|
|
|
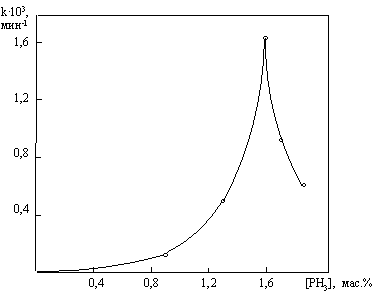
Рис. 10.
Зависимость константы скорости разложения пероксида водорода при катализе
хромитом меди от концентрации добавок фосфина
|
Теория активной поверхности объясняет влияние каталитических
ядов тем, что, размещаясь на неоднородной поверхности различным образом, одно и
то же вещество может оказывать отравляющий или промотирующий эффект. Например,
добавки фосфина экстремально изменяют активность хромита меди в реакции
разложения пероксида водорода (рис. 10).
Электронная теория катализа на полупроводниках
Ф.Ф. Волькенштейна
Основные положения теории:
катализатор - ионный кристалл, часть электронов которого
переходит при температуре выше 0К к поверхности, образуя положительные и
отрицательные свободные валентности;
доля валентностей при каждой температуре постоянна и
определяет адсорбционную способность поверхности кристалла.
При осуществлении катализа на полупроводниках наблюдается три
типа взаимодействия кристалла и молекулы адсорбата:
) затягивание валентных электронов адсорбата в поверхностную
зону кристаллической решетки - «слабая» связь;
) образование связи донорно-акцепторного типа - «сильная»
связь;
) ионная связь, образующаяся за счет полного перехода электрона
к адсорбенту от адсорбата (или наоборот).
В зависимости от реализации того или иного типа
взаимодействия происходит большая или меньшая активация молекул.
Катализ на слоистых кристаллах, цеолитах и
ионитах
Эти катализаторы отличаются тем, что для молекул адсорбата
доступен практически весь объем катализатора. Слоистые кристаллы (графит,
дисульфиды ряда металлов) дают возможность молекулам адсорбата проникать в
межслоевое пространство. Цеолиты - высокопористые каркасные конструкции,
получаемые термообработкой алюмо- и силикагелей, адсорбируют молекулы по всей
поверхности пор. Кроме того, они обладают ситовым эффектом, т.е. «отбирают» для
реакции только те молекулы, которые по своим размерам способны проникать в
поры. Иониты - органические полимеры, имеющие в своем составе ионогенные группы
(катиониты и аниониты). Реакция может протекать не только на поверхности, но и
в массе полимера, особенно в случае жидкофазных процессов, когда наблюдается
явление набухания материала ионита в жидкости.
Катализ на сложных катализаторах
Промышленные катализаторы практически всегда являются
многокомпонентными и во многих случаях многофазными. Создание сложных
катализаторов преследует три цели:
повышение активности и селективности катализатора введением
промоторов;
повышение активности, селективности и стабильности
катализатора при использовании различных носителей;
улучшение свойств катализатора, непосредственно не связанных
с его химической активностью.
Последняя операция называется модифицированием.
Модифицирование катализаторов часто проводят с целью сохранения активной фазы
катализатора в ходе проведения каталитического процесса. Промотирование
катализаторов (введением 0,1-5 мас.% добавок) существенно изменяет
каталитические свойства основного компонента. Механизмы промотирования
разнообразны. Это и образование новых электронных уровней (изменение
донорно-акцепторных свойств катализаторов), и перестройка кристаллической
решетки катализатора, и стабилизация соотношения различных окисленных форм
основного компонента, и изменение ряда других свойств катализатора и носителя.
Среди многофазных катализаторов можно выделить в отдельную группу
полифункциональные катализаторы, на которых можно проводить превращения,
неосуществимые на исходных компонентах. Например, если платину или платину с
промоторами нанести на γ-оксид алюминия, то при
риформинге бензиновых фракций арены образуются по реакции дегидрирования
нафтеновых углеводородов на платине, а парафины изомеризуются на катализаторе
кислой природы. Иногда применяют каталитические системы, полученные
механическим смешением различных катализаторов без их химического
взаимодействия.
Взаимодействие катализатора и реакционной среды
При проведении гетерогенно-каталитических реакций катализатор
подвергается воздействию не только со стороны целевых реагентов, но и со
стороны реакционной среды в целом. Под воздействием среды в катализаторе могут
изменяться:
только поверхностные свойства;
химический состав всего объема катализатора без образования
новых фаз;
химический состав с образованием новых фаз.
Изменение каталитических свойств поверхности происходит уже
при самом акте адсорбции реагентов. Изменение химического состава без
образования новых фаз заметно сказывается на каталитической активности. В
оксидных полупроводниковых катализаторах это связано с обогащением или
обеднением поверхности кислородом по сравнению со стехиометрическим составом. В
кислотно-основных катализаторах − с изменением степени гидратации
поверхностных атомов или молекул. Изменение фазового состава катализатора большей
частью вызвано окислением или восстановлением каталитических систем. При этом
каталитические активности образующихся фаз могут различаться достаточно сильно.
Выяснено, что если реакции превращения фаз катализатора не
являются промежуточными стадиями каталитической реакции и скорость превращения
фаз значительно меньше скорости основного процесса, то активность катализатора
зависит от того, приближается ли температура реакции к рабочему значению со
стороны более высоких или со стороны более низких температур. Это явление
называют гистерезисом каталитической активности.
Иногда взаимодействие катализатора с реакционной средой
приводит к существенному понижению и даже к полному исчезновению каталитических
свойств. Это явление может наблюдаться или при отравлении катализатора, или при
блокировке поверхности катализатора. Под отравлением катализатора понимают
специфическое действие небольшого количества некоторых веществ (каталитических
ядов) по отношению к данному катализатору и данной реакции, которое приводит к
существенному понижению скорости процесса, а в пределе и к его прекращению.
Блокировка же приводит к недоступности поверхности катализатора для реагентов в
результате либо отложения на ней примесей, либо при сильной хемосорбции одного
из реагентов. В связи с этим блокировка неспецифична ни по отношению к
катализатору, ни по отношению к реакции. Часто блокировка является обратимым
процессом. Так, если на поверхности катализатора отлагаются углеродистые
соединения, то их, чаще всего, можно удалить простым выжиганием. При отравлении
часто наблюдается изменение структуры катализатора под действием яда. При
ионном катализе яд связывает расположенные на поверхности катализатора активные
ионы в малореакционные соединения. Например, при кислотном катализе все щелочные
соединения являются ядами, так как их воздействие приводит к нейтрализации
поверхностных кислотных центров. Более сложный механизм отравления наблюдается
в случае электронного катализа на металлах и полупроводниках. Металлы, особенно
благородные, более чувствительны к действию ядов, чем оксидные
полупроводниковые катализаторы. Каталитические яды по их действию на металлы
обычно подразделяют на три группы:
молекулы с неподеленными электронными парами;
металлы или их ионы с неподеленными валентными d-электронами;
молекулы с кратными связями.
Яды первой группы образуют с металлом прочную хемосорбционную
связь, выключая его из каталитического акта. Яды второй группы специфичны по
отношению к металлам подгруппы платины и благородным металлам из других (за
исключением восьмой) групп. Они образуют при адсорбции прочные
интерметаллические соединения. Яды третьей группы являются ядами лишь при
добавке их к основным реагентам как конкурентные адсорбаты. Кроме того,
адсорбированные яды могут изменять структуру поверхностной кристаллической
решетки катализатора. Полупроводниковые катализаторы достаточно устойчивы к
действию ядов, что можно частично объяснить тем, что в этих катализаторах уже
имеется достаточное количество примесей и некоторое изменение их количества не
приводит к существенным изменениям поверхностных и структурных свойств.
Отравление катализатора иногда используют для увеличения его селективности,
поскольку действие яда неодинаково сказывается на основных и побочных реакциях.
В процессе эксплуатации катализатора может происходить его
дезактивация, не связанная с процессами отравления и блокировки, например, при
спекании частиц активного металлического компонента, нанесенного на носитель,
при рекристаллизации активной фазы или при потере активных компонентов
катализатора (например, унос компонентов газовым потоком с поверхности).
1.3
Кинетическое моделирование гетерогенных процессов
Кинетические модели гетерогенных реакций могут быть построены
только на базе эксперимента, проведенного в кинетической области. Требования к
кинетическим моделям в принципе отвечают требованиям, предъявляемым к моделям
гомогенных реакций, однако часть из них применялась к последним «по умолчанию»,
тогда как в более сложных случаях они должны быть сформулированы в явном виде.
Итак, кинетические модели гетерогенных реакций должны обладать полнотой,
однозначностью и автономностью. Сущность этих требований:
Полнота - модель должна описывать скорость реакции во
всей области изменения параметров процесса в промышленном реакторе.
Однозначность - в модели должны отсутствовать
закоррелированные параметры. Для этого параметры модели должны определяться в
широком интервале начальных условий проведения процесса.
Автономность - параметры модели не должны зависеть от
текущего состояния катализатора (отравления, блокировки, других изменений
активности). Для создания автономной модели состояние катализатора должно быть
описано в зависимости не от текущего времени, а от параметров процесса
(концентрации реагентов, давления и температуры в аппарате и т.п.).
Все реакции гетерогенного катализа являются многостадийными.
Их теория базируется на принципе стационарности. Для элементарных стадий обычно
принимают закон, аналогичный закону действующих масс для гомогенных процессов,
- закон действующих поверхностей (поверхностных концентраций). Исходные
вещества могут превращаться в продукты реакции различными путями, проходящими
через разные элементарные стадии. Каждый такой путь называется маршрутом
реакции. Совокупность всех возможных маршрутов - базис маршрутов. Правильность
выбора маршрута определяется соответствием кинетической модели, полученной на
его основе, эксперименту. Число возможных независимых маршрутов часто
определяют с помощью правила Хориути: число независимых маршрутов (N) равно разности числа
стадий реакции (Np) и числа независимых промежуточных соединений (Ns).
N=Np-Ns.
Анализ маршрутов и числа независимых маршрутов и
промежуточных соединений определяется с помощью методов матричной алгебры.
После выбора маршрута реакции составляется отвечающая ему
кинетическая модель, построенная на основе законов адсорбции и закона
действующих поверхностей с привлечением принципа стационарности. Формы изотерм
и их уравнения сильно различаются в зависимости от вида адсорбции (физическая
или химическая), от свойств поглощаемых веществ и от условий процесса. Вид
изотермы и ее параметры для изучаемых веществ и катализатора желательно
устанавливать в предварительных опытах и в дальнейшем использовать при
кинетическом моделировании.
Стационарный режим процесса осуществляется тогда, когда
величины, характеризующие параметр, постоянны в исследуемый промежуток времени,
т.е. свойства системы практически не меняются. Если процесс протекает в
стационарном режиме, наблюдается постоянство концентраций стабильных веществ и
активных промежуточных соединений и малое время существования последних по
сравнению со стабильными веществами.
Применение изотерм адсорбции при кинетическом
моделировании гетерогенных реакций
Изотермы адсорбции применяют в предположении быстрой
адсорбции и медленной химической реакции. В этом случае равновесие адсорбции
наступает быстрее, чем адсорбированное вещество расходуется в последующих
реакциях.
Пусть мы имеем превращение 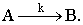 Если адсорбция вещества А на поверхности катализатора описывается
изотермой Ленгмюра, то доля занятой веществом А поверхности составит
Если адсорбция вещества А на поверхности катализатора описывается
изотермой Ленгмюра, то доля занятой веществом А поверхности составит
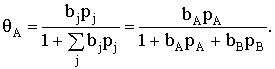
Если принять, что скорость реакции, например rB, пропорциональна концентрации
сорбированного реагента А на поверхности катализатора, то
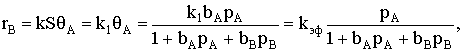
где S − реакционная поверхность; k −
истинная константа скорости химической реакции.
Наблюдается дробно-линейная зависимость скорости от рА
и торможение реакции продуктом. Проанализируем поведение уравнения скорости в
зависимости от свойств реакционной системы.
