно з електронами обох сусідніх атомів. Виникаючий при цьому p-зв’язок “нелокалізований”, бо він для кожного атома С
здійснюється і з “лівим”, і з “правим” електронами. Оскільки всі атоми карбону
рівноправні, зв’язок має місце між усіма атомами карбонового кільця у молекулі
бензолу. Така “делокалізація” p-зв’язку
приводить до його зміцнення. Таким чином енергія дисоціації “нелокалізованого”
зв’язку значно перевищує енергію дисоціації у випадку чергування трьох
одинарних і трьох подвійних зв’язків. Крім того, електрони, які приймають
участь у “нелокалізованому” p-зв’язку,
дуже рухливі. Вони здатні переміщуватися за рахунок тунельного ефекту від
одного атома вуглецю до іншого. “Нелокалізований” p-зв’язок у молекулі бензолу може служити моделлю так
званого металічного зв’язку характерного для кристалів, металів і деяких інших
речовин [7].
Розгляд полімерних матеріалів показав, що міжатомна взаємодія в них і
низькомолекулярних речовинах за своїм характером не має принципової різниці.
1.2
Виникнення і розвиток поняття макромолекула
Молекула полімера - це об’єднання найпростіших елементів
структури [8]. З’єднання великої кількості малих молекул або атомів за рахунок
хімічних зв’язків приводить до утворення гігантської ланцюгової молекули. Ця
ланцюгова молекула полімера називається макромолекулою. Об’єднання найпростіших
елементів полягає в тому, що спостерігається повторення однієї і тієї ж
структурної одиниці або чергування різних структурних одиниць. Ці найпростіші
елементи структури називаються мономерами або ланками, що повторюються.
З’єднання мономерів у макромолекулу полімера відбувається в результаті хімічних
реакцій, що протікають за законом ланцюгових чи ступінчастих процесів. Степінь
полімеризації визначається числом мономерних ланцюгів в одній макромолекулі і
визначає молекулярну масу полімера, яка складає десятки, сотні, а інколи й
мільйони одиниць. Молекулярна маса полімера дорівнює вихідній масі мономера,
помноженій на степінь полімеризації:
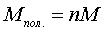 ,
(1.4)
,
(1.4)
де n - степінь полімеризації, М - маса мономера.
Макромолекули полімерів утворюються в результаті процесів
полімеризації, або поліконденсації [9, 10, 11]. У самому загальному випадку
вони є лінійні, розгорнуті, або сітчасті системи, які складаються з однакових
(гомополімери) або різних (сополімери) елементів структури (мономер них або
ланок, що повторюються), що мають певну автономність - кінцеве число ступенів
вільності. Ця часткова автономність проявляється не дивлячись на те, що
мономерні ланки зв’язані між собою хімічними або змішаними зв’язками.
Лінійна макромолекула утворюється в тому випадку, коли у вихідному
стані мономер має подвійну функціональність, тобто дві вільні валентності, що
забезпечує можливість полімеризації, або дві автономні групи - здатність до
конденсації [12]. Прикладом полімеризації може служити утворення поліетилену з
етилену:
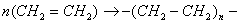
Прикладом поліконденсації - утворення поліефіру з біфункціонального
спирту і дикарбоксильної кислоти:
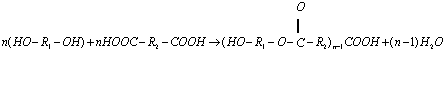 .
.
Тут R1 і R2 - аліфатичні або ароматичні радикали. При полімеризації
вихідний мономер, хоч і втрачає функціональність проте зберігає свою хімічну
формулу, однак при поліконденсації формула ланки, яка повторюється, відрізняється
від хімічних формул вихідних компонентів. Це вірно і у тих випадках, коли
вихідний мономер здатний конденсуватися “на себе”.
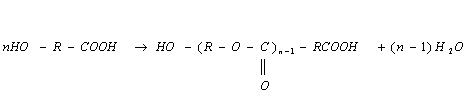
Тому термін ланка, що повторюється, точніший, ніж мономерна ланка. Але
для сополімерів, де мономерні ланки можуть чергуватися без видимого порядку,
він також є не повним. Враховуючи ці фактори, використовують термінм ланка.
Розгалужені макромолекули утворюються, якщо в процесі сополімеризації
приймає участь невелика кількість трифункціонального мономера, а також у
результаті випадкових хімічних реакцій.
При деякій критичній концентрації три- або мультифункціонального мономера
замість розгалуженого полімера утворюється неперервна сітка - плоска або
просторова. Ступінь вільності в такій сітці визначається довжинами
нерозгалужених ділянок, які обчислюються кількістю вхідних у ці ділянки ланок,
тобто ступенем полімеризації, між поперечними зв’язками або вузлами сітки. Чим
вужчі ці ділянки, тим менше ступенів вільності у ланок і, завдяки однотипності
зв’язків, тим ближчі властивості полімера до властивостей твердого тіла.
Абсолютно правильна просторова сітка не відрізняється від ковалентного кристалу
типу алмаз і може плавитися одночасно з полімеризацією.
Перш ніж склалися уявлення про те, що молекули полімера є
довгими ланцюгами ланок, наука пройшла тривалий і тернистий шлях. На початку XX
століття хіміки працювали над питанням кристалізації чистих хімічних з’єднань.
На основі великої кількості експериментів цей факт було доведено. Проте, деякі з
них зосередили свої зусилля на вивченні властивостей колоїдів. Одним з них був
Семюел Шродер Піклс, який працював у Лондонському Імператорському інституті
[13]. Вивчаючи властивості каучуку, його взаємодію з Br2, яка призводять до
розкриття подвійних зв’язків, він прийшов до висновку, що такі речовини не
можуть бути асоціацією малих молекул, а повинні бути великими молекулами, де
ланки зв’язані ковалентними зв’язками. Опублікована стаття Піклса з цих питань
не була визнана. У цей час пануючим уявленням про специфічні властивості
полімерів залишалось те, що вони є складними колоїдами, а відсутність кінцевих
груп відносили до існування “міцел”, що складаються з малих кілець. На початку
20-х років XX століття, з’являються роботи німецького фізика-хіміка Ентера
Германа Штаудінгера (1881-1965), в яких на основі аналізу експериментів було
висунуто ідею про ланцюгову будову полімерів. Спочатку ці ідеї викликали
недовіру. Але відкритий у тридцяті роки і доведений Полем Дж. Флорі (1910-1985)
принцип рівної хімічної реакційної здатності груп і рентгенівські дослідження
частково кристалічних полімерів, проведені Куртом Х. Мейєром і Германом Ф.
Марком, підтвердили у загальних рисах гіпотезу Штаудінгера про ланцюгову будову
молекул. Проте відкритим залишалося питання, про велику молекулярну масу
полімера. Рентгенівські дослідження показували, що довжина кристалітів невелика
- всього 30-60 нм. Якщо вважати, що молекула не може бути довшою, ніж
кристаліт, то молекулярна маса повинна складати 5000-10000 одиниць. Проте і це
питання відпало, коли в 1957 р. було відкрито складення ланцюгів, цей факт
вказував на те, що розміри кристалітів можуть бути набагато менші за розміри
макромолекул. В. Геллер вже у 1931 р. показав, що ланцюги не такі короткі, як
передбачав Штаудінгер. Швейцарець Вернер Кун зробив висновок про те, що за
рахунок обертання навколо зв’язків, макромолекула повинна мати форму клубка, а
не стержня.
Слід зазначити, що ідея існування макромолекул була розвинута
в біохімії, не залежно від відповідних ідей в органічній і фізичній хімії,
Джейсом Самнером (1887-1955). Експерименти проведені шведським професором
Теодором Сведбергом (1884-1971) в 1924-1927 рр. з визначення молекулярної маси
ензиму (ферменту) уреазу на ультрацентрифузі і шведським вченим Арне Тізеліусом
(1902-1971) в 1938 р., який показав, що ензими сильно відрізняються від
поведінки неорганічних колоїдів, дали змогу підтримати ідею макромолекулярної
будови ферментів.
Ідея ланцюгової будови молекул полімерів остаточно завоювала
світ після другої світової війни. В 1953 р. Штаудінгер був нагороджений
Нобелевською премією з хімії після більше ніж 30 років роботи.
Таким чином, основи теорії макромолекулярної будови
полімерних матеріалів і їх властивостей була розроблена в 30-ті роки ХХ ст.
фундаторами сучасної фізики полімерів: С. Марком, Гутом, Т. Куном, П. Кобеко,
Я. Френкелем, С. Бреслерем. Подальший розвиток сучасних уявлень в області
фізико-хімії полімерів отримано в роботах П. Флорі, М. Волькенштейна,
В.Каргіна.
Для введення основних геометричних констант макромолекул
потрібно врахувати, що фізичні властивості макромолекул визначаються їх
конфігурацією і конформацією. Під конфігурацією макромолекули слід розуміти
просторовий розподіл атомів (підсистем) у макромолекулі, що визначається
довжинами відповідних зв’язків і значенням валентних кутів. Іншими словами ці
властивості визначаються станом електронних оболонок; до яких відносяться
оптичні властивості, електропровідність, надпровідність полімерів. Конфігурацію
макромолекули можна змінити тільки проведенням хімічної реакції, яка призводить
до зміни перестановки зв’язків або валентних кутів. Макромолекули полімерів, на
відміну від простих молекул, навіть якщо вони мають однаковий склад і число
ланок, індивідуальні. Індивідуалізовані навіть окремі досить протяжні ділянки
макромолекул так, що на них вже записана деяка комбінаторна інформація. Із
моменту виникнення макромолекули система має здатність до самоускладнення і
самовдосконалення. Інформацію про макромолекулу можна розділити на два дуже
різні поняття:
· структурна інформація - інформація
спільна для значної групи макромолекул, наприклад, хімічна формула ланки;
· конфігураційна інформація (дискретна)
- індивідуальна інформація кожної макромолекули, яка отримується за рахунок
комбінації на певних ділянках ланцюга деяких конкретних положень атомів або
груп атомів ланки [14].
Конформацією макромолекул називається взаємне розміщення її
ланок у просторі [15]. У кожний момент часу всі атоми, а отже і ланки,
макромолекули займають певні положення в просторі, але в результаті теплового
руху ці положення весь час змінюються, тобто миттєві конформації переходять
одна в одну. Коли мова йде про конформацію макромолекули, то мають на увазі усю
сукупність миттєвих конформацій, які приймає макромолекула в певних умовах. Особливістю
макромолекул є їх здатність знаходитися в різних конформаціях. Конформаційні
властивості визначають основні особливості як біологічних, так і синтетичних
полімерів. Слід зауважити, що конформаційні явища в макромолекулах відбуваються
на просторово-часових масштабах, які значно перевищують атомні. У результаті
внутрішньо-молекулярного (мікробраунівського) теплового руху ланок або ж під
впливом зовнішніх динамічних силових полів полімерний ланцюг здатний змінювати
свою конформацію. Ця здатність макромолекули змінювати свою форму називається
гнучкістю. Гнучкість макромолекули зумовлена тим, що мономерні ланки при
тепловому чи будь-якому іншому русі обертаються навколо простих зв’язків
основного ланцюга. Будь-яка зміна конформації макромолекули пов’язана із серією
таких елементарних поворотів. Обмеженість кута повороту зумовлена взаємодією
бокових груп сусідніх ланок (рис.1.6).
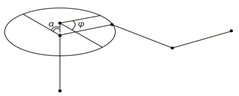
Рис.1.6. Схема, що пояснює внутрішнє обертання в полімерному
ланцюгу. a - кут, який доповнює валентний
Однією з граничних форм, яких може набувати макромолекула, є
витягнута форма. Для прояву такої форми макромолекули необхідна наявність сил,
які не дозволяють макромолекулі проявити свою гнучкість. У залежності від
конкретного хімічного складу полімера ця конформація може бути плоским
транс-зигзагом (з простими С-С зв’язками і не масивними боковими групами) або
спіраллю (у макромолекулах з масивними боковими групами) (рис. 1.7).
Макромолекули, в яких малі крутильні коливання, можуть
знаходитися у витягнутій конформації. Витягнуту конформацію такої макромолекули
можна спостерігати в розбавленому розчині, але ступінь полімеризації їх повинна
бути такою малою, щоб характерна макромолекулі гнучкість не встигала
проявитися. Якщо полімерний ланцюг проявляє гнучкість, то більшість миттєвих
конформацій, які приймає макромолекула, будуть згорнутими і їх сукупність дасть
статистичний клубок. Суттєвою особливістю такого клубка є те, що він є сильно
флуктуаційною і дуже неоднорідною системою і його густина зменшується із ростом
довжини макромолекули.
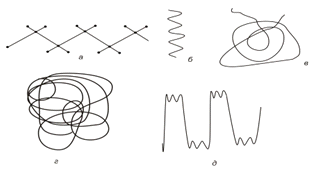
Рис. 1.7. Форми лінійних макромолекул: витягнута
конформація(а,б): а - транс-зигзаг; б - спіраль; в - статистичний клубок; г -
статистична глобула; д - складчаста конформація.
Якщо густина у згорнутому, переплетеному стані макромолекули
постійна в усьому об’ємі, тобто флуктуації відсутні, то для макромолекули
характерна конформація, яка називається статистичною глобулою. Потрібно
зауважити, що у випадку такої конформації, її густина більша за густину
статистичного клубка. Конформацію статистичного клубка макромолекули набувають
як у розведених, так і концентрованих розчинах, а також в аморфних полімерах.
Глобули спостерігаються у випадку поганих розчинників при дуже малих концентраціях
полімера і бінарних сумішах несумісних полімерів.
Ще однією конформацією макромолекули є складчаста. Таку форму
можна уявити як витягнутий ланцюг зі зламами (рис. 1.7. д.). Взаємодія між
витягнутими ділянками макромолекули при такій конформації відбувається
безпосередньо без допомоги інших макромолекул.
.3 Макромолекула, як об’єкт запису і перетворення інформації
Як неодноразово підкреслювалось, індивідуальну інформацію, яка міститься у кожній макромолекулі, не враховують
при розробці технології виготовлення і переробки, що приводить до перетворення
даної технології в "антитехнологію". Успіхи молекулярної біології
все-таки змусили звернути увагу на "індивідуальні" інформаційні
характеристики макромолекул. Виявилося, що дуже багато макроскопічних властивостей
біологічних систем залежать від будови і конфігурації однієї або декількох
конкретних макромолекул. Більше того, індивідуальні властивості конкретних
макромолекул значно важливіші для всієї біологічної системи, ніж
середньостатистичні властивості набору макромолекул. Так, генетичний код -
індивідуальна інформація в макромолекулі, і загальний для усіх макромолекул ДНК
фазовий перехід - плавлення.
Розглядаючи макромолекулу навіть такого "простого" полімеру, як
поліпропілен (ПП), відмітимо, що всі його ланцюги індивідуалізовані не
дивлячись на те, що вони можуть, на приклад, складатися з однакової кількості
ланок. Так, метильний радикал СН3 може розміщуватися по обидві сторони від
площини, в якій розміщений основний ланцюг. Тому, якщо ланцюг містить n ланок і немає особливих обмежень для
вибору напрямів радикала СН3 при приєднанні мономеру до кінця ланцюга, який
росте, то можна отримати 2" різних за характером розміщення цього радикала
у ланцюгу з однаковою хімічною формулою.
Виходячи з комбінаторного підходу до поняття інформація, можна відмітити,
що описаний ланцюг, що складається з ланок, містить максимально п бітів
інформації [16].
Якщо дослідити будь-яку просту речовину визначеного хімічного складу, то
його молекули не можуть бути використані в якості системи знаків (символів),
оскільки вони не відрізняються. Тобто записати і зберегти будь-яку інформацію
на системі з простих молекул неможливо. Звичайно, прості молекули можуть
відрізнятися, наприклад, ступенем збудження, але ці відмінності не зберігаються
в часі. Зовсім інша справа - макромолекули полімерів. Навіть якщо вони мають
однаковий склад і кількість ланок, вони все рівно індивідуалізовані: їх можна
відрізнити одну від одної, причому в наслідок особливої форми конденсації (всі
зв'язки між ланками ковалентні) ці відмінності практично "вічні".
Індивідуалізовані навіть окремі досить видовжені ділянки макромолекул, так, що
і на них записана деяка комбінаторна інформація. Тому акт полімеризації -
значно більше фундаментальна подія, ніж звичайний фазовий перехід, оскільки з
моменту виникнення макромолекули система набуває здатність до самоускладнення і
самовдосконалення [17-23]. Інформацію про макромолекулу ми
розділимо на два різних класи:
- структурна інформація, інформація загальна для значної групи
макромолекул, наприклад хімічна формула ланки;
- конфігураційна інформація (дискретна) - індивідуальна інформація
кожної макромолекули, яка отримується за рахунок комбінацій на певних ділянках
ланцюга деяких конкретних початкових положень атомів і груп атомів ланки.
Приклад конфігураційної інформації, "закодованої" у вініловому
ланцюзі, що має дві можливі конфігурації сусідів, - ізо (І) і синдіо (С):
І І СІСІССІ І ІССІССІСССІ І ІССІ ІСССІССС…
або
СССІ І ІСССІ І ІСССІ І ІССІ ІССССІССССІ ІС…
Тут "кодонами", які несуть інформацію, будуть групи ІІС, ІСС, III і ін. В принципі, за аналогією з
азбукою Морзе, "літерою" можна вважати будь-які поєднання невеликої
кількості послідовних ланок.
У сучасній науковій практиці існує ряд підходів до визначення інформації.
Про статистичний (шенноновий) найбільше відомий підхід ми вже згадували, в
якості ж наступного варіанту згадуємо логіко-семантичні теорії інформації,
початок яких покладено в роботах Карнапа і Бар-Хилела. В даних теоріях один з
основних моментів - визначення "вислови", які повністю описують одну
з можливих альтернатив в даній мові. Найважливішим є також поняття "опису
стану" і логічної або індуктивної ймовірності. Опис стану є кон'юнкція, що
містить в якості компонентів для кожного атомарного (тобто неподільного на
більш дрібні частини) пропозицію саму або її заперечення (але не обидва разом)
і що не містить ніяких інших пропозицій. Опис стану можна виразити наступним
чином:
(±)Р1(а1)Λ(±)Р2(а1)Λ… Λ(±)Рn(а1)Λ;
Λ(±)Р1(а2)Λ(±)Р2(а2)Λ…
Λ(±)Рn(а2)Λ
Λ(±)Р1(а3)Λ(±)Р2(а3)Λ…
Λ(±)Рn(а3)Λ
Λ(±)Р1(аn)Λ(±)Р2(аn)Λ… Λ(±)Рn(аn)Λ,
де Λ - знак кон’юнкції, тобто пропозиційна зв’язка «і», Р1,Р2,Р3…Рn - одномісні незалежні предикати,
а1,а2,а3…аn - індивіди, а символ (±) означає, що
вираз, який за ним стоїть , може мати або не мати від’ємний знак. Зміст опису
полягає в тому, що він є висловом, який повністю описує одну з можливих
альтернатив у даній мові, а клас описаний станом - всі логічні можливості
універсуму відносно даних властивостей індивідів. Повертаючись до опису макромолекул,
бачимо, що на базі семантичного підходу можна описати стан макромолекулярних
систем на будь-якому заданому рівні: враховуючи лише хімічну структуру; хімічну
структуру і конфігурацію; хімічну структуру, конфігурацію і набір конформацій і
т.п. Необхідно лише пам'ятати, що для кожного з цих варіантів буде побудована
мова опису з набором альтернативних станів.
У молекулярній кібернетиці найбільш перспективний алгоритмічний підхід до
теорії інформації, розвинутий А.М.Колмогоровим. У цьому випадку інформація,
об'єкта А відносно об'єкта В визначається складність (довжиною) програми
(алгоритму), що описує послідовність дій, необхідних для перетворення об'єкта В
в об'єкт А. Очевидно, що описані таким чином макромолекули, наприклад
поліпропілену з різними конфігураціями відносно молекул пропілену або атомів
вуглецю і водню, вже містять інформацію, якої досить для відтворення конкретних
конфігурацій молекул. При цьому різними інформаційними значеннями будуть
описуватися варіанти ізо-, синдіо- і атактичного полімеру. Важливо відмітити,
що інформація про структуру макромолекул поліпропілену може суттєво допомогти у
технології. Так, у випадку синдіо- або ізотактичного полімеру останній здатний
кристалізуватися і утворювати досить міцні волокна,, а полімер атактичний вже
при невисоких температурах каучукоподібний і ніяких волокон утворювати не може
[24-27].
