|
Условия спекания
|
50 об. % ГА-50 об. % ZrO2
|
40 об. % ГА-60 об. % ZrO2
|
|
Тведрость, ГПа
|
Изломо-стойкость, МПа×м1/2
|
Тведрость, ГПа
|
Изломо-стойкость, МПа×м1/2
|
|
1400оC, 2ч
|
2,217±0,03
|
2,217±0,11
|
3,127±0,13
|
3,057±0,33
|
|
1450оC, 1ч
|
2,687±0,03
|
2,047±0,32
|
3,747±0,16
|
3,457±0,53
|
|
1450оC, 2ч
|
3,587±0,08
|
2,647±0, 20
|
4,537±0,28
|
4,377±0,54
|
|
1500оC, 1ч
|
4,217±0,10
|
2,597±0,15
|
5,017±0,23
|
4,257±0,64
|
Интересные результаты получили при изучении влияния примесей
Ag на характеристики керамики ГА [81,82]. Поскольку температура плавления Ag
(962оС) меньше температуры спекания керамики ГА (1100-1300оС),
то спекание ГА происходило с образованием жидкой прослойки. Это обеспечивало
большую однородность образцов; а поскольку частицы Ag являются препятствиями на
пути распространения трещин, то увеличивалась изломо (трещино-) стойкость
материала. Было также обнаружено, что добавки Ag приводят к уменьшению размеров
частиц, плотности, твердости и повышению прочности ГА (рис.1.25 и 1.26).
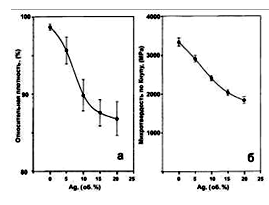
Рис.1.25. Плотность (а) и твердость по Кнупу (б) спеченной
керамики ГА с различным содержанием примеси Ag [81].
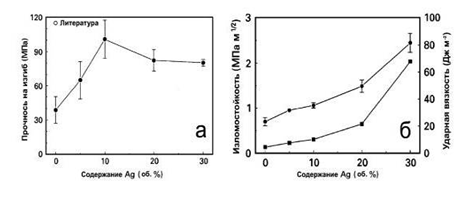
Рис.1.26. Прочность на изгиб (а), изломостойкость и ударная
вязкость (б) спеченной керамики ГА с различным содержанием примеси Ag [82].
Влияние примесей Si на процесс спекания керамики ГА
исследовано в работе [83]. Обнаружено, что примеси Si в концентрации 0-1,6 вес.
% уменьшают рост частиц ГА и замедляют начало спекания по сравнению с керамикой
ГА без примесей (рис.1.27).
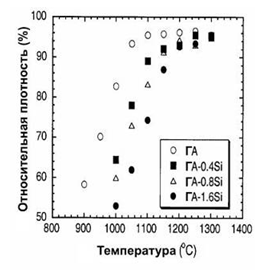
Рис.1.27. Кривые спекания образцов ГА при различных
концентрациях примеси Si [83].
В работе [84] исследовано влияние примесей F на процесс
спекания ГА. Было показано, что наличие примесей F в концентрации до 10 вес. %
замедляет процесс спекания и ухудшает механические свойства керамики ГА. В
работах [85,86] изучали влияние примесей F на функциональные характеристики
керамики ГА в интервале концентраций 0-2 ат. %. Установлено, что добавки
примесей F приводят к увеличению размеров кристаллитов в керамике, уменьшению
плотности, увеличению изломостойкости и твердости (рис.1.28 и 1.29).
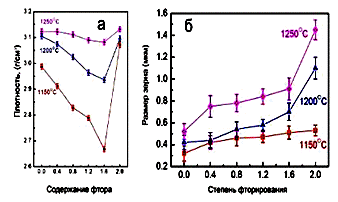
Рис.1.28. Плотность (а) и размер кристаллитов керамики ГА (б)
при различном содержании примеси F [85].
