Технология переработки полимеров
Технология
переработки полимеров
Содержание
Контрольная работа № 1
. Факторы, влияющие на
гибкость макромолекулы
. Радикальная и ионная
полимеризация
. Равновесная и неравновесная
поликонденсация
Контрольная работа № 2
. Деформационно-прочностные
свойства аморфных и кристаллических полимеров
. Природа вынужденной
высокоэластичности
. Интервал температур
плавления в полимерах
Контрольная работа № 3
. Термическое воздействие на
полимер
. Ионизирующие излучения на
полимер
. Реакции, протекающие при
сдвиговых деформациях
. Сшивание эластомеров серой
. Расчетная задача №1
. Расчетная задача №2
Список литературы
полимер эластомер поликонденсация
Контрольная работа № 1
1.Факторы, влияющие на гибкость макромолекулы
Важнейшим свойством макромолекул является
гибкость. Гибкость макромолекул - это способность полимерных цепей изменить
свою конфигурацию, т. е. способность изменять свою форму под влиянием теплового
движения звеньев или движения внешнего поля, в которое полимер помещен. Это
свойство макромолекул связано с внутренним вращением отдельных частей молекулы
относительно друг друга. Различают два понятия гибкости цепи полимера -
термодинамическую и кинетическую гибкость (или жесткость).
Термодинамическая гибкость характеризует
способность цепи изменяться под действием внутреннего теплового движения. О
термодинамической гибкости цепи судят на основании изучения конформации,
которые макромолекулы принимают в равновесном состоянии. Поэтому
термодинамическая гибкость называется равновесной. Она реализуется в очень
разбавленных растворах, в которых цепи находятся в изолированном состоянии, при
отсутствии каких-либо внешних воздействий. Термодинамическая гибкость связана с
размерами макромолекулы, рассчитываемыми для цепи со свободным вращением.
В реальной полимерной цепи вращение в той или
иной степени заторможено. Чем больше заторможено внутреннее вращение, т.е чем
меньше угол вращения, тем больше отношение среднеквадратичных расстояний между
концами реальной цепи. Среднеквадратичные расстояния между концами цепи
полимера определяются экспериментально, исследуя очень разбавленные растворы в
растворителях, которые не оказывают влияние на конформацию макромолекул.
Кинетическая гибкость отражает скорость перехода
макромолекулы в силовом поле из одной конформации в другую и определяется
величиной кинетического сегмента, т.е. той части макромолекулы, которая
отзывается на внешнее воздействие как единое целое. В отличие от
термодинамического сегмента, он определяется температурой и скоростью внешнего
воздействия. С повышением температуры растут кинетическая энергия и гибкость
макромолекулы и уменьшается величина кинетического сегмента. В условиях, когда
время действия силы больше, чем время перехода из одной конформации в другую,
кинетическая гибкость высока, а кинетический сегмент по величине приближается к
термодинамическому сегменту. При быстрой деформации кинетический сегмент близок
к гидродинамической длине макромолекулы, и даже термодинамически гибкая цепь
ведет себя как жесткая. Кинетическая гибкость изолированной макромолекулы
определяется по вязкоупругим свойствам сильно разбавленных растворов с
последующей их экстраполяцией к нулевой коцентрации. Макромолекулы гибкоцепного
аморфного полимера имеют клубкообразную форму как в изолированном виде, так и в
массе. При этом структура полимера не похожа на структуру «молекулярного
войлока», в котором макромолекулы перепутаны хаотически, как считали ранее.
.Радикальная и ионная полимеризация
Существуют два основных метода синтеза полимеров
- цепные (полимеризация) и ступенчатые (поликонденсация) реакции.
Цепные реакции реализуются при раскрытии кратных
(двойных или тройных) связей или циклов молекул мономеров и проходят в три
стадии: инициирование, рост цепи и обрыв цепи. Радикальная и ионная
полимеризации относятся к цепным реакциям.
Свободно-радикальная полимеризация- это
полимеризация при которой свободные радикалы, генерированы тепловым, световым
воздействиями, но они малоэффективны или сопровождают побочными явлениями.
Ионная полимеризация сопровождается координацией
мономера на поверхности катализатора и отличается от радикальной реакции тем,
что: растущие частицы (ионы) более активны; инициаторы каталитические
(позволяют получать стереорегулярные полимеры); суммарная энергия активации
меньше (позволяет снизить температуру); среда растворитель с сольватирующим
действием на ионы.
.Равновесная и неравновесная поликонденсация
Поликонденсацией называется процесс образования
полимеров из би- или полифункциональных мономеров с выделением побочных
низкомолекулярных продуктов (вода, спирты и др.), поэтому элементный состав их
звеньев не соответствует составу мономеров.
Ступенчатый синтез полимеров
включает равновесные (обратимые) и неравновесные (необратимые) процессы.
Особенность равновесных процессов, напримерсинтез полиамидов при нагревании
дикарбоновых кислот с диаминами, - протекание обратных реакций с
низкомолекулярным продуктом, приводящих к распаду полимерных цепей.