Пусть продукт реакции сорбируется значительно слабее, чем исходное
вещество, т.е. bb<<bA: 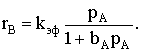 Скорость реакции даже при больших степенях превращения реагента А
зависит практически только от его парциального давления. Этому же уравнению
подчиняется и начальная скорость реакции, если проводить ее в отсутствии
добавок продукта. Если bb>>bA, то
Скорость реакции даже при больших степенях превращения реагента А
зависит практически только от его парциального давления. Этому же уравнению
подчиняется и начальная скорость реакции, если проводить ее в отсутствии
добавок продукта. Если bb>>bA, то 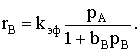 Фактически наблюдается первый порядок по веществу А даже при его
малой конверсии и торможение продуктом В. При очень низкой сорбции обоих
веществ bj<<1 адсорбция будет описываться
изотермой Генри и реакция будет подчиняться закону первого порядка. То же самое
произойдет и при очень низких парциальных давлениях веществ А и В:
Фактически наблюдается первый порядок по веществу А даже при его
малой конверсии и торможение продуктом В. При очень низкой сорбции обоих
веществ bj<<1 адсорбция будет описываться
изотермой Генри и реакция будет подчиняться закону первого порядка. То же самое
произойдет и при очень низких парциальных давлениях веществ А и В: 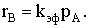
Рассмотрим реакцию с участием двух реагентов: 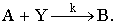
Если считать, что скорость реакции пропорциональна поверхностным
концентрациям адсорбированных реагентов, то
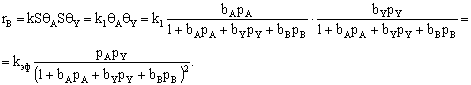
Скорость реакции с уравнением, содержащим квадратичную зависимость
в знаменателе, часто проходит через максимум при повышении начальных
концентраций реагентов за счет более быстрого роста знаменателя, чем числителя.
Бывает, что реакция происходит за счет взаимодействия сорбированной молекулы
одного реагента (А) с молекулой второго вещества (Y), поступающей из объема. Тогда
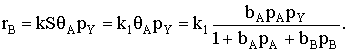
Вклад адсорбции Y падает до
нуля, что отражается на знаменателе. Так как скорость реакции зависит от
концентрации вещества А на поверхности и от парциального давления реагента Y в объеме, то знаменатель не возводится в квадрат.
Все полученные зависимости по форме совпадают с уравнением
Михаэлиса-Ментен. Это связано с тем, что и при металлокомплексном, и при
гетерогенном катализе существуют активные и неактивные формы катализатора. В
первом случае речь идет о несвязанных и связанных в комплекс молекулах, во
втором - о занятых и незанятых молекулами адсорбата активных центрах
поверхности.
Заметим, что далеко не всегда при кинетическом моделировании
гетерогенных процессов можно непосредственно использовать изотермы адсорбции,
полученные при исследовании сорбции веществ в отсутствии химической реакции.
Это связано с тем, что не все адсорбционные центры одновременно являются
каталитически активными. В таком случае адсорбционные коэффициенты, описывающие
изотермы адсорбции, не совпадут с коэффициентами, входящими в кинетическое
уравнение.
Кинетическое моделирование гетерогенных реакций с
использованием метода стационарности
В том случае, если скорость адсорбции сравнима со скоростями
превращений реагентов, для построения кинетической модели используют
стационарность системы, которая обеспечивается в открытых системах (например,
при использовании реакционного аппарата, близкого к модели РИВ или РИС-Н). В
таком случае устанавливается равенство скоростей всех последовательных стадий
процесса. При этом чем выше константы скоростей реакций, тем ниже концентрации
участников этих реакций. Стадии могут быть равновесными и неравновесными.
Особенности механизма гетерогенных процессов таковы, что неравновесность
реакций наблюдается вследствие того, что равновесие в этих стадиях
устанавливается очень медленно и скоростью обратной реакции можно пренебречь по
сравнению с прямой. Принято считать, что все обратимые стадии - быстрые,
необратимые - медленные. Если необратимая стадия только одна, то она
оказывается лимитирующей.
Рассмотрим пример построения кинетических моделей, исходя из
условия стационарности. Пусть из вещества А образуется продукт В через две
последовательные обратимые стадии с образованием промежуточного соединения АZ на активном центре Z поверхности твердого катализатора:
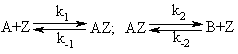
По условию стационарности количество занятых промежуточным
соединением активных центров постоянно, как и общее число активных центров.
Тогда скорости изменения их количества равны нулю (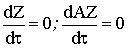 ). Запишем эти скорости исходя из закона
действующих поверхностей.
). Запишем эти скорости исходя из закона
действующих поверхностей.
rZ=-k1cAqZ+k-1qAZ+k2qAZ-k-2cBqZ=0; (1)
rАZ=k1cAqZ-k-1qAZ-k2qAZ+k-2cBqZ=0; (2)
qAZ+qZ=1; qAZ=1-qZ. (3)
Скорость накопления продукта В равна: rВ= k2qAZ-k-2cBqZ.
Определим qZ,
решая систему алгебраических уравнений (1) и (3):
k1cAqZ-k-2cBqZ+(k-1+k2)(1-qZ)=0;
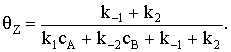
Аналогично, решая систему уравнений (1) и (2), получим для qAZ:
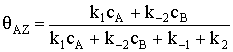
и подставим их в выражение скорости rВ:
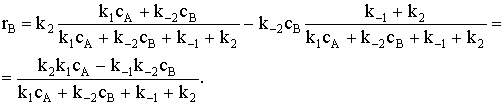
Рассмотрим вариант, когда вторая стадия реакции окажется
необратимой:
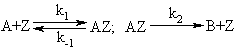
Уравнение скорости примет следующий вид при k-2=0:
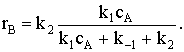
При условии k2<<k-1 (так как скорость необратимых реакций намного ниже
скорости установления равновесия) уравнение превратится в 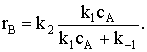 Разделим числитель и знаменатель на k-1:
Разделим числитель и знаменатель на k-1:
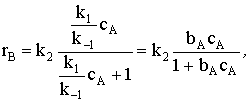
т.е. мы пришли к виду кинетического уравнения на основе доли
занятой поверхности, описывающейся изотермой Ленгмюра, которая предполагает
быстрое установление равновесия между адсорбированным на однородной поверхности
веществом А и веществом А, находящимся в газовой фазе.
1.4
Математическое моделирование процессов гетерогенного катализа
В отличие от гомогенных химических процессов гетерогенные
включают не только стадии химического превращения, но и стадии массопереноса.
Общая скорость расходования вещества в таком случае может определяться любой из
макро- и микростадий. В связи с этим задача математического моделирования
гетерогенной кинетики намного более сложна, чем для гомогенных систем, и не
всегда доступна даже для применения современных вычислительных средств. По этой
причине закономерности гетерогенных процессов изучают, принимая ряд допущений,
которые сильно упрощают решение задачи, но и огрубляют ее. Тем не менее, такие
упрощения оправданы, так как сохраняют принципиальные особенности гетерогенной
кинетики и позволяют оценить поведение системы в тех или иных условиях.
Мы будем изучать реакции, проводящиеся в стационарных и
изотермических условиях при следующих упрощениях:
перенос вещества в ПГС и порах осуществляется молекулярной
диффузией согласно закону Фика (диаметр пор настолько велик, что кнудсеновская
диффузия не имеет места);
к задачам применим локальный подход, т.е. в каждом рассматриваемом
сечении наблюдаемые закономерности не зависят от того, как они протекают в
других сечениях, и от того, как процесс протекал в другие моменты времени;
частицы твердого тела имеют малую кривизну, т.е. их можно
считать плоскими;
к задачам применим квазистационарный подход, т.е. скорость
изменения свойств катализатора и реакционной среды намного меньше скорости
перехода процесса к новым условиям проведения.
Все рассматриваемые катализаторы - пористые тела,
лимитирующей стадией процесса может быть любая из микро- или макростадий.
Заметим, что в условиях стационарного процесса скорости всех последовательных
стадий одинаковы. Поэтому в данном случае лимитирующей считается стадия, в
которой сосредотачивается наибольшее сопротивление протеканию процесса. Только
в этом смысле можно говорить о быстрых и медленных стадиях.
Гетерогенно-каталитический процесс на внешней
поверхности катализатора
Рассмотрим процесс, который включает перенос исходного вещества из
ядра потока через ПГС к поверхности и реакцию превращения 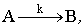 протекающую на внешней поверхности
твердого катализатора (рис. 11).
протекающую на внешней поверхности
твердого катализатора (рис. 11).
|

|
|
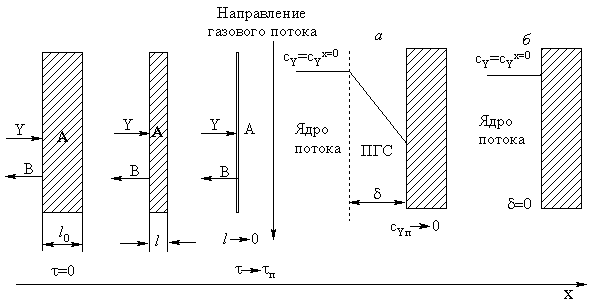
Рис. 11.
Перенос вещества А из ядра потока с концентрацией сАх=0
к поверхности катализатора, у которой устанавливается концентрация сАп
|
Перенос осуществляется молекулярной диффузией потоком FA, направленным по нормали
к плоской поверхности частицы катализатора из ядра газового потока. Процесс
протекает в стационарных условиях в проточном реакторе, поэтому в ядре потока
устанавливается постоянная в данном сечении концентрация исходного вещества сАх=0,
на поверхности формируется концентрация сАп за счет подвода вещества
потоком FA и расходования его по реакции. Скорости подвода вещества к
поверхности катализатора за счет диффузии (приход) и его расходования за счет
химической реакции равны, иначе на поверхности наблюдалось бы накопление или
расходование вещества А, что противоречило бы условию стационарности. По тому
же условию величина потока постоянна. Запишем уравнение закона Фика:
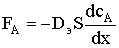
где FА - мольный поток реагента А к поверхности катализатора; Dэ
- коэффициент эффективной диффузии реагента А в ПГС; S - внешняя поверхность
катализатора; 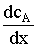 - градиент концентрации реагента А вдоль
выбранного направления; x - координата, направленная по нормали из потока газа
к поверхности катализатора;
- градиент концентрации реагента А вдоль
выбранного направления; x - координата, направленная по нормали из потока газа
к поверхности катализатора;
Проинтегрируем дифференциальное уравнение:
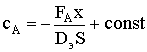 .
.
Воспользуемся начальными и граничными условиями:
x = 0; cА = cАx=0; const = cАx=0; = δ; cА = cАп; и 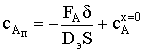 .
.
Для стационарных условий величина потока FA будет равна скорости
химической реакции на поверхности раздела фаз rx.п. Для реакции первого
порядка получим:
FА = k сАп S; 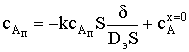 ; и
; и
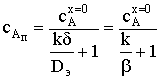 .
.
Здесь 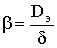 - коэффициент массоотдачи. Таким образом,
уравнение для расчета скорости химического процесса rх.п будет
иметь следующий вид:
- коэффициент массоотдачи. Таким образом,
уравнение для расчета скорости химического процесса rх.п будет
иметь следующий вид:
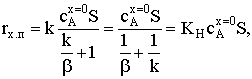
где 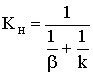 − наблюдаемая константа скорости
гетерогенного процесса.
− наблюдаемая константа скорости
гетерогенного процесса.
Полученное уравнение формально имеет вид кинетического
уравнения, описывающего необратимую реакцию первого порядка. Проведем анализ
полученного уравнения. Для этого рассмотрим два крайних случая:
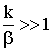 и
и 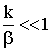 .
.
) ;
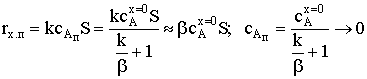 .
.
В этом случае самая медленная стадия − перенос вещества
через ПГС. Иначе - лимитирующей стадией является внешняя диффузия, и говорят,
что процесс протекает во внешнедиффузионной области.
2) ;
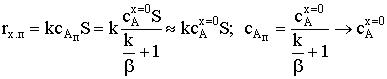 .
.
В этом случае самой медленной стадией является химическая
реакция. Иначе - химическая реакция оказывается лимитирующей стадией, и
говорят, что процесс протекает в кинетической области, или лимитируется
кинетикой.
Если изменить соотношение величин k и b, можно перевести процесс
из внешнедиффузионной области во внешнекинетическую и наоборот. Коэффициент
массоотдачи при неизменных свойствах среды (постоянный коэффициент Dэ) зависит от толщины ПГС d. При d®0 b®¥ и лимитирующей стадией
становится химическая реакция. Толщина ПГС падает при увеличении линейной
скорости газового потока через сечение реактора, поэтому увеличение расхода
газа через аппарат уменьшает диффузионное торможение процесса (рис. 12).
|

|
|
Рис. 12.
Толщина ПГС d и изменение
концентрации реагента на поверхности частицы сАп при
различных линейных скоростях потока газа
|
Концентрация сА в ПГС описывается уравнением
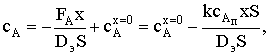 где
где 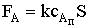 .
.
Тогда
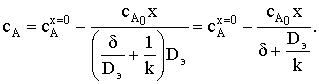
При любой толщине ПГС концентрация вещества А падает в нем
линейно, но тангенс угла наклона прямой увеличивается с уменьшением d.