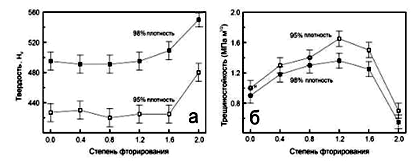
Рис.1.29. Твердость (а) и трещиностойкость керамики ГА (б)
при различном содержании примеси F [86].
Выводы к
разделу 1
1. Процесс кристаллизации ГА в водных растворах
происходит с образованием промежуточной фазы. Это аморфный фосфат кальция
(АФК), который со временем трансформируется в ГА. Согласно первоначальным
представлениям, АФК состоит из сферических кластеров, размеры которых около
9,5Е. Образование ГА объясняется растворением АФК и последующей кристаллизацией
ГА из раствора. Однако в последних работах, выполненных с применением техники
атомного разрешения, утверждается, что кристаллизация ГА происходит путем
структурной реорганизации кластеров внутри АФК и образования в нем дальнего
порядка. Противоречивы также рентгеновские данные об АФК (неопределенность в
положении диффузного максимума). К тому же, не установлен его химический состав:
из АФК на ранних стадиях синтеза образуются различные продукты после
термического отжига.
2. Показано, что наличие примесей СО32-,
Cd2+, Mg2+, Y2+, F-, Sr2+ в
образцах ГА приводит к изменению параметров решетки. Примеси карбонатных групп
при замещениях СО32-®РО43 - приводят
к уменьшению параметра решетки а и возрастанию параметра с, а при
замещениях СО32-®ОН - к
увеличению параметра решетки а и уменьшению параметра с. Примеси
Cd2+, Mg2+, Y2+, F - уменьшают
параметр решетки а и содействуют увеличению размера кристаллитов
при спекании. Примеси Sr2+ увеличивают оба параметра а и с.
Изменение параметров решетки объясняется различием ионных радиусов примесей и
ионов кристаллической решетки ГА. В тоже время практически не изучено влияние
примесей H2O, NH4+, HPO42 - и
NO3-, изначально присутствующих в растворе при
кристаллизации ГА. Имеющиеся сведения о влиянии карбонатных ионов на параметры
кристаллической решетки ГА относятся к отдельным интервалам температур, что не
позволяет представить общую картину изменения параметров после отжига в
интервале температур от комнатной до 1200оС.
3. Спекание образцов ГА при введении примесей Al2O3,
ZrO2, Ag, Si, F приводит к упрочнению образующейся керамики.
Примеси Al2O3 в концентрации до 40 об. %, а также ZrO2
в концентрации до 20-60 об. %, увеличивают плотность, трещиностойкость,
прочность на изгиб, твердость. Примеси Ag в концентрации до 25 об. %
увеличивают прочность и уменьшают плотность, твердость. Примеси Si в
концентрации до 0.16 вес. % и F в концентрации до 10 вес. % понижают
температуру начала спекания. При этом примеси F увеличивают трещиностойкость,
твердость и уменьшают плотность. Считается, что улучшение прочностных свойств
происходит благодаря тому, что примесные частицы создают препятствия для
распространения трещин в спеченных образцах ГА. Вместе с тем, практически
отсутствуют работы, посвященные влиянию примесей, происхождение которых связано
с условиями синтеза ГА. Особый интерес представляют решеточные замещения
комплексными ионами, которые могут разлагаться в процессе отжига и влиять на
вакансионную систему спекающегося тела. Практически отсутствуют работы, в
которых описывается механизм спекания образцов ГА. Таким образом, в литературе
отсутствуют систематические исследования, посвященные зависимости структурных
характеристик образцов фосфатов кальция, содержащих примеси, от условий синтеза
при различных температурах, а также отжигах в интервале температур 20-1200оС.
Для решения этой задачи следовало:
· Исследовать структурные изменения в
процессе кристаллизации ГА;
· Установить природу и происхождение
примесных соединений и решеточных замещений в образцах ГА;
· Изучить структурные изменения в образцах
ГА нестехиометрического и стехиометрического состава после отжиге в интервале
температур 20-1200оС;
· Выяснить влияние примесей на спекание в
образцах ГА.