Выделяющийся низкомолекулярный продукт (вода из диамина) может реагировать с
амидными группами, и в результате гидролиза образуются исходные структуры или
выделяютсяиз макромолекул низкомолекулярные фрагменты. Синтез
фенолформальдегидных смол сетчатого строения является примером неравновесной
реакции. Выделяющиеся вода и формальдегид не могут вновь реагировать с простыми
эфирными связями или метиленовыми группами между фенольными ядрами
соответственно, и равновесие реакции практически полностью сдвинуто в сторону
образования сетчатого полимера. Кроме того, сама сетчатая структура полимера
способствует сдвигу реакции вправо, так как система становится нерастворимой и
неплавкой. Поэтому ее функциональные группы даже в тех случаях, когда они могут
реагировать с низкомолекулярными компонентами, недоступны для них, и обратная
реакция практически не протекает.
Низкомолекулярный продукт
линейной поликонденсации дигалогенуглеводородов и полисульфида натрия, не
способный к реакции с функциональ-ными группами в макромолекулах
полисульфидного эластомера, является также причиной неравновесности реакции,
даже если система не теряет своей растворимости и плавкости:
nCl-R-Cl+nNa2Sx®-(-R-Sx-)n-+2nNaCl.
Выделение низкомолекулярного
компонента в газообразном состоянии на границе раздела фаз мономеров, не
смешивающихся друг с другом, также обеспечивает неравновесность реакции. При
синтезе полиамидов из дихлорангидридов кислот и диаминов реакция образования
полимера также проходит в узкой области - на границе раздела фаз двух
несмешивающихся растворов мономеров. Образующийся в виде тонкой пленки полиамид
можно непрерывно удалять механическим путем, что позволяет провести реакцию
практически в неравновесных условиях до полного исчерпания мономеров.
Обратимые и необратимые реакции
синтеза полимеров по ступенчатому механизму количественно оценивают константой
равновесия - отношением констант скоростей прямой и обратной реакций:
Кр=Кпрям/Кобратн.
Считают реакцию синтеза
полимера равновесной при Кр не более 102 и неравновесной
при Кр более 103. При промежуточных значениях Кр
равновесность оценивают по условиям проведения реакции: для обратимых реакций-
малые скорости и большая энергия активации (80-170кДж/моль), а необратимых -
высокие скорости и малая энергия активации (8-42кДж/моль).
При поликонденсации на границе раздела фаз
несмешивающихся растворов каждого из мономеров образующуюся пленку полимера
постоянно удаляют с границы до полного завершения реакции. Так получают
полиэфиры, полиамиды, полиуретаны.
Контрольная работа № 2
.Деформационно-прочностные свойства аморфных и
кристаллических полимеров
Структурным стеклованием - это переход полимера
в стеклообразное состояние при охлаждении и означает фиксацию определенной
структуры и определенного ближнего порядка, которые не меняются при дальнейшем
охлаждении. Фиксация структуры делает стеклообразный полимер неравновесным, что
приводит к зависимости Тс от скорости охлаждения. При медленном
охлаждении сегменты успевают перемещаться даже при приближении к Тс,
и требуется сильно охладить полимер для предотвращения вязких перестроек
структуры. Излом на кривой зависимости удельного объема от температуры
(рис.2.15) сместится в область более низких температур. Так, выдерживая
поливинилацетат при каждой температуре в одном опыте 0,02 ч, а в другом - 100
ч, получим значения Тс соответственно 32 и 23оС. В случае
потери способности полимера к высокоэластической или вязкотекучей деформации
при большой скорости действия силы (механическое стеклование), которая
наступает при критерии Деборы D=1,
структура полимера не фиксируется, и тепловое движение сегментов не
прекращается. Однако скорость теплового движения оказывается меньше скорости
действия силы, и заметные деформации не успевают развиваться. Чем больше
скорость действия силы, тем выше Тс при механическом стекловании.
Чем выше скорость охлаждения, тем выше Тс при структурном стекловании.
Это значит, что стеклование является не фазовым (структурным), а релаксационным
переходом. Стеклование определяется не перестройкой надмолекулярной структуры,
а величиной отклика системы на внешнее воздействие. Этим оно отличается от
известных фазовых переходов - кристаллизации или плавления.
К первой группе способов определения температуры
стеклования Тс относятся методы оценки свойств полимера
(термомеханические кривые и температурные зависимости теплоемкости, удельного
объема и показателя преломления). Вторая группа включает методы определения
температурных изменений подвижности сегментов (радиотермолюминесцентный, ЯМР и
диэлектрический). При малых степенях полимеризации Тс растет с
увеличением ММ до определенного предела, когда начинают перемещаться не
молекулы, а сегменты. Размер сегмента не зависит от ММ, поэтому Тс
остается постоянной. Повышается Тс с ростом полярности и плотности
сшивания полимера, а также гидростатического давления, когда оно уменьшает
свободный объем. Пластификаторы понижают Тс пропорционально их
объемной доле. Несмотря на отсутствие достаточного свободного объема,
стеклообразный полимер деформируется без разрушения на сотни процентов под
действием механических напряжений, увеличивающих свободный объем при неизменной
температуре, но не сокращается самопроизвольно после снятия нагрузки.