Повертаючись до опису інформаційного змісту в макромолекулах, нагадаємо
зауваження відомих японських вчених Тюдзе і Каваі. Вони зазначають, що якщо
порівнювати між собою інформаційні властивості так званих біополімерів, які
зустрічаються в природі, і синтетичних полімерів, то структура останніх в
значній мірі детермінована імовірнісними процесами. Виходячи з алгоритмічного
представлення інформації, можна сказати, що утворення синтетичних полімерів
зумовлене все-таки алгоритмом технології у відповідності з основним визначенням
цього поняття: "алгоритм - це процес послідовної побудови величин, який
проходить в дискретному часі таким чином, що в початковий момент часу задається
вихідна кінцева система величин, а в кожний наступний момент система величин
отримується за певним законом (програмою) із системи величин в попередній
момент часу". А біополімери виникли завдяки не алгоритмічному, а
евристичному - процесу послідовної побудови величин, що проходить в дискретному
часі таким чином, що в початковий момент часу задається деяка система величин,
а в кожний наступний момент система за одним, а за рядом певних законів із
існуючих в попередній момент часу. Ці закони можуть змінюватися під час
евристичного процесу". У випадку біополімерів така евристика - еволюційний
процес. У цьому і полягає різниця між полімерами синтетичними і біо; перші
виготовляються за певним технологічним алгоритмом, інші отримані у результаті
деякої історичної випадковості і зовсім не оптимальному. А інші отримані в
результаті багатовікової еволюційної самоорганізації. Хотілося б відмітити, що,
виходячи з принципів молекулярної кібернетики, як раз і не можливо вважати
сучасні технології отримання полімерів або виробів з них оптимальними. Ніхто не
заперечить те, що значним досягненням буде проведення такого ж направленого
еволюційного відбору технологій для синтетичних полімерів. Хоча для еволюції
біополімерів необхідні були мільйони років, а нам чекати ніколи. Тут на
допомогу молекулярної кібернетики прийде кібернетика звичайна. Останнім часом
дуже активно розвивається галузь останньої - еволюційне моделювання складних
систем на ЕОМ. Розвинуті багаточисленні еволюційні алгоритми і програми, що
забезпечують оптимальну самоорганізацію складних систем [28, 29].
РОЗДІЛ ІІ. ОБ’ЄКТИ ТА МЕТОДИ ДОСЛІДЖЕНЬ
.1 Лінійні аморфні полімери та неорганічні наповнювачі.
Отримання зразків
Як основний об’єкт досліджень був вибраний лінійний гнучколанцюговий полімер вінілового
ряду - полівінілхлорид. Важливо відмітити, що ПВХ, як один із представників
аморфних полімерів, цікавий як "модельний матеріал", оскільки володіє
досить широким спектром змін структурної організації на різних рівнях.
Досліджували ПВХ суспензійної полімеризації марки С - 65, очищений
переосадженням з розчину, що дозволило позбутися низькомолекулярних домішок,
залишків ініціатора, емульгаторів і захисних колоїдів.
Молекулярна маса переосадженого полімера, визначена віскозиметрично в
циклогексаноні на візкозиметрі Освальда-Уббелоде при 295 К і розрахована за
емпіричним рівнянням Марка-Куна-Хувинка, склала 1,4×105.
Основні фізико-механічні характеристики блочного ПВХ представлені в
таблиці. 2.1.
Як наповнювач для ПВХ використовували високодисперсний, промисловий
порошок вольфраму (W) з переважаючим
розміром частинок 7 мкм. Розмір частинок визначали седиментометричним і
мікроскопічним аналізом. Основні фізико-хімічні характеристики вольфраму
представлені в таблиці 2.2.
Зразки для екпериментальних досліджень готували методом механічного
змішування ПВХ з порошком вольфраму, попередньо обробленим CCl4, з подальшим формуванням в Т-р
режимі у вигляді пластин товщиною 5 мм і діаметром 25 мм. ПКМ готували двох
типів:
1 без накладання ЕП в процесі
формування;
2 з накладанням ЕП в процесі
формування;
з однаковим об¢ємним вмістом наповнювача (j) в системах.
Таблиця 2.1
Основні фізико-механічні характеристики ПВХ
|
Показники
|
Значення
|
|
1
|
2
|
|
Густина при 293К, кг/м3×10-3
|
1,35 ¸ 1,43
|
|
Твердість по Брінелю, МН/м2
|
130 ¸ 160
|
|
Міцність, МН/м2
|
|
|
при розтязі
|
40 ¸ 60
|
|
при згині
|
80 ¸ 120
|
|
Модуль пружності при
розтязі, ГН/м2
|
3 ¸ 4
|
|
Ударна питома в’язкість,
кДж/м2
|
2 ¸ 5
|
|
Відносне видовження при
розриві, %
|
5 ¸ 10
|
|
Смуги в IЧ-спектрі, см-1
|
2968 ¸ 310
|
|
Температура склування, К
|
353
|
|
Температура текучості, К
|
453
|
|
Питома теплоємність, кДж×/(кг×К)
|
0,85 ¸ 2,14
|
|
Теплопровідність, Вт×/(м×К) при температурі 90 ¸ 340К
|
0,125 ¸ 0,175
|
|
Коефіцієнт об’ємного
термічного розширення, К-1×104 при температурі 293 ¸ 357 К
|
3 ¸ 4,2
|
|
Температурний коефіцієнт лінійного
розширення, К-1×105 при температурі 293 ¸ 357 К
|
6 ¸ 10
|
|
Питомий об’єм, при тиску 20
МПа і температурі 229 ¸ 373 К, м3/кг
|
0,704 ¸ 0,730
|
|
Теплостійкість по Мартенсу,
К
|
323 ¸ 353
|
|
Питомий електричний опір
при 293 К
|
|
|
об’ємний, ГОм×м
|
10 ¸ 1000
|
|
поверхневий, ТОм×м
|
10 ¸ 100
|
|
Діелектрична проникність
|
3 ¸ 5
|
|
Тангенс кута діелектричних
втрат
|
0,03 ¸ 0,05
|
|
Показник заломлення
|
1,5
|
|
Водопоглинання за 24 год, %
|
0,4 ¸ 0,6
|
|
Морозостійкість, К
|
213 ¸ 258
|
|
Молекулярна маса, кг/моль×103
|
62,5
|
|
Енергія активації при
термодеструкції по методу Фрімена-Керола, кДж/моль
|
68
|
|
Енергія терморозпаду,
кДж/моль
|
100 ¸ 110
|
|
Електрична міцність кВ/мм
при 293 К при 413 К
|
35 ¸ 45 < 5
|
Таблиця 2.2
Фізико-хімічні характеристики вольфраму.
|
ПоказникиЗначення
|
|
|
Атомна маса
|
183,86
|
|
Густина, кг/м3×10-3
|
18,6 ¸ 19,1
|
|
Атомний радіус, м×10-10
|
1,41
|
|
Модуль пружності, ГН/м2
|
342 ¸ 400
|
|
Модуль зсуву, ГН/м2
|
88 ¸ 215
|
|
Твердість по
Брінелю, МН/м2
|
1960 ¸ 2450
|
|
Часовий опір на розрив, МН/м2
|
699 ¸ 809
|
|
Питома теплоємність, Дж/(кг×К)
|
136
|
|
Теплопровідність при 300 К,
Вт/(м×К)
|
130
|
|
Питомий електричний опір при 300 К, нОм×м
|
55
|
|
Температурний коефіцієнт
лінійного розширення при 373 К, К-1×10-5
|
4,3
|
|
Питома поверхня, м2/кг
|
51
|
|
Тип кристалічної гратки
|
ОЦК
|
|
Здатність до окислення на
повітрі при кімнатній температурі
|
не окислюється
|
Зразки нагрівали до температури 403 К і під тиском 10 МПа
охолоджували до 293 К з накладанням ЕП, напруженістю 1кВ/мм і 5 кВ/мм для ПВХ,
та 1 кВ/мм для композицій ПВХ+W, та без нього. Швидкість охолодження зразків становила 4 кВ/хв, час дії
електричного поля 30 хв, градієнт постійного електричного поля співпадав з
напрямком прикладання силового поля.
2.2 Використання методів розсіювання світла для визначення
макромолекулярних констант
Оптичні властивості полімерів характеризують взаємодію з
електромагнітним випромінюванням оптичного діапазону, який включає
ультрафіолетову, видиму і інфрачервону область. Ультрафіолетовій області
відповідають електромагнітні випромінювання з довжиною хвилі 1,0·10-8м÷3,8·10-7м, видимій - 3,8·10-7м-7,6·10-7м,
інфрачервоний - 7,6·10-7м-1,0·10-3м . Інфрачервона область ділиться на три
діапазони: ближній(7,6·10-7м÷2,5·10-6м), середній
(2,5·10-6м-5,0·10-5м), дальній (5,0·10-5м-1,0·10-3м). Ультрафіолетову область
поділяють на діапазони вакуумного (1,0·10-8м-1,7·10-7м) і нормального
(1,7·10-7м-3,8·10-7м) ультрафіолетового випромінювання. Частоти оптичного
діапазону спектра електромагнітних хвиль знаходяться в інтервалі 3,0·1016 до
3,0·1011 Гц.
Оскільки полімерні системи є ізотропними і анізотропними
середовищами, то до оптичних властивостей відносять відбивання і поглинання
світла такими середовищами, розсіювання і деполяризація розсіюваного світла,
оптичну обертаючу здатність, а також явища що виникають в таких системах під
дією силових полів. Ці макроскопічні явища знаходять свої пояснення на основі
уявлень про структуру полімерів(молекулярно-кінетичний підхід).
При рекомбінації зарядів можлива люмінесценція. Розрізняють в
залежності від способу збудження фотолюмінісценцію (збудження видимим світлом),
катодолюмінісценцію (катодними променями), рентгенолюмінесценцію
(рентгенівськими променями), радіолюмінісценцію (радіохвилями),
термолюмінісценцію (тепловим полем), електролюмінісценцію (електричним полем),
хемілюмінесценцію (хімічними реакціями).
Характерною особливістю радіотермолюмінісценції полімерів є
те, що максимуми інтенсивності свічення на температурній залежності знаходяться
в інтервалі тих температур, де знаходять прояв різноманітні кінетичні і
структурні переходи, зумовлені рухливістю структурних елементів макромолекул, а
також молекулярним рухом в аморфних і кристалічних областях. Інтенсивність
свічення різко зростає, коли виникає рухливість окремих частинок макромолекул.
Для аморфних полімерів на температурній залежності
інтенсивності максимуми з’являються в області релаксаційних переходів, а
кристалічних - в областях фазових переходів і поліморфних перетворень. Оскільки
максимуми розміщені в температурній області, що відповідає релаксаційним
переходам, то це дає можливість визначати енергію активації їх. Метод
радіотермолюмінісценції дозволяє досліджувану ступінь однорідності
двокомпонентних сумішей полімерів і їх сумісність, проводити аналіз процесів
структуроутворення системи полімер-наповнювач , полімер-полімер-наповнювач.
При поширенні світла і переході з одного середовища в інше
можливий випадок, коли електромагнітна хвиля поширюється за напрямками
відмінними від напрямків поширення відбитої та заломленої хвиль. Такий процес
називається розсіюванням світла і розрізняють два його види: класичне (релеєвське)
та комбінаційне. Класичне розсіювання світла відбувається без зміни довжини
хвилі світла. З оптичної точки зору, полімери можна розділити на два класи: а)
сильно розсіюючі (оптично мутні), оптичні властивості яких можна описати в
моделі багаторазового розсіювання скалярних хвиль у випадково неоднорідному
середовищі з поглинанням; б) слабко розсіюючі (прозорі), оптичні властивості
яких описуються в моделі одноразового розсіювання упорядкованого середовища із
щільним розміщенням розсіювачів, які містять центри поглинання. У розсіюючому
середовищі векторний характер хвиль виявляється як виникнення поляризації у
падаючому неполяризованому пучку світла чи як деполяризація при поширенні в
середовищі поляризованого пучка.
Розрізняють два режими опромінення полімерів: неперервний і з
розділенням по часу. Режим з розділенням по часу реалізується при опроміненні
полімерів короткими лазерними імпульсами (10-9 ÷
10-11 с) і
прийманням розширених імпульсів розсіюваного випромінювання, так і шляхом
опромінення модульованим за інтенсивністю світлом на частотах в діапазоні 100
МГц÷10 ГГц і реєстрації глибини модуляції розсіюваного
випромінювання і відповідного зсуву фаз на частотах модуляції. Основою режиму з
розділенням по часу є збудження у сильнорозсіюючому середовищі спектра хвиль
фотонної густини, що описується в рамках нестаціонарної теорії переносу
випромінювання, а режим неперервного випромінювання описується в рамках
стаціонарної теорії [30]. Згідно зі стаціонарною теорією переносу
випромінювання отримують математичний опис процесу поширення немодульованого
світла в розсіюючому середовищі, що містить ансамбль віддалених один від одного
розсіювачів (наповнені полімерні системи). Для монохроматичного світла
стаціонарне рівняння можна записати у такому вигляді:
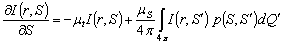 ,(2.1)
,(2.1)
де 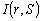 - променева інтенсивність в точці r в напрямку S;
- променева інтенсивність в точці r в напрямку S; 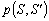 - фазова функція розсіювання,
- фазова функція розсіювання, 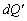 - одиничний тілесний кут в напрямку
- одиничний тілесний кут в напрямку 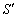 ;
; 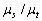 -
альбедо одиничного розсіювання, при відсутності внутрішніх джерел
випромінювання.
-
альбедо одиничного розсіювання, при відсутності внутрішніх джерел
випромінювання.
На практиці використовується не сама функція , а інтеграли від неї за деякими областями фазового
простору 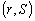 . Фазова функція описує
розсіюванні властивості середовища і є функцією густини ймовірності для
розсіювання в напрямку - фотона, що рухається в напрямку S, тобто характеризує елементарний акт розсіювання. У випадку
симетричного розсіювання фазова функція залежить від кута
. Фазова функція описує
розсіюванні властивості середовища і є функцією густини ймовірності для
розсіювання в напрямку - фотона, що рухається в напрямку S, тобто характеризує елементарний акт розсіювання. У випадку
симетричного розсіювання фазова функція залежить від кута 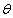 між напрямками
між напрямками 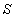 і
і
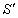 :
: 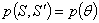 .
.
При відсутності просторової кореляції (випадкове розміщення часток
наповнювача в полімері) умова нормування набуває вигляду:
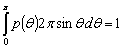 .(2.2)
.(2.2)
У багатьох випадках 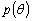 апроксамується
функцією Хеньї-Грінштейна:
апроксамується
функцією Хеньї-Грінштейна:
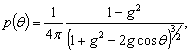 (2.3)
(2.3)
де g - середнє значення косинуса кута розсіювання (параметр анізотропії розсіювання)
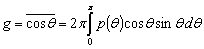 .(2.4)
.(2.4)
де g=0 відповідає випадку ізотропного (релеєвського) розсіювання, g=1 -
повному розсіюванню (розсіюванню µi на великих частинках).
Інтегро-диференціальне рівняння спрощується, коли розв’язок подано у
вигляді сферичних гармонік. Таке спрощення призводить до системи (N+1)2
зв’язаних диференціальних рівнянь в частинних похідних. Для N=1 одержують
чотири зв’язаних рівняння, які зводяться до єдиного рівняння дифузійного типу,
яке у випадку ізотропного середовища має вигляд:
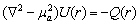 ,
(2.5)
,
(2.5)
де 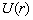 - повна освітленість в точці,
- повна освітленість в точці, 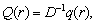 q(r) - число фотонів виникаючих
в одиниці об’єму,
q(r) - число фотонів виникаючих
в одиниці об’єму, 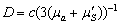 - коефіцієнт дифузії фотонів;
- коефіцієнт дифузії фотонів; 
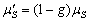 -
транспортний коефіцієнт розсіювання, с - швидкість світла,
-
транспортний коефіцієнт розсіювання, с - швидкість світла, 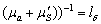 - середня транспортна довжина пробігу фотонів
(довжина, на якій фотон втрачає свій початковий напрям). Дифузійна теорія добре
працює, коли g=0,1, а
- середня транспортна довжина пробігу фотонів
(довжина, на якій фотон втрачає свій початковий напрям). Дифузійна теорія добре
працює, коли g=0,1, а 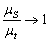 .
.
Більш коректний розв’язок рівняння переносу отримується методом дискретних
ординат (багатопотокова теорія), коли рівняння (2.1) перетворюється в матричне
диференціальне рівняння для освітленості за багатьма дискретними напрямками
(кутами). При збільшенні кількості кутів розв’язок є більш точним.
Важливе значення при розгляді взаємодії світла з полімерами як
розсіюючим середовищем має когерентність. Когерентність світла важлива при
селекції фотонів, що не зазнають або мало зазнають розсіювання, а також при
отриманні спекл-модульованих полів від розсіюючих фазових об’єктів з одноразовим
чи багаторазовим розсіюванням. Такі підходи важливі для когерентної томографії,
дифрактометрії, голографії, фотон-кореляційної спектроскопії і
спекл-інтерферометрії блочних полімерів і розчинів.
Експериментальні методи визначення оптичних параметрів розсіювання
світла поділяють на дві групи: безпосередні і непрямі. До прямих методів
відносяться ті, в основі визначення яких лежать базові поняття, зокрема закон
Бугера-Беєра, фазова функція одноразового розсіювання для тонких зразків, або
ефективна глибина проникнення світла для об’ємного середовища. Параметрами, що
вимірюються безпосередньо є колімоване пропускання, індикатриса розсіювання для
тонких зразків (полімерних плівок), освітленість всередині об’ємного
середовища. Переваги цих методів полягають в нескладній обробці отриманих
даних, а недоліки пов’язані з необхідністю чіткого виконання умов експерименту,
що відповідає використовуваним моделям, виключення впливу поляризації світла і
заломлення на межі зразків, виключенням впливу джерел опромінювання на
детектори реєстрації освітленості.
Непрямі методи передбачають розв’язок оберненої задачі розсіювання на
основі використання тієї чи іншої теоретичної моделі поширення світла в
середовищі. Розрізняють непрямі не ітераційні методи, в яких оптичні властивості
визначаються через параметри, безпосередньо пов’язані з вимірювальними
величинами (модель Кубелки-Мунка, багато потокова модель) і непрямі ітераційні
методи, в яких оптичні властивості виражаються уявно через параметри, що
вимірюються. Величини, що визначають оптичні властивості розсіюючого
середовища, оцінюються до тих пір, поки розрахункові значення, що
характеризують явища відбивання і пропускання світла не будуть з заданою
точністю співпадати з виміряними. Останні методи базуються на моделях більш
складних, зокрема дифузійній теорії, інверсному методі додавання - подвоєння,
інверсійному методі Монте-Карло.
При використанні прямих і непрямих методів, як джерела світла
використовуються лазери імпульсної і неперервної дії. Завдяки їх використанню
створюється інтенсивний, монохроматичний пучок поляризованого світла, що майже
не розходиться. Вони особливо зручні при динамічних випробуваннях, де потрібні
короткі експозиції [31]. Схему такої установки подано на рис. 2.1.
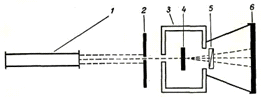
Рис. 2.1. Схема дифрактометра:1- джерело світла (лазер); 2- діафрагма;
3- термокамера; 4- зразок; 5- аналізатор; 6- фіксатор світла.