Раздел 2.
Материалы и методы исследования
2.1 Получение
порошка гидроксилапатита
ГА осаждали из водного раствора (по Ярчо) [87]. Метод основан
на химической реакции:
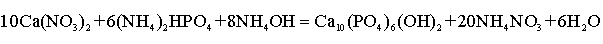 (2.1)
(2.1)
Раствор (NH4) 2HPO4 вводили
капельным образом в раствор Ca (NO3) 2 в течение 40 мин
(скорость 20 мл/мин), тщательно перемешивая реагирующую смесь; при этом pH
смеси поддерживали вблизи значения 11 путем добавления NH4OH. После
прокапывания коллоидный раствор при перемешивании выдерживали в течение суток
для формирования ГА (так называемая стадия созревания).
При выборе масс реагентов исходили из того, что стехиометрическое
атомное отношение Ca/P для ГА равно 1,67. Температура синтеза в большинстве
опытов была равна комнатной (вблизи 20оС). Такие температуры
химических реакций условно называют низкими. Для исследований, посвященных
изучению решеточных изменений в ГА в процессе отжига и расчетов параметров
решетки, синтез проводили при высокой температуре (96оС).
Осадок в реакторе появлялся сразу после смешивания исходных
растворов. Отделение осадка от маточного раствора производили путем
центрифугирования при 2800 об/мин в течение 20 мин. Очистку от примесей
проводили путем промывки осадка в дистиллированной воде. Для этого осадок
диспергировали в дистиллированной воде при помощи мешалки, и образовавшийся
коллоидный раствор центрифугировали. Последующие промывки и центрифугирования
проводили аналогичным образом. Всего проводили 4 цикла очистки. После
завершения процедуры очистки осадок сушился при комнатной температуре в течение
3 суток. Для получения порошка после сушки продукт растирали в ступке пестиком
и просеивали через 100 мкм сито.
.2 Получение
аморфного фосфата кальция
Процедура состояла из нескольких этапов:
а) На первом этапе проводили собственно синтез АФК. Для
этого готовили растворы нитрата кальция и гидрофосфата аммония. Как и прежде,
массовое соотношение компонентов выбирали из такого расчета, чтобы молярное
отношение Са/Р в реакционной смеси составляло 1,67. Синтез проводили путем
быстрого смешивания растворов в реакторе при непрерывном перемешивании
получаемой смеси. Время смешивания 1мин, рН равно 12, температура синтеза 21оС.
б) На втором этапе полученный маточный раствор с осадком
замораживали при - 23оС в морозильной камере. Процедура
замораживания необходима для отделения осадка от раствора.
в) На данном этапе твердый осадок размораживали. Время
размораживания составляло около 4 часов. После полного размораживания воду
аккуратно сливали, остатки воды удаляли фильтровальной бумагой. Температуру
внутри стакана поддерживали вблизи 1оС.
г) После удаления воды влажный осадок вновь
замораживали и выдерживали при - 23оС в морозильной камере в течение
1-3 недель. Это позволило удалить остатки воды путем возгонки льда при низкой
температуре (лиофильная сушка).
д) В результате получали сухой, рассыпчатый порошок.
Для исследования особенностей состава и структуры АФК на
ранних стадиях кристаллизации раствор, полученный на стадии a), был разделен на
несколько частей. Первая порция была помещена в морозильную камеру примерно
через 2 минуты после начала синтеза. Остальные порции замораживались с
интервалом в 2 часа. Последняя порция в этом ряду содержала осадок через 16
часов после начала синтеза. Для сравнения результатов с полученным при
синтезе кристаллическим ГА (подраздел 2.1), часть раствора оставляли в закрытой
емкости для созревания в течение месяца. Порошки из всех частей получали в
соответствии с пунктами а) - д).
.3 Методы
исследования
Рентгеноструктурный
анализ
Исследования проводили с помощью дифрактометра ДРОН-2.0 в
режиме съемки образца на отражение с фокусировкой пучка по методу
Брегга-Брентано. Использовали медное Ka излучение со средней
длиной волны l=1,54 Е. Kb излучение отфильтровывали при помощи Ni фильтра.