Перейдем к более сложным случаям. Пусть скорость реакции 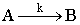 зависит от квадрата поверхностной
концентрации исходного вещества А.
зависит от квадрата поверхностной
концентрации исходного вещества А.
Поверхностная концентрация может быть найдена из квадратного
уравнения
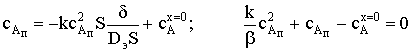 ;
;
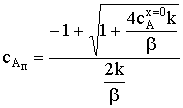 .
.
После подстановки в уравнение скорости химического процесса
получим:
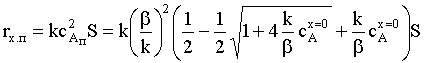
зависимость, весьма далекую от уравнения второго порядка.
Проанализируем выражение rx.п. при разных соотношениях константы
скорости химической реакции k и коэффициента
массоотдачи b.
) . В выражении для сАп в числителе можно пренебречь
единицей под корнем по сравнению с дробью и -1 по сравнению с корнем. Тогда
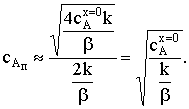
Подстановка этого значения сАп в уравнение скорости
химического процесса даст
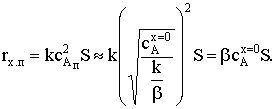
) . Концентрация вещества А на поверхности
выражается отношением двух бесконечно малых величин (неопределенность вида
0/0). Применим правило Лопиталя, продифференцировав выражение для сАп по (k/b):
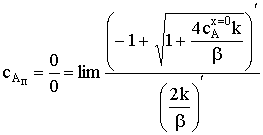 ;
; 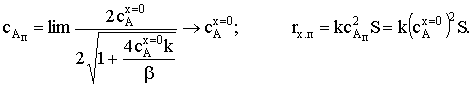
Таким образом, и в этом случае скорость химического процесса
определяется скоростью наиболее медленной стадии.
3) Рассмотрим вариант, когда кинетика реакции подчиняется
уравнению изотермы Ленгмюра: 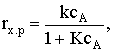 где произведение KсА
сравнимо с единицей.
где произведение KсА
сравнимо с единицей.
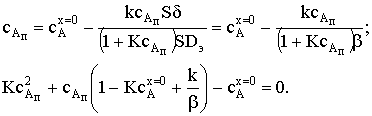
Также получили квадратное уравнение с положительным корнем
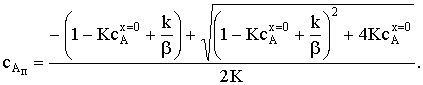
Подставив это выражение в уравнение скорости процесса, получим
общее выражение. Проанализируем, однако, зависимость величины сАп от соотношения констант.
) k/b>>1. Вернувшись к квадратному уравнению, увидим, что всеми слагаемыми
в скобках, кроме k/b, можно пренебречь. Поскольку концентрация вещества А на
поверхности очень низка, то ее квадрат тем более мал по сравнению с остальными
слагаемыми. Тогда 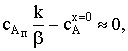 откуда
откуда
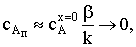 скорость процесса
скорость процесса 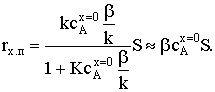
Очевидно, что b¤k<<1 и KcAх=0b¤k<<1. Тогда скорость химического гетерогенного процесса
лимитируется скоростью диффузии.
) k/b<<1. Квадратное уравнение приобретает вид:
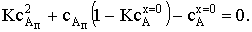
Его положительный корень сАп=сАх=0.
Скорость химического процесса 
В табл. 2 и на рис. 13 представлена зависимость концентрации
реагента на поверхности катализатора при различных соотношениях k/b и cАх=0=1 моль/л для реакций 1, 2 порядка и дробно-линейной функции (K=2).
Таблица 2. Концентрация реагента на поверхности частицы при
различных соотношениях константы скорости k и коэффициента массоотдачи b
|
сАп, моль/л
|
|
k/b
|
1 порядок
|
2 порядок
|
Дробно-линейная
функция
|
|
|
|
|
|
0,01
|
0,990
|
0,990
|
0,997
|
|
0,1
|
0,909
|
0,916
|
0,967
|
|
0,5
|
0,667
|
0,732
|
0,843
|
|
1
|
0,500
|
0,618
|
0,707
|
|
2
|
0,333
|
0,500
|
0,500
|
|
5
|
0,167
|
0,358
|
0,225
|
|
10
|
0,091
|
0,270
|
0,108
|
|
100
|
0,010
|
0,095
|
0,010
|
|

|
|
|
Рис. 13.
Зависимость поверхностной концентрации реагента А от соотношения k/b для реакций различных порядков: ♦ − 1 порядок; ■
− 2 порядок; ´ −
дробно-линейная функция
|
|
|
|
|
|
|
Таким образом, из приведенных примеров видно, что во
внешнедиффузионной области наблюдаемая скорость процесса подчиняется закону
первого порядка по реагенту, т.е. описывается уравнением диффузии. В том
случае, если речь идет о кинетической области, то скорость процесса подчиняется
кинетике химической реакции. В переходной области при сравнимых скоростях
реакции и массообмена зависимость скорости процесса от концентрации реагента
описывается сложными функциями, в которых трудно распознать как исходное
кинетическое уравнение, так и уравнение массопередачи.
Гетерогенно-каталитический процесс в порах
катализатора
Если катализатор представляет собой пористое тело, а условия
процесса таковы, что объем пор доступен для реагентов, то основное количество
вещества перерабатывается именно внутри пор, так как их поверхность на порядки
превышает внешнюю поверхность частицы. Реальные поры в частице извилисты, имеют
разную длину и переменный диаметр, поэтому кинетическое описание процесса в
таких порах затруднено. Введем некоторые допущения, которые упростят задачу
математического описания химического процесса, но сохранят его основные
закономерности. Пусть превращение стационарно протекает по реакции первого порядка в гладкой
цилиндрической поре диаметром d. В каждом
сечении поры S концентрация сA реагента А постоянна, а по длине поры
0<l<L падает по мере протекания реакции. Следовательно, в направлении
радиуса поры материальный поток отсутствует, а в направлении от входа в пору к
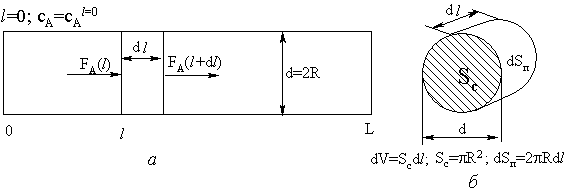
ее днищу происходит перенос вещества (рис. 14).
|
|
|
Рис. 14. Потоки
веществ в гладкой цилиндрической поре (а) и элементарный объем поры (б)
|
Величины потока вещества А слева и справа от выбранного на
длине l элемента объема dV площадью Sc и толщиной dl различны. Выходящий
поток справа от него меньше входящего в него слева на элементарный поток,
обусловленный расходованием вещества А на цилиндрической поверхности dSп. Таким образом, если
принять rx.p пропорциональной концентрации реагента А в
первой степени, то:
FA(l) = FA(l+dl) + rx.pdSп;
dFA = FA(l+dl) - FA(l)= -rx.pdSп.
Применим закон Фика к элементарному потоку:
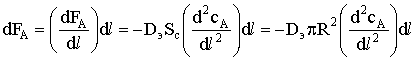
и приравняем dFA к
скорости химического процесса:
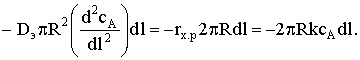
Преобразуем это выражение к виду:
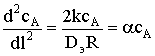 .
.
Полученное дифференциальное уравнение второго порядка имеет
следующее общее решение:
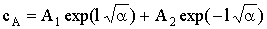 .
.
Найдем постоянные интегрирования А1 и А2,
анализируя граничные условия на входе в пору и у ее днища.
l = 0; сА
= сАп - в начале
поры концентрация вещества А совпадает с его концентрацией на внешней
поверхности;
= L; 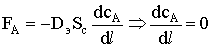 - поток вещества через днище поры
отсутствует.
- поток вещества через днище поры
отсутствует.
Применив эти соотношения к общему решению, получим
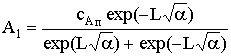 ;
; 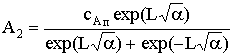 .
.
Подставив постоянные интегрирования в общее решение
дифференциального уравнения, получим:
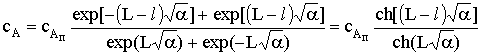 .
.
Это уравнение показывает, как изменяется концентрация
реагента А по мере его диффузии в пору катализатора за счет реакции на
поверхности поры. Скорость химической реакции будет равна
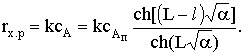
Скорость химического процесса на элементе поверхности составит,
как и в рассмотренном случае реакции на внешней поверхности, drx.п=rx.pdSп или для
нашей реакции rx.п=kсA.dSп.
Подставим полученное значение сА в выражение скорости
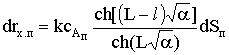
и проинтегрируем его по поверхности поры, чтобы получить общую
скорость процесса:
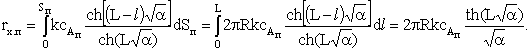
Умножим и разделим последнее выражение на L. Тогда в уравнении скорости химического процесса можно выделить
полную поверхность поры Sп:
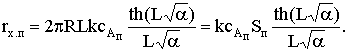 (4)
(4)
Величину, стоящую в знаменателе и под знаком гиперболического
тангенса 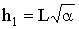 называют модулем Тиле. Индекс «1» говорит
о том, что модуль получен для реакции, скорость которой пропорциональна
концентрации реагента в первой степени. Он отражает отношение скоростей химического
превращения и диффузии реагента в порах катализатора:
называют модулем Тиле. Индекс «1» говорит
о том, что модуль получен для реакции, скорость которой пропорциональна
концентрации реагента в первой степени. Он отражает отношение скоростей химического
превращения и диффузии реагента в порах катализатора:

а отношение 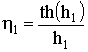 , называемое степенью использования
внутренней поверхности катализатора, условно показывает отношение поверхности,
на которой протекает химическая реакция, к общей поверхности поры.
, называемое степенью использования
внутренней поверхности катализатора, условно показывает отношение поверхности,
на которой протекает химическая реакция, к общей поверхности поры.
Удостоверимся в этом. Допустим, скорость химической реакции
намного меньше, чем скорость диффузии в порах. Тогда по всей длине поверхности
установится постоянная концентрация реагента А, равная его концентрации в устье
поры, и скорость процесса будет равна
Отношение скоростей (4) к (5) как раз
и будет h1.
Степень использования внутренней поверхности поры лежит в пределах
от 0 до 1: 0<h1<1. Покажем это. Пусть h1<<1. Тогда th(h1)»h1 (по свойству функции тангенса) и 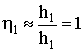 . При h1>>1 th(h1)®1 и
. При h1>>1 th(h1)®1 и 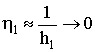 .
.
Скорость химического процесса при 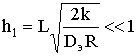 , т.е. при очень низкой константе скорости по сравнению с
коэффициентом диффузии составит
, т.е. при очень низкой константе скорости по сравнению с
коэффициентом диффузии составит 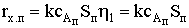 . Реакция идет в кинетической области,
. Реакция идет в кинетической области,
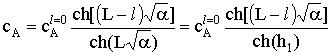 .
.
При h1<<1 ch(h1)®1. Тогда
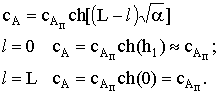
По всей длине поры устанавливается концентрация сА»сАп, h1=1.
При 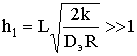 ch(h1)®¥, т.е. при
намного более высокой скорости химической реакции по сравнению со скоростью
массопередачи, можно ожидать, что реагенты не успеют глубоко проникнуть в пору,
так как будут израсходованы по химической реакции, процесс протекает во
внутридиффузионной области, а степень использования внутренней поверхности пор h1®0. Скорость химического процесса подчиняется уравнению
ch(h1)®¥, т.е. при
намного более высокой скорости химической реакции по сравнению со скоростью
массопередачи, можно ожидать, что реагенты не успеют глубоко проникнуть в пору,
так как будут израсходованы по химической реакции, процесс протекает во
внутридиффузионной области, а степень использования внутренней поверхности пор h1®0. Скорость химического процесса подчиняется уравнению
 .
.
Распределение концентраций по длине поры:
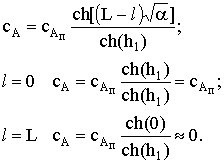
Таким образом, по мере продвижения в глубину поры концентрация
вещества А падает, причем скорость ее снижения связана со степенью
использования внутренней поверхности катализатора h1 и зависит от модуля Тиле. Зная величину модуля Тиле, можно
оценить эффективную глубину проникновения вещества в пору, т.е. среднее
расстояние, которого могут достичь молекулы исходного вещества, прежде чем
прореагирует их подавляющее большинство. Для этого модуль Тиле представляют в
следующем виде:
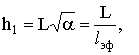
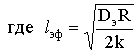 - эффективная глубина проникновения.