Дослідження розсіювання світла під великими кутами дає змогу визначити
розміри макромолекул, молекулярну масу, конформації, розгалуження,
полідисперсність, взаємодію з розчинником, а малокутове розсіювання світла в
полімерах використовується для аналізу кінетики утворення, розмірів, форми
зародків при кристалізації, процесів формування надмолекулярних утворень. При орієнтації
плівок і волокон малокутове розсіювання дозволяє досліджувати процеси
деформації і руйнування, крім того цей метод використовується для вивчення
утворення в полімерах пустот, областей різної густини чи кристалічності,
областей різної анізотропії, анізотропних областей різної орієнтації. Для
розчинів полімерів вивчаються процеси утворення довгоживучих асоціатів і
молекулярних комплексів, сіток в гелях, при фазовій сегрегації.
Важливим методом дослідження полімерів є метод комбінаційного
розсіювання світла, який базується на модуляції розсіяного світла власними
коливаннями молекул.
В експериментах по спонтанному комбінаційному розсіюванню промінь
лазера з частотою 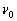 направляється на зразок і розсіюване світло розкладається
на частотні полоси
направляється на зразок і розсіюване світло розкладається
на частотні полоси 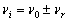 . Найбільш сильне випромінювання спостерігається на
частоті падаючого променя, що пов’язане з пружним розсіюванням (релеєвським).
Полоси комбінаційного розсіювання розміщені симетрично відносно лінії і з
низькочастотного боку називаються стоксовими, а з високочастотного - антистокcовими. Втрати енергії падаючим фотоном внаслідок непружної взаємодії з
молекулою призводить до збудження коливального руху останньої. Стоксові лінії
більш інтенсивні в порівнянні з антистоксовими, що зумовлено різною заселеністю
молекул в основному і першому збудженому стані відповідно до розподілу
Больцмана.
. Найбільш сильне випромінювання спостерігається на
частоті падаючого променя, що пов’язане з пружним розсіюванням (релеєвським).
Полоси комбінаційного розсіювання розміщені симетрично відносно лінії і з
низькочастотного боку називаються стоксовими, а з високочастотного - антистокcовими. Втрати енергії падаючим фотоном внаслідок непружної взаємодії з
молекулою призводить до збудження коливального руху останньої. Стоксові лінії
більш інтенсивні в порівнянні з антистоксовими, що зумовлено різною заселеністю
молекул в основному і першому збудженому стані відповідно до розподілу
Больцмана.
Висока ступінь поляризації і малий діаметр лазерного променя дають
можливість отримувати поляризацію спектри комбінаційного розсіювання для
молекул в орієнтованому і кристалічному станах. Використання подвійних і
потрійних монохроматорів дозволяє отримувати спектри комбінаційного розсіювання
в низькочастотній області поблизу основної лінії збудження. Блок-схема експериментальної
установки для дослідження комбінаційного розсіювання світла подано на рис. 2.2.

Рис. 2.2. Блок-схема установки для дослідження комбінаційного
розсіювання світла.
Використання спектроскопії комбінаційного розсіювання світла
для вивчення структури полімерів зумовлене простотою методу, а також його
високою інформативністю. Показано, що із спектрів комбінаційного розсіювання
світла можливо отримати кількісну інформацію про коефіцієнти розкладу функції
розподілу орієнтації, що є однією з найважливіших характеристик структури і
властивостей анізотропних полімерів. Використання абсолютних інтенсивностей
ліній комбінаційного розсіювання приводить до необхідності введення додаткової
операції нормування спектрів. Більш коректним і зручним способом є вимірювання
деполяризаційних відношень. Такий підхід не пов’язаний з втратою інформації і
також використовується при вивченні орієнтаційного порядку в полімерах. У
спектрах комбінаційного розсіювання світла виявляються лінії, що відповідають
коливанням різноманітних конформерів і локальних угрупувань. Тому для
розрахунків спектрів комбінаційного розсіювання використовують функції
розподілу орієнтації певних розсіюючих одиниць, таких як кристаліти,
різноманітні конформери, кінцеві чи бокові групи за відношенням до осі
макромолекули [32].
У випадку структурного елемента макромолекули вважається, що
він як розсіююча одиниця, світло розсіює некогерентно. Тоді інтенсивність
комбінаційного розсіювання світла описується виразом:
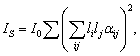 (2.6)
(2.6)
де αіj - компонента тензора похідної
поляризованості за нормальною координатою розсіюючої одиниці в системі
координат зразка xyz, li і lj - направляючі косинуси збуджуючого і розсіюючого
випромінювання в тій же системі координат, 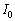 -
постійна, що залежить від інтенсивності падаючого світла і параметрів
експериментальної установки. Сумування здійснюється за всіма розсіюючими
одиницями, що дають вклад в інтенсивність, що вимірюється. У системі координат
xyz тензор поляризованості визначається як:
-
постійна, що залежить від інтенсивності падаючого світла і параметрів
експериментальної установки. Сумування здійснюється за всіма розсіюючими
одиницями, що дають вклад в інтенсивність, що вимірюється. У системі координат
xyz тензор поляризованості визначається як:
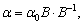 (2.7)
(2.7)
де α0 - тензор поляризованості за осями симетрії
розсіюючої одиниці, В - матриця кутів Ейлера Ө, ψ,
φ.
У випадку довільної орієнтації макромолекул полімерного зразка
спектроскопія комбінаційного розсіювання дає можливість визначити
співвідношення між коефіцієнтами розкладу функції розподілу орієнтації:
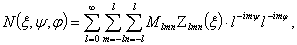 (2.8)
(2.8)
де 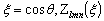 - узагальнена функція Лежанура,
- узагальнена функція Лежанура,  - коефіцієнт розкладу, а в деяких випадках і значення
окремих коефіцієнтів. Це дає можливість створювати теоретичні моделі еволюції
структури полімерів при деформації, а також ефективно контролювати зміну властивостей
при отриманні орієнтованих матеріалів. Якщо функція розподілу орієнтації має
деякі елементи симетрії за аналізом спектрів комбінаційного розсіювання світла,
є можливість більш точно визначити параметри орієнтації макромолекул і характер
міжмолекулярної взаємодії.
- коефіцієнт розкладу, а в деяких випадках і значення
окремих коефіцієнтів. Це дає можливість створювати теоретичні моделі еволюції
структури полімерів при деформації, а також ефективно контролювати зміну властивостей
при отриманні орієнтованих матеріалів. Якщо функція розподілу орієнтації має
деякі елементи симетрії за аналізом спектрів комбінаційного розсіювання світла,
є можливість більш точно визначити параметри орієнтації макромолекул і характер
міжмолекулярної взаємодії.
Під дією світла здійснюється процеси, які призводять до утворення
макромолекул. Ці процеси називаються фотополімеризацією. Процеси
фотополімеризації можуть протікати як за полімеризаційним, так і за
поліконденсаційним механізмами. Для здійснення фотоініційованої ланцюгової
полімеризації кванти світла служать як ініціатори процесу, а при
поліконденсації - для фотохімічних реакцій.
РОЗДІЛ ІІІ. МАКРОМОЛЕКУЛЯРНІ КОНСТАНТИ ЛІНІЙНИХ АМОРФНИХ
ПОЛІМЕРІВ ТА ЇХ СИСТЕМ
.1 Основні геометричні константи макромолекул лінійних
аморфних полімерів
Найпростішою характеристикою просторового розміру
макромолекули є середньоквадратична відстань між її кінцями. Оцінюють
середньоквадратичну відстань макромолекули, використовуючи модельні уявлення.
Розглядають ідеальну макромолекулу, що є ланцюгом, який складається з N
послідовно шарнірно з’єднаних безтілесних жорстких ланок (сегментів) довжиною l
кожна (рис. 3.1). Такий ланцюг носить назву вільно-сполученого ланцюга [33].
Рис. 3.1. Вільно-сполучений ланцюг
Нехай вектор 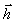 з’єднує кінці макромолекули. Знаходять
середньоквадратичну відстань між кінцями вільно-сполученого ланцюга. У
загальному випадку вектор знаходиться як:
з’єднує кінці макромолекули. Знаходять
середньоквадратичну відстань між кінцями вільно-сполученого ланцюга. У
загальному випадку вектор знаходиться як:
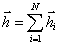 .(3.1)
.(3.1)
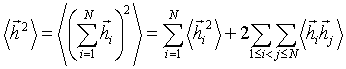 (3.2)У
вільно-сполученому ланцюгу
(3.2)У
вільно-сполученому ланцюгу 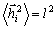 ,
а
,
а 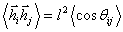 . Оскільки кут між векторами
. Оскільки кут між векторами 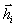 і
і 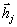 лежить
у межах від 0 до 2p, то середнє значення
лежить
у межах від 0 до 2p, то середнє значення 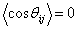 . Отже, остаточно отримують:
. Отже, остаточно отримують:
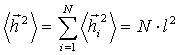 (3.3)
(3.3)
Виходячи із співвідношення (3.3), говорять про те, що середній розмір
макромолекули 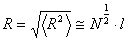 . Такий результат, при N>>1, показує, що середні
розміри макромолекули менші за повну довжину Nl, виміряну вздовж контуру
ланцюга. Отже, серед усієї сукупності конформацій, імовірність існування
витягнутої форми дуже мала.
. Такий результат, при N>>1, показує, що середні
розміри макромолекули менші за повну довжину Nl, виміряну вздовж контуру
ланцюга. Отже, серед усієї сукупності конформацій, імовірність існування
витягнутої форми дуже мала.
У випадку моделі ланцюга, відмінного від вільно-сполученого, скалярний
добуток 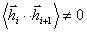 , тому що напрямок різних ділянок макромолекули
скорельований [33]. Кількісною мірою цієї кореляції, яка визначає гнучкість
ланцюга, є середнє значення косинуса кута між різними ділянками макромолекули.
Оскільки функція cos q має властивість
мультиплікативності (якщо на ланцюгу є два сусідніх відрізки з довжинами L і L¢, тобто
, тому що напрямок різних ділянок макромолекули
скорельований [33]. Кількісною мірою цієї кореляції, яка визначає гнучкість
ланцюга, є середнє значення косинуса кута між різними ділянками макромолекули.
Оскільки функція cos q має властивість
мультиплікативності (якщо на ланцюгу є два сусідніх відрізки з довжинами L і L¢, тобто 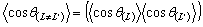 ),
то
),
то
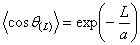 ,
(3.4)
,
(3.4)
де множник перед експонентного рівний 1, тому, що при L = 0 cos q(L = 0) = 1, а а - константа для кожного даного
полімера. Ця константа визначає основну кількісну міру гнучкості і називається
персистентною довжиною. Виходячи із співвідношення (3.4), визначають, що
персистентна довжина а - це проекція нескінченно довгого ланцюга неперервної
кривизни (відпадає модель шарнірів) на напрямок орієнтації першої ланки:
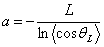 .
(3.5)
.
(3.5)
Аналіз співвідношення (3.5) показує, що у випадку, коли 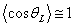 тоді контурна довжина ланцюга L набагато менша за
персистентну довжину а (L<<a).
тоді контурна довжина ланцюга L набагато менша за
персистентну довжину а (L<<a).
Це означає, що на ділянці короткій у порівнянні з персистентною
довжиною, гнучкість не проявляється, і така ділянка поводить себе як жорсткий
стержень (qL » 0). У
другому граничному випадку, коли 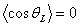 ,
отримують, що (L>>a), тобто кут qL з
рівною ймовірністю набуває значень від 0 до 2p і гнучкість ланцюга призводить до повної незалежності
ділянок макромолекули, розділених довжиною більшою за персистентну. Іншими
словами можна сказати, що на довжині порядку персистентної, втрачається пам’ять
про напрямок ланцюга. Тобто на великих довжинах флуктуації згину руйнують
пам’ять про напрямок ланцюга.
,
отримують, що (L>>a), тобто кут qL з
рівною ймовірністю набуває значень від 0 до 2p і гнучкість ланцюга призводить до повної незалежності
ділянок макромолекули, розділених довжиною більшою за персистентну. Іншими
словами можна сказати, що на довжині порядку персистентної, втрачається пам’ять
про напрямок ланцюга. Тобто на великих довжинах флуктуації згину руйнують
пам’ять про напрямок ланцюга.
Оцінка персистентних довжин реальних молекул показує, що вони можуть
бути різними, наприклад для полістиролу а » 1,0 ¸1,4
нм, а для подвійної спіралі ДНК а » 50 нм [34].
Для характеристики ступеня гнучкості макромолекули крім персистентної
довжини використовують величину ефективного сегмента Куна. Розглянемо реальний
клубок (рис. 3.2)
Рис. 3.2. Схема розбиття реального клубка на статистичні сегменти Куна.
Цьому клубку можна протиставити еквівалентну
шарнірну модель, де стержні вільно обертаються навкруги кульових шарнірів.
Кожний такий стержень відповідає S ланкам реального ланцюга, а умови
еквівалентності зводяться до того, що довжина стержня А стає такою, при якій
орієнтація ланки і + 1 не залежить від орієнтації і - ланки. Така ланка
довжиною А називається статистичним сегментом Куна.
Порівнюючи величини а і А, слід відмітити, що вони мають один і той же
порядок і тому обидві можуть бути використані як характеристики гнучкості
полімерного ланцюга. Слід зазначити, що довжину сегмента Куна простіше
визначити експериментально, а персистентна довжина має безпосередній
мікроскопічний зміст. Для визначення відстані між кінцями ланцюга, потрібно
обчислити ефективний сегмент для кожного конкретного механізму гнучкості.
Розглядають персистентну модель з ізотропною гнучкістю. Нехай
конформація персистентного ланцюга довжиною L задається векторм 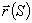 . Вводять
. Вводять 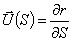 -
одиничний вектор напрямку ланцюга в точці, що відстає на відстань S вздовж
контура макромолекули від її початку. Тоді вектор можна записати у вигляді:
-
одиничний вектор напрямку ланцюга в точці, що відстає на відстань S вздовж
контура макромолекули від її початку. Тоді вектор можна записати у вигляді:
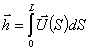 .(3.6)
.(3.6)
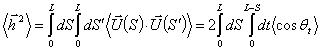 ,
(3.7)
,
(3.7)
де t = S - S¢.
Використовуючи співвідношення (3.3), отримують:
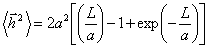 .(3.8)
.(3.8)
Аналіз цього співвідношення показує, що у випадку короткої (L
<< a) і дуже довгої (L >> a) макромолекули, отримують:
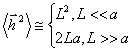 .(3.9)
.(3.9)
Порівняння цього співвідношення з середньоквадратичною відстанню для
ідеальної макромолекули 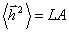 дає можливість говорити про те, що сегмент Куна для
персистентної моделі у двічі перевищує персистентну довжину
дає можливість говорити про те, що сегмент Куна для
персистентної моделі у двічі перевищує персистентну довжину
А = 2а .(3.10)
З іншого боку рівність (3.9) означає, що коротка макромолекула майже
немає гнучкості і відстань між її кінцями практично дорівнює контурній довжині
R @ L.
Врахування ролі квантовомеханічних факторів, що забороняють зближення
бокових радикалів R понад деяку критичну відстань і, тим самим визначаючи
значення “дозволеного” кута, дає можливість знайти 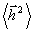 у вигляді:
у вигляді:
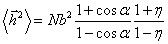 ,
(3.11)
,
(3.11)
де a - кут, доповнений до валентного; b - довжина
ланки; h - середній косинус кута обертання, що
визначає гнучкість і може бути визначений так:
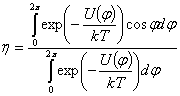 .
(3.12)
.
(3.12)
Тут U(j) - потенціальна енергія внутрішнього
обертання, яка набуває дискретних значень у поворотно-ізомерній моделі М.В.
Волькенштейна [35], або одне єдине значення в стрічковій моделі напівжорсткого
ланцюга С.Е. Бреслера і Я.І. Френкеля [36]; k - константа Больцмана і T -
абсолютна температура. В останньому випадку U(j)>>kT, що означає високу жорсткість, і формулу
(3.11) записують у вигляді:
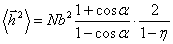 .
(3.13)
.
(3.13)
Для конкретних молекул типу (- СH2 - CR2 -)n і ізотактичних молекул
типу (- СH2 - CRR¢ -) з незалежними конформаціями мономерних
одиниць, з врахуванням різних валентних кутів при групах ізомера - СH2 - і -
CRR¢ -, а також можливою різницею умов обертання
зв’язків (СH2 - CRR¢ і
CRR¢ - СH2) отримані середньоквадратичні відстані
за допомогою методу наближених поворотно-ізомерних станів [35].
Для оцінки розмірів незбурених клубків гнучколанцюгових макромолекул
вважають, що вони гаусові. Пояснюється це тим, що така конформація
макромолекули може бути замінена еквівалентною їй за розмірами і гідродинамічними
властивостями вільно-сполученим ланцюгом так, що кореляції між орієнтаціями
достатньо віддалених ділянок не має. Тобто макромолекула є N-статистичних
сегментів довжиною А, орієнтація яких у просторі незалежна. При цьому задача
визначення зводиться до так званої дифузійної задачі, що
розв’язується в теорії броунівського руху. При N >> 1, 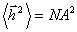 , а ймовірність того, що розміри такого ланцюга
знаходяться в інтервалі від h до h + dh описується гаусівською функцією:
, а ймовірність того, що розміри такого ланцюга
знаходяться в інтервалі від h до h + dh описується гаусівською функцією:
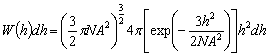 .(3.14)
.(3.14)
Найбільш імовірне і середнє значення відповідно дорівнюють:
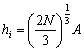 ;
;
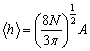 .(3.15)
.(3.15)
Звідси слідує, що відносна дисперсія відстані між кінцями
макромолекули, дорівнює 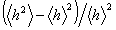 , відмінна від нуля і не залежить від N. Значить
флуктуаційні розміри статистичного клубка мають той же самий масштаб, що і самі
розміри полімерного клубка.
, відмінна від нуля і не залежить від N. Значить
флуктуаційні розміри статистичного клубка мають той же самий масштаб, що і самі
розміри полімерного клубка.
Кількісною мірою геометричних розмірів полімерного ланцюга є значення
радіуса інерції Ri, який залежить від відстані ri кожного з N однакових
елементів масою m0 до центра тяжіння макромолекули. Внаслідок зміни конформації
макромолекули, змінюється відстань елементів масою m0 до центра тяжіння, тобто
величина ri ¹ const. У цьому випадку потрібно розглядати
середньоквадратичний радіус інерції:
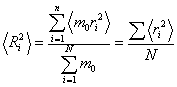 .(3.16)
.(3.16)
Для гаусівських ланцюгів співвідношення між 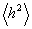 і
і 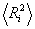 має
вигляд:
має
вигляд:
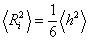 .
(3.17)
.
(3.17)
Мірою гнучкості служить і ступінь згорнутості макромолекули b = h/L. Величина b як безпосередня конформаційна характеристика
розподілена за законом Максвелла і найбільш імовірне значення дорівнює:
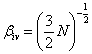 ,(3.18)
,(3.18)
де N - число статистичних елементів ланцюга.
Слід зауважити, що полімерні макромолекули, для яких b £ 1/3,
називають гаусівськими. Відхилення від гаусівського розподілу спостерігається
при підвищеній жорсткості, сильній взаємодії з розчинником, малій молекулярній
масі, вибірковій взаємодії між ланками в сополімерах.
Експериментально середньоквадратичні розміри макромолекул визначаються
на основі визначення гідродинамічних характеристик полімера і молекулярної
маси. Характеристичну в’язкість і коефіцієнт поступального тертя можна
визначити на основі рівнянь Ейнштейна і Стокса виразити через :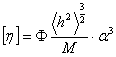 , (3.19)
, (3.19)
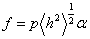 .(3.20)
.(3.20)
де Ф - параметр Флорі і залежить від гнучкості ланцюга і характеру
статистичного розподілу їх розмірів; Р - другий параметр Флорі, який беруть
рівним 5,1.
Крім гідродинамічних методів розміри клубків визначаються за даними
розсіювання світла чи малокутового розсіювання рентгенівських променів.
Останній метод дозволяє також безпосередньо визначити персистентну довжину
[37].