Ускоряющее напряжение 30 кВ, ток трубки 10 мА. Использовали щели первичного
пучка 0.5 мм, отраженного - 0,25 мм и щели Соллера с расходимостью 0,25о.
Углы дифракции 2θ измеряли в интервале 4-70о
в зависимости от поставленной задачи. Для определения фазового состава образцов
расчета параметров кристаллической решетки использовали непрерывный режим
съемки; при этом скорости составляли 1/4о/мин и 1/32о/мин,
соответственно. Исследование профилей дифракционных линий проводили в
дискретном режиме регистрации дифракционной картины с шагом 0.01о 2θ и экспозицией 10сек. Для коррекции смещения нуля дифрактометра
образцы снимали с внутренним стандартом, в качестве которого использовали
химически чистый Al (99,99%).
Контрольные дифрактограммы получали в Германии (университеты
Бохума и Дуйсбург-Эссена) на рентгеновском аппарате Bruker AXS D8 Advance
(Siemens, Germany) в режиме съемки образца на отражение (фокусировка пучка по
Бреггу-Брентано). Использовали графитовый монохроматор (медное Ka1 излучение с длиной волны
l=1,5056
Е). Ускоряющее напряжение 40 кВ, ток трубки 30 мА. Диапазон углов дифракции
20-70о. Съемку проводили в непрерывном режиме с шагом 0,0143о и
временем экспозиции 2 сек.
ИK-спектроскопия
Инфракрасные спектры образцов снимали на спектрометре SPECORD
75IR (Germany) в интервале волновых чисел 400-4000 см-1 в режиме
просвечивания. Образцы готовили путем прессования смеси 1 мг исследуемого
порошка и 100 мг порошка KBr (давление 1.6 ГПа в течение 1 мин). В качестве
эталона при записи спектров использовали полистирол с характеристическими
полосами поглощения 1602 и 3028 см-1. Разрешение спектрометра 4 см-1.
Полученные ИК - спектры сравнивали со стандартными [88-92].
Отжиг
образцов
Образцы отжигали в муфельной электропечи СУОЛ-044/12-М2-У4.2
с высокоточным регулятором температуры ВРТ-2, позволяющим контролировать нагрев
образцов и заданную температуру с точностью ±0,5о.
Максимальная температура отжига образцов 1250oC. Скорость нагрева 5о/мин.
Спекание образцов
Из полученных порошков готовили прессовки путем прессования в
стальной пресс-форме под давлением 100 МПа. Они имели циллиндрическую форму
диаметром 7 мм и высотой 4 мм. Прессовки отжигали в воздухе в интервале
температур от 600 до 1200оС в течение часа при каждой заданной
температуре.
Дериватография
Исследования потери массы образцов в процессе нагрева
(термогравиметрия, ТГ) и дифференциальный термический анализ (ДТА) проводили на
дериватографе Q-DERIVATOGRAPH (Hungary). Порошок в количестве 0,5 г помещали в
корундовый тигель. В качестве эталона использовали керамику ГА, спеченную при
1150оС. Образцы нагревали от комнатной температуры до 1000оС
при скорости нагрева 5 о/мин.
Масс-спектрометрия
Десорбцию газов из порошков в процессе нагрева исследовали с
помощью масс-спектрометра МХ-7304. Диапазон массовых чисел 1-200, порог
чувствительности по Ar - 10-9 Па, наибольшее рабочее давление в
камере масс-спектрометра 10-3 Па. Образец помещали в ампулу из
нержавеющей стали, один конец которой был связан с системой напуска СНА-2.
Образцы нагревали со скоростью 5 о/мин.
Растровая электронная микроскопия
Морфологические особенности частиц порошка и сколов керамики
изучали на сканирующем электронном микроскопе LEO 1530 Gemini SEM (Germany).
Для обеспечения стока электронов с поверхности, на образцы напыляли тонкую
пленку золота с палладием. Наибольшее увеличение 200000´.