На первой стадии деформации (рис.1.1) полимер
растягивается упруго за счет увеличения межмолекулярных расстояний или малого
смещения узлов флуктуационной сетки на доли процента или несколько процентов
(область I). На второй стадии
(область II) сегменты
перемещаются и ориентируются в направлении действия силы в вершине какого-либо
микродефекта. На образце возникает шейка, а на кривой - максимум, после чего
напряжение в образце несколько снижается. При дальнейшем растяжении весь
образец постепенно переходит в шейку, и этот процесс сопровождается выделением
тепла. При этом напряжение остается постоянным, на кривой возникает
горизонтальный участок, а величина деформации достигает сотен процентов. Степень
ориентации сегментов в шейке оказывается высокой. При освобождении из зажимов
образец не сократится самопроизвольно, исчезнет только упругая деформация (доли
процентов), однако при нагревании выше Тс он сократится до длины,
близкой к исходной. Таким образом, при растяжении стеклообразного полимера
возникает ориентация сегментов, частичное разворачивание макромолекулярных
клубков, а при нагревании макромолекулы свертываются. Способность
стеклообразных полимеров к большим деформациям называют явлением вынужденной
эластичности, а сами деформации - вынужденно-эластическими. После формирования
шейки процесс переходит в третью стадию (область III),
а величина деформации остается как на первой стадии.

Рис.1.1. Кривая напряжение-деформация для
стеклообразных полимеров (а)
и изменение ее формы под влиянием температуры
(б): T1<T2<T3<T4<T5;
-приT1<Тxp
(хрупкое разрушение); 5-приТ5>Тc
(без образования шейки).
Перемещение сегментов при
вынужденно-эластической деформации полимера происходит под действием
напряжения, однако определенный запас тепловой энергии в нем имеется и при
Т<Тс. С ростом температуры в области ниже Тс запас
тепловой энергии сегментов увеличивается, и требуется меньше внешней
механической энергии для перемещения сегментов, так как предел вынужденной
эластичности (предел текучести σт)
уменьшается (рис.2.16б). При понижении температуры увеличивается предел
вынужденной эластичности, а сама кривая вырождается, становится неполной.
Разрушение образца может произойти даже до достижения предела вынужденной
эластичности (кривая 1), при очень малых деформациях (доли процента), т.е.
полимер ведет себя как хрупкий, не способный к вынужденно-эластическим
деформациям. Для стеклообразного состояния решающим является вклад механической
энергии в релаксационные процессы и важную роль играет способность полимеров
выдерживать длительное воздействие механической нагрузки.
Кривая напряжение-деформация кристаллического
полимера делится также на участки, отражающие стадии процесса растяжения
(рис.1.2). Первая стадия (линейный участок I)
характеризует полностью обратимую деформацию неразрушенной структуры
кристаллитов, которая приводит к увеличению свободного объема полимера и
достигает нескольких или нескольких десятков процентов. Вторая стадия (участок II)
начинается после достижения предела текучести σт,
совпадающего с моментом микроразрушения в наиболее дефектном месте, проходит
через все стадии образования шейки при неизменном напряжении с выделением тепла
за счет внутреннего трения и сопровождается перестройкой кристаллической
структуры. По форме кривой эта стадия напоминает пластическую деформацию
металлов, поэтому напряжение в образце и называют пределом текучести (σт
).
Формирование шейки на образце проходит через стадии рекристаллизации сегментов,
частичного разрушения кристаллитов и пластической деформации кристаллитов и
сферолитов. Третья стадия (участок III)
включает деформацию сформировавшейся шейки вплоть до разрыва образца. Предел
текучести увеличивается с ростом скорости деформации или со снижением
температуры, что может привести к разрушению образца на второй стадии
деформации до завершения формирования шейки (кривая 2), и даже к хрупкому
разрушению при σт
ниже предела прочности (кривая 1). Кривая 4 имеет S-образную
форму и может быть получена и для частично кристаллического полимера
(полиэтилена низкой плотности) при малой скорости деформации, и дляэластомеров,
кристаллизующихся при деформации (вулканизатов НК). В первом случае
кристаллический полимер, подобно аморфному эластомеру (рис.2.16), растягивается
без образования шейки вплоть до разрыва, а во втором - при достижении
определенного удлинения (около 400%) вследствие интенсивной кристаллизации
резко возрастает напряжение. Вблизи точки разрыва напряжение в вулканизате кристаллизующегося
эластомера может в несколько раз (иногда на порядок) превышать напряжение в
некристаллизующемся эластомере с сохранением S-образной
формы кривой.
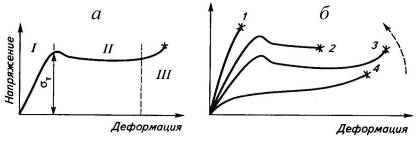
Рис.1.2. Кривая напряжение-деформация
кристаллического полимера (а),
и изменение ее формы под влиянием температуры и
скорости деформации (б): стрелка - направление уменьшения температуры или роста
скорости деформации; звездочки - точки разрыва образцов.
По релаксации напряжения и ползучести
кристаллические полимеры при температурениже Тпл и напряжениях нижеσт,
когда шейка не может образоваться, аналогичны стеклообразным полимерам.