.2 Частотний спектр структуроутворень гетерогенних полімерних систем
У вихідних аморфних лінійних полімерах питання про самовільні
необоротні процеси розповсюдження коливальними рухами їх структурної
організації потребує визначення потенціальної енергії взаємодії і сили
взаємодії структурного елемента (атомної групи) з усіма можливими структурними
елементами. Для визначення такої енергії необхідно знайти суму потенціальної
енергії взаємодії за всіма можливими парами атомних груп, які складають пару з
даним, а потім результат помножити на число структурних елементів.
Подамо потенціал взаємодії структурних елементів макромолекули в вигляді
потенціалу Леннард-Джонса. Тоді сила з якою даний структурний елемент
макромолекули діє на віддалений від нього на відстань r структурний елемент, складає:
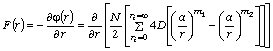 . (3.21)
. (3.21)
З
іншого боку, силу взаємодії, в гармонійному наближенні можемо записати як:
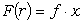 , (3.22)
, (3.22)
де
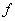 - коефіцієнт квазіпружної сили;
- коефіцієнт квазіпружної сили; 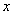 - зміщення структурного елементу від положення
рівноваги;
- зміщення структурного елементу від положення
рівноваги; 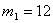 ,
, 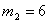 ,
, 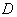 - енергія міжатомного зв’язку;
- енергія міжатомного зв’язку; 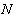 - число структурних елементів, розміщених на відстані
- число структурних елементів, розміщених на відстані 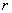 від даного структурного елемента.
від даного структурного елемента.
Враховуючи,
що багатоступінчастий структурний підхід до розгляду поведінки макромолекул
потребує використання фізичної кінетики, будемо враховувати рухливість
структурних елементів. Будемо вважати, що власний час життя структурного
елементу суттєво не змінюється під дією зовнішніх факторів, а час його
"осілого" життя в коливальному чи трансляційному режимі змінюється.
Для визначення діапазону частот коливань структурних елементів макромолекули,
визначимо максимальне значення сили, використовуючи умову екстремуму 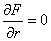 . Тоді
. Тоді
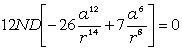 . (3.23)
. (3.23)
Позначивши
віддаль між структурними елементами при 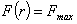 через
через 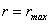 , зайдемо, що
, зайдемо, що 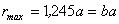 .
Значить, при деформації макромолекули максимальна сила взаємодії між
струкутрними елементами рівна
.
Значить, при деформації макромолекули максимальна сила взаємодії між
струкутрними елементами рівна
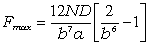 . (3.24)
. (3.24)
Оскільки
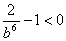 - це сила притягання, а величину розривної сили
визначимо як:
- це сила притягання, а величину розривної сили
визначимо як:
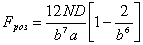 . (3.25)
. (3.25)
Виходячи
з вище сказаного, в співвідношенні (3.22) величина може змінюватися від 0 до 0,245а. Тоді величина
зміщення структурних елементів від
рівноважного стану пропорційна 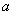
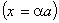 , а відстань між структурними елементами макромолекули
також пропорційна
, а відстань між структурними елементами макромолекули
також пропорційна 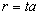 . До того ж величину
. До того ж величину 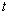 можна
виразити через
можна
виразити через 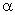 як:
як:
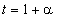 . (3.26)
. (3.26)
Коефіцієнт квазіпружної складової сили взаємодії може бути
визначений як:
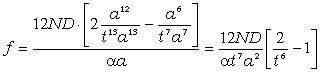 . (3.27)
. (3.27)
Використовуючи рівняння руху структурних елементів
макромолекули у вигляді:
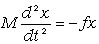 , (3.28)
, (3.28)
визначимо частоту їх коливань:
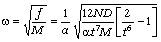 . (3.29)
. (3.29)
З
врахуванням виразу (3.26) для виразимо
частоту коливань як:
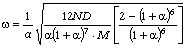 . (3.30)
. (3.30)
Аналіз
співвідношення (3.30) показує, що частота коливань структурних елементів
макромолекул визначається енергією взаємодії , масою
структурного елементу 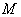 і коефіцієнтом , що
визначає ступінь відхилення від положення рівноваги. Оскільки для даних
структурних елементів величини і за впливу зовнішніх факторів залишаються незмінними
(крім хімічних перетвотень) дослідимо залежність частоти коливань від a. Визначимо при якому значенні a частота коливань
буде рівна нулеві (
і коефіцієнтом , що
визначає ступінь відхилення від положення рівноваги. Оскільки для даних
структурних елементів величини і за впливу зовнішніх факторів залишаються незмінними
(крім хімічних перетвотень) дослідимо залежність частоти коливань від a. Визначимо при якому значенні a частота коливань
буде рівна нулеві (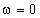 )
)
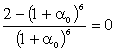 (3.31)
(3.31)
Зі
співвідношення (3.31) слідує, що 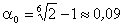 .
Оскільки значення
.
Оскільки значення 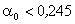 , то проаналізуємо зміну частоти спектру в діапазоні
зміни величини
, то проаналізуємо зміну частоти спектру в діапазоні
зміни величини 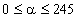 . При
. При 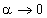 , частота
коливань структурних елементів
, частота
коливань структурних елементів 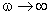 . Це
означає, що макромолекулу можна розглядати як жорсткий стержень (жорсткість
зв’язків дуже велика) на який нанизана система бусинок (атомних груп), з
коефіцієнтом квазіпружної сили
. Це
означає, що макромолекулу можна розглядати як жорсткий стержень (жорсткість
зв’язків дуже велика) на який нанизана система бусинок (атомних груп), з
коефіцієнтом квазіпружної сили 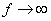 . Зміна
величини a від 0 до 0,09 викликана дією між структурними
елементами сили притягання, а діапазон зміни a від 0,09 до
0,245 зумовлений дією зовнішніх сил, які намагаються розтягнути макромолекулу
(макромолекула в зовнішньому динамічному полі). Така дія зовнішніх сил може
привести до руйнування макромолекули і порушення зв’язків між структурними
елементами за ланцюгом головних валентностей, при значенні зовнішньої сили
. Зміна
величини a від 0 до 0,09 викликана дією між структурними
елементами сили притягання, а діапазон зміни a від 0,09 до
0,245 зумовлений дією зовнішніх сил, які намагаються розтягнути макромолекулу
(макромолекула в зовнішньому динамічному полі). Така дія зовнішніх сил може
привести до руйнування макромолекули і порушення зв’язків між структурними
елементами за ланцюгом головних валентностей, при значенні зовнішньої сили 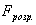 .
.
Проаналізуємо
характер зміни частотного спектру для макромолекули ПВХ, знаючи, що енергія
взаємодії вздовж головного ланцюга валентностей складає прорядку 42×10 -20 Дж, а при реалізації міжмолекулярних зв’язків
боковими групами порядку 3,5×10 -20 Дж. Відстані між
структурними елементами складають 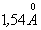 (для С-С
зв’язку);
(для С-С
зв’язку); 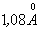 (для Н-Н),
(для Н-Н), 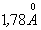 (для Н-Сl).
Частотний спектр коливань структурних елементів показано на рис 3.3. Як
видно з даного рисунка при реалізації С-С зв’язку за ланцюгом головних
валентностей в усьому діапазоні зміни a частота коливань
структурних елементів вища, ніж у випадку реалізації Н-Н зв’язку чи Н-Сl
зв’язку. Особливо значні розходження спостерігаються при значеннях a®0 і a® 0,245.
(для Н-Сl).
Частотний спектр коливань структурних елементів показано на рис 3.3. Як
видно з даного рисунка при реалізації С-С зв’язку за ланцюгом головних
валентностей в усьому діапазоні зміни a частота коливань
структурних елементів вища, ніж у випадку реалізації Н-Н зв’язку чи Н-Сl
зв’язку. Особливо значні розходження спостерігаються при значеннях a®0 і a® 0,245.
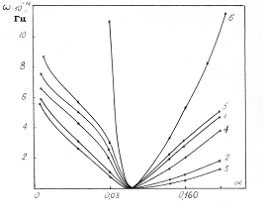
Рис.
3.3. Залежність w від a для потенціалу Леннард-Джонса:
1) С-С -зв’язок; 2) Н-Н -зв’язок; 3) Н-Сl -зв’язок; 4)
H-Bi -зв’язок; 5) Cl-Bi -зв’язок; 6) H-F -зв’язок
Розглядаючи взаємодію поверхневих атомів наповнювача (ФГ; ФГBi, ФГHg; ФГPb) з структурними
елементами макромолекули лініійного аморфного полімера визначали частотний
спектр коливань цих елементів на межі поділу фаз полімер-наповнювач.
Враховуючи, що на поверхні наповнювача існує 1-2% активних центрів, якими
можуть бути атоми металу, або реалізовуватися водневі зв’язки брали відповідні
значення і .
Енергія взаємодії складає 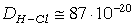 Дж,
Дж, 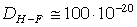 Дж,
Дж,
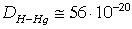 Дж,
Дж, 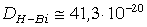 Дж,
Дж,
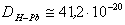 Дж, а міжструктурні відстані у випадку реалізації
зв’язків Me-Cl -
Дж, а міжструктурні відстані у випадку реалізації
зв’язків Me-Cl -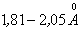 ; H-Hg; H-Bi, H-Pb відповідно
; H-Hg; H-Bi, H-Pb відповідно 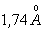 ,
, 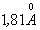 ,
,
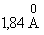 , а для H-F -зв’язку
, а для H-F -зв’язку 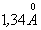 . За
результатами даних по
. За
результатами даних по  і
і 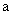 згідно
співвідношення (3.30) проводили розрахунки частотного спектру коливань
структурних елементів на межі поділу фаз полімер-наповнювач. Як слідує з рис.
3.3, частота коливань структурних елементів на межі поділу фаз
полімер-наповнювач вища, чим структурних елементів макромолекул вихідного ПВХ.
згідно
співвідношення (3.30) проводили розрахунки частотного спектру коливань
структурних елементів на межі поділу фаз полімер-наповнювач. Як слідує з рис.
3.3, частота коливань структурних елементів на межі поділу фаз
полімер-наповнювач вища, чим структурних елементів макромолекул вихідного ПВХ.
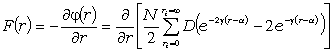 , (3.32)
, (3.32)
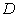 -
енергія зв’язку між частинками,
-
енергія зв’язку між частинками, 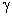 -
постійна пов’язана з значенням амплітуди коливань взаємодіючих структурних
елементів.
-
постійна пов’язана з значенням амплітуди коливань взаємодіючих структурних
елементів.
При
розгляді взаємодії між двома сусідніми структурними елементами макромолекули
отримаємо, що
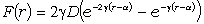 (3.33)
(3.33)
Як
і при використанні потенціалу Леннард-Джонса, визначимо максимальне відхилення
структурних елементів від положення рівноваги. З умови екстремуму функції 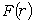 слідує, що
слідує, що
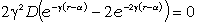 (3.34)
(3.34)
або
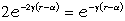 (3.35)
(3.35)
звідки
максимальна відстань між структурними елементами рівна
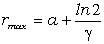 (3.36)
(3.36)
а
максимальне відхилення 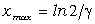 .
.
Якщо
вважати, що в гармонійному наближенні сила взаємодії між структурними
елементами змінюється згідно закону Гука то отримаємо:
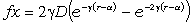 (3.37)
(3.37)
з
даного рівняння визначимо квазіпружну константу взаємодії
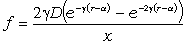 (3.38)
(3.38)
Не
обмежуючи загальності, будемо вважати що параметр оберненопропорційний амплітуді коливань структурних
елементів 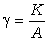 .
.
Якщо
максимальне відхилення від положення рівноваги рівне відстані між структурними
елементами 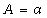 , а
, а 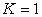 , то
, то 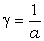 . Отже максимальне значення сили взаємодії між
структурними елементами буде рівне при
. Отже максимальне значення сили взаємодії між
структурними елементами буде рівне при 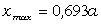 , а
значення квазіпружної константи
, а
значення квазіпружної константи
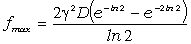 (3.39)
(3.39)
При
, частота коливань при максимальній діючій силі буде
рівна:
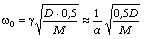 (3.40)
(3.40)
В
загальному випадку амплітуда коливань 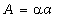 і нехай
дорівнює зміщенню
і нехай
дорівнює зміщенню 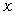 . Отже значення
. Отже значення  . В цьому
випадку в співвідношенні (3.38)
. В цьому
випадку в співвідношенні (3.38)  і
і 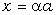 , отримаємо, що
, отримаємо, що
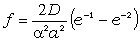 , (3.41)
, (3.41)
а частота коливань структурних елементів рівна:
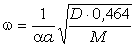 (3.42)
(3.42)
Аналіз
співідношення (3.42) показує, що при 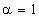 ,
,  і крім того ні при якому
і крім того ні при якому 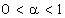 значення
значення  . При
. При  ,
,  і
характер залежності в діапазоні зміни
і
характер залежності в діапазоні зміни 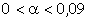 близький,
до характеру залежності частотоного спектру, що описується співвідношенням (3.33).
близький,
до характеру залежності частотоного спектру, що описується співвідношенням (3.33).
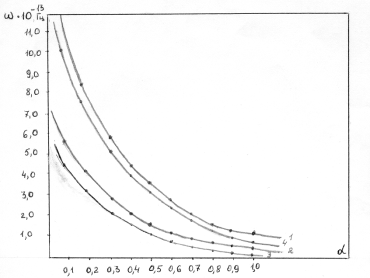
Рис.
3.4. Залежність w від a для потенціалу Морзе: 1) С-С
-зв’язок; 2) Н-Н -зв’язок; 3) Н-Сl -зв’язок; 4) H-Bi -зв’язок; 5) Cl-Bi -зв’язок; 6) H-F
-зв’язок
На рис.3.4. показано залежності частотного спектру структурних
елементів макромолекули ПВХ (ПВБ) при реалізації С-С зв’язку за ланцюгом
головних валентностей і при реалізації водневих зв’язків структурних елементів
як між собою, так і з активним центром наповнювача. Як видно з даного рисунка
найбільш “жорсткий” спектр мають ті структурні елементи макромолекули, які приймають
участь в реалізації внутрішньомолекулярної взаємодії. Співставлення результатів для частотного спектру
коливань структурних елементів макромолекул отриманих на основі потенціалу
Леннард-Джонса і потенціалу Морзе показує, що в першому випадку значення частот
коливань для однакових значень вищі. Це
вказує на те, що потенціал Леннард-Джонса описує більш “жорсткі” взаємодії, а
потенціал Морзе більш “м’які”.Порівняємо відповідні значення частот коливання
структурних елементів для випадку коли 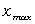 обчислене
за потенціалом Леннард-Джонса, рівна тому ж значенню зміщення знайденому з
використанням потенціалу Морзе. Тоді
обчислене
за потенціалом Леннард-Джонса, рівна тому ж значенню зміщення знайденому з
використанням потенціалу Морзе. Тоді
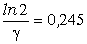 . (3.43)
. (3.43)
Звідки
знайдемо 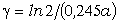 . Підставляючи це значення в співвідношення (3.30)
знайдемо що:
. Підставляючи це значення в співвідношення (3.30)
знайдемо що:
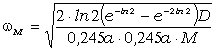 , (3.44)
, (3.44)
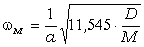 . (3.45)
. (3.45)
Відповідно за співвідношенням (3.33)
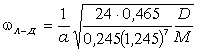 (3.46)
(3.46)
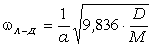 (3.47)
(3.47)
Як
слідує з результатів обчислень, за даних умов, відношення 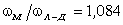 , тобто значення частот близькі між собою.
, тобто значення частот близькі між собою.
Введення
в полімерну матрицю металевих наповнювачів змінює характер взаємодії на межі
поділу фаз. Зумовлено, це перш-за все, процесами структуроутворенння на межі
поділу полімер-метал. На відміну від атомів в об’ємі, поверхневі атоми
наповнювача (для більшості металів) частково гідратовані наявністю
гідроксильних груп. Однак при цьому на поверхні металевого наповнювача існує
1-2% активних центрів, які вступають у взємодію з структурними елементами. Якщо
ж поверхня металевого наповнювача не покрита окисною плівкою (наприклад для
вольфраму), то характер взаємодії на межі поділу фаз полімер-метал стає більш
складним. Якщо бокові групи макромолекули здатні утримувати елементарний заряд
(наприклад Cl-), а поверхня наповнювача заряджена зарядом q1
(наприклад внаслідок тертя при введенні його в полімерну матрицю) то крім сил
хімічної природи, міжмолекулярних, виникають сили електростатичного характеру.
Відомо, що така взаємодія на близьких відстанях між поверхнею і зарядженим
іоном не описується законом Кулона. Оскільки металеві наповнювачі мають форму
кулі, а заряджену бокову групу можна вважати точковим зарядом (рис. 3.6), то
визначимо електростатичний потенціал jел(l)
Якщо
розглядати заряджену бокову групу, як точковий заряд q2, то тоді
маємо взаємодію зарядженої кулі радіуса 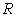 і
точкового заряду. Відстань від центра кулі до точкового заряду позначимо як
і
точкового заряду. Відстань від центра кулі до точкового заряду позначимо як 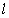 . З геометричних міркувань цю відстань запишемо, як
. З геометричних міркувань цю відстань запишемо, як 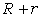 , де
, де 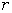 - відстань від поверхні кулі до точкового заряду. В
цьому випадку в кулі виникає лише одне зображення, і тому електростатичний
потенціал знаходиться в кінцевому виді:
- відстань від поверхні кулі до точкового заряду. В
цьому випадку в кулі виникає лише одне зображення, і тому електростатичний
потенціал знаходиться в кінцевому виді:
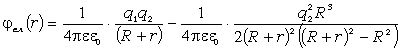 (3.48)
(3.48)
Електорстатична сила, яка виникає в такій системі визначиться, як
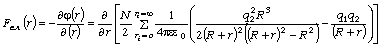 (3.49)
(3.49)
При розгляді взаємодії тільки однієї частинки з поверхнею
наповнювача отримаємо:
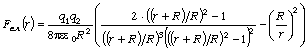 (3.50)
(3.50)
Аналіз співвідношення (3.50) показує, що у випадку рівності зарядів 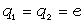 при відношенні
при відношенні 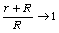 електрична
сила є силою притягання навіть коли заряди
електрична
сила є силою притягання навіть коли заряди 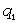 і
і
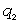 одноіменні. Тобто при наближенні зарядженої бокової
групи на близькі відстані до поверхні наповнювача, електростатичне
відштовхування змінюється притяганням. Оцінимо
числове значення сили електростатичної взаємодії при введення наповнювача у
вигляді вольфраму. Радіус вольфраму становить
одноіменні. Тобто при наближенні зарядженої бокової
групи на близькі відстані до поверхні наповнювача, електростатичне
відштовхування змінюється притяганням. Оцінимо
числове значення сили електростатичної взаємодії при введення наповнювача у
вигляді вольфраму. Радіус вольфраму становить 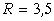 мкм, а
відстань між іоном хлору і атомом вольфраму -
мкм, а
відстань між іоном хлору і атомом вольфраму - 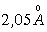 ,
діелектрична проникливість становить 3,3, тоді значення
,
діелектрична проникливість становить 3,3, тоді значення 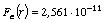 Н.
Н.