Рентгеновский фазовый анализ
Качественный рентгеновский фазовый анализ выполняли путем
сравнения дифрактограмм исследуемого образца со стандартными из международной
базы данных рентгеновской дифракции порошковых образцов ICDD [93]. Для
идентификации чистых фаз использовали стандарты: ГА - PDF № 9-432, b-ТКФ - № 9-169, a-ТКФ - №9-348.
Определение параметров решетки
Параметры решетки определяли при помощи компьютерной
программы DICVOL06 [94]. Они также вычислялись по двум дифракционным отражениям
[95] плоскостей (002) и (300):
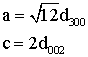 , (2.2)
, (2.2)
где d - соответствующие межплоскостные расстояния. Точность
определения параметров решетки Dа=±0.003
Е, Dс=±0.003 Е.
Определение размеров областей когерентного рассеяния рентгеновских
лучей
Размеры областей когерентного рассеяния рентгеновских лучей (ОКР)
определяли по формуле Селякова-Шеррера [96]:
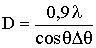 , (2.3)
, (2.3)
где q - угол
дифракции, l - длина волны рентгеновского излучения, Dq - полуширина дифракционной линии на
половине высоты. Значение Dq
находили путем аппроксимации дифракционного профиля функцией Коши:
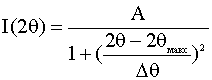 , (2.4)
, (2.4)
где I (2q) -
интенсивность дифракционной линии, А - интенсивность дифракционной линии в
максимуме, 2qмакс - угловое положение максимума дифракционной линии, Dθ - ширина дифракционной линии на половине
высоты. Уширение дифракционной линии, связанное с малостью размеров ОКР (Dq), связано с инструментальным уширением (Dθинстр) и полным уширением дифракционной линии (Dθполн) соотношением:
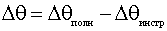 (2.5)
(2.5)
Аппроксимацию профилей дифракционных линий осуществляли при помощи
компьютерной программы Fityk [97]. Инструментальное уширение дифракционной
линии определяли из ширины дифракционной линии на половине высоты для
стандарта. В качестве стандарта использовали керамику ГА, полученную спеканием
при 1300оС в течение часа.
Выводы к
разделу 2
1. Синтез ГА методом осаждения из водных растворов
позволяет получить высокодисперсные порошки стехиометрического и
нестехиометрического состава, которые состоят из наночастиц.
2. Использование масс-спектрометрии газов, выделяющихся
в процессе отжига образцов ГА, а также ИК-спектроскопии образцов ГА позволяет
определить примесные соединения NH4NO3, Ca (NO3)
2, которые присутствуют в образцах ГА в виде отдельных фаз, а также
примесные частицы Н2О, НРО42-, СО32-,
расположенные в кристаллической решетке ГА.
. Термогравиметрия образцов ГА дает возможность
установить температурные интервалы, в которых происходит разложение примесных
соединений и частиц в решетке ГА, а также оценить содержание этих примесей в
образцах ГА.
. Использование методов структурного анализа позволяет
определить параметры решетки, фазовый состав и степень совершенства
кристаллической структуры образцов ГА.
. Растровая и просвечивающая микроскопия образцов ГА
позволяет определить средние размеры частиц и их морфологию.
Раздел 3.
Структурные изменения в процессе кристаллизации гидроксилапатита
Как отмечалось в разделе 1, при формировании ГА из водных
растворов образуется промежуточная фаза - аморфный фосфат кальция (АФК) [6],
которая со временем переходит в ГА. Эта фаза характеризуется диффузным
максимумом на рентгенограммах и полосами поглощения ионов PO43-,
которые не разрешаются на ИК-спектрах [7-29, 98]. Процесс перехода
АФК в ГА занимает несколько часов [15] в зависимости от условий синтеза.