- эффективная глубина проникновения.
Очевидно, lэф/L =1/h1. Это гиперболическая функция на участке 1£h1£¥. При h1<1 lэф/L>1,
что означает беспрепятственное проникновение вещества на всю глубину пор при
малых значениях модуля Тиле. Практически можно считать, что при h1<0,5 скорость процесса определяется скоростью химической
реакции, процесс протекает во внутрикинетической области, при h1>2 определяющую роль играет скорость диффузии, что
обусловливает внутридиффузионную область протекания. В пределах h1=0,5-2 осуществляется внутрипереходная область. Заметим, что даже
во внутридиффузионной области, в отличие от внешнедиффузионной, скорость
процесса зависит от величины константы скорости химической реакции k. Рассмотрим, как наблюдаемая константа скорости меняется с
температурой при протекании процесса в различных областях на примере реакции,
скорость которой прямо пропорциональна концентрации реагента А.
Внешнедиффузионная область
rх.п.=bсАх=0S. Коэффициент массоотдачи настолько слабо зависит от
температуры, что его можно считать постоянным в исследуемом интервале.
Следовательно, наблюдаемая константа скорости также не меняется с температурой.
Переходная область
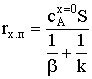 .
.
Температурная зависимость наблюдаемой константы не подчиняется
уравнению Аррениуса.
Внешнекинетическая область
rх.п=kсАх=0S. Константа скорости подчиняется аррениусовской зависимости
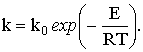
Внутридиффузионная область
Если реакция протекает при лимитировании скорости процесса стадией
диффузии внутри пор, то можно считать, что молекулы реагентов достигают
поверхности частицы беспрепятственно, толщина ПГС d®0 и сАп=сАх=0. Тогда
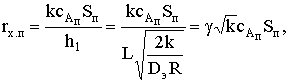
где 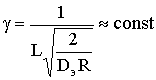 при слабо зависящем от температуры
эффективном коэффициенте диффузии. Тогда
при слабо зависящем от температуры
эффективном коэффициенте диффузии. Тогда
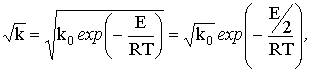
т.е. наблюдаемая энергия активации эффективной константы скорости
будет вдвое ниже, чем энергия активации той же реакции во внешнекинетической
области.
Внутрипереходная область при сАп=сАx=0:
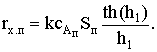
Изменение наблюдаемой константы скорости с температурой не
отвечает уравнению Аррениуса даже формально.
Внутрикинетическая область при сАп=сАx=0:
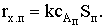 Аррениусовская зависимость наблюдаемой константы скорости с энергией
активации, равной Е.
Аррениусовская зависимость наблюдаемой константы скорости с энергией
активации, равной Е.
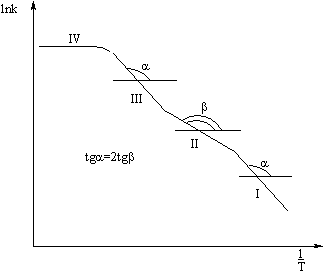
Если представить графически зависимость константы скорости от
температуры во всех рассмотренных областях в координатах Аррениуса, то получим
график, называемый кривой Зельдовича (рис. 15).
Судя по рис. 15, одна и та же реакция, проводимая при различных
температурах, может протекать в различных областях. Конечно, на примере одного
и того же процесса нельзя видеть весь спектр переходов от внешнедиффузионной до
внешнекинетической области, практически невозможно провести одну реакцию в
столь широком температурном интервале, но перемещения между соседними областями
весьма вероятны.
|
|
|
Рис. 15. Кривая
Зельдовича. Области протекания процесса: I - внутрикинетическая; II -
внутридиффузионная; III - внешнекинетическая;
IV - внешнедиффузионная
|
Переход из области в область возможен и за счет изменения
условий массопереноса. Например, из внешнедиффузионной области во
внешнекинетическую можно перейти, увеличивая скорость газового потока. Это
приведет к уменьшению толщины ПГС и увеличению коэффициента массотдачи, так как
b=Dэ/d.
Переход из внутридиффузионной области во внутрикинетическую
происходит с увеличением радиуса пор и уменьшением размера частиц катализатора,
что облегчает доступ молекул газа к внутренней поверхности пор катализатора.
Вообще, при проведении лабораторных исследований
гетерогенно-каталитических реакций надо стремиться к проведению работы в
кинетических областях, когда можно достоверно оценить вид кинетического
уравнения и его активационные параметры.
2. Некаталитические гетерогенные процессы в
системе газ−твердое тело
гетерогенный катализатор реакционный кинетический
В химической технологии весьма распространены процессы, в
которых участвуют твердые и газообразные реагенты. К таким процессам относятся
горение твердого топлива, газификация, реакции, происходящие при
коксообразовании, формирование пленок углерода на поверхностях и многие другие.
Часто реакции, в которых принимает участие, как минимум, одно твердое исходное
вещество или один твердый продукт, называют топохимическими. Эти реакции
протекают на границе раздела фаз, как и в случае гетерогенно-каталитических
реакций, но они обладают и рядом важных особенностей. Главная из них -
расходование или накопление твердого тела по мере протекания реакции, которое
сопровождается и изменением физико-химических свойств его поверхности. В связи
с этим в большинстве случаев невозможно считать процесс стационарным по твердой
фазе. Скорость ее превращения всегда намного выше, чем скорость изменения
концентраций газообразного реагента в потоке. Более того, если речь идет о
превращении неплоских частиц конечного размера, их линейный размер быстро
снижается. Это приводит к еще более быстрому уменьшению величины реакционной
поверхности. Таким образом, мы приходим к задаче, в которой всегда присутствуют
две независимые переменные - связанный со временем реакции размер частиц и
расстояние между ядром конвективного потока реагента и реакционной
поверхностью. Заметим, что на практике часто применяют некоторые допущения и
упрощения, позволяющие избежать применения уравнений в частных производных.
Следует заметить, что есть реакции твердых веществ с
образованием твердых или газообразных и жидких продуктов, которые протекают во
всем объеме твердого тела, например, термическая деструкция каменного угля без
образования пластического слоя. Такие реакции, по-видимому, не следует относить
к топохимическим. При математическом моделировании процессов такого рода
применяют модель постепенного превращения. Она предусматривает превращение
твердого реагента сразу по всему объему частицы. Если температура частицы
одинакова по объему, то концентрация реагента в выбранный момент времени также
постоянна по объему и падает во времени от начального значения до нуля (рис.
16). Время полного превращения частицы обозначим tп. За это время в твердой
фазе не останется исходного вещества, и она будет состоять только из твердого
продукта. Размер частицы может меняться относительно мало, если образуется
немного летучих продуктов, или снижаться до нуля, если твердых продуктов не
образуется.
|
|
|
Рис. 16.
Эволюция твердой частицы согласно модели постепенного превращения
|
Заметим, что формально сходные с термодеструкцией твердых тел
процессы могут осуществляться как топохимические. Например, графитация
карбонизованного материала или полиморфное превращение «графит-алмаз». В этих
процессах в твердом материале могут присутствовать одновременно две разные
кристаллические фазы, и превращение веществ протекает на границе раздела двух
твердых фаз.
Общая последовательность протекания топохимической реакции с
образованием твердого продукта такова: сначала на активных центрах поверхности
исходного вещества происходит взаимодействие реагента, например газообразного,
с твердым телом. Активный центр, как и в случае катализатора при гетерогенном
катализе, обладает бóльшим запасом энергии,
чем остальная поверхность. Такой активный центр часто называют зародышем.
Реакция приводит к изменению структуры твердого вещества вокруг зародыша, при
этом возникают новые активные центры (ядро продуктов реакции). Газообразные
реагенты диффундируют через образовавшийся слой продуктов реакции к поверхности
(границе) раздела двух твердых фаз и там вступают в химическое взаимодействие.
В результате этого ядра продуктов реакции растут по всем направлениям, хотя и с
разной скоростью.
По мере протекания процесса поверхность раздела двух твердых
фаз увеличивается, что приводит к росту скорости химического превращения.
Спустя некоторый промежуток времени растущие ядра продуктов реакции начинают
соприкасаться, а затем и перекрывать друг друга. Скорость роста поверхности
раздела фаз при этом уменьшается (хотя сама поверхность и увеличивается), что
приводит к менее интенсивному росту скорости химического процесса. Наконец,
наступает момент, когда вся поверхность твердой частицы покрыта слоем продуктов
реакции. Поверхность становится практически однородной, и пока размер частиц не
начинает заметно сокращаться, скорость реакции остается почти постоянной. Можно
считать, что в этот период процесс протекает в квазистационарных условиях.
Заметим, что при эпитаксиальных синтезах размер частиц меняется мало и
стационарность процесса сохраняется до его окончания. Если же исходное вещество
расходуется, то через некоторое время почти постоянная скорость процесса
начинает снижаться до нуля после исчерпания исходной твердой фазы. Эволюция
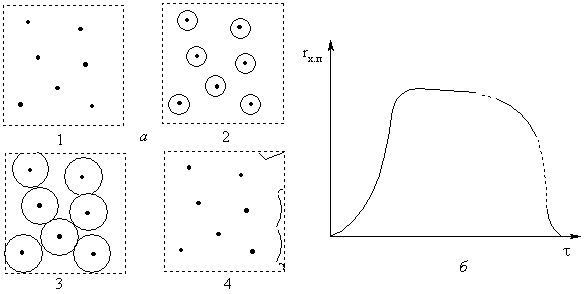
ядер продуктов и изменение скорости реакции представлены на рис. 17, а,б.
При построении математических моделей реакций «газ−твердое»
считают, что взаимодействие происходит на некоторой поверхности раздела фаз
(фронт реакции), которая перемещается в пространстве по мере расходования
твердого реагента. Фронт реакции расположен либо между твердым и газообразным
реагентами, либо между твердым реагентом и твердым продуктом, если он прочно
связан с поверхностью реагирующей частицы. Иногда в случае взаимодействия
твердых веществ с газами применяют модель постепенного превращения, но это вряд
ли можно считать оправданным.
|
|
|
Рис. 17.
Начальные стадии топохимической реакции (а): 1 -активные центры на
участке поверхности; 2 - образование ядер продуктов; 3 - начало слияния ядер;
4 - полное покрытие поверхности продуктом и изменение скорости химического
процесса во времени (б)
|
Мы будем строить математические модели топохимических реакций
для стационарной области химического процесса, протекающего в изотермических
условиях, для плоских и сферических образцов. Примером химической реакции
выберем необратимое взаимодействие твердого вещества А с газообразным реагентом
Y, протекающее в
соответствии с законом 1 порядка Атв + Yг ® В при константе скорости
химической реакции k.
2.1 Реакции без образования твердых продуктов
Реакция подчиняется уравнению  . К таким реакциям относятся, например, горение малозольного
твердого топлива или газификация кокса.
. К таким реакциям относятся, например, горение малозольного
твердого топлива или газификация кокса.
Плоские частицы
Модель плоской частицы отвечает таким процессам, как стравливание
пленок графита с алмазных поверхностей при эпитаксиальном синтезе алмаза или
разрушение (абляция) углеродистых поверхностей летательных аппаратов при
движении их в плотных слоях атмосферы. Эволюцию частицы можно представить
следующим образом (рис. 18):
|
|
|
Рис. 18.
Эволюция плоской частицы и распределение концентраций газообразного реагента
при реакции без образования твердых продуктов, протекающей в диффузионном (а)
и кинетическом (б) режимах
|
Внешнедиффузионная область
Самая медленная стадия процесса - перенос молекул реагента Y через ПГС толщиной d. Перенос осуществляется
молекулярной диффузией, стационарный поток в направлении пластины после
интегрирования выражения закона Фика составит:
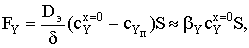
где S - площадь поверхности образца.
Пусть концентрация реагента в газовой фазе практически не меняется
по высоте образца. Обозначим скорость химического процесса расходования
реагента А, составляющего твердую фазу, выразив ее через мольный поток
реагента: 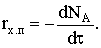 Приравняем количество вещества Y, приносимое диффузионным потоком в единицу времени, к
скорости химического процесса:
Приравняем количество вещества Y, приносимое диффузионным потоком в единицу времени, к
скорости химического процесса: 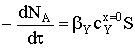 и решим дифференциальное уравнение с разделяющимися переменными
при следующих начальных условиях: t=0; NA=NA0. Тогда
и решим дифференциальное уравнение с разделяющимися переменными
при следующих начальных условиях: t=0; NA=NA0. Тогда
NA=NA0-bYcYx=0St.