Повышенная доля аморфной части увеличивает при релаксации в условиях Т>Тс
падение напряжения, а при Т>Тпл, когда полимер расплавлен,
напряжение может быстро упасть до нуля. Предельная деформация ползучести так же
больше, чем в аморфных полимерах, а при превышении некоторого предела
напряжения ползучесть становится незатухающей. Этот процесс аналогичен
растяжению без образования шейки. В нагруженном кристаллическом полимере со
временем может произойти разрастание наиболее опасного микродефекта с
образованием шейки, и он может внезапно быстро удлиниться в десятки раз, но не
разрушиться полностью. Полимер при этом упрочняется, и ползучесть прекращается,
однако самопроизвольное удлинение детали или конструкции под нагрузкой
равносильно ее разрушению. Ползучесть в кристаллизующемся эластомере
определяется тем, достигает ли общая эластическая деформация величины,
необходимой для начала кристаллизации, или нет. В первом случае ползучесть
развивается как в некристаллизующемся эластомере, а во втором - кристаллиты
прекращают дальнейшее ее развитие. Циклические деформации, как и в
стеклообразных полимерах, выявляют ряд максимумов потерь в области Т<Тс,
которые дают важную информацию о структуре кристаллических полимеров. Максимум
при Т=Тс показывает долю аморфной части, а широкий размытый максимум
в области Тс<Т<Тпл вблизи Тпл связан с
нарушениями кристаллической решетки и наличием проходных цепей между ламелями.
Высокоэластическая деформация эластомеров,
вынужденно-эластическая деформация стеклообразных полимеров и пластическая
деформация кристаллических полимеров приводят к развертыванию макромолекулярных
клубков и ориентации макромолекул в направлении действия силы. В
ориентированных эластомерах можно охлаждением до температуры ниже Тс
зафиксировать это состояние. Все ориентированные полимеры имеют одно общее
свойство: их прочность и модуль упругости при растяжении в направлении
ориентации намного больше, а в перпендикулярном направлении - намного меньше,
чем у исходного неориентированного полимера.
.Природа вынужденной высокоэластичности
При нагружении стеклообразного полимера,
например полистирола, мгновенно увеличивается длина образца за счет развития
упругой деформации. Далее реализуется замедленная упругость, характеризующая
развитие вынужденно-эластической деформации, которая качественно аналогична
развитию высокоэластической деформации. При этом возможны два случая: либо
деформация перестает расти (затухающая ползучесть), либо она развивается
непрерывно (незатухающая ползучесть) как за счет истинно необратимой, так и за
счет замедленной вынужденно-эластической деформации без образования шейки.
Полимер может применяться как конструкционный материал в том случае, если под
действием заданной нагрузки в нем развивается только затухающая ползучесть,
позволяющая обеспечить относительное постоянство размеров детали в условиях
эксплуатации.
При исследовании стеклообразных полимеров,
например полиметилмет-акрилата, в условиях циклических деформаций обнаружили
релаксационные α-, β- и
γ-переходы,
на которые затрачивается много механической энергии, переходящей в теплоту за
счет внутреннего трения сегментов.При охлаждении до T<Tcсегменты
теряют подвижность, и потери уменьшаются. На повороты эфирных групп (комнатная
температура) идет меньше энергии, а дальнейшее охлаждение «замораживает» их.
При температуре около минус 267оС наблюдается γ-переход,
связанный с вращением метильных подвесок в сложноэфирных группах полимера. Эти
переходы называются вторичными релаксационными переходами и существенно влияют
на механические свойства полимера, особенно его хрупкость и стойкость к ударным
нагрузкам.
Все стеклообразные полимеры более стойки к удару
и менее хрупки по сравнению с силикатными стеклами. Хрупкость - это свойство
стеклообразных полимеров разрушаться при малых деформациях (до 1%). Полимер
будет хрупким, когда время до разрушения много меньше, чем время релаксации,
чтоисключает перегруппировку сегментов под действием силы. Хрупкость оценивают
по величине температуры хрупкости Тхр, которую определяютпо точке
пересечения зависимостей σт
и σр
от
температуры (рис.2.1). Интервал вынужденной эластичности (Тс-Тхр)
является областью безопасного применения пластмасс как конструкционного
материала, а охлаждение полимера ниже Тс может привести к переходу в
хрупкое состояние. Температура хрупкости,как и Тс,зависит от
молекулярной массы. Из-за малой молекулярной массы Тс и Тхролигомеров
совпадают. С ростом молекулярной массы увеличиваются температурные интервалы
вынужденной эластичности (Тс-Тхр) и высокоэластичности (Тт-Тс),
но ухудшается способность полимеров к необратимым деформациям, что выражается в
росте Тт.
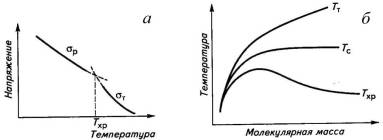
Рис.2.1. Температурные зависимости прочности и
вынужденной эластичности полимера (а) и взаимосвязь между молекулярной массой
полимера и температурами его текучести, стеклования и хрупкости (б).
.Интервал температур плавления в полимерах
Наличие интервала температур плавления, который
составляет градусы и даже десятки градусов. Сначала расплавится часть полимера,
а при дальнейшем нагревании - другая, более упорядоченная его часть, что
связано с неоднородностью структуры и разным размером кристаллитов.
Температура плавления Тпл превышаетна
несколько градусов и даже десятков градусов в зависимости от скорости
нагревания или охлаждения температуру кристаллизации Ткр полимеров.
Кривые охлаждения и нагревания кристаллического полимера образуют петлю,
похожую на петлю гистерезиса при намагничивании и размагничивании железного
сердечника. Несовпадение температур и гистерезис кристаллизации - следствие
релаксационных процессов при создании кристаллической структуры (рис.3.1).