Якщо
врахувати, що крім електростатичної взаємодії існують і інші види взаємодії які
описуються потенціалом Леннард-Джонса, то результуючу силу запишемо як:
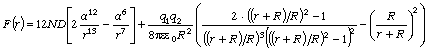 (3.51)
(3.51)
Так
як електорстатична сила на близьких відстанях, це сила притягання, яка не
приводить до різкого збільшення амплітуди коливань, а отже і зміщення
структурних елементів, від положення рівноваги, як і в попередніх випадках для
визначення частотного спектру коливань структурних елементів макромолекули
аморфного лінійного полімера на межі поділу фаз полімер-металевий наповнювач
використаємо гармонійне наближення. Тоді частота коливань структурних елементів
складає
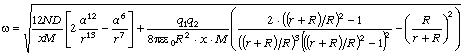 (3.52)
(3.52)
Оцінимо
частоту коливань зарядженого структурного елементу макромолекули, припустивши,
що між частинкою металевого наповнювача і структурним елементом діє лише сила
електростатичного притягання (відштовхування). В цьому випадку співвідношення
(3.52) запишеться в такому вигляді:
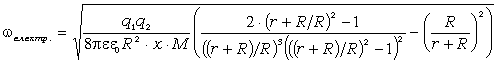 (3.53)
(3.53)
Як
і при розгляді попередніх випадків припустимо що зміщення
, тоді 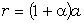 , а
заряди відповідно виразимо через величину електричного заряду
, а
заряди відповідно виразимо через величину електричного заряду 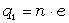 ,
, 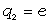 . Тоді
співвідношення (3.53) набуває такого вигляду
. Тоді
співвідношення (3.53) набуває такого вигляду
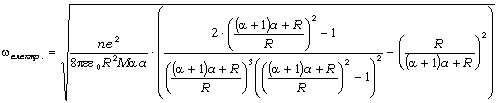 (3.54)
(3.54)
Аналіз
даного виразу показує, що оскільки 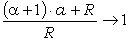 , то
другий співмножник прямує за даної умови до значення
, то
другий співмножник прямує за даної умови до значення 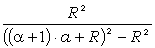 .
.
В цьому випадку отримаємо, що
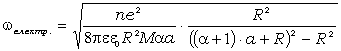 (3.55)
(3.55)
Оскільки
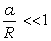 , то
, то 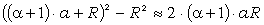
В кінцевому випадку отримаємо, що:
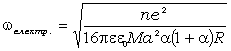 (3.56)
(3.56)
Оскільки
для даного полімера і наповнювача величини  ; М;
; М;  і є
незмінними, то з співвідношення (3.56) слідує, що частота коливань структурних
елементів, яка зумовлена електростатичною взаємодією визначається величиною
заряду, який розміщений на поверхні металевого наповнювача і ступенем
відхилення зарядженого структурного елемента макромолекули від положення
рівноваги. Оцінимо частоту коливань атомної групи макромолекули ПВХ (СН2Сl)
заряд якої складає , яка взаємодіє з зарядженою вольфрамовою сферою,
заряд якої
і є
незмінними, то з співвідношення (3.56) слідує, що частота коливань структурних
елементів, яка зумовлена електростатичною взаємодією визначається величиною
заряду, який розміщений на поверхні металевого наповнювача і ступенем
відхилення зарядженого структурного елемента макромолекули від положення
рівноваги. Оцінимо частоту коливань атомної групи макромолекули ПВХ (СН2Сl)
заряд якої складає , яка взаємодіє з зарядженою вольфрамовою сферою,
заряд якої  . В цьому випадку при ;
. В цьому випадку при ; 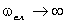 і це означає, що структурний елемент “жорстко”
прикріплений до поверхні сфери. Якщо у вираз (3.56) підставити відповідні
значення для ПВХ і W то отримаємо,
що:
і це означає, що структурний елемент “жорстко”
прикріплений до поверхні сфери. Якщо у вираз (3.56) підставити відповідні
значення для ПВХ і W то отримаємо,
що:
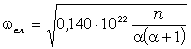 (3.57)
(3.57)
При 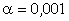 ;
; 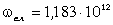 Гц,
а при
Гц,
а при 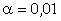 і
і 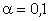 відповідні
значення частоти коливань структурного елементу складають 3,723×1011 Гц і 1,128×1011 Гц.
відповідні
значення частоти коливань структурного елементу складають 3,723×1011 Гц і 1,128×1011 Гц.
В загальному випадку, якщо заряд сферичного металевого наповнювача
відмінний від елементарного заряду (n¹1),
будемо мати 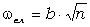 , при фіксованих значеннях a. Графічно ця залежність показана на рис. 3.5.
, при фіксованих значеннях a. Графічно ця залежність показана на рис. 3.5.
Аналіз даних залежностей показує, що чим більший заряд знаходиться на кульовій
поверхні металевого наповнювача, то тим жорсткіший частотний спектр структурних
елементів макромолекул лінійних полімерів в гармонійному наближенні.
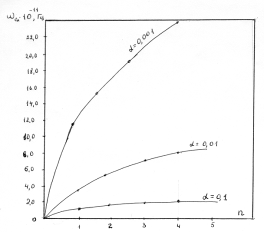
Рис. 3.5. Залежність 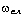 від
n для систем ПВХ + W
від
n для систем ПВХ + W
Оцінимо частотний спектр структурного елемента макромолекули в випадку
прояву дії молекулярних сил (хімічних, міжмолекулярних) і електростатичних.
Згідно співвідношення (3.52) отримаємо з врахуванням (3.56) отримаємо
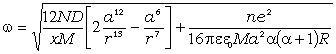 (3.58)
(3.58)
З врахуванням значення ,
отримаємо, що
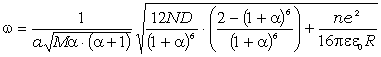 (3.59)
(3.59)
Таблиця
3.1
Частотний спектр структурних елементів макромолекул для систем ПВХ + W , ПВХ + Cu, ПВХ + Al
|
 , Гц
при n=1 , Гц
при n=1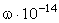 , Гц
при n=10, Гц
при n=100, Гц
при n=1000, Гц
при n=1, Гц
при n=10, Гц
при n=100, Гц
при n=1000 , Гц
при n=10, Гц
при n=100, Гц
при n=1000, Гц
при n=1, Гц
при n=10, Гц
при n=100, Гц
при n=1000
|
|
|
|
|
|
|
|
|
|
|
ПВХ + W (
для зв’язку Me - Cl)
|
ПВХ + Cu
( для зв’язку Cu - Cl)
|
|
0,001
|
5,916
|
5,916
|
5,917
|
5,928
|
0,001
|
32,869
|
32,869
|
32,870
|
32,871
|
|
0,01
|
1,871
|
1,871
|
1,871
|
1,875
|
0,01
|
10,390
|
10,390
|
10,391
|
10,393
|
|
0,1
|
0,141
|
0,141
|
0,142
|
0,145
|
0,1
|
0,845
|
0,845
|
0,845
|
0,845
|
|
ПВХ + Al (Al - Cl)
|
ПВХ + Cu ( для зв’язку Cu - H)
|
|
0,001
|
20,432
|
20,432
|
20,433
|
20,435
|
0,001
|
25,917
|
25,917
|
25,918
|
25,919
|
|
0,01
|
6,461
|
6,461
|
6,462
|
6,466
|
0,01
|
8,196
|
8,196
|
8,197
|
8,200
|
|
0,1
|
0,525
|
0,525
|
0,525
|
0,525
|
0,1
|
0,667
|
0,667
|
0,667
|
0,668
|
Використовуючи
співвідношення (3.59) знайдемо значення частотного спектру структурних
елементів для систем: ПВХ+W, коли реалізується зв’язок типу W-CH2Cl; ПВХ+Cu при реалізації зв’язків Cu-Cl, Cu-H;
ПВХ +Al при реалізації зв’язку Al-Cl. Радіус частинок наповнювача вибирався
однаковим і становив 3,5 мкм, заряд наповнювача змінювався від 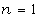 до
до 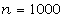 ,
відповідні дані для зразків були вибрані наступні: W-CH2Cl,
відстань зв¢язку , енергія
- 4,98×10-20 Дж; утворенння хімічних зв¢язків типу Cu-Cl і зв¢язку типу Cu-H
характеризується наступними даними: 150×10-20 Дж;
,
відповідні дані для зразків були вибрані наступні: W-CH2Cl,
відстань зв¢язку , енергія
- 4,98×10-20 Дж; утворенння хімічних зв¢язків типу Cu-Cl і зв¢язку типу Cu-H
характеризується наступними даними: 150×10-20 Дж; 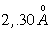 ; 48×10-20 Дж;
; 48×10-20 Дж; 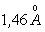 ;
хімічний зв¢язок Al-Cl характеризується енергією дисоціації 63,456×10-20 Дж і відстанню
;
хімічний зв¢язок Al-Cl характеризується енергією дисоціації 63,456×10-20 Дж і відстанню 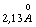 . Як видно з даних поданих в таблиці 3.1
електростатична взаємодія не вносить суттєвих змін в частотний спектр
структурних елементів ПВХ для яких реалізуються хімічні зв¢язки з поверхнею наповнювача. Найбільш помітні зміни в
зміщенні спектру коливань структурних елементів спостерігаються у випадку
реалізації міжмолекулярної взаємодії з поверхнею наповнювача і при зростанні
величини заряду на поверхні наповнювача.
. Як видно з даних поданих в таблиці 3.1
електростатична взаємодія не вносить суттєвих змін в частотний спектр
структурних елементів ПВХ для яких реалізуються хімічні зв¢язки з поверхнею наповнювача. Найбільш помітні зміни в
зміщенні спектру коливань структурних елементів спостерігаються у випадку
реалізації міжмолекулярної взаємодії з поверхнею наповнювача і при зростанні
величини заряду на поверхні наповнювача.
Розглянемо структурний елемент макромолекули, як диполь з дипольним моментом
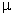 , який потрапляє в електричне поле частинки
наповнювача, або знаходиться в зовнішньому електричному полі. В цьому випадку
на диполь буде діяти сила, яка і зумовлюватиме коливання структурного елементу.
Якщо високодисперсний металічний наповнювач вводити в полімер, який знаходиться
в зовнішньому електричному полі, буде відбуватися деформація зовнішнього
електричного поля і силові лінії поля є ортогональними провідній поверхні
наповнювача. Оскільки наповнювач є провідною кулею, який знаходиться в
зовнішньому електричному полі, то для знаходження потенціалу зовнішнього поля
, який потрапляє в електричне поле частинки
наповнювача, або знаходиться в зовнішньому електричному полі. В цьому випадку
на диполь буде діяти сила, яка і зумовлюватиме коливання структурного елементу.
Якщо високодисперсний металічний наповнювач вводити в полімер, який знаходиться
в зовнішньому електричному полі, буде відбуватися деформація зовнішнього
електричного поля і силові лінії поля є ортогональними провідній поверхні
наповнювача. Оскільки наповнювач є провідною кулею, який знаходиться в
зовнішньому електричному полі, то для знаходження потенціалу зовнішнього поля 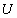 (постійний на поверхні кулі
(постійний на поверхні кулі 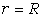 ), потрібно знайти розв¢язок рівняння Лапласа в сферичних координатах [3].
), потрібно знайти розв¢язок рівняння Лапласа в сферичних координатах [3].
 ,
(3.60)
,
(3.60)
для зовнішньої області  ,
який зрівнюважував би зміни
,
який зрівнюважував би зміни  при
. Враховуючи, що
при
. Враховуючи, що 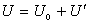 і
і
 при знайдемо
як потенціал поля диполя, орієнтованого по осі oz і
локалізованого в початку координат:
при знайдемо
як потенціал поля диполя, орієнтованого по осі oz і
локалізованого в початку координат:
 (3.61)
(3.61)
Так як 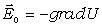 , то проекція напруженості поля в сферичних
координатах матиме вигляд:
, то проекція напруженості поля в сферичних
координатах матиме вигляд:
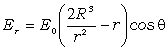 (3.62)
(3.62)
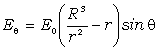
Таким
чином на структурний елемент макромолекули, як диполь з боку зовнішнього
електричного поля буде діяти сила
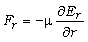 , (3.63)
, (3.63)
або
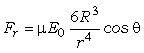 (3.64)
(3.64)
З
іншого боку силу дії подамо у вигляді (гармонійне наближення) як 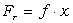 . З врахуванням того, що
. З врахуванням того, що 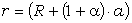 і , будемо
мати
і , будемо
мати
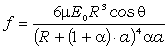 . (3.65)
. (3.65)
Звідки
частота коливань диполя в електричному полі визначається як:
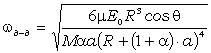 . (3.66)
. (3.66)
В
більшості випадків радіус частинки наповнювача 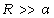 , тоді
, тоді 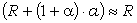 і співвідношення (3.66) набуває вигляду
і співвідношення (3.66) набуває вигляду
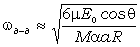 (3.67)
(3.67)
Аналіз
співвідношення (3.67) показує, що частота коливань структурного елементу
макромолекули визначається не тільки його дипольним моментом і масою, а також
орієнтацією структурного елемента в електричному полі частинки наповнювача. У
випадку розміщення структурного елемента, як диполя перпендикулярно до силових
ліній електричного поля (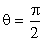 ), такий структурний елемнт коливатися не буде, коли ж
диполь орієнтується вздовж поля (
), такий структурний елемнт коливатися не буде, коли ж
диполь орієнтується вздовж поля (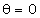 ) будемо
мати
) будемо
мати
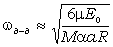 (3.68)
(3.68)
На
основі співвідношення (3.68) розрахуємо частотний спектр структурних елементів
макромолекули ПВХ. Кожний структурний елемент макромолекули будемо
характеризувати дипольним моментом і масою
М. Так атомна група СНСl має дипольний момент 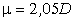 , а атомна група СН2 відповідно
, а атомна група СН2 відповідно 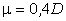 . Розглядаючи їх в електричному полі частинки
наповнювача вольфрама мкм і напруженості поля
. Розглядаючи їх в електричному полі частинки
наповнювача вольфрама мкм і напруженості поля 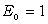 кВ/м оотримаємо залежність
кВ/м оотримаємо залежність  від .
Як видно з даних поданих на рис.3.6. частотний спектр СНСl групи вищий за частотний спектр СН2 групи. Наприклад при відхиленні структурного
елемента від рівноважної відстані на відповідні
частоти складають 2,645×109 Гц і 2,236×109 Гц. Якщо, при формуванні такої композиції,
накладається зовнішнє електричне поле
від .
Як видно з даних поданих на рис.3.6. частотний спектр СНСl групи вищий за частотний спектр СН2 групи. Наприклад при відхиленні структурного
елемента від рівноважної відстані на відповідні
частоти складають 2,645×109 Гц і 2,236×109 Гц. Якщо, при формуванні такої композиції,
накладається зовнішнє електричне поле
(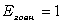 кВ/м), то як показано напруженість поля біля поверхні
наповнювача становить
кВ/м), то як показано напруженість поля біля поверхні
наповнювача становить 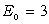 кВ/м і відповідний частотний спектр коливань
структурних елемнтів складає 4,583×1010
Гц і 3,873×1010 Гц.
кВ/м і відповідний частотний спектр коливань
структурних елемнтів складає 4,583×1010
Гц і 3,873×1010 Гц.
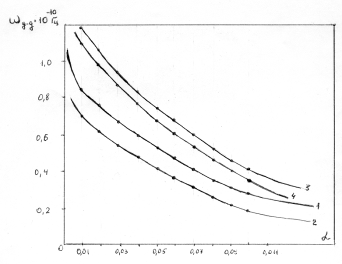
Рис. 3.6. Залежність wд-д від a для систем ПВХ + W: 1 - структурного елементу СНCl; 2 - СН2 для систем сформованих без
поля; 3 - НСl; 4 - СН2 - для систем сформованих в
зовнішньому електричному полі
Як слідує з поданих результатів такі напруженості електричних
полів частинок наповнювача не дають суттєвого зсуву частотного спектру
структурних елементів макромолекул лінійних полімерів. Основний вклад в
частотний спектр структурних елементів вносить ковалентний зв¢язок, який виникає на межі
поділу фаз полімер-наповнювач, або міжмолекулярна взаємодія, яка виникає на
даній межі.
Визначимо, якою повинна бути напруженість поля частинки наповнювача,
щоб частота коливань диполя була співмірною з частотою коливань структурного
елемнта у випадку реалізації міжмолекулярної взаємодії з поверхнею наповнювача.
Для систем ПВХ+W частота коливань структурного елемента при складає w = 1,410×10 13 Гц (таблиця 3.1).
Тоді напруженість поля частинки наповнювача буде рівна:
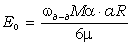 (3.69)
(3.69)
Підставивши відповідні значення, отримаємо що 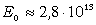 В/м, що значно перевищує електричну міцність ПВХ.
В/м, що значно перевищує електричну міцність ПВХ.
При розгляді частотного спектру вихідних лінійних полімерів та
композицій на їх основі потрібно з¢ясувати
можливості рухливості (коливання) макромолекул як єдиного цілого і частинок
наповнювача. Кожну макромолекулу будемо розглядати як підсистему
надмолекулярних утворень, а кожну частинку наповнювача, як підсистему
просторової структури, яку утворює сукупність частинок наповнювача.
У наповнених полімерних композиціях утворюються частинками наповнювача
просторові структури двох типів, в залежності від вмісту наповнювача. Якщо
вміст наповнювача (jн) менший за критичний (jкр), то у випадку відсутності сегрегації частинки
наповнювача не знаходяться у вузлах регулярної структури, займають випадкові
положення в просторі. В першому наближенні таку гетерогенну систему можна
моделювати в вигляді квазіодномірної гратки з хаотичними відстанями між
частинками. При цьому кооперативного руху частинок наповнювача не відбувається,
тому що не виникає квазіпружна складова сили взаємодії на межі поділу фаз.
Частинки наповнювача, які знаходяться в полімерній матриці, зазнають
індивідуального впливу, що виникає при спільній дії сил тертя і опору
середовища. При цьому інтенсивність їх броунівського руху визначається
температурою термостата, розмірами частинок наповнювача, в¢язкістю середовища.
Під дією зовнішньої сили частинки
наповнювача будуть рухатися незалежно від руху сусідніх, як колоїдні частинки в
середовищі з великою в¢язкісю.
В міру зростання вмісту високодисперсного наповнювача в системі все
більша частина полімерної матриці переходить в межовий шар, який “витісняє”
полімер в об¢ємі. При jн ³ jкр трьохкомпонентна гетерогенна система вироджується в
двохкомпонентну типу наповнювач-межовий шар. У випадку jн = jкр можна вже
говорити про деяке просторове упорядкування системи, оскільки рух частинок
наповнювача залежить від руху сусідніх.
У випадку просторової коагуляційної структури гетерогенної системи
частинки наповнювача утворюють просту кубічну гратку як можливий тип будови
тіла при мінімальному об¢ємі.
Період такої гратки рівний 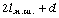 (
(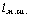 - товщина межового шару;
- товщина межового шару; 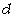 - діаметр частинки наповнювача). Її вузли (частинки
наповнювача) занурені в полімерну матрицю, яка знаходиться в стані межового
шару. Силове внутрішнє поле матеріалізоване полімерним “в¢яжучим”, через яке здійснюється взаємодія між
частинками наповнювача. Будемо моделювати полімерне “в¢яжуче” пружними зв¢язками з константою пружності в поздовжньому напрямку b, а в поперечному - c. При цьому будемо вважати, що в такій системі
частинки наповнювача будуть коливатися в ансамблі як поздовжньому, так і в
поперечному напрямку
- діаметр частинки наповнювача). Її вузли (частинки
наповнювача) занурені в полімерну матрицю, яка знаходиться в стані межового
шару. Силове внутрішнє поле матеріалізоване полімерним “в¢яжучим”, через яке здійснюється взаємодія між
частинками наповнювача. Будемо моделювати полімерне “в¢яжуче” пружними зв¢язками з константою пружності в поздовжньому напрямку b, а в поперечному - c. При цьому будемо вважати, що в такій системі
частинки наповнювача будуть коливатися в ансамблі як поздовжньому, так і в
поперечному напрямку
Для визначення товщини межового шару, використаємо рівняння
адсорбційної ізотерми. Використання даного рівняння, дає можливість визначити
масу межового шару mм.ш. в розрахунку на полімер:
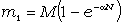 ,
(3.70)
,
(3.70)
де a - коефіцієнт пропорційності; N - число частинок наповнювача; М - маса полімерної матриці. Розглядаючи межовий шар
як кульовий ефективної товщиини lм.ш.
визначимо ефективний об¢єм його межового
шару:
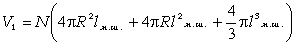 (3.71)
(3.71)
Порівняння співвідношень (3.70) і (3.71) дає можливість знайти лінійні
розміри межового шару:
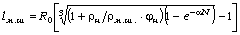 .
(3.72)
.