По ходу протекания процесса толщина частицы падает со временем.
Эта связь может быть выражена аналитически.
Количество вещества А, содержащееся в исходной частице: NA0=cA0Sl0. В произвольный момент времени в ходе реакции это количество
составит: NA=cA0Sl. Концентрация вещества А в твердой фазе cA0 по ходу реакции не меняется, количество вещества снижается только
за счет уменьшения толщины пластины. Подставим значения NA0 и NA в полученное выше уравнение материального
баланса для вещества А:
cA0Sl=cA0Sl0-bYcYx=0St,
откуда
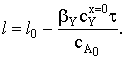
Пусть время полного превращения твердого реагента t=tп.
За это время толщина пластины l обратится в
нуль. Найдем tп из этого соображения 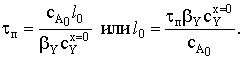 Теперь можно связать конверсию исходного
твердого вещества А и время реакции:
Теперь можно связать конверсию исходного
твердого вещества А и время реакции:
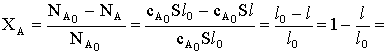
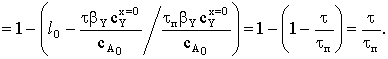
Таким образом, конверсия меняется со временем линейно.
Кинетическая область
Скорость процесса определяется скоростью химической реакции между
молекулами газообразного регента Y и твердого
вещества А на его поверхности. Тогда скорость превращения реагента А можно
представить следующим образом:
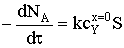 .
.
После решения дифференциального уравнения получим:
A=NA0-kcYx=0St.
Выразим связь между временем проведения процесса, толщиной частицы
и степенью превращения реагента А.
сA0Sl=сA0Sl0-kсYx=0St,
откуда
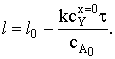
Время полного превращения частицы при снижении толщины пластины до
нуля
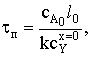
а степень превращения в произвольный момент времени
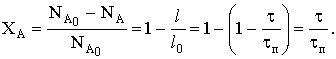
Конверсия также меняется со временем линейно.
Сферические частицы
Модель сферических частиц более употребительна при описании
процессов технологии природных энергоносителей и углеродных материалов. Она
удовлетворительно описывает явления, наблюдаемые при взаимодействии с газами
многих кусковых материалов даже неправильной формы, так как они достаточно
быстро принимают форму, близкую к сферической, в ходе реакции. Эволюцию
сферической частицы можно представить следующим образом (рис. 19).
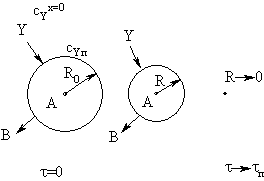
|
Рис. 19.
Эволюция сферической частицы при реакции без образования твердых продуктов
|
|
Размер частицы по мере протекания реакции падает, сокращается
реакционная поверхность, поэтому снижается во времени и скорость процесса.
Построим математическую модель процесса во внешнедиффузионной и кинетической
областях. Такая модель носит название модели сжимающейся сферы.
Внешнедиффузионная область
Самая медленная стадия процесса - перенос молекулярной
диффузией реагента Y к сферической реакционной поверхности из ядра потока. Уравнение
баланса потоков веществ представим в виде
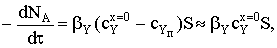
где S=4pR2 - площадь сферической поверхности частицы
радиусом R, которая снижается по мере протекания
реакции.
Пусть концентрация реагента в газовой фазе сY0 практически постоянна в реакционном
объеме.
Количество вещества А в одной частице объемом V к некоторому моменту времени t будет равно
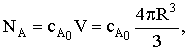
а в начальный момент времени:
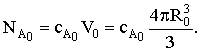
Элементарное количество вещества dNA в объеме сферического слоя толщиной dR составит после дифференцирования
количества реагента А в объеме V:
dNA=сА0×4pR2dR.
Подставив выражения dNA и
реакционной поверхности в соотношение баланса потоков, получим дифференциальное
уравнение с разделяющимися переменными:
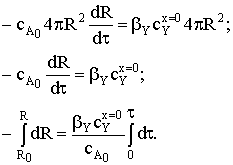
После решения уравнения найдем зависимость радиуса частицы от
времени протекания процесса:
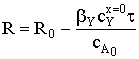 .
.
Радиус сферической частицы линейно снижается во время реакции.
Время полного превращения при достижении нулевого радиуса будет равно:
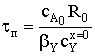 .
.
Степень превращения определяется радиусом частицы или временем
протекания реакции:
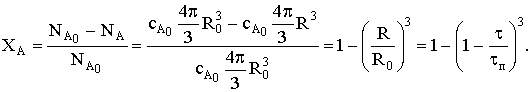
Кинетическая область
Скорость процесса определяется скоростью реакции 1 порядка при
условии равенства концентраций газообразного реагента у реакционной поверхности
и в ядре потока:
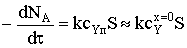 .
.
При подстановке соотношений между геометрическими размерами
сферических частиц и количеством вещества в них получим:
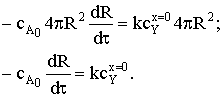
Меняющийся по ходу реакции радиус частицы составит
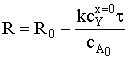 .
.
Время полного превращения будет равно:
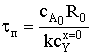 .
.
Степень превращения определяется радиусом частицы или временем
протекания реакции:
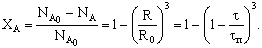
Таким образом, для частиц любой формы, принимающих участие в
реакции 1 порядка по реагенту, находящемуся в газовой фазе, математическая
модель имеет одинаковый вид независимо от области протекания процесса. Параметр
модели имеет смысл константы скорости в кинетической области и коэффициента
массоотдачи в диффузионной.
2.2 Реакции с образованием твердых продуктов
В реакциях с образованием твердых продуктов последние могут легко
осыпаться с поверхности реагента или оставаться прочно связанными с ней. В
первом случае кинетика реакции хорошо описывается моделью плоской частицы с
падающей толщиной или моделью сжимающейся сферы. Во втором случае эти
рассмотренные выше модели оказываются непригодными, и для моделирования
реальных систем принимаются более сложные аппроксимации.
Обсудим примеры таких аппроксимаций для плоских и сферических
частиц. Допустим, что протекает реакция твердого вещества А с газообразным
реагентом Y, продуктом оказывается твердое вещество
В, прочно связанное с поверхностью вещества А:

Плоские частицы
Рассмотрим плоскую частицу вещества А, омываемую потоком
газообразного реагента Y, концентрация
которого в ядре потока равна сYх=0.
Процесс протекает в изотермических и стационарных условиях, т.е. концентрация сYх=0 постоянна во времени и по высоте пластины. Эволюция
плоской частицы представлена на рис. 20.
|
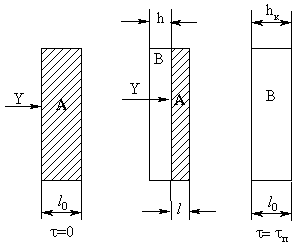 Рис. 20. Эволюция плоской частицы при
реакции с образованием твердого продукта Рис. 20. Эволюция плоской частицы при
реакции с образованием твердого продукта
|
|
Очевидно, что газообразный реагент должен преодолеть
диффузионный подслой в газовой фазе и пройти к поверхности слоя твердого продукта
(золы), а затем сквозь слой золы дойти до реакционной поверхности, которая
представляет собой границу между твердыми слоями золы и исходного вещества.
Таким образом, молекулы реагента Y должны преодолевать два диффузионных сопротивления. В
результате на внешней поверхности частицы создается концентрация реагента сYп, на реакционной поверхности − концентрация сYх.р (сYх=0³сYп³сYх.р). Принято считать, что перенос
газообразного реагента через ПГС осуществляется внешней диффузией, а через слой
золы - внутренней. Обычно зола является высокопористым веществом, через
развитые и достаточно широкие поры молекулы газа диффундируют к поверхности
твердого реагента намного более свободно, чем внутри пор исходного твердого
вещества. Оба диффузионных процесса описываются законом Фика или его аналогом.
Распределение концентраций сY вдоль оси, перпендикулярной реакционной
плоскости, в зависимости от соотношения скоростей реакции и диффузии
представлено на рис. 21.
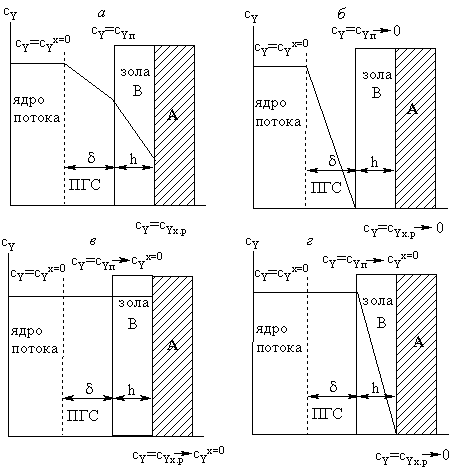
|
|
|
Рис. 21.
Распределение концентраций газообразного реагента при топохимической реакции
с образованием твердого продукта: а -переходная область; б -
внешнедиффузионная область; в - кинетическая область; г -
внутридиффузионная область
|
Внешнедиффузионная область
Наиболее медленная стадия химического процесса - перенос
вещества Y
через ПГС молекулярной диффузией. На поверхности твердого тела (на слое золы) и
на реакционной поверхности (поверхность раздела между твердым реагентом и
золой) устанавливается близкая к нулю концентрация реагента Y.
Составим баланс по потокам веществ:
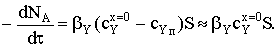
Реакционная поверхность S=const, поэтому скорость процесса неизменна во
времени и количество вещества А, найденное после интегрирования, падает со
временем линейно:
NA=NA0-bYсYx=0St.
Установим связь между временем проведения процесса, толщиной
частицы и степенью превращения вещества А. NA=сA0Sl, тогда
сA0Sl=сA0Sl0-bY сYx=0St, откуда

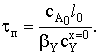
Теперь можно связать конверсию исходного твердого вещества А и
время реакции:
Таким образом, конверсия меняется со временем линейно.
Кинетическая область
Скорость процесса определяется скоростью химической реакции между
молекулами газообразного регента Y и твердого
вещества А на его поверхности под слоем золы. Оба диффузионных торможения почти
отсутствуют, самая медленная стадия процесса - химическая реакция. Тогда
скорость превращения реагента А можно представить следующим образом:
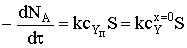 ,
,
где k - константа скорости химической реакции.
После решения дифференциального уравнения получим:
NA=NA0-kсYх=0St.
Выразим связь между временем проведения процесса, толщиной частицы
и степенью превращения реагента А. cA0Sl=cA0Sl0-kcYх=0St, откуда
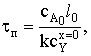
а степень превращения в произвольный момент времени
Таким образом, для плоской частицы во внешнедиффузионной и
кинетической областях математические модели химического процесса одинаковы
независимо от наличия или отсутствия слоя золы на ее поверхности.
Внутридиффузионная область
Реакция также протекает на границе раздела слоя исходного вещества
А и золы В, но самой медленной стадией оказывается перенос вещества Y через слой золы толщиной h. Толщина слоя h постоянно
возрастает по мере протекания процесса, а потому нарастает и диффузионное
торможение по мере увеличения конверсии А. Толщина ПГС d остается постоянной.
Допустим, что перенос вещества Y через золу осуществляется по закону, сходному с законом Фика: 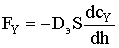 . Составим уравнение баланса потоков,
считая, что скорость расходования твердого вещества (мольный поток) совпадает с
мольным потоком газообразного реагента Y, приносимого за счет диффузии:
. Составим уравнение баланса потоков,
считая, что скорость расходования твердого вещества (мольный поток) совпадает с
мольным потоком газообразного реагента Y, приносимого за счет диффузии:
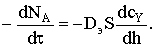
Видно, что левая часть уравнения зависит от времени, а правая - от
линейной координаты. Это значит, что в принципе, мы должны записывать это
уравнение в частных производных  , а такое уравнение невозможно решить непосредственным
интегрированием. Однако решение можно сильно упростить, если принять, что
процесс протекает в квазистационарных условиях. Скорость перемещения фронта
реакции намного меньше скорости диффузии газообразного реагента, поэтому
скорость превращения вещества А можно принять практически постоянной по
отношению к скорости массопереноса
, а такое уравнение невозможно решить непосредственным
интегрированием. Однако решение можно сильно упростить, если принять, что
процесс протекает в квазистационарных условиях. Скорость перемещения фронта
реакции намного меньше скорости диффузии газообразного реагента, поэтому
скорость превращения вещества А можно принять практически постоянной по
отношению к скорости массопереноса 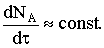 В этом случае можно вернуться к полным производным, разделить
переменные и проинтегрировать получившиеся уравнение. Пределы интегрирования
будут следующими:
В этом случае можно вернуться к полным производным, разделить
переменные и проинтегрировать получившиеся уравнение. Пределы интегрирования
будут следующими:
h=0 сYп»сYx=0 (на внешней поверхности частицы концентрация газообразного
реагента равна его концентрации в ядре потока);
h=h сYх.р»0
(концентрация реагента на поверхности фронта реакции близка к нулю).