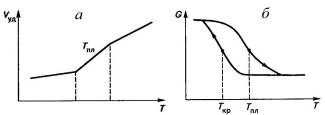
Рис.3.1. Температурная зависимость удельного
объема (а) и модуля упругости (б) полимера в области плавления (нагревание и
охлаждение показаны стрелками).
Протяженность интервала температур, в котором
происходит плавление, зависит от Ткр. Чем выше Ткр и
ближе она к Тпл, тем медленнее идет кристаллизация, меньше возникает
дефектов в кристаллической структуре и однороднее по размерам оказываются
возникшие кристаллиты. Все это обуславливает сужение интервала температур плавления
с ростом Ткр, то есть с уменьшением переохлаждения
кристаллизующегося расплава. Отсюда - необходимость правильного выбора метода
их определения.
Контрольная работа № 3
1.Термическое воздействие на полимер
Скорости реакций радикальной полимеризации и
деполимеризации возрастают с температурой и при некоторой температуре могут
стать равными. Это можно установить по равенству вязкости растворов продуктов
полимеризации и деструкции, что свидетельствует об одинаковой их молекулярной
массе. Установлена важная зависимость характера продуктов деструкции от теплоты
полимеризации. Деполимеризуются в основном до мономера полимеры с четвертичными
атомами углерода в цепи и низкой теплотой полимеризации (поли-α-метилстирол;
39,7 кДж/моль). Если полимер содержит в цепях вторичные и третичные атомы
углерода и имеет высокое значение теплоты полимеризации (выше 60 кДж/моль), то
при термической деструкции мономер практически не образуется, а процесс
заканчивается образованием устойчивых макромолекул пониженной молекулярной
массы.
Цепной радикальный процесс термической
деструкции включает в себя стадии инициирования, роста, передачи и обрыва
реакционной цепи. Радикальные реакции передачи цепи идут за счет отрыва
водорода от макромолекулы полимеров, содержащих атом водорода у третичного
атома углерода или α-метиленовые
группы у двойных связей. Такие полимеры, например полиметилакрилат без двойных
связей на концах макромолекул, почти не дают мономеров при термическом распаде:
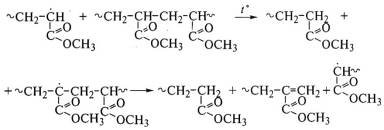
Частный случай цепной деструкции - цепная
деполимеризация путем последовательного отщепления мономерных звеньев от конца
цепей вплоть до полного перехода полимера в мономер. При этом молекулярная
масса полимера последовательно уменьшается. Так идет термическая деструкция
полиметилметакрилата с двойными связями на концах макромолекул - продукта
полимеризации при обрыве цепи диспропорционированием:
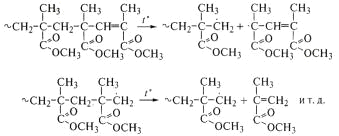
Таким образом, реакцонная способность радикала,
образующегося при разрыве макромолекулы, и легкость отрыва атома водорода
определяют дальнейшее направление деструкции. Из-за невозможности передачи цепи
термодеструкция поли-α-метилстирола
и политетрафторэтилена проходит с высоким выходом их мономеров. Чтобы это
исключить, применяют метод сополимеризации подобных мономеров с мономером,
склонным к реакции передачи цепи, например метилметакрилата с 2% акрилонитрила.
.Действие ионизирующих излучений на полимер
Глубокие химические изменения происходят в
полимерах при действии радиационных излучений - рентгеновских и γ-лучей,
быстрых и медленных нейтронов, быстрых электронов, α-частиц,
протонов и других продуктов ядерных реакций. Энергия этих излучений значительно
больше энергии химических связей в полимерах, поэтому они способны вызвать
разрыв связей в цепи. Под действием излучений высоких энергий происходят
деструкция и сшивание цепей,образование двойных связей, циклизация, отщепление
боковых функциональных групп и разрушение кристаллических структур.
Макромолекулы полимера ионизируются и возбуждаются, распадаясь на два свободных
радикала, что и будет актом деструкции: Р → Р+ + е-;
Р+ + е-→ Р*; Р*→R1+R2,
где Р-полимерная макромолекула, Р+-ионизованная макромолекула, Р*-возбужденная
макромолекула, е- - электрон. Выделяющийся при радиолизе вторичный
электрон с относительно низкой скоростью может не только рекомбинировать с
образовавшимся ионом полимера (реакция в «клетке»), но и реагировать с другими
молекулами (выход из «клетки»), образуя новые ионы. Указанные изменения
происходят очень быстро (10-12с). Время жизни полимерных ионов или
радикалов зависит от подвижности макромолекул и при низких температурах
составляет недели и месяцы.
При облучении линейного полиэтилена выделяется
до 99% водорода, авозникновение разветвленности сопровождается образованием
бутана:
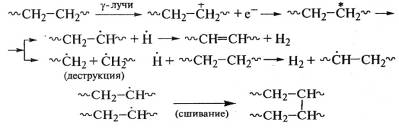
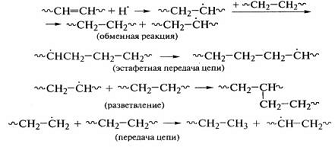 .