(3.72)
Коефіцієнт визначає міру активності наповнювача і визначається
на основі “стрибка” теплоємності для наповненого  і
ненаповненого
і
ненаповненого  зразків з умови, що
зразків з умови, що 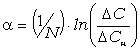 .
.
При виконанні умови jн = jкр і виходячи з припущення, що частинки наповнювача
рівномірно дисперговані в об’ємі полімера, отримуємо:
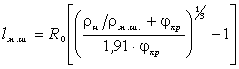 .
(3.73)
.
(3.73)
Тоді період супергратки частинок наповнювача буде рівний
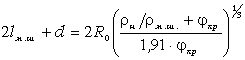 .
(3.74)
.
(3.74)
Для розрахунків частотного спектру коливань такої супергратки,
припустимо, що кожна частинка наповнювача через полімерне “в’яжуче” взаємодіє
тільки з двома сусідніми, до того ж в першому наближенні повертаюча сила
пропорційна зміщенню. Тоді рівняння руху частинок наповнювача матиме вигляд:
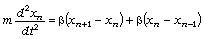 ,
(3.75)
,
(3.75)
де
 - маса частинки наповнювача;
- маса частинки наповнювача; 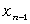 ,
, 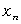 ,
, 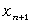 - її зміщення від положення рівноваги.
- її зміщення від положення рівноваги.
Розв’язок
рівняння (3.75) буде:
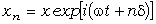 , (3.76)
, (3.76)
що
відповідає періодичному в просторі і часі процесу. Тут - деяка стала (амплітуда), w - циклічна частота, d - зсув фаз. Зсув за фазою між
частинками наповнювача, що знаходяться одна від одної на відстані 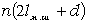 рівний
рівний 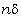 . У
випадку
. У
випадку 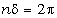 ,
, 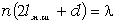 . Тоді
. Тоді
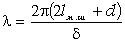 , (3.77)
, (3.77)
і
після підстановки в (3.75) отримаємо
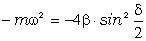 , (3.78)
, (3.78)
або
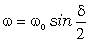 , де
, де 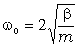 .
.
Оскільки
довжина ультразвукової хвилі 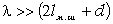 (див.
табл. 3.2), будемо мати, що
(див.
табл. 3.2), будемо мати, що 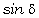 ~d і
~d і
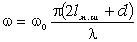 , (3.79)
, (3.79)
Оскільки
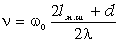 і
і 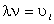 ,
отримаємо
,
отримаємо
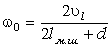 . (3.80)
. (3.80)
Використаємо
співвідношення (3.80) для систем ПВХ+ФГ, ПВХ+Cu. Як було показано,
критичний вміст наповнювача в цих системах за даними температурної залежності
питомої теплоємності  відповідно складає 52 об.% і 65 об.%. Розрахунки
показують (табл. 3.2), що частота коливань частинок наповнювача складає w0 = 0,34×109 Гц для систем ПВХ+52 об.%ФГ і 0,20×109 Гц - ПВХ+65 об.%. Cu, при діаметрі
частинок наповнювача відповідно 10 мкм і 13 мкм.
відповідно складає 52 об.% і 65 об.%. Розрахунки
показують (табл. 3.2), що частота коливань частинок наповнювача складає w0 = 0,34×109 Гц для систем ПВХ+52 об.%ФГ і 0,20×109 Гц - ПВХ+65 об.%. Cu, при діаметрі
частинок наповнювача відповідно 10 мкм і 13 мкм.
Таблиця
3.2
Лінійні
розміри і частотний спектр макрогратки наповеювача і надмолекулярних утворень в
ПВХ - системах.
|
 , об. % , об. %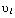 , м/с , м/с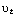 , м/с, мкм , м/с, мкм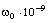 , Гц , Гц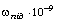 , Гц , Гц 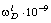 , Гц , Гц 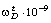 , Гц , Гц 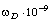 , Гц , Гц 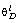 , К , К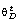 , К , К
|
|
|
|
|
|
|
|
|
|
|
|
ПВХ + ФГ
|
|
52
|
2400
|
1470
|
2,14
|
0,34
|
1,761
|
1,310
|
0,802
|
0,886
|
0,01
|
0,006
|
|
ПВХ + Cu
|
|
65
|
1850
|
1000
|
5,24
|
0,20
|
0,544
|
0,395
|
0,214
|
0,238
|
0,003
|
0,002
|
З іншого боку квазіпружну силу взаємодії між частинками наповнювача можна
оцінити виходячи з таких міркувань. На межі поділу фаз полімер-наповнювач
структурні елементи макромолекул взаємодіють з структурними елементами поверхні
наповнювача. Якщо вважати, що на поверхні наповнювача структурні елементи
(атоми, молекули) мають щільну упаковку, то кількість структурних елементів на
поверхні рівна:
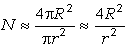 , (3.81)
, (3.81)
де
- лінійні розміри структурного елемента наповнювача.
Тоді знаючи, що на поверхні наповнювача існує 1-2% структурних елемнтів, які
взаємодіють з структурними елементами макромолекул, можна знайти рівнодійну
всіх сил, які діють на частинку:
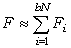 , (3.82)
, (3.82)
де
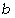 - визначає частку структурних елементів поверхні які
взаємодіють з структурними елементами макромолекул; Fi - значення
одиничної сили взаємодії. В гармонійному наближенні частота коливань частинок
наповнювача в такій гратці складатиме:
- визначає частку структурних елементів поверхні які
взаємодіють з структурними елементами макромолекул; Fi - значення
одиничної сили взаємодії. В гармонійному наближенні частота коливань частинок
наповнювача в такій гратці складатиме:
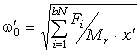 , (3.83)
, (3.83)
де
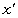 - зміщення частинки наповнювача від положення
рівноваги.
- зміщення частинки наповнювача від положення
рівноваги.
Для
системи ПВХ+ФГ, маса частинки наповнювача складає 1,294×10 -11 кг, зміщення приймемо
рівне відстані між структурними елементами макромолекули і поверхнею
наповнювача. Тоді частота коливань буде рівною 2,539 ·109 Гц, а для систем
ПВХ+65об.%Cu (Мr =6,318·10-11 кг) -2,482·109 Гц.
Враховуючи, що умови формування наповненої полімерної системи
передбачають взаємодію з поверхнею дисперсного наповнювача не тільки
структурного елемента макромолекули, але й певної ділянки макромолекули,яка
входить в склад надмолекулярних утворень різних типів визначимо частоту
коливань надмолекулярних утворень, які закріплені на поверхні двох сусідніх часток
наповнювача. Оскільки ступінь полімеризації ПВХ, який використовується в даній
роботі рівний 2240, то оцінки лінійних розмірів макромолекули, яка може
перебувати в різних конформаціях показують, що у випадку конформації клубка в
гаусовій моделі середня відстань між кінцями макромолекули рівна 0,02 мкм.
Використання моделі макромолекули у вигляді вільнозв’язаного
просторово-шарнірного ланцюга, при довжині сегменту Куна А=29,6 середньостатистична відстань між кінцями макромолекули
рівна 0,148 мкм. Якщо ж молекула перебуває в конформації витягнутого ланцюга,
то довжина макромолекули рівна 0,385 мкм. Таким чином лінійні розміри
макромолекул значно менші за відстань між частинками наповнювача. Розглядаючи
коливання надмолекулярних утворень будемо вважати, що їхні поперечні розміри
значно менші за 2lм.ш. В цьому випадку коливання таких
надмолекулярних утворень можна розглядати за аналогією з вимушеними коливаннями
струни. Тоді рівняння руху закріпленої структурної підсистеми полімера матиме
вигляд:
середньостатистична відстань між кінцями макромолекули
рівна 0,148 мкм. Якщо ж молекула перебуває в конформації витягнутого ланцюга,
то довжина макромолекули рівна 0,385 мкм. Таким чином лінійні розміри
макромолекул значно менші за відстань між частинками наповнювача. Розглядаючи
коливання надмолекулярних утворень будемо вважати, що їхні поперечні розміри
значно менші за 2lм.ш. В цьому випадку коливання таких
надмолекулярних утворень можна розглядати за аналогією з вимушеними коливаннями
струни. Тоді рівняння руху закріпленої структурної підсистеми полімера матиме
вигляд:
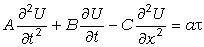 ,
(3.84)
,
(3.84)
де
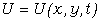 , а граничні умови матимуть вигляд:
, а граничні умови матимуть вигляд: 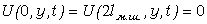 і початкові умови
і початкові умови 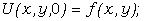
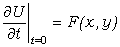 .
.
Розв’язок
рівняння (3.84) для основного тону коливань підсистеми буде мати вигляд
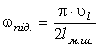 . (3.85)
. (3.85)
Порівняння
частоти коливань частинок наповнювача і надмолекулярних утворень показує, що
останні мають дещо вищу частоту коливань. Якщо макромолекули ПВХ знаходилася б
у конформації витягнутого ланцюга, то частота її коливань за аналогією з
коливаннями струму дорівнювала б 1,957·1010 Гц, що значно перевищує частоту
коливань суперрешітки частинок наповнювача і надмолекулярних утворень.
При
розгляді макромолекули лінійного полімера, як вільно з’єднаного
просторово-шарнірного ланцюга, використання співвідношення (3.85) дає
значення частоти 5,092·1010 Гц, а сегмент Куна буде мати чатоти коливань, при
використанні такої аналогії, 2,546·1012 Гц.
Оцінки
характеристичного спектру структурних елементів можна провести, використовуючи
континуальні моделі, в яких не аналізуються особливості взаємодії між
структурними елементами макромолекул, макромолекул і структурними елементами наповнювача
чи пластифікатора.
Згідно
теорії Дебая обмежуюча частота спектру коливань структурних елементів
знаходиться згідно співвідношення:
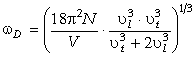 .
(3.86)
.
(3.86)
Якщо врахувати, що для частотних віток позвовжніх і поперечних хвиль
існує не одна і таж обмежуюча частота, як в теорії Дебая, а різні обмежуючі
частоти, то густину спектрального розподілу потрібно розбивати на дві частини
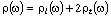 , (3.87)
, (3.87)
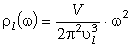 і
і 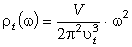
і
нормувати окрему частину на N:
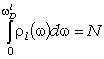 і
і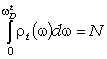 . (3.88)
. (3.88)
Тоді обмежуючі частоти 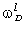 і
і
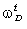 рівні
рівні
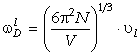 , (3.89)
, (3.89)
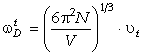 . (3.90)
. (3.90)
Відповідні характеристичні температури знаходяться як
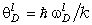 ;
; 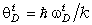 .
.
Як
видно з даних таблиці 3.3. в міру зміни концентрації мінерального наповнювача
змінюються і значення і . Так для
системи ПВХ+ФГ в області концентрації наповнювача 0,1-10,0 об.% ФГ максимальне
значення спостерігається при jн = 1,0 об.% ФГ. Частота зростає в
діапазоні концентрацій 2,0 - 20,0 об.% ФГ. Для систем наповнених модифікованими
фолрмами фосфогіпсу максимальне значення обмежуючої поздовжньої частоти
характерне для області концентрацій 10 - 20 об.%, а залежність від j аналогічна до
залежності системи ПВХ+ФГ. Співставлення характеристичних частот наповнених
полімерних систем з відповідними частотами для вихідних полімерів ПВХ (ПВБ)
показує, що ,вищі у гетерогенних полімерних систем (ПВХ+ФГ, ПВХ+ФГPb,
ПВХ+ФГBi, ПВХ+ФГHg). Слід також зауважити, що модифікація поверхні ФГ
солями важких металів приводить до зростання характеристичних частот в
порівнянні з системою ПВХ+ФГ.
Таблиця
3.3
Концентраційна
залежність характеристичного частотного спектру для ПВХ і ПВБ -систем
наповнених мінеральних наповнювачів і пластифікованих Т = 293 К.
|
, об. %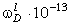 , Гц , Гц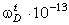 , Гц , Гц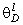 , К , К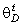 , К, об. %, Гц, Гц, К, К , К, об. %, Гц, Гц, К, К
|
|
|
|
|
|
|
|
|
|
|
ПВХ + ФГ
|
ПВХ + ФГHg
|
|
0,1
|
2,155
|
0,980
|
165
|
75
|
0,1
|
3,081
|
1,323
|
236
|
101
|
|
0,5
|
2,203
|
0,941
|
168
|
72
|
0,5
|
2,970
|
1,349
|
228
|
103
|
|
1,0
|
2,204
|
1,000
|
168
|
76
|
1,0
|
3,023
|
1,343
|
232
|
103
|
|
2,0
|
2,192
|
0,999
|
167
|
76
|
2,0
|
3,127
|
1,382
|
240
|
106
|
|
3,0
|
2,115
|
1,020
|
162
|
78
|
3,0
|
3,063
|
1,374
|
235
|
105
|
|
5,0
|
2,141
|
1,020
|
163
|
78
|
5,0
|
3,060
|
1,413
|
235
|
108
|
|
10,0
|
2,150
|
1,105
|
165
|
84
|
10,0
|
3,200
|
1,475
|
245
|
113
|
|
20,0
|
2,386
|
1,250
|
182
|
107
|
20,0
|
3,253
|
1,615
|
249
|
124
|
|
ПВХ + ФГPb
|
ПВХ + ФГBi
|
|
0,1
|
3,204
|
1,357
|
245
|
104
|
0,1
|
3,093
|
1,334
|
237
|
102
|
|
0,5
|
2,996
|
1,299
|
229
|
0,5
|
3,010
|
1,347
|
231
|
103
|
|
1,0
|
3,020
|
1,305
|
231
|
100
|
1,0
|
3,197
|
1,358
|
245
|
104
|
|
2,0
|
3,141
|
1,329
|
241
|
102
|
2,0
|
3,142
|
1,372
|
241
|
105
|
|
3,0
|
3,073
|
1,356
|
235
|
104
|
3,0
|
3,024
|
1,393
|
232
|
107
|
|
5,0
|
3,178
|
1,404
|
243
|
106
|
5,0
|
3,165
|
1,418
|
242
|
109
|
|
10,0
|
3,169
|
1,481
|
243
|
113
|
10,0
|
3,313
|
1,474
|
254
|
113
|
|
20,0
|
3,541
|
1,654
|
271
|
127
|
20,0
|
3,081
|
1,510
|
236
|
116
|
|
ПВХ + ДБФ
|
ПВБ + ДБФ
|
|
0,5
|
2,084
|
1,018
|
159
|
78
|
0,5
|
1,556
|
0,712
|
119
|
55
|
|
1,0
|
2,088
|
1,009
|
159
|
77
|
1,0
|
1,565
|
0,714
|
120
|
55
|
|
3,0
|
2,056
|
0,980
|
157
|
75
|
3,0
|
1,527
|
0,705
|
117
|
54
|
|
5,0
|
2,014
|
0,963
|
154
|
74
|
5,0
|
1,520
|
0,688
|
116
|
53
|
|
10,0
|
1,920
|
0,901
|
147
|
69
|
10,0
|
1,494
|
0,684
|
114
|
52
|
|
15,0
|
1,815
|
0,836
|
139
|
64
|
15,0
|
1,404
|
0,617
|
107
|
47
|
|
20,0
|
1,739
|
0,771
|
133
|
59
|
20,0
|
1,338
|
0,651
|
102
|
42
|
При
введенні в ПВХ і ПВБ низькомолекулярного пластифікатора ДБФприводить до того,
що коливальний спектр систем в порівнянні з вихідними полімерами змінюється.
Співставлення розрахункових значень ,для пластифікованого ПВХ з відповідними даними
вихідного полімера показують, що вони дещо вищі для ПВХ. Для систем ПВБ+ДБФ в
області концентрації 0,5 ¸ 3 об.% 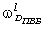 <
<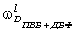 .
.
Аналіз
концентраційної залежності характеристичних частот показує, що в області
пластифікатора 0 ¸ 1,0 об.% значення ,практично не змінюються. В діапазоні концентрацій
пластифікатора 3,0 ¸ 20,0 об.%
значення ,зменшуються. Порівняння відповідних обмежуючих частот
для системи ПВХ+ДБФ і ПВБ+ДБФ показує, що в усьому діапазоні вмісту
пластифікатора обмежуючі частоти першої системи значно вищі за відповідні
значення частот другої системи.
Для
полімер-полімерних систем типу ПВХ+ПВБ (табл. 3.4)
концентраційна залежність характеризується максимумом при jн = 12 об.% ПВБ. Найбільш суттєві зміни
характеристичних частот спостерігаються в області концентрацій ПВБ 12 - 23
об.%.
Таблиця
3.4
Концентраційна
залежність характеристичного частотного спектру для систем ПВХ + ПВБ, ПВХ + ПВБ
+ ДБФ при Т = 293 К
|
, об. %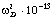 Гц Гц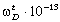 , Гц, К, К, об. % , Гц, К, К, об. %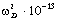 , Гц , Гц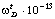 , Гц, К, К , Гц, К, К
|
|
|
|
|
|
|
|
|
|
|
ПВХ + ПВБ
|
ПВХ + 6 об. % ПВБ + ДБФ
|
|
0
|
2,114
|
0.952
|
165
|
75
|
0,5
|
2,063
|
0,995
|
157
|
76
|
|
6
|
2,018
|
0,872
|
154
|
67
|
1,0
|
2,052
|
1,016
|
157
|
78
|
|
12
|
2,103
|
0,880
|
161
|
67
|
3,0
|
1,996
|
0,971
|
156
|
74
|
|
16
|
2,030
|
0,950
|
155
|
73
|
5,0
|
1,899
|
0,937
|
152
|
72
|
|
23
|
1,707
|
0,808
|
130
|
62
|
10,0
|
1,839
|
0,875
|
141
|
67
|
|
32
|
1,826
|
0,745
|
140
|
57
|
15,0
|
1,767
|
0,818
|
135
|
63
|
|
100
|
1,513
|
0,683
|
116
|
52
|
20,0
|
1,691
|
0,744
|
129
|
59
|
|
ПВХ + 12 об. % ПВБ + ДБФ
|
ПВХ + 6 об. % ПВБ + ДБФ
|
|
0,5
|
1,970
|
0,967
|
151
|
74
|
0,5
|
2,028
|
0,952
|
155
|
73
|
|
1,0
|
2,000
|
0,966
|
154
|
74
|
1,0
|
2,030
|
0,943
|
155
|
72
|
|
3,0
|
1,967
|
0,940
|
150
|
78
|
3,0
|
1,971
|
0,930
|
151
|
71
|
|
5,0
|
1,965
|
0,912
|
150
|
70
|
5,0
|
1,913
|
0,895
|
146
|
68
|
|
10,0
|
1,790
|
0,864
|
137
|
66
|
10,0
|
1,818
|
0,846
|
139
|
65
|
|
15,0
|
1,720
|
0,785
|
132
|
60
|
15,0
|
1,702
|
0,781
|
130
|
60
|
|
20,0
|
1,650
|
0,719
|
127
|
55
|
20,0
|
1,590
|
0,704
|
122
|
54
|
|
ПВХ + 23 об. % ПВБ + ДБФ
|
ПВХ + 32 об. % ПВБ + ДБФ
|
|
0,5
|
1,919
|
0,923
|
149
|
71
|
0,5
|
1,770
|
0,890
|
143
|
68
|
|
1,0
|
1,890
|
0,917
|
145
|
70
|
1,0
|
1,919
|
0,888
|
147
|
68
|
|
3,0
|
1,879
|
0,903
|
144
|
69
|
3,0
|
1,833
|
0,870
|
140
|
67
|
|
5,0
|
1,839
|
0,825
|
141
|
68
|
5,0
|
1,826
|
0,852
|
140
|
65
|
|
10,0
|
1,676
|
0,807
|
128
|
62
|
10,0
|
1,680
|
0,796
|
129
|
61
|
|
15,0
|
1.611
|
0,753
|
123
|
58
|
15,0
|
1,620
|
0,733
|
124
|
56
|
|
20,0
|
0,698
|
114
|
53
|
20,0
|
1,514
|
0,677
|
116
|
52
|
Характеристичний
спектр для пластифікованих полімер-полімерних систем характеризується
максимальним значенням в області концентрацій ДБФ 0,5 - 1,0 об.%. Збільшення
вмісту пластифікатора (j ³ 1 об.%) приводить до зменшення і .