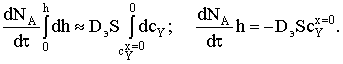
Определим, как изменяется толщина частицы в зависимости от времени
проведения процесса. На этом этапе решения задачи проводим интегрирование
изменения количества твердого реагента по времени реакции, что позволяет получить
зависимость толщины пластины от времени.
Толщина пластины связана с толщиной слоя золы: l=l0-h. Отсюда
dl= −dh.
NA=сA0Sl; dNA=сA0Sdl=-cA0Sdh;
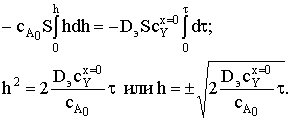
Очевидно, что физический смысл имеет положительный корень
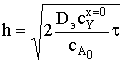 .
.
Определим время полного превращения, исходя из условия, что за это
время вся частица будет состоять из продуктов реакции h=l0.
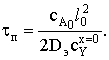
Толщина пластины меняется по закону 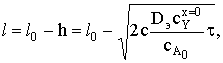 а степень превращения
а степень превращения 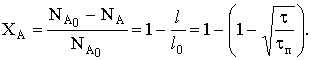
Сферические частицы
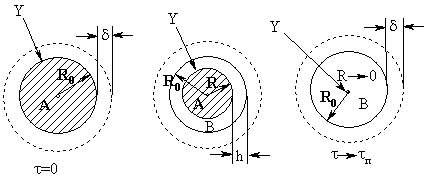
Эволюцию частицы можно представить следующим образом (рис. 22):
|
|
|
Рис. 22.
Эволюция сферической частицы в топохимической реакции с образованием слоя
золы
|
По ходу протекания реакции размер частицы R0 не меняется, однако на
поверхности образуется слой золы, толщина которого увеличивается вслед за
продвижением фронта реакции радиусом R. Как и в случае плоской частицы, по мере
накопления твердого продукта увеличивается сопротивление диффузии газообразного
реагента в слое золы за счет роста его толщины. Реакция также может протекать в
кинетической, внутри- и внешнедиффузионной областях, но реакционная поверхность
будет сокращаться с ростом конверсии реагента А. Такая модель называется
моделью сжимающегося ядра. В данном случае под ядром понимают объем сферы,
занимаемый непрореагировавшим исходным веществом.
Внешнедиффузионная
область
Составим баланс по потокам реагентов А и Y с условием почти нулевой
концентрации газообразного реагента на поверхности золы и на реакционной
поверхности:
Несмотря на то, что поверхность фронта реакции постоянно
уменьшается за счет его перемещения вглубь частицы, внешняя поверхность
остается постоянной. Соответственно, не меняется и геометрия ПГС. Поскольку
основное сопротивление процессу сосредоточено в пленке газового диффузионного
подслоя, оно остается постоянным в течение всего превращения твердого тела, а
площадь сечения, через которое переносится поток реагента Y, составит S=S0=4πR02.
Тогда 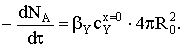
Исходное количество вещества А: 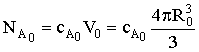 ;
;
текущее количество вещества А: 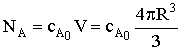 - объем сферы, содержащей исходный реагент, постоянно
уменьшается, причем концентрация вещества А в твердой фазе не меняется до
окончания процесса.
- объем сферы, содержащей исходный реагент, постоянно
уменьшается, причем концентрация вещества А в твердой фазе не меняется до
окончания процесса.
Элементарное количество твердого реагента в произвольный момент
времени в сферическом слое составит: dNA=сA0×4pR2dR, тогда
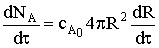 .
.
Приравняем поток расходования вещества А и поток газообразного
реагента Y через внешнюю поверхность частицы:
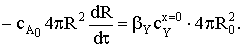
Разделим переменные и проинтегрируем выражение:
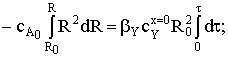
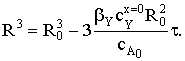
Найдем время полного превращения твердого вещества А tп при достижении нулевого радиуса ядра частицы: 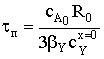 и степень превращения ХА:
и степень превращения ХА:
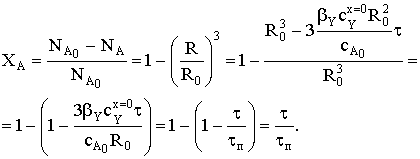
И для модели сжимающегося ядра во внешнедиффузионной области
конверсия реагента А - линейная функция времени процесса.
Кинетическая
область
Скорость процесса равна скорости химической реакции на
поверхности сжимающейся сферы при постоянной концентрации газообразного
реагента, равной его концентрации в ядре потока сYх=0:
Составим баланс по потокам веществ А иY:
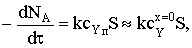 где S=4pR2. Тогда
где S=4pR2. Тогда 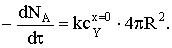
Запишем выражения для текущего количества вещества А, его
элементарного приращения и величины реакционной поверхности (площадь границы
раздела двух твердых фаз):
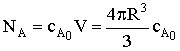 ; dNA=cA0×4pR2dR
; dNA=cA0×4pR2dR
и подставим их в уравнение баланса потоков:
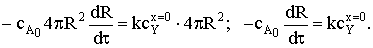
После разделения переменных и интегрирования получим:
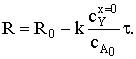
Время полного превращения при достижении нулевого радиуса ядра:
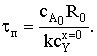
Степень превращения твердой фазы:
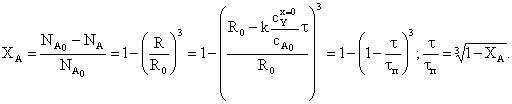
Внутридиффузионная
область
Как и в случае плоской частицы, мольный поток расходования
твердого вещества A равен мольному потоку диффузионного
переноса газообразного реагента Y через слой
золы толщиной h=R0−R. Запишем соответствующее дифференциальное уравнение 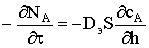 и перейдем к полным производным, приняв
постоянной скорость перемещения фронта реакции по сравнению со скоростью
переноса газообразного реагента диффузией из тех же соображений, что и при
плоской частице. Тогда для квазистационарных условий процесса Надо заметить, что в случае сферической
частицы квазистационарность будет существовать ограниченное время, так как рано
или поздно радиус частицы и реакционная поверхность станут столь малыми, что
скорость процесса будет определяться скоростью химической реакции, и процесс
перейдет в кинетическую область. При наличии квазистационарности можно
вернуться к полным производным
и перейдем к полным производным, приняв
постоянной скорость перемещения фронта реакции по сравнению со скоростью
переноса газообразного реагента диффузией из тех же соображений, что и при
плоской частице. Тогда для квазистационарных условий процесса Надо заметить, что в случае сферической
частицы квазистационарность будет существовать ограниченное время, так как рано
или поздно радиус частицы и реакционная поверхность станут столь малыми, что
скорость процесса будет определяться скоростью химической реакции, и процесс
перейдет в кинетическую область. При наличии квазистационарности можно
вернуться к полным производным
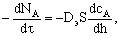
разделить переменные и проинтегрировать получившееся уравнение.
Пределы интегрирования будут следующими:
h=0 (R=R0) сYп»сYx=0
(на внешней поверхности частицы концентрация газообразного реагента равна его
концентрации в ядре потока);
h=h (R=R) сYх.р»0
(концентрация реагента на поверхности фронта реакции близка к нулю).
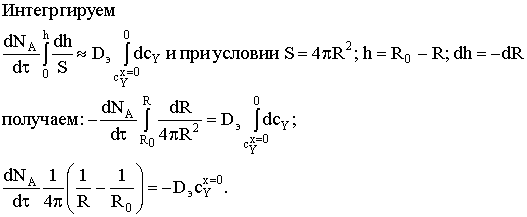
Определим, как изменяется размер частицы в зависимости от времени
проведения процесса. На этом этапе решения задачи проводим интегрирование
изменения количества твердого реагента по времени реакции, что позволяет
получить зависимость радиуса сферы от времени. Текущее количество вещества и
его элементарное приращение в ходе реакции за бесконечно малый промежуток
времени:
; и dNA=cA0×4pR2dR.
Подставим это соотношение в уравнение баланса потоков веществ А и Y:
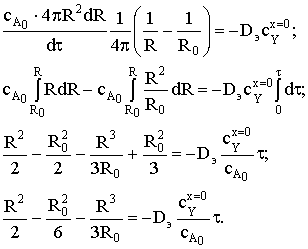
Время полного превращения при R=0 составит
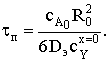
Степень превращения зависит от радиуса частицы
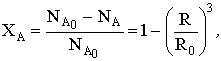 откуда
откуда 
Преобразуем уравнение, в неявном виде связывающее радиус
частицы со временем реакции, к виду:
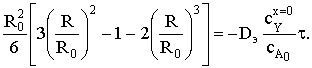
Заменив комплекс постоянных величин на время полного
превращения, получим в неявном виде зависимость конверсии твердого реагента от
относительного времени превращения:
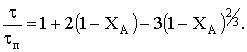
Представим связь между степенью превращения и временем
реакции 1 порядка для рассмотренных случаев в виде табл. 3.
Таблица 3. Конверсия твердой частицы ХА и время ее
полного превращения tп в зависимости от области протекания
гетерогенного процесса
|
Область
протекания
|
Вид частицы
|
|
плоская
|
сферическая
|
|
Без образования
золы
|
|
Внешнедиффузионная
|
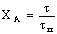 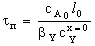 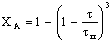 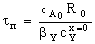
|
|
|
|
|
Кинетическая
|
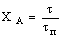 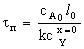 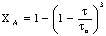 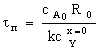
|
|
|
|
|
С образованием
золы
|
|
Внешнедиффу-зионная
|
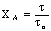 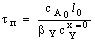 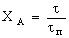 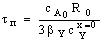
|
|
|
|
|
Кинетическая
|
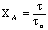 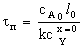 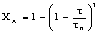
|
|
|
|
|
Внутридиффузионная
|
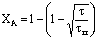 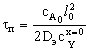 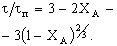 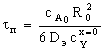
|
|
|
|
Из данных таблицы следует, что при реакции без образования
твердого продукта время полного превращения определяется при прочих равных
условиях коэффициентом массоотдачи или константой скорости в зависимости от
времени протекания реакции. При этом конверсия частицы зависит от времени
реакции линейно для плоских частиц и по закону кубической параболы для
сферических. Очевидно, для плоских частиц существует полная аналогия с
реакциями гетерогенного катализа, так как реагирующая поверхность пластин
остается неизменной в течение почти всего процесса, как и поверхность
гетерогенного катализатора. В случае сфер процесс замедляется по мере снижения
площади реакционной поверхности в равной степени и в кинетической, и в
диффузионной области.
При образовании золы для плоской частицы уравнения для
внешнедиффузионной и кинетической областей совпадают с аналогичными
зависимостями для процессов без образования твердых продуктов. Это понятно, так
как увеличение толщины слоя золы в данном случае не влияет на геометрию твердой
частицы и ПГС, в котором сосредоточено диффузионное сопротивление процессу.
В кинетической области протекания процесса образование золы
не влияет на его скорость, так как образование твердой фазы изменяет только
условия диффузии, а не кинетическое уравнение реакции.
Достаточно сложные математические модели реакций, протекающих
во внутридиффузионной области, связаны с тем, что условия массопередачи в ходе
процесса изменяются за счет увеличения слоя золы.
2.3 Топохимические реакции с участием двух
твердых фаз
Последовательность основных стадий подобных реакций сходна с
начальными стадиями реакций в системах «газ−твердое», т.е. первоначально
происходит образование зародышей, вокруг которых формируются увеличивающиеся в
размерах ядра. Однако единого фронта реакции с равномерными свойствами
поверхности не возникает: каждое из ядер постепенно вовлекает в реакцию
прилежащий к нему объем твердого тела, создавая новые ядра. Реакция
заканчивается, когда исчерпывается вся масса твердого тела. Кинетика таких
превращений описывается специфическими уравнениями, наиболее известно из
которых уравнение Ерофеева−Аврами.
Рассмотрим принцип построения математических моделей
топохимических реакций подобного типа. Очевидно, что скорость расходования
твердого исходного вещества или накопления твердого продукта будет определяться
двумя факторами:
) скоростью и законом образования ядер;
) скоростью и законом превращения вещества в продукт на
поверхности ядер (граница раздела фаз).