.
Реакции деструкции и сшивания протекают
одновременно с преобладанием одной из них в зависимости от химического строения
полимера. В полипропилене могут протекать реакции деструкции:
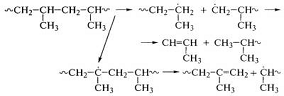 .
.
Деструкции подвергаются α-замещенные
этиленовых углеводородов и другие полимерыс низкими значениями теплоты полимеризации
(полиметил-метакрилат, полиизобутилен, поли-α-метилстирол,
полиэтилентерефталат, бутилкаучук, целлюлоза и др.), а при их пиролизе
образуется большое количество мономера. Радиолиз полиизобутилена, например,
идет по схеме:
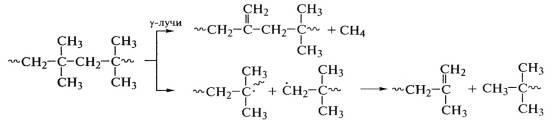 .
.
Быстрая рекомбинация в «клетке» легко
образовавшихся радикалов часто затруднена из-за стерического фактора. Поэтому
они успевают выйти из «клетки», закрепляя акт деструкции макромолекулы
полиметилметакрилата:
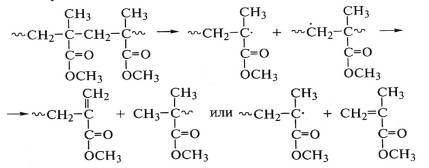 .
.
Полимеры с высокой теплотой полимеризации, малым
выходом мономера при пиролизе, без четвертичных атомов углерода в цепи, при
действии излучений в основном сшиваются (полиэтилен, полистирол,
поливинилацетат, полиизопрен, полибутадиен, полиметилакрилат). Разрывы цепей
при облучении происходят по случайному закону, а число разрывов или сшивок
пропорционально дозе облучения и не зависит от его интенсивности. В присутствии
кислорода при облучении часто развивается процесс окисления полимера. Стойкость
к облучению увеличивается при наличии в структуре ароматических колец, что
связано с значительным рассеянием энергии и называется «эффектом губки». Это
явление используют для защиты полимеров от нежелательного действия излучений
при радиационном старении, а вещества, препятствующие развитию деструктивных
процессов, называются антирадами. Они содержат в структуре ароматические кольца
(например, N.N!-дифенил-n-фенилендиамин,
1,4-нафтохинон, 2-нафтиламин и др.). Сшивание макромолекул при облучении
используют для радиационной вулканизации полиизопрена, полибутадиена,
бутадиен-стирольного и бутадиен-нитрильного сополимеров, полиэтилена,
полиамидов и полиакрилонитрила. Химические связи интенсивно образуются после
перехода из стеклообразного состояния, в котором полимер подвергался
облучению,в высокоэластическое, облегчающее подход сегментов друг к другу на
расстояния, равные длине химической связи между атомами углерода соседних
макромолекул. Сшивание при облучении облегчается и тем, что возникший при
отрыве водорода свободный радикал передает неспаренный электрон вдоль цепи,
отчего увеличивается вероятность его нахождения по соседству с таким же
свободным радикалом другой макромолекулы. Минимальная доза облучения, при
которой образуется единая сетчатая структура полимера, называется дозой
гелеобразования Dги
выражается формулой:
Dг
= 1/αМ(2-β/2),
где α-константа
скорости сшивания; М-ММ исходного полимера; β-константа
деструкции.
Таким образом, основная часть низкомолекулярных
соединений (до 90%), выделяющихся при радиолизе углеводородных полимеров,
составляет водород. При радиолизе политетрафторэтилена выделяется и CF4,
а полиакрилонитрила - HCN,
что следует учитывать при эксплуатации изделий из этих полимеров. Анализ
превращений полимеров при термическом и радиационном воздействиях позволяет
выявить в них существенные различия. При термическом воздействии у полиэтилена
и полиамидов преобладает деструкция, у полиизопрена - деполимеризация, у
полиакрилонитрила - циклизация, а при радиационном воздействии все ониструктурируются.
Ряд полимеров эксплуатируется в атомной промышленности и космосе, где действуют
интенсивные потоки различных радиоактивных излучений, и от поведения их в этих
условиях зависят сроки эксплуатации изделий.
3.Реакции, протекающие при сдвиговых деформациях
При перемешивании воды или бензола в какой-либо
емкости никакие химические реакции не идут. Протекание химических реакций при
воздействии механических напряжений характерно только для макромолекул и
связано с превышением суммарной энергии слабых физических взаимодействий между
их звеньями над энергией химической связи в главной цепи. Величины энергии и
длины различных типов связей позволяют сделать вывод, что суммарная энергия
даже слабых ван-дер-ваальсовых физических связей на протяжении 100-150 звеньев
макромолекулы малополярных полимеров превысит энергию ковалентной С-С-связи в
основной цепи. Очевидно, при сближении отдельных участков макромолекулы в поле
механических напряжений возрастает суммарная энергия межмолекулярного
взаимодействия их друг с другом (рис.3.1). Когда суммарная энергия связей
сблизившихся участков цепи превысит энергию ковалентной С-С-связи, последняя
разорвется.