Проведені
розрахунки для вихідного ПВХ і ПВХ -систем (табл. 3.5) показують, що
в міру зростання температури , мають тенденцію до зменшення. При цьому характерно,
що в усьому діапазоні температур < 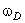 <. Це
означає, що степеням вільності структурних елементів макромолекул лінійних
полімерів відповідає більший інтервал частот, ніж в теорії Дебая. Зменшення
величини при збільшенні температури вказує на зростання прояву
ангармонійних ефектів і послаблення міжмолекулярної взаємодії між структурними
елементами не тільки макромолекул, але й на межі поділу фаз полімер-наповнювач,
полімер-полімер.
<. Це
означає, що степеням вільності структурних елементів макромолекул лінійних
полімерів відповідає більший інтервал частот, ніж в теорії Дебая. Зменшення
величини при збільшенні температури вказує на зростання прояву
ангармонійних ефектів і послаблення міжмолекулярної взаємодії між структурними
елементами не тільки макромолекул, але й на межі поділу фаз полімер-наповнювач,
полімер-полімер.
Таблиця
3.5
Температурна
залежність характеристичного частотного спектру для ПВХ - систем
|
Т,К
|
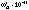 , Гц , Гц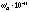 , Гц, К, КТ,К, Гц, Гц, К, КТ,К, Гц, Гц, К, К , Гц, К, КТ,К, Гц, Гц, К, КТ,К, Гц, Гц, К, К
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
ПВХ
|
ПВХ + 3 об. ФГ
|
ПВХ + 5 об. % БЖ
|
|
293
|
2,11
|
0,952
|
162
|
73
|
293
|
2,115
|
1,020
|
162
|
78
|
293
|
1,700
|
0,820
|
130
|
63
|
|
303
|
2,02
|
0,943
|
155
|
72
|
303
|
1,999
|
0,986
|
153
|
75
|
303
|
1,689
|
0,786
|
129
|
60
|
|
313
|
1,92
|
0,924
|
147
|
71
|
313
|
1880
|
0,940
|
144
|
72
|
313
|
1,642
|
0,775
|
126
|
59
|
|
323
|
1,83
|
0,878
|
141
|
67
|
323
|
1,743
|
0,922
|
133
|
70
|
323
|
1,450
|
0,775
|
111
|
59
|
|
333
|
1,75
|
0,833
|
134
|
64
|
333
|
1,643
|
0,885
|
126
|
68
|
333
|
1,378
|
0,766
|
106
|
59
|
|
343
|
1,72
|
0,824
|
131
|
63
|
343
|
1,533
|
0,840
|
117
|
64
|
343
|
1,258
|
0,755
|
96
|
58
|
|
353
|
1,60
|
0,779
|
122
|
59
|
353
|
1,432
|
0,785
|
109
|
60
|
353
|
1,186
|
0,755
|
91
|
58
|
В
таблиці 3.6 наведені результати залежності і від
прикладеного зовнішнього тиску під час формування зразків композицій. Як слідує
з даних, в міру зростання тиску пресування змінюється значенння і . В
області тисків 10 ¸ 60 МПа для сиситем ПВХ+
20об.%Cu, ПВХ+6 об.% W зростання величин , незначне. Найбільш суттєві зміни спостерігається в області титсків пресування 60 ¸ 120 МПа. Результати розрахунків показують, що при
тисках більших за 60 МПа збільшення і для систем ПВХ+W, ПВХ+Cu
дещо вищі ніж у вихідного ПВХ. Це вказує на те, що підвищення тиску пресування
сприяє процесам структуроутворення в напрямку зростання жорсткості підсистем, а
також додатковому утворенню зв¢ків на межі поділу фаз
полімер-металевий наповнювач.
Таблиця
3.6
Залежність
характеристичного частотного спектру для ПВХ - систем від тиску пресування при
Т = 293 К
|
Р, МПа
|
, Гц, Гц, К, К, Гц, Гц, К, К
|
|
|
|
|
|
|
|
|
ПВХ + 20 об. %
Cu
|
ПВХ + 5 об. % W
|
|
10
|
1,860
|
0,995
|
142
|
76
|
1,890
|
0,972
|
145
|
74
|
|
60
|
1,953
|
1,023
|
150
|
78
|
1,980
|
0,999
|
152
|
77
|
|
120
|
1,232
|
1,116
|
171
|
85
|
1,098
|
1,089
|
172
|
83
|
|
200
|
1,279
|
1,125
|
175
|
86
|
1,107
|
1,107
|
176
|
85
|
|
300
|
1,297
|
1,141
|
176
|
87
|
1,116
|
1,116
|
179
|
85
|
|
|
|
|
|
|
|
|
|
|
Накладання
електричного поля в процесі формування композицій (табл. 3.7)
приводить до зростання характеристичних частот в порівнянні з системами, які
сформовані без дії електричного поля. Це вказує на те, що прикладене електричне
поле створює можливості для переходу в межовий шар в наповнених системах
більшого числа структурних елементів і утворенню більшого числа координаційних
зв¢язків на межі поділу фаз. Підтвердженням цього є
збільшення концентрації межових шарів, в системах, які формуються в електричних
полях. Зростання характеристичних частот , як в вихідному ПВХ сформованому в електричному полі,
так і в наповнених ПВХ - системах можна пояснити також дією додаткової сили з
боку поля на диполі і їх часткового упорядкування.
Таблиця
3.7
Вплив
електричного поля на характеристичний частотний спектр ПВХ - систем при Т = 293
К
|
, об. %, Гц, Гц, К, К, Гц, Гц, К, К
|
|
|
|
|
|
|
|
|
|
ПВХ + W (без поля)
|
ПВХ + W (Е = 106 В/м)
|
|
0
|
2,114
|
0,952
|
156
|
74
|
2,222
|
0,988
|
170
|
76
|
|
0,1
|
2,160
|
0,959
|
157
|
70
|
2,214
|
0,968
|
169
|
74
|
|
0,5
|
2,029
|
0,945
|
148
|
69
|
2,073
|
0,985
|
159
|
75
|
|
1,0
|
2,057
|
0,928
|
150
|
68
|
2,115
|
0,960
|
162
|
73
|
|
3,0
|
2,057
|
0,918
|
150
|
67
|
2,071
|
0,948
|
158
|
73
|
|
5,0
|
1,890
|
0,972
|
151
|
68
|
1,992
|
0,907
|
152
|
69
|
|
|
|
|
|
|
|
|
|
|
Якщо вважати, що значення енергії макрорешітки квантовані, то
виходячи із значень поздовжньої і поперечної швидкостей ультразвуку в системі,
визначимо значення характеристичних частот поздовжніх, поперечних коливань і
характеристичну частоту Дебая:
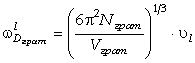 ;
;
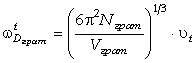 ;
;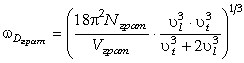 (3.91)
(3.91)
Як
видно з даних характеристичний спектр макрорешітки лежить в області частот ~ 109 Гц, що добре узгоджується з значенням отриманими згідно
співвідношень (3.80) і (3.83). При цьому характерно, що для систем ПВХ+ФГ значення
частот дещо вищі ніж для систем ПВХ+Cu. Це в першу чергу, зумовлено
тим, що у вузлах макрорешітки системи ПВХ+Cu знаходяться
більш масивні частинки наповнювача, ніж в системі ПВХ+ФГ.
Значення
, дають
можливість визначити характеристичні температури Дебая, оскільки має місце
квантування енергії, а хвильове число зумовлене величиною 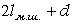 . Проведені розрахунки показали, що значення
характеристичних температур Дебая близькі до нуля (табл.3.3). Отже,
макрорешітка практично не має власного коливального руху. Однак з цього не
випливає, що внутрішня енергія частинок наповнювача, який утворює вузли макрорешітки,
також відповідає нульова температура. В них температура дорівнює температурі
термостата.
. Проведені розрахунки показали, що значення
характеристичних температур Дебая близькі до нуля (табл.3.3). Отже,
макрорешітка практично не має власного коливального руху. Однак з цього не
випливає, що внутрішня енергія частинок наповнювача, який утворює вузли макрорешітки,
також відповідає нульова температура. В них температура дорівнює температурі
термостата.
Таке
значення характеристичних температур дає змогу зробити висновок про те, що
вклад макрорешітки в теплоємність полімерних систем у порівнянні з їх величиною
для полімерної матриці і межового шару незначний і при проведенні практичних
розрахунків ним можна знехтувати.
Якщо
вважати, що характеристична частота коливань відповідає
частоті коливань структурних елементів макромолекул, які взвємопов’язані
внутрішньомолекулярним зв’зком вздовж ланцюга головних валентностей, то для
вихідного ПВХ значення характеристичної поздовжньої частоти співпадає з
значенням отриманим на основі співвідношення (3.33) при ступені
відхилення структурного елемента від положення рівноваги 0,08 <a< 0,09 і a = 0,1. Використання потенціалу Морзе і порівнянння з
відповідними даними для 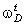 дає значення a рівні 0,087 ¸ 0,088 і 0,11 ¸ 0,12 при
використанні потенціалу Леннард-Джонса. Використання для порівняння з частотою спектру розрахованого згідно співвідношення
(3.42) дає значення a = 0,55 ¸ 0,60 у випадку реалізації H-Cl зв¢язку 0,70-0,74 при реалізації Н-Н зв¢язку.
дає значення a рівні 0,087 ¸ 0,088 і 0,11 ¸ 0,12 при
використанні потенціалу Леннард-Джонса. Використання для порівняння з частотою спектру розрахованого згідно співвідношення
(3.42) дає значення a = 0,55 ¸ 0,60 у випадку реалізації H-Cl зв¢язку 0,70-0,74 при реалізації Н-Н зв¢язку.
Співставлення
теоретично розрахованого спектру структурних елементів макромолекули ПВХ з
експериментальними даними по інфрачервоній спектрометрії даного полімера дають
змогу визначити ступінь відхилення рівноважного положення. В таблиці 3.8
наведені результати розрахунків a для частотного спектру ПВХ при використанні
потенціалів взаємодії Леннард-Джонса і Морзе. Для аналізу валентних коливань
СН, СН2, ССl, С-С груп використовували залежність С-С зв¢язку, а для деформаційних коливань відповідно Н-Н зв¢язок і Н-Сl зв¢язок. Як видно з отриманих результатів значення a близькі як при використанні потенціалу Леннард-Джонса так і потенціалу
Морзе. Виняток становить лише процес коливання бокових груп, які зв¢язані через Н…Сl зв¢язок. Отримані результати розрахунків і їх
співставлення з експериментом підтверджєують, що в області відхилень
структурних елементів 0 £ a £ 0,09
використання даних потенціалів дають значення частотного спектру, які близькі
до експериментальних.
Таблиця
3.8
Співставлення
експериментального частотного спектру ПВХ з теоретичними розрахунками
 Гц
Гц
|
(експеримент)Тип
коливаньСтупінь відхилення a (за потенціалом
Леннард-Джонса)Ступінь відхилення a (за потенціалом
Морзе)
|
|
|
|
|
5,5954
|
Валентні коливання n (СН)
|
0,038
|
0,040
|
|
5,5201
|
Валентні коливання n (СН2)
|
0,039
|
0,041
|
|
2,7016
|
Деформаційні коливання
водневих зв’язків d (СН2)
|
0,048 (C-Cl зв’язок) 0,056
(Н-Н зв’язок)
|
0,054 0,060
|
|
2,5980
|
Позаплощинні деформаційні
коливання gm (CH2)
|
0,080
|
0,083
|
|
2,3418
|
Деформаційні коливання
водневих зв’язків d (СН)
|
0,056 (C-Cl зв’язок) 0,064
(Н-Н зв’язок)
|
0,059 0,066
|
|
2,2665
|
Позаплощинні деформаційні
коливання gm (CH2)
|
0,083
|
0,086
|
|
2,1063
|
Валентні коливання n (С-С)
|
0,084
|
0,088
|
|
1,3000
|
Валентні коливання n (С-Сl)
|
0,088
|
0,091
|
|
0,6858
|
Деформаційні коливання d (СCl)
|
0,080
|
0,084
|
|
0,1206
|
Деформаційні коливання d (СH…Cl)
|
0,089
|
0,501
|
Порівняння
розрахункових значень граничних частот з експериментальними значеннями спектру
ПВХ свідчить, що значення 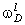 близькі до спектру регулярного сіндіотактичного
ланцюга (відповідно 2,114×1013 Гц і 1,907×1013 Гц), а
значення - до спектру деформацій скелетного ланцюга (відповідно
0,952×1013 Гц і 1,283×1013 Гц ).
Крім того розраховані значення близькі
до експериментальних значень частоти коливань груп при Н-Сl
взаємодії. Таким чином, оскільки при j ³ jкр частинки
дисперсного наповнювача взаємодіють між собою через полімерне в¢яжуче і коливання частинок залежать від руху їх
сусідів, тому найбільш доцільно аналізувати поведінку таких складних систем, як
едине ціле. При такому підході коливальний процес в макрорешітці є
суперпозицією елементарних коливальних рухів частинок наповнювача, атомних груп
на межі поділу фаз полімер-наповювач і структурних елементів макромолекул, що
перебувають в об¢ємі полімерної матриці, які
розповсюджуються в макрорешітці у вигляді хвиль певної довжини.
близькі до спектру регулярного сіндіотактичного
ланцюга (відповідно 2,114×1013 Гц і 1,907×1013 Гц), а
значення - до спектру деформацій скелетного ланцюга (відповідно
0,952×1013 Гц і 1,283×1013 Гц ).
Крім того розраховані значення близькі
до експериментальних значень частоти коливань груп при Н-Сl
взаємодії. Таким чином, оскільки при j ³ jкр частинки
дисперсного наповнювача взаємодіють між собою через полімерне в¢яжуче і коливання частинок залежать від руху їх
сусідів, тому найбільш доцільно аналізувати поведінку таких складних систем, як
едине ціле. При такому підході коливальний процес в макрорешітці є
суперпозицією елементарних коливальних рухів частинок наповнювача, атомних груп
на межі поділу фаз полімер-наповювач і структурних елементів макромолекул, що
перебувають в об¢ємі полімерної матриці, які
розповсюджуються в макрорешітці у вигляді хвиль певної довжини.
Оскільки
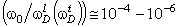 , вкладом коливальних процесів "вузлів"
макрогратки в теплофізичні властивості систем можна знехтувати. Однак,
відношення частот внутрішньомолекулярної і міжмолекулярної взаємодії складає
порядку 10-1. Відповідно, внутрішня енергія частинок наповнювача як сума
енергій міжмолекулярної і внтрішньомолекулярної взаємодії близька до
внутрішньої енергії полімерної матриці. Вклад полімерного в¢яжучого, який при j ³ jкр знаходиться в стані межового шару, в динамічні і
теплофізичні властивості макрогратки в порівнянні з її "вузлами" буде
домінуючий. В цьому випадку енергія структурних елементів для різних типів
коливань, які лежать в діапазоні частот 108 ¸ 1014 Гц, в різних анпрямках макрорешітки вирівнюється
шляхом зміни пружних констант за середній час релаксації t » 10
-9 с. Однак, на межі поділку фаз полімер-наповнювач час накладання і зняття
напружень зсуву рівний t1 » 10 -14 с. тому тут має місце обмін енергією між
"теплими" і "холодними" ділянками системи, і відбуваються
втарти енергії в області активних центрів наповнювач-полімер.
, вкладом коливальних процесів "вузлів"
макрогратки в теплофізичні властивості систем можна знехтувати. Однак,
відношення частот внутрішньомолекулярної і міжмолекулярної взаємодії складає
порядку 10-1. Відповідно, внутрішня енергія частинок наповнювача як сума
енергій міжмолекулярної і внтрішньомолекулярної взаємодії близька до
внутрішньої енергії полімерної матриці. Вклад полімерного в¢яжучого, який при j ³ jкр знаходиться в стані межового шару, в динамічні і
теплофізичні властивості макрогратки в порівнянні з її "вузлами" буде
домінуючий. В цьому випадку енергія структурних елементів для різних типів
коливань, які лежать в діапазоні частот 108 ¸ 1014 Гц, в різних анпрямках макрорешітки вирівнюється
шляхом зміни пружних констант за середній час релаксації t » 10
-9 с. Однак, на межі поділку фаз полімер-наповнювач час накладання і зняття
напружень зсуву рівний t1 » 10 -14 с. тому тут має місце обмін енергією між
"теплими" і "холодними" ділянками системи, і відбуваються
втарти енергії в області активних центрів наповнювач-полімер.
З
врахуванням того, що частота теплових коливань “вузлів” макрогратки значно
нижча частоти теплових коливань структурних елементів ПВХ, доцільно вважати, що
гармонійні пружні хвилі поширюються в однорідному середовищі. В випадку
структурних елементів макромолекул, які безпосередньо взаємодіють з активними
центрами наповнювача, ці дві частоти (nт » nструк.ел)
близькі між собою, тому вони можуть бути додатковими джерелами дисипації.
В
міру зростання дисперсності наповнювача при j ³ jкр можна за рахунок зміни кількості активних центрів
на межі поділу фаз полімер-наповнювач направлено регулювати число ступенів
вільності структурних елементів полімерної системи. Це дозволяє реалізувати
умови для зміни динаміки і теплофізичних властивостей макрорешітки.
Таким
чином, при j ³
jкр макрорешітку можна використати як вихідну модель
для дослідження поведінки гетерогенних полімерних систем в полях різної природи
з врахуванням, що межа поділу фаз полімер-наповнювач за рахунок енергії
міжмолекулярної взаємодії зосереджена в цій області і є додатковим джерелом
дисипативних втрат енергії, а сама макрогратка - генератором чи трансформатором
енергії в області частот, недосяжних для низькомолекулярних кристалів.
3.3 Макромолекулярні константи і дефект модуля зсуву в гетерогенних
полімерних системах
Досліджуючи дисипативні втрати енергії в полімерних композиційних
матеріалах (ПКМ), було показано, що, використовуючи експоненціальним закон
розподілу густини сегментів в адсорбційному шарі, середня величина роботи
(W(l)), яку необхідно затратити для того, щоб зблизити центри двох структурних
підсистем на певну відстань, залежить від типу наповнювача, стану полімерної
матриці та інших факторів [38].
У свою чергу, це обумовлює втрати механічної енергії та зміну модуля
пружності для хвилі напружень, що поширюється в ПКМ, в якому мають місце
закріплення структурних елементів полімера активними центрами високодисперсного
наповнювача [39, 40].
Щоб з'ясувати це питання використовують закон Ньютона для опису закону руху
такого структурного елементу:
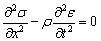 .(3.92)
.(3.92)
Вважають, що деформація ε
складається з
двох частин - пружної деформації εn та деформації εc
структурного
елементу, який може брати участь у русі під дією прикладених напружень, таким
чином:
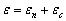 .(3.93)
.(3.93)
Пружна деформація визначається згідно з теорією пружності:
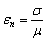 .(3.94)
.(3.94)
Середнє зміщення структурного елементу знаходять як:
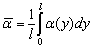 ,(3.95)
,(3.95)
де у - координата вздовж лінії орієнтації структурного елементу.
Отже, якщо L - загальна довжина структурних елементів, які можуть брати
участь у русі під дією періодично діючої сили, то
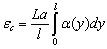 .(3.96)
.(3.96)
Математична форма рівняння руху закріпленого активними центрами
структурного елементу полімеру має вигляд:
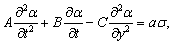 (3.97)
(3.97)
де α=α(х, у, t). Граничні умови мають вигляд: α(х, 0, t)=α(x, 1,
t)=0, α - зміщення елементу
від його положення рівноваги; у - просторова координата цього структурного
елементу; А - ефективна маса на одиницю довжини; 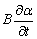 -
демпферуюча сила на одиницю довжини;
-
демпферуюча сила на одиницю довжини; 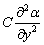 -
сила на одиницю довжини, що здійснює натяг структурного елементу; а σ -
сила, що діє на одиницю довжини
за рахунок зовнішньої напруги зсуву. Сталі коефіцієнти визначаються за
співвідношенням: A=πρa2; C=2μa2/π(1-ν), де ρ - густина, μ - модуль зсуву, а - ефективні розміри статистичного
елементу Куна, ν
- коефіцієнт Пуассона.