Принято считать, что на практике осуществляются два типа
формирования ядер топохимической реакции, скорости которых подчиняются закону
экспоненциального роста ядер:
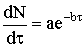
и закону степенного роста ядер:
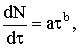
где 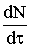 − скорость роста ядер, 1/с; t − время; а и b − постоянные для данных условий реакции параметры.
− скорость роста ядер, 1/с; t − время; а и b − постоянные для данных условий реакции параметры.
В самом общем виде можно сказать, что экспоненциальный рост
ядер наблюдается тогда, когда скорость роста ядер пропорциональна удельной
скорости процесса образования ядер (количество новых ядер, образующихся на
одном ядре в единицу времени), а степеннóй − когда эта
скорость определяется числом имеющихся в данный момент времени ядер.
Допустим, реакция протекает на интервале времени 0 − t. Выберем на этом участке момент времени t¢. В этот момент времени наблюдается
скорость роста ядер 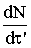 . Скорость реакции накопления твердого
продукта В для отдельного ядра обозначим как r′(t,t¢) и найдем суммарную скорость реакции r (моль/с) как интегральную величину
. Скорость реакции накопления твердого
продукта В для отдельного ядра обозначим как r′(t,t¢) и найдем суммарную скорость реакции r (моль/с) как интегральную величину
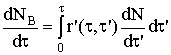 .
.
Это выражение включает и вид функции скорости реакции на ядре, и
вид функции скорости роста самих ядер. Его интегрирование и подстановка
полученного значения скорости в уравнение реакции накопления продукта приводит
к следующим зависимостям, если ядро имеет сферическую форму: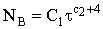 − при степеннóм законе роста числа зародышей;
− при степеннóм законе роста числа зародышей;
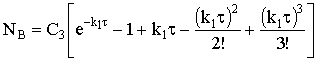 − при экспоненциальном законе,
− при экспоненциальном законе,
где k1, С1, С2, С3 − постоянные
для данных условий реакции параметры.
На практике чаще всего пользуются упомянутым выше уравнением
Ерофеева-Аврами, которое предполагает степенной закон скорости роста ядер, записанный
в форме
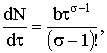
где s − целое
число, зависящее от свойств зародыша ядра.
Интегрирование этого выражения при сферической форме ядра приводит
к простой формуле накопления продукта реакции В из исходного количества реагента
А NA0:
 ,
,
которую и называют уравнением Ерофеева−Аврами.
В этом уравнении k и n − параметры, зависящие от механизма реакции и геометрии
зародышей. Величина n тем больше, чем выше размерность
зародышей (точка, линия или объемное тело различных форм).
Недостатком уравнения является то, что практически любую кривую
накопления продукта топохимической реакции (или расходования исходного
вещества) можно аппроксимировать данной функцией, меняя значения параметров,
т.е. его нельзя использовать для выяснения, например, механизма реакции. В то
же время его легко применять для формального описания конкретной топохимической
реакции, протекающей в фиксированных условиях тепло- и массообмена.
3. Процессы, протекающие в газожидкостных
системах
Будем называть процессы, протекающие в системах «газ (или
жидкость)−жидкость», гетерофазными. Они имеют много общего с процессами в
системах «газ (или жидкость)−твердое тело». Как и в случае гетерогенных
реакций, между фазами имеется поверхность раздела, и реакции, протекающие в
них, сопровождаются явлениями массопереноса. Однако математическое описание
гетерофазного химического процесса практически всегда оказывается намного более
сложным, чем гетерогенного. Это связано с двумя обстоятельствами. Первое из них
то, что площадь поверхности раздела фаз − величина непостоянная, она
сильно зависит от интенсивности перемешивания фаз, и второе − то, что
реакция крайне редко идет именно на границе раздела. Чаще всего реагенты
взаимодействуют либо в пленке некоторой толщины вблизи границы раздела, либо
вообще в объеме одной или обеих фаз. В таком случае молекулы реагентов
преодолевают не одно диффузионное сопротивление ПГС, а как минимум, два −
в пленках двух взаимодействующих фаз.
Газожидкостные процессы часто встречаются в химической
технологии. Обычно их можно подразделить на две основные группы: массообменные
процессы, не сопровождающиеся химической реакцией (абсорбция), и химические
реакции в системах «газ−жидкость», когда реагенты находятся в разных
фазах. В таком случае наблюдается сначала переход реагентов между фазами, после
чего протекает химическая реакция (в одной или в обеих фазах). Очевидно,
массообмен в присутствии химической реакции подчиняется тем же физическим
законам, что и без нее. Тогда математическая модель переноса вещества,
полученная для случая абсорбции, не осложненной реакцией, может быть применена
и для описания массообмена в гетерофазных реакциях. В связи с этим
предварительно рассмотрим математическую модель абсорбции, а затем перейдем к
более сложным случаям. Для определенности будем обсуждать в дальнейшем только
газожидкостные системы.
3.1 Массопередача между газом и жидкостью
Пусть имеется система «газ−жидкость», состоящая из
газовой фазы, в которой присутствует компонент А с парциальным давлением рА,
и жидкости, в которой растворено это же вещество с концентрацией сА.
Будем считать, что на границе раздела фаз соотношение между концентрацией
растворенного вещества и его парциальным давлением в газе отвечает закону
Генри, то есть достигнуто состояние равновесия между растворенным и
газообразным веществом А. Представим распределение концентраций и парциальных
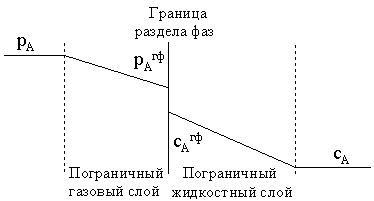
давлений вещества А вблизи поверхности раздела фаз следующим образом (рис. 23):
|
|
|
Рис. 23.
Распределение парциальных давлений и концентраций вещества А в случае
физической абсорбции
|
На удалении от границы раздела фаз парциальное давление
компонента А в объеме газовой фазы практически постоянно, а по толщине ПГС оно
снижается за счет перемещения части вещества в жидкую фазу. Аналогично,
концентрация вещества А в объеме жидкой фазы практически постоянна, а в
пограничном жидкостном слое (ПЖС) она повышается к границе раздела за счет
подпитки веществом из газовой фазы. Согласно закону Генри, можно записать:
рАгф = НАсАгф,
где рАгф − парциальное давление
вещества А на границе раздела фаз со стороны газа, Па; сАгф
− концентрация вещества А на границе раздела фаз со стороны жидкости,
моль/л; НА − константа Генри, Па∙л/моль.
Выберем два потока, за счет которых осуществляется
массообмен. Они направлены последовательно − перенос вещества А из
газовой фазы к границе раздела FAг и перенос его от границы раздела в жидкую фазу: FAж. В случае стационарного
процесса эти потоки равны FAг=FAж=FA. Движущей силой потоков являются разность
парциальных давлений со стороны газа и разность концентраций со стороны
жидкости.
 .
.
Эти уравнения содержат две не определяемые экспериментально
величины − парциальное давление и концентрацию вещества А на границе
раздела фаз. Чтобы исключить одну из них, приравняем величины потоков:
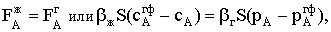
воспользуемся уравнением Генри:
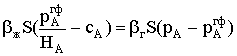
и найдем неизвестное парциальное давление
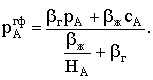
Подстановка этого соотношения в величину потока вещества из
газовой фазы к поверхности раздела приведет к выражению
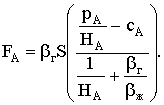
Последнюю формулу можно преобразовать к следующему виду:
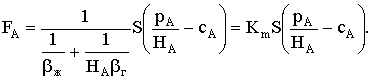
Величину 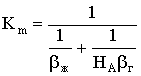 называют коэффициентом массопередачи, а
мольный поток вещества А, отнесенный к поверхности массообмена, скоростью
массопередачи.
называют коэффициентом массопередачи, а
мольный поток вещества А, отнесенный к поверхности массообмена, скоростью
массопередачи.
Из приведенного уравнения следует, что перенос вещества А из
газовой в жидкую фазу будет осуществляться до тех пор, пока его концентрация в
жидкости не достигнет равновесной величины 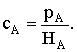 При этом движущая сила процесса (разность концентраций на границе
раздела и в объёме жидкости) обратится в нуль.
При этом движущая сила процесса (разность концентраций на границе
раздела и в объёме жидкости) обратится в нуль.
Каждое из слагаемых, стоящих в знаменателе выражения Km, характеризует сопротивление
массопереносу в одной из фаз, в частности, 1/(НАbг) − сопротивление ПГС, а 1/bж −
ПЖС. Очевидно, что возможно существование двух крайних случаев, когда одним из
сопротивлений можно пренебречь. Допустим, газ обладает высокой растворимостью в
жидкости. Тогда константа Генри мала, и скорость абсорбции будет определяться
величиной коэффициента массоотдачи от газа к жидкости bг, и величина потока вещества примет вид: 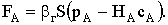 Примером подобных систем могут служить
газообразный аммиак и вода. Обратная картина наблюдается при слабой
растворимости газа и высокой константе Генри (например, кислород и
углеводородный растворитель). В этом случае величина потока зависит от сопротивления
массопередаче в жидком слое:
Примером подобных систем могут служить
газообразный аммиак и вода. Обратная картина наблюдается при слабой
растворимости газа и высокой константе Генри (например, кислород и
углеводородный растворитель). В этом случае величина потока зависит от сопротивления
массопередаче в жидком слое:
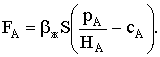
В любом случае скорость абсорбции можно увеличить за счет
повышения площади поверхности массообмена. Для этого необходимо повышать
интенсивность перемешивания фаз либо за счет повышения линейной скорости
движения газа в аппарате, либо путем введения специальных перемешивающих
устройств.
3.2 Химическая реакция, сопровождающаяся абсорбцией
Реакции в гетерофазной системе могут протекать в разных зонах
жидкой фазы − от границы раздела с газом до полного объема жидкости. Доля
реакционной зоны в жидкости определяется соотношением скоростей массопередачи
(диффузии газа к поверхности раздела и диффузии растворенного вещества в
жидкости) и химической реакции. Рассмотрим крайние случаи протекания
химического процесса в гетерофазной системе:
) скорость реакции очень велика по сравнению со скоростью
массопереноса, тогда реакция протекает в пограничной пленке жидкости;
) скорость реакции намного меньше скорости массопереноса, тогда
реакция осуществляется в объеме жидкой фазы при концентрации реагента в
жидкости, определяющейся условиями его переноса из газовой фазы.
Гетерофазная химическая реакция, протекающая в пограничном
слое жидкости
Пусть протекает быстрое взаимодействие с единичной стехиометрией
между реагентом Y, растворенным в жидкости, и газообразным
веществом А:
А + Y ® В.
Очевидно, что химической реакции предшествует стадия абсорбции
газообразного реагента. Пусть при этом на границе раздела фаз осуществляется
равновесие между растворенным и газообразным компонентом А. Реагенты
диффундируют навстречу друг другу в пограничной пленке жидкости толщиной d и мгновенно реагируют на реакционной
поверхности, параллельной границе раздела фаз. Расстояние до этой поверхности
от границы раздела х0 определяется соотношением скоростей диффузии
реагентов А и Y в жидкости. Площади реакционной
поверхности и поверхности раздела одинаковы.
Распределение парциальных давлений и концентраций реагентов
представлено на рис. 24. Рассмотрим три мольных потока веществ: 1) перенос
газообразного реагента А к границе раздела; 2) перенос растворенного реагента А
к поверхности реакции; 3) перенос растворенного реагента Y от внутренней границы пограничного слоя к поверхности
реакции. Запишем величины этих потоков, которые равны друг другу в условиях
стационарного процесса:
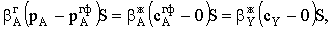
где bАг, bАж, bYж − коэффициенты массоотдачи реагента А в газовой и жидкой
фазах и реагента Y в жидкости; рА −
парциальное давление реагента А в газовой фазе; рАгф −
парциальное давление реагента А на границе раздела фаз; сY − концентрация реагента Y в жидкой фазе; S − площади поверхности массообмена и
реакционной поверхности.
Концентрации реагентов на поверхности реакции мгновенно падают до
нуля в результате быстрого взаимодействия между ними.
|

|
|
Рис. 24.
Распределение концентраций реагентов в случае быстрой химической реакции в
пленке жидкости
|
Коэффициенты массоотдачи для веществ А и Y в отсутствии химической
реакции при тех же гидродинамических условиях определяются как
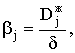
где Djж − эффективный коэффициент диффузии реагента j в жидкости;
d − толщина ПЖС.