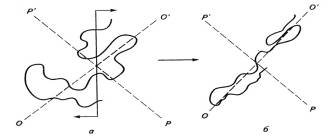
Рис.2.1. Конформация макромолекулярного клубка
исходного аморфного полимера (а) и при разрушении флуктуационной сетки (б) в
поле сдвиговых напряжений (растяжение по оси О-О` и сжатие по оси Р-Р`).
Механодеструкция приводит не только к снижению
ММ полимера до некоторой величины, которая определяется соотношением суммарной
энергии физических межмолекулярных взаимодействий и энергии химической связи в
основной цепи. Выравниваются размеры макромолекул до этой величины, т.е.
изменяется вид кривой молекулярно-массового распределения (рис.3.2). Молекулы
малых размеров участвуют лишь в механическом перемешивании.
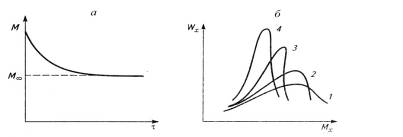
Рис.2.2. Снижение молекулярной массы (а) и
изменение ММР (б) полимера в результате деструкции от времени механической
обработки τ: 1-исходное ММР;
2-4-ММР после пластикации на вальцах при различной продолжительности τ
(τ2> τ3>
τ4).
Процессы механодеструкции протекают при
переработке полимеров на вальцах, в экструдере или резиносмесителе, где в поле
сдвиговых напряжений интенсивно снижаются ММ и вязкость, что облегчает их
последующую переработку. Эти процессы ускоряются реакциями макрорадикалов с
низкомолекулярными акцепторами свободных радикалов, специально вводимых в
полимерную матрицу или присутствующих в ней в виде примесей. Предельно низкое
значение ММ (М∞), достигаемое при механической обработке,
называется пределом механодеструкции. Полимеры сильно различаются по
эффективности механодеструкции, которая тесно связана с плотностью энергии
когезии.
На разрушение физических связей влияет
температура, и от неесильно зависит эффект механодеструкции. При низких
температурах механическим силам труднее преодолевать силы взаимодействия между
макромолекулами полимера из-за отсутствия проскальзывания между ними, а с
повышением температуры эффект скольжения макромолекул возрастает.
Следовательно, механодеструкция имеет отрицательный температурный коэффициент,
т.е. число актов разрывов химических связей в главных цепях растет с понижением
температуры. Наиболее существенные факторы, влияющие на эффективность
механодеструкции полимеров, суммированы.
.Сшивание эластомеров серой
Молекулярная сера является главным вулканизующим
агентом в составе большинства серно-ускорительных групп, ареакции сшивания
ненасыщенных каучуков серой наиболее интересны. Дисульфиды, в качестве
ускорителей применяют и другие вещества. В отсутствие серы они не способны
сшивать эластомеры, нообразуют с серой промежуточные активные полисульфиды,
которые легко распадаются и сшивают каучук:
RSSR+S8→RSS8SR+КаН→КаSxSR+RS8-xH→
КаSx*+RS*+RS8-xH;
КаSx*+RS*+КаН→КаSx*+Ка*+RSH→КаSxКа
(полисульфидная сшивка);
RSH+S8→ RSxH→RS*x-y+S*yH;
КаН+Ка*+S*yH→
КаS*y-1+Ка*+H2S→
КаSy-1Ка
(полисульфиднаясшивка).
Промежуточные гидрополисульфиды могут также
присоединяться к двойным связям полидиенов, химически модифицируя
макромолекулы. Из таких ускорителей наиболее распространены меркаптобензотиазол
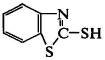
и получаемые на его основе сульфенамидные
производные:
циклогексил-бензотиазил-сульфенамид ,
,
трет-бутил-бензотиазил-сульфенамид ,
,
морфолил-бензотиазил-сульфенамид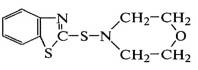 .
.
В присутствии ускорителей диэтилдитиокарбамата
цинка и дифенилгуанидина:
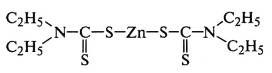
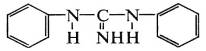
протекают ионные реакции с гетеролитическим
распадом кольца серы по схеме S8→-S-S6-S+.
В присутствии оксидов или жирнокислотных солей металлов реакции распада
ускорителей и получения активных промежуточных продуктов протекают на их
поверхности и имеют топохимический характер, что сказывается на сетчатой
структуре вулканизата:
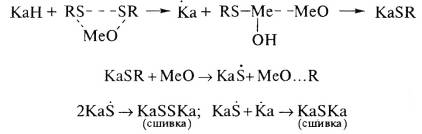
Выделяют три стадии сшивания серы:
1.взаимодействие серы, ускорителей, оксида или
соли цинка (или другого металла) между собой с образованием активных
промежуточных соединений полисульфидной природы -мономерные полисульфиды;
.взаимодействие промежуточных соединений с
макромолекулами эластомера по его реакционным центрам с образованием
реакционноспособных участков (подвесков) на макромолекулах- полимерные
полисульфиды;
.реакции активных центров между собой или с
активными центрами других макромолекул с образованием поперечных связей -
тетрафункциональных узлов сетки различнойсульфидности.