-
сила на одиницю довжини, що здійснює натяг структурного елементу; а σ -
сила, що діє на одиницю довжини
за рахунок зовнішньої напруги зсуву. Сталі коефіцієнти визначаються за
співвідношенням: A=πρa2; C=2μa2/π(1-ν), де ρ - густина, μ - модуль зсуву, а - ефективні розміри статистичного
елементу Куна, ν
- коефіцієнт Пуассона.
Співвідношення (3.92) - (3.97) можна подати у вигляді системи інтегро-диференціальних
рівнянь:
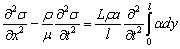 ,(3.98)
,(3.98)
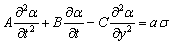
з граничними умовами для α.
Перше рівняння є збуреним хвильовим рівнянням. Особливий
інтерес викликає випадок, коли σ є періодичною функцією часу і
не залежить від у. У цьому випадку
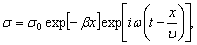 (3.99)
(3.99)
і тоді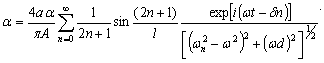 ,
,
де 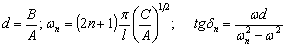 .
.
Якщо частота зовнішньої періодично діючої сили дорівнює одній з
непарних гармонік, виразом для членів більш високих порядків можна знехтувати
(зрозуміло, якщо величина деформації незначна). Тому надалі використовують лише
перший член ряду. Співвідношення для σ і α задовольняє систему рівнянь (3.97), якщо
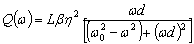
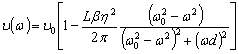 ,(3.100)
,(3.100)
де 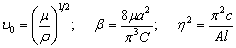 .
.
Отже, величина поглинання та швидкість хвилі напруження в ПКМ залежить
від частоти. Характерно, що величину зміни швидкості υ(ω) -
υ0(υ(ω) - швидкість
в ПКМ, а υ0-
швидкість в полімерній матриці)
вносить лише та компонента зміщення структурного елементу, яка перебуває у фазі
з прикладеним напруженням. Компонента зміщення, яка не співпадає за фазою з
прикладеною напругою, обумовлює поглинання Q(ω) енергії за період. Відповідно зміна модуля в
залежності від частоти дії зовнішнього механічного поля має вигляд:
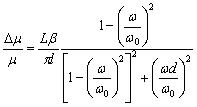 ,(3.101)
,(3.101)
Аналіз отриманих співвідношень показує, що тут можливі такі
випадки дисипативних втрат енергії в ПКМ, зокрема:
) залежність Q(ω) при L~const для малих значень
демпферування (ω0<<d) буде лінійна майже до резонансної
частоти, потім проходить через max при ω=ω0 і далі зменшується як 1/ω3.
У випадку (ω>>d)
- великих
демпферувань - початкова залежність лінійна аж до Q(ω΄0)
- max, який
виникає при (ω΄0<ω0). У діапазоні резонансної
частоти Q(ω) змінюється обернено пропорційно ω
і далі падає як ω3.
) Q - max при ω=ωо для малих демпферувань і
при ω=ω0/d - для великих. Оскільки Q=f(ω,
L, α), то
для ПКМ max Q(α) може бути "розмитим" із
врахуванням того, що розміри структурного елементу не мають одну і ту ж
довжину, а має місце розподіл їх по довжині в залежності від концентрації
активних центрів на поверхні високодисперсного наповнювача, жорсткості
макромолекул, концентрації та дисперсності наповнювача тощо. У випадку
неупорядкованого розподілу структурних елементів на активних центрах
наповнювача, коли функція їх розподілу має вигляд:
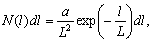 (3.102)
(3.102)
тоді декремент для такого розподілу буде:
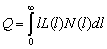 .(3.103)
.(3.103)
Враховуючи значення С виражають відносну зміну модуля зсуву через
коефіцієнт Пуассона:
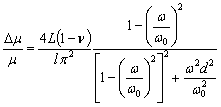 .(3.104)
.(3.104)
Оскільки ступінь полімеризації ПВХ рівний 2240, то оцінки лінійних
розмірів макромолекули, яка може перебувати в різних конформаціях показують, що
у випадку конформації клубка в гаусовій моделі відстань між кінцями
макромолекули рівна 0,02 мкм. Використання моделі макромолекули у вигляді
вільноз’єднаного просторово-шарнірного ланцюга, при довжині сегмента Куна а =
29,6 середьостатистична відстань між кінцями макромолекули
рівна 0,148 мкм. Якщо ж макромолекула перебуває в конформації витягнутого
ланцюга, то довжина макромолекули рівна 0,385 мкм. Таким чином лінійні розміри
макромолекул значно менші за відстань між частинками наповнювача, яка для розглянутих
систем, при даній дисперсності ФГ складає
середьостатистична відстань між кінцями макромолекули
рівна 0,148 мкм. Якщо ж макромолекула перебуває в конформації витягнутого
ланцюга, то довжина макромолекули рівна 0,385 мкм. Таким чином лінійні розміри
макромолекул значно менші за відстань між частинками наповнювача, яка для розглянутих
систем, при даній дисперсності ФГ складає 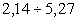 мкм.
Резонансні частоти коливань у випадку конформації витягнутого ланцюга складають
1,957×10 10 Гц. Відповідні значення резонансних
частот для сегмента Куна - 2,546×10 12 Гц,
а вільно-з’єднаного просторово-шарнірного ланцюга - 5,092×10 10 Гц.
мкм.
Резонансні частоти коливань у випадку конформації витягнутого ланцюга складають
1,957×10 10 Гц. Відповідні значення резонансних
частот для сегмента Куна - 2,546×10 12 Гц,
а вільно-з’єднаного просторово-шарнірного ланцюга - 5,092×10 10 Гц.
Якщо під дією зовнішньої періодичної сили будуть коливатися атомні
групи макромолекул, розміри яких складають 1,54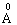 ,
а частота коливань лежить в межах
,
а частота коливань лежить в межах 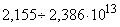 Гц,
то відносна зміна динамічного модуля зсуву складає порядку 0,07×10-4 (таблиця 3.9), тобто є дуже малою
величиною. У випадку дії періодичної сили значно вищих амплітуд, коли в процес
втягуються сегменти Куна величина
Гц,
то відносна зміна динамічного модуля зсуву складає порядку 0,07×10-4 (таблиця 3.9), тобто є дуже малою
величиною. У випадку дії періодичної сили значно вищих амплітуд, коли в процес
втягуються сегменти Куна величина 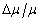 зростає.
Максимальні зміни модуля зсуву спостерігаються при рухливості макромолекул, що
знаходяться в конформації витягнутого ланцюга. В цьому випадку складає порядку
зростає.
Максимальні зміни модуля зсуву спостерігаються при рухливості макромолекул, що
знаходяться в конформації витягнутого ланцюга. В цьому випадку складає порядку 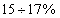 .
.
Таблиця
3.9
Концентраційна
залежність коефіцієнта Пуассона і дефекту модуля зсуву для систем ПВХ+ФГ при
частоті дослідження ω=2,516·106Гц.
|
j, об.%
|
n
|
Дефект модуля зсуву
Dm/m, при різних ступенях рухливості
|
|
|
атомні групи
|
сегмент Куна
|
конформація
Гаусовий клубок
|
вільно-з’єднаного
ланцюга
|
витягнутого ланцюга
|
|
0,1
|
0,37
|
0,066×10-4
|
1,26×10-4
|
8,51×10-4
|
63,0×10-4
|
163,8×10-4
|
|
0,5
|
0,37
|
0,066×10-4
|
8,51×10-4
|
63,0×10-4
|
163,8×10-4
|
|
1,0
|
0,36
|
0,067×10-4
|
1,28×10-4
|
8,65×10-4
|
64,0×10-4
|
166,4×10-4
|
|
2,0
|
0,36
|
0,067×10-4
|
1,28×10-4
|
8,65×10-4
|
64,0×10-4
|
166,4×10-4
|
|
3,0
|
0,35
|
0,068×10-4
|
1,30×10-4
|
8,79×10-4
|
65,0×10-4
|
169,0×10-4
|
|
5,0
|
0,35
|
0,068×10-4
|
1,30×10-4
|
8,79×10-4
|
65,0×10-4
|
169,0×10-4
|
|
10,0
|
0,33
|
0,070×10-4
|
1,34×10-4
|
9,06×10-4
|
67,0×10-4
|
174,2×10-4
|
|
20,0
|
0,33
|
0,070×10-4
|
1,34×10-4
|
9,06×10-4
|
67,0×10-4
|
174,2×10-4
|
Дефект модуля зсуву спостерігається завжди, незалежно від
того, чи закріплений структурний елемент активним центром, чи ні. Це знаходить
своє підтвердження при дослідження ПКМ у випадку низького рівня напружень, коли
мають місце гістерезисні процеси, що обумовлюють дисипативні втрати, які
лінійно зростають з ростом s.
Зроблені висновки знаходять своє підтвердження при
дослідженні і розгляді концентраційної і температурної залежності al і at (рис. 3.7; 3.8) ПКМ. Слід
відмітити, що в усьому діапазоні концентрацій наповнювача величини al і at систем ПВХ + ФГ менші ніж
для вихідного ПВХ.
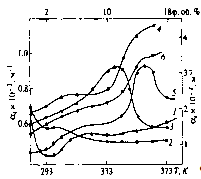
Рис. 3.7. Концентраційна залежність al (1), at (2) для систем ПВХ+ФГ при Т=293 К
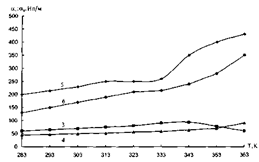
Рис. 3.8. Температурна залежність al (3, 4), at (5,6) для композицій ПВХ (3,5);
ПВХ+ФГ (4,6)
Враховуючи, що експериментальні значення al складаються з втрат на розсіювання від межі частинок
наповнювача і власних дисипативних втрат у самій матриці та межовому шарі,
проводять теоретичні розрахунки втрат на розсіювання для полімерних композицій
в релеєвському наближенні [41]:
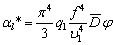 ,(3.105)
,(3.105)
де al = 18,4;  - діаметр частинки наповнювача, al -
частота.
- діаметр частинки наповнювача, al -
частота.
Проведені розрахунки показують, що значення al* не
суттєві і основну роль в процесі дисипації енергії відіграє полімерне в'яжуче.
Аналіз температурної залежності al і at ПВМ показує, що максимум дисипативних втрат енергії
елементами структури композицій спостерігається в області температурного α - переходу.
ВИСНОВКИ
макромолекула
аморфний полімер
На основі аналізу літературних джерел, результатів
експериментальних досліджень та моделювання лінійних аморфних полімерів
показано:
. Макромолекула лінійних аморфних полімерів є складною
системою, яка включає підсистеми - структурні елементи (атомні групи, сегменти,
мономірні ланки). Це об’єкт запису, перетворення і передачі ентропійної та
енергетичної інформації, яка може бути записана на стадії формування
макромолекули, або внаслідок дії на систему зовні полів.
. На основі аналізів процесів розсіювання світла
(релеєвського і комбінаційного) в полімерних системах та моделювання взаємодії
електромагнітної хвилі з структурними елементами макромолекул можна
досліджувати еволюцію структури полімерів, а також визначити параметри
орієнтації ланцюгових молекул і характер міжмолекулярної взаємодії.
. Моделюванням макромолекул, з використанням під
системного підходу, дає можливість визначити макромолекулярні геометричні
параметри, зокрема, середньоквадратичну відстань, середнє значення косинуса
кута між різними ділянками, персистентну довжину ланцюга, статистичний сегмент
Куна, радіус інерції, ступінь згорнутості макромолекул.
. Для лінійних аморфних полімерів ПВХ і ПВБ, та їх
систем визначені геометричні макромолекулярні константи та резонансні частоти
коливань зазначених структуроутворень, що складають 1,957*1010 Гц - 5, 092*1010
Гц.
. Вплив рухливості структуроутворень макромолекул на
дефект модуля зсуву геометричних полімерних систем.
ЛІТЕРАТУРА
1. Физика микромира [маленькая енциклопедия/ глав.ред. Ширков Д.В.] - Москва: Советская энциклопедия, 1980. - 528 с.
2. Соколов А.А. Квантовая механика/ Соколов
А.А., Тернов И.М., Жуковский В.Ч. - Москва: Наука, 1979. - 528 с.
. Блохинцев Д.И. Основы квантовой механики/ Д.И. Блохинцев.
- Москва: Наука, 1983. - 664 с.
4. Вихман Э. Квантовая фізика: Учеб.руководство/ Э.
Вихман; пер.с англ.под ред. А.И. Шальникова и А.О. Вайсенберга. - Моска: Наука,
1986. - 382 с.
5. Мултановский В.В. Курс теоретической физики:
Квантовая механика: Учебное пособие для студентов физ.-мат. фак. пед. ин-тов/ В.В. Мултановский, А.С.
Василевский. - Москва: Просвещение, 1991 - 320 с.
6. Суханов А.Д. Лекции по квантовой физике/ А.Д. Суханов -
Москва: Наука, 1991. - 383 с.
7. Фейнман Р. Фейнмановские лекции по физике. В 9
випусках./ Р. Фейнман, Р. Лейтон, М. Сэндс; пер.с. англ. И.Г. Копылова, под. ред. Я.А. Смородинского. - Москва: Мир, 1978. - Вып. 8,9.
Квантовая механика.
. Френкель С.Я. Макромолекула/ С.Я. Френкель//
Энциклопедия полимеров - Москва: Советская
энциклопедия, 1974. - Т.2. - С. 100-133 с.
. Бартенев Г.М. Физика и механика полимеров: Учеб.
пособие для вузов/ Г.М. Бартенев, Ю.В. Зеленев. - Москва: Высшая школа, 1983. -
381 с.
. Кузнецев В.Н. Химия и физика полимеров: Учебник для
хим.-технол. вузов/ В.Н. Кулезнев, В.А. Шершнев. - Москва: Высш.школа, 1988. -
312 с.
. Элиас Г.-Г. Мегамолекулы/ Элиас Г.-Г.; пер. с
англ./Под ред. С.Я. Френкеля - Ленинград: Химия, 1990. - 272 с.
. Щур А.М. Высокомолекулярные соединения/ А.М. Щур. -
Москва: Высшая школа, 1981. - 656 с.
. Боэчко Ф.Ф. Основи хімії полімерів/ Ф.Ф. Боєчко. - К.: Рад. Шк..,
1988. - 199 с.
. Френкель С.Я. Молекулярная кибернетика/ Френкель
С.Я., Цыгельный И.М., Колупаев Б.С. - Львов: Свит, 1990. - 168 с.
. Гросберг А.Ю. Физика в мире полимеров/ А.Ю.
Гросберг, А.Р. Хохлов. - Москва: Наука, 1989. - 208 с.
16. Куни Ф.М. Статистическая физика и термодинамика. - М.: Наука. - 1981. -
352 с.
17. Нечаев С.К. Проблемы вероятносной топологии:
статистика узлов и некоммутативных случайных блужданий // Успехи физических
наук. - 1998. - Т. 168,
№4. - С. 369-405.
18. Бордюк М.А., Колупаєв Б.С., Іваніщук С.М.,
Ліпатов Ю.С.,
Нікітчук В.І. Прояви гармонійних та ангармонійних ефектів в пластифікованих
полімер-полімерних системах // Фізика конденсованих високомолекулярних систем.
Наукові записки Рівненського педінституту. - 1998. - Випуск 5. - С. 12-17.
. Колупаев Б.С. Энергообменные процессы в
металонаполненых гибкоцепных полимерах. Диссертация на соискание ученой степени
д.х.н. спец. 01.04.19 - физика полимеров. Ровно - 1982. - 264 с.
20. Федорченко А.М. Теоретична фізика. Т.2.
Квантова механіка, термодинаміка і статистична фізика. - Київ: Вища шк., 1993.
- 415 с.
. Болеску Р. Равновесная и неравновесная
статистическая механика. - М.: Мир, 1978. - 405 с.
. Рейф Ф. Статистическая физика - М.: Наука, 1986. -
336 с.
. Лифшиц И.М., Гредескул С.А., Пастур Л.А. Введение в
теорию надпорядоченных систем. - М.: Наука, 1982. - 246 с.
. Цветков В.Н. Жесткоцепнные полимерные молекулы. -
Л.: Наука, 1985. - 380 с.
. Цветков В.Н., Эскин В.Е., Френкель С.Я. Структура
макромолекул в растворах. - М.: Наука, 1964. - 270 с.
26. Frenkel S. Thermokinetics of formation of
polymeric mesomorphous phases in blockopolymers and polymer mixture //I.
Polym.Symp. - 1977 - №61. -Р. 327-350.
. Frenkel S.Ya., Vilesov A.D. Agamalyan M.M.
Pecularities of thermal motion in block copolymeric supercristals //Acta
polymerica. - 1982. - №33. - Р. 421-425.
. Frenkel S.Ya., Vilesov A.D. Multiple
phasetransition in block copolymers //Macromol. Chem. - 1984. - №86. - Р. 93-106.
29. Бартенев Г.М., Саидитов Д.С. Релаксационные процессы
в стеклообразных системах. - Новосибирск: Наука, 1986. - 238 с.
30. Тучин В.В. Исследование биотканей методами
светорассеяния/ В.В. Тучин// Успехи физическмх наук. - 1997. - Т.167, №5. -
С.517-539.
31. Волков Т.И. Малоугловое рассеяние
поляризационного света аморфнокристаллическими полимерными системами. Обзор. в
кн. Новое в методах исследования полимеров./ Т.И. Волков, Б.Г. Баранов. -
Москва: Мир, 1968. - С.7-54.
32. Николаева Г.Ю. Возможности спектроскопи
комбинационного рассеяния света для количественного анализа ориентации
макромолекул поликристаллических полимеров// Высокомолекулярные соединения. -
1988. - Т.40(Б), №10. - С.1695-1705.
33. Гросберг А.Ю. Статистическая физика макромолекул/
А.Ю. Гросберг, А.Р. Хохлов. - Москва: Наука, 1989. - 344 с.
. Фрайфелдер Д. Физическая биохимия/ Фрайфелдер Д.; пер. с англ. Е.С. Громовой и С.В. Яроцкого под ред. З.А.
Шабаровой. - Москва: Мир, 1980 - 582 с.
. Бирштейн Т.М. Конформации макромолекул/ Т.М.
Бирштейн, О.Б. Птицын под ред. М.В. Волькенштейна. - Москва: Наука, 1964. - 391
с.
. Френкель Я.И. Кинетическая теория жидкостей/
Френкель Я.И. - Ленинград: Наука, 1975. - 592 с.
. Рентгенографические методы изучения полимерных
систем/ Ю.С. Липатов, В.В. Шилов, Ю.П. Гомза, Н.Е. Кругляк. - К.: Наук. думка, 1982. - 296 с.
38. Уэллс А. Структурная неорганическая химия. В 3 т./ А. Уэллс;
пер. с англ. В.А. Долгих, Ф.М. Путилиной, С.И. Троянова под ред. М.А.
Порай-Кошица. - Москва: Мир, 1987. - 696 с.
. Дослідження процесів дисипації механічної
енергії в гетерогенних системах на основі гнучко ланцюгових лінійних полімерів/
М.А. Бордюк, Т.М, Бордюк, О.М. Самонюк [та ін.]// Журнал фізичних досліджень. -
2002. - Т.6, №3. - С.317-323.
. Колупаєв Б.С. Внутрішнє тертя та дефект
модуля зсуву в ПКМ/ Б.С. Колупаев// Фізика конденсованих високомолекулярних
систем. - 1997. - Вип.3. - С.3-5.
. Красильников В.А. Введение в физическую
акустику/ В.А. Красилников, В.В. Крілов. - Москва: Наука, 1984. - 400 с.