Однако в данном случае молекулы каждого реагента преодолевают
только часть толщины ПЖС, в частности молекулы вещества Y − d−x0, молекулы вещества А − х0. Тогда наблюдаемые на
практике коэффициенты bjж можно
определить следующим образом:
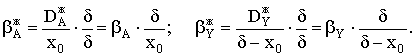
Теперь уравнения баланса потоков можно записать как
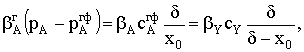
где bАг − коэффициент массоотдачи через ПГС.
В уравнениях имеются две неизвестные величины: отношение х0/d и концентрация сАгф,
связанная с парциальным давлением уравнением Генри сАгф=рАгф/НА.
Найдем отношение х0/d из
последнего равенства потоков:
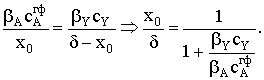
Подставив это соотношение в первое равенство с учетом уравнения
Генри, получим:
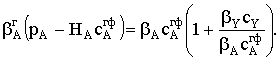
Теперь можно выразить концентрацию реагента, переходящего из
газовой фазы, на поверхности раздела через известные величины:
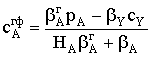
и окончательно определить положение реакционной поверхности:
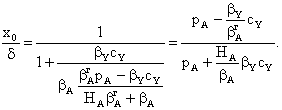
Очевидно, если парциальное давление газообразного реагента намного
больше, чем вторые слагаемые в числителе и знаменателе дроби, отношение х0/d становится равным единице и фронт реакции
перемещается на внутреннюю границу ПЖС (на рисунке вправо). Если же при
увеличении концентрации реагента Y в растворе
величина 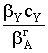 становится равной парциальному давлению
компонента А в газовой фазе, фронт перемещается на границу раздела фаз.
становится равной парциальному давлению
компонента А в газовой фазе, фронт перемещается на границу раздела фаз.
Найдем скорость химического процесса как скорость абсорбции газа
жидкостью в присутствии быстрой химической реакции:

В результате скорость химического процесса будет подчиняться
следующему уравнению:
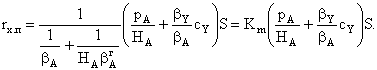
Если сравнить это выражение с уравнением, описывающим скорость
физической абсорбции, то можно видеть, что в обоих случаях сопротивление
массопереносу одинаково (знаменатель дроби, стоящей в начале выражения), но в
присутствии химической реакции значительно увеличивается градиент концентраций
реагента, переходящего из газовой фазы в жидкость. Возрастает и движущая сила
процесса − разность концентраций газообразного реагента на границе
раздела фаз и в объеме жидкости.
Гетерофазная химическая реакция, протекающая в объеме
жидкости вне пограничной пленки
Рассмотрим реакцию газообразного реагента А с растворенным в
жидкости веществом Y:  в случае относительно быстрой массопередачи молекул вещества А из
ядра газового потока к поверхности раздела и сравнимых скоростей химической
реакции в жидкой фазе и переноса компонента А в пограничной пленке жидкости.
При этом массоперенос растворенного компонента происходит быстро по сравнению с
химической реакцией, соответственно, концентрация реагента оказывается
постоянной и по объему жидкости, и в пограничной пленке (рис. 25).
в случае относительно быстрой массопередачи молекул вещества А из
ядра газового потока к поверхности раздела и сравнимых скоростей химической
реакции в жидкой фазе и переноса компонента А в пограничной пленке жидкости.
При этом массоперенос растворенного компонента происходит быстро по сравнению с
химической реакцией, соответственно, концентрация реагента оказывается
постоянной и по объему жидкости, и в пограничной пленке (рис. 25).
|
 Рис. 25. Распределение концентраций
реагентов в случае медленной химической реакции вне ПЖС Рис. 25. Распределение концентраций
реагентов в случае медленной химической реакции вне ПЖС
|
|
Пусть скорость химической реакции отвечает закону первого
порядка по газообразному реагенту, растворенному в жидкости, а порядок по
реагенту Y
будет произвольным:
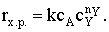
Поток вещества А (и скорость химического процесса) будет равен
произведению скорости химической реакции на объем жидкой фазы V, так как rx.p − скорость поглощения вещества А единицей объема раствора, а
диффузионный поток (и скорость химического процесса) равен скорости поглощения
вещества всем объемом реакционной массы:
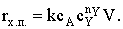
Составим баланс потоков компонента А:
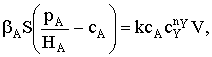
где bА − коэффициент массопередачи
вещества А в жидкой фазе.
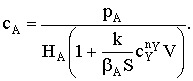
Теперь скорость химического процесса запишется так:
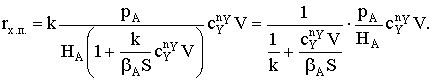
Из приведенного уравнения следует, что в переходной области
протекания химического процесса наблюдаемый порядок реакции даже по тому
реагенту, который не участвует в массообменных процессах, не совпадает с
истинным и может меняться от 0 до nY. Проанализируем изменение наблюдаемого вида скорости процесса в
зависимости от соотношения величин слагаемых в знаменателе дроби:
а) 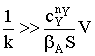 (константа скорости реакции очень мала по
сравнению с величиной, обратной сопротивлению массопереноса −
кинетический режим):
(константа скорости реакции очень мала по
сравнению с величиной, обратной сопротивлению массопереноса −
кинетический режим):
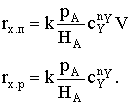
Скорость химического процесса определяется скоростью химической
реакции и обнаруживает первый порядок по компоненту А, количество которого
выражено через парциальное давление в газовой смеси, и истинный порядок nY по реагенту Y;
б) 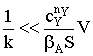 (константа скорости реакции очень велика
по сравнению с величиной, обратной сопротивлению массопереноса −
диффузионный режим):
(константа скорости реакции очень велика
по сравнению с величиной, обратной сопротивлению массопереноса −
диффузионный режим):
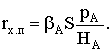
Скорость химического процесса определяется скоростью переноса
вещества А в пограничной пленке, обнаруживает первый порядок по компоненту А и
не зависит от концентрации реагента Y.
Даже если скорость химической реакции не отвечает закону первого
порядка по реагенту А, то в диффузионном режиме наблюдаемый порядок будет
первым, а в кинетическом режиме он будет совпадать с истинным порядком.
Исключением из этого правила будет случай нулевого порядка по газообразному
реагенту. Рассмотрим этот вариант. Пусть уравнение скорости химической реакции
имеет вид: 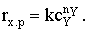
Баланс потоков:
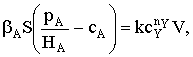
откуда
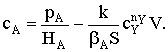
Подставим последнее соотношение в уравнение скорости химического
процесса:
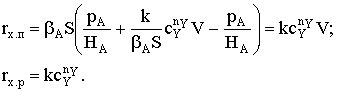
Как видно, в данном случае скорость процесса не зависит от
скорости переноса реагента из газовой фазы. Фактически реакция протекает в
кинетическом режиме независимо от величины коэффициента массоотдачи и
поверхности массообмена.
Гетерофазная химическая реакция, сопровождающаяся
взаимодействием веществ в пограничной пленке
Кратко рассмотрим случай, промежуточный между описанными выше,
т.е. скорость реакции намного меньше, чем скорость мгновенного взаимодействия
на реакционной поверхности внутри пленки, но намного больше, чем скорость
реакции без учета взаимодействия в пленке. В таком случае вещества А и Y взаимодействуют в пограничном слое, но реакция в нем
проходит не полностью. После того, как непрореагировавшая часть вещества А
проходит через ПЖС, реагент равномерно распределяется в объеме, и далее реакция
между растворенными компонентами протекает как гомогенный процесс.
Распределение концентраций представлено на рис. 26. Концентрации жидкого реагента
Y и растворенного газообразного вещества А
в пограничной пленке снижаются нелинейно за счет химической реакции,
протекающей параллельно диффузии реагентов.
|
 Рис. 26. Распределение концентраций в
гетерофазном процессе с учетом реакции в ПЖС Рис. 26. Распределение концентраций в
гетерофазном процессе с учетом реакции в ПЖС
|
|
Скорость диффузии подчиняется закону Фика:

где DA − эффективный коэффициент диффузии вещества А в
жидкой фазе.
Выберем в пограничном слое элементарный объем dV=Sdx (рис.
27),
где S − поверхность контакта фаз; dx − толщина элементарного объема.
|

|
|
Рис. 27. Потоки
вещества А через элементарный объем ПЖС
|
Входящий в элементарный объем поток FA(x) и выходящий из него FA(x+dx) отличаются на величину
элементарного потока dFA. Применив к нему закон Фика, получим:
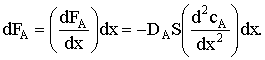
Потоки отличаются на величину химической реакции в элементарном
объеме, т.е.
dFA=−rx.pdV=−rx.pSdx,
откуда

Обозначим a=k/DA,
тогда дифференциальное уравнение второй степени примет вид:
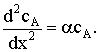
Аналогичное уравнение было решено в разделе, посвященном
гетерогенному катализу. В частности, его общее решение имеет вид:
 .
.
Граничные условия:
х=0 сА=РА/НА=сАгф;
х=d сА=сАж.
Отсюда находим постоянные интегрирования, и после их подстановки в
общее решение и преобразований получаем закон изменения концентрации в
пограничной пленке:
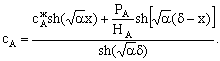
Поток вещества А, поглощенного из газовой фазы, будет равен
диффузионному потоку на границе раздела фаз (при х=0):
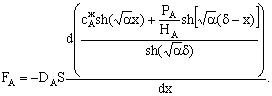
Определим скорость химического процесса как преобразованное выражение
диффузионного потока:
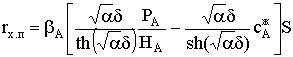
или
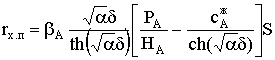 .
.
Если константа скорости химической реакции близка к нулю по
отношению к коэффициенту диффузии, то это выражение превратится в уравнение
физической абсорбции вещества А жидкостью. Следовательно, поведение
газообразного реагента при большом избытке последнего не будет зависеть от вида
кинетического уравнения и вообще от присутствия химической реакции. При условии
быстрого установления равновесия между газообразным и растворенным реагентом
его концентрация в реакционной массе будет величиной постоянной во времени и в
объеме (при интенсивном перемешивании жидкости), а скорости расходования
реагента Y и накопления продуктов будут определяться
скоростью химической реакции. Если газообразный реагент имеется в количестве,
сравнимом с количеством растворенного компонента реакции, то его количество в
любой момент времени может быть рассчитано из соотношений материального
баланса. Таким образом, скорость его расходования также будет определяться
скоростью химической реакции. В газожидкостной системе в целом будет реализован
кинетический режим протекания химического процесса. Заметим, что при указанном
выше соотношении константы скорости и коэффициента диффузии обе последние
модели приводят к одинаковому результату.
С увеличением константы скорости и возрастанием a будет возрастать кривизна линии,
отражающей изменение концентрации реагента в пограничной пленке за счет
химической реакции, т.е. увеличится движущая сила процесса. Это приведет к его
ускорению по сравнению с физической абсорбцией. Математическая модель процесса,
не учитывающая химическую реакцию в ПЖС, также приводит к подобному выводу.
Фактически различие между двумя моделями заключается в том, что криволинейный
профиль концентрации реагента А, по-видимому, более точно описывающий реальную
картину, аппроксимируется прямой линией. Последнее допущение, вероятно,
приемлемо в случае многих реакций, протекающих в течение нескольких минут, и
тем более, часов. Более быстрые процессы требуют более сложных моделей.
Заключение
Рассмотренные примеры построения моделей показывают, что задача
математического моделирования гетерогенных и гетерофазных процессов существенно
усложняется по сравнению с исследованием и моделированием гомогенных реакций.
Представленные зависимости получены при высокой степени упрощения реальной
физической картины химического взаимодействия веществ на фоне массообменных
явлений даже в изотермических условиях. Тем не менее, полученные уравнения
достаточно надежно демонстрируют поведение реальных объектов в широком
интервале изменения физических условий протекания реакций. Часто они могут
использоваться не только как иллюстрация теоретических представлений о
гетерогенных и гетерофазных процессах, но и для установления, например,
протекает ли изучаемая реакция в диффузионной или кинетической области, или для
расчета размеров химических реакторов при условии соблюдения заданного
гидродинамического режима.
Рекомендательный список литературы
1
Бесков
В.С. Общая химическая технология: учебник для вузов/В.С. Бесков − М.: ИКЦ
«Академкнига», 2005. − 452 с.
2
Крылов
О.В. Гетерогенный катализ: учеб. пособие для вузов/О.В. Крылов − М.: ИКЦ
«Академкнига», 2004. − 679 с.
3
Данквертс
П. Газожидкостные реакции./П. Данквертс: пер. с англ.: под. ред. И.А.
Гильденблата. − М.: Химия, 1973. − 296 с.