Расчетная задача №1
Рассчитать параметры пространственной сетки
ненаполненого вулканизата натурального каучука методом равновесного напряжения,
если известна зависимость напряжения σ от
деформации а при комнатной температуре
Решение
При одноосном растяжении ненаполненного
вулканизата зависимость напряжения от деформации хорошо описывается уравнением
Муни-Ривлина:
 Или после
преобразованиий:
Или после
преобразованиий:
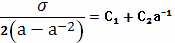
Константы С1 и С2 могут
быть определенны графическим способом. Соответствующие расчеты приведены в
таблице
|
а
|
σ
|
а-1
|
а2
|
а-2
|
а(а-а-2)
|
σ/2(а-а-2)
|
|
1,25
|
10,5
|
0,80
|
1,56
|
0,64
|
1,22
|
|
1,50
|
15,5
|
0,67
|
2,25
|
0,44
|
2,12
|
7,31
|
|
1,75
|
21,5
|
0,57
|
3,06
|
0,33
|
2,84
|
7,57
|
|
2,00
|
25,5
|
0,50
|
4,00
|
0,25
|
3,50
|
7,29
|
|
2,50
|
32,0
|
0,40
|
6,25
|
0,16
|
4,68
|
6,84
|
|
3,00
|
37,5
|
0,33
|
9,00
|
0,11
|
5,78
|
6,49
|
|
3,50
|
42,5
|
0,29
|
12,25
|
0,081
|
6,84
|
6,21
|
|
4,00
|
49,5
|
0,25
|
16,00
|
0,063
|
7,87
|
6,29
|
|
4,50
|
55,0
|
0,22
|
20,25
|
0,049
|
8,90
|
6,17
|
|
5,00
|
64,0
|
0,20
|
25,00
|
0,040
|
9,92
|
6,45
|
Из графика зависимости σ/2(а-а-2)
от а-1 находим константу высоко-эластичности;  =5,50 кгс/см2
=5,50 кгс/см2
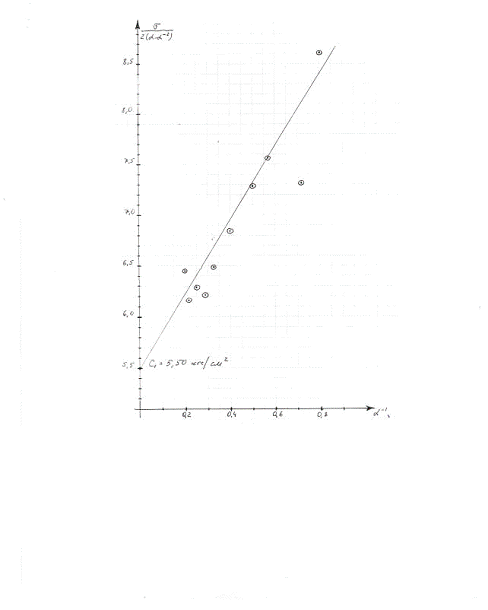
Вычислим параметры пространственной
сетки вулканизата:
В формулах:
 - плотность натурального каучука
();
- плотность натурального каучука
();
R -
универсальная газовая постоянная (84,8 кгс·см/град·моль);
Т - температура, К;
Ответ:
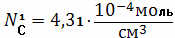
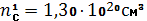
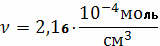
Расчетная задача № 2
Расчитать параметры пространственной
сетки ненаполненого вулканизата на основе известного каучука с известными
значениями равновесной степени набухания (Qр) и
относительного содержания равновесной золь-фракции (а) в известном
растворителе.
Тип каучука СКС-30АРК, тип
растворителя - бензол
Qр =7,0;
а=0,16;
Рк=0,94 г/см3;
Рр=0,879 г/см3;
V0=89,5 см3/моль;
μ=0,37
Решение
Зависимость между величиной
равновесного набухания и густотой сетки вулканизационных узлов описывается
уравнением Флори-Ренера:
где 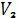 - объемная доля каучука в набухшем
геле
- объемная доля каучука в набухшем
геле
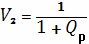
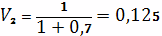
Отсюда Мс=10689 гс/моль
Концентрация активных цепей в
единице объема вулканизата составит:
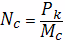
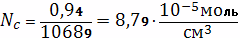
Абсолютная концентрация цепи в
единице объема вулканизата найдем по формуле:
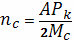
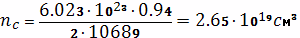
Найдем концентрацию узлов
пространственной сетки в единицу объема вулканизата:
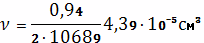
Степень сшивания найдем по формуле
Чарльсби:
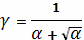
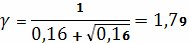
Вычислим молекулярную массу по
формуле:
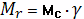
Вычисляем долю активных цепей:

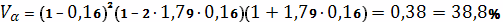
Ответ:
Мс=10689 гс/моль
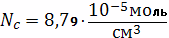
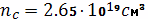
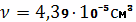
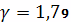
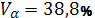
Литература
1. Никитин Ю.Н. Курс лекций по
химии и физики полимеров: Учеб. Пособие. Омск: Изд-во филиала ГОУ ВПО «МГУТУ
им. К.Г. Разумовского» в г. Омске, 2011.-158с.
2. Тагер А.А. Физикохимия
полимеров. М., Химия, 1978.
.Тугов И.И., Кострыкина Г.И. Химия и
физика полимеров. М.: Химия,1989