Существует несколько мнений о происхождении, составе и структуре частиц АФК и о
том, каким образом осуществляется переход AФК в ГА. Ранее считалось, что этот
переход идет по механизму растворения - перекристаллизации [6, 99]. Согласно
другим работам [34], АФК рассматривается источником ионов кальция и фосфора. Из
этих ионов АФК на более поздних стадиях синтеза образуется ГА, т.е. это процесс
внутреннего структурного упорядочения в АФК. Наиболее распространенной моделью
строения частиц АФК является модель Познера [25], в соответствии с которой
частицы АФК состоят из сферических кластеров размерами около 1нм. Однако
современные исследования показывают [28-30], что картину дифракции, характерную
для аморфных тел, могут давать кристаллические вещества, размеры частиц в которых
порядка 1 нм. Другими словами, по рентгенограммам невозможно однозначно
определить в каком состоянии находится образец: аморфном или
нанокристаллическом. Таким образом, вопросы формирования ГА из водных
растворов, а также роли АФК на различных стадиях синтеза остаются
невыясненными.
Изучение природы AФК и его перехода в ГА имеет первостепенное
значение для понимания процессов биоминерализации и создания новых
биоматериалов, основанных на AФК.
3.1 Процессы
на начальной стадии кристаллизации ГА
В течение первых 6 часов осаждения на рентгенограммах
наблюдается диффузный максимум в диапазоне углов 29-30.8о. Такой
максимум обычно связывают с присутствием в осадках AФК [6,11,15,100-102].
Определение положения диффузного максимума методом полнопрофильного анализа
[103] показало тенденцию к увеличению угла дифракции во время синтеза в течение
8часов (рис.3.2а). Первые слабые дифракционные максимумы
нанокристаллического апатита регистрируются для образцов, извлеченных из
раствора через 8часов после начала синтеза. Наиболее интенсивные дифракционные
линии ГА от плоскостей (211), (112) и (300) расположены в интервале углов 31-33о
[93]. Однако они не разрешаются из-за наноразмерности и (или) дефектности
кристаллитов [6,104,105]. С увеличением времени синтеза интенсивности
дифракционных максимумов от образцов увеличиваются, а площадь под диффузным
максимумом уменьшается. Эта зависимость хорошо коррелирует с данными,
приведенными в литературе относительно перехода AФК в ГА [6, 11].
На ИК-спектрах образцов для первых 6ч синтеза можно
обнаружить следующие полосы поглощения (рис.3.3). Широкая полоса в интервале
2800-3700 см-1 и более острая - при 1630 см-1 связаны с
присутствием в образцах адсорбированной и кристаллизационной воды [69]. Широкая
полоса поглощения ионов NH4+ в интервале 3100-3190 см-1,
а также острая и интенсивная полоса колебаний NO3 - групп
при 1380 см-1 отражают присутствие в образцах побочного продукта
синтеза NH4NO3. Наличие этого продукта
объясняется тем, что образцы сушились при низкой температуре без
предварительной очистки от примесей [141]. Интенсивные полосы поглощения при
1040 см-1 (υ3) и 580 см-1 (υ4), связанные с колебаниями PO43 - групп, не
разрешались. Как отмечалось, этот факт является характерной особенностью
ИК-спектров AФК [6, 98]. Заметное разрешение полос колебаний PO43
- групп было обнаружено для образцов после 8ч синтеза, и это разрешение
коррелирует с увеличением интенсивности дифракционных отражений (рис.3.1).
Кроме того, были обнаружены два слабых пика (плеча) при 630 и 3570 см-1
от колебаний ОН - групп, интенсивность которых увеличивалась с
течением времени синтеза.
Термогравиметрия образца, полученного через 2 мин после
начала синтеза, показала, что после отжига в интервале от комнатной температуры
до 950єС образцы теряют около 41,5% массы (рис.3.4). Данный интервал температур
нагревания можно разделить на четыре участка: комнатная - 200, 200-300, 300-600
и 600-930єC.
Результаты масс-спектрометрии показали (рис.3.5), что потеря
массы в каждом из указанных участков связана с различными факторами. В процессе
нагревания образца в вакууме фиксировали выделения таких газообразных
продуктов: H2O (наиболее интенсивные пики М/Z=18 и 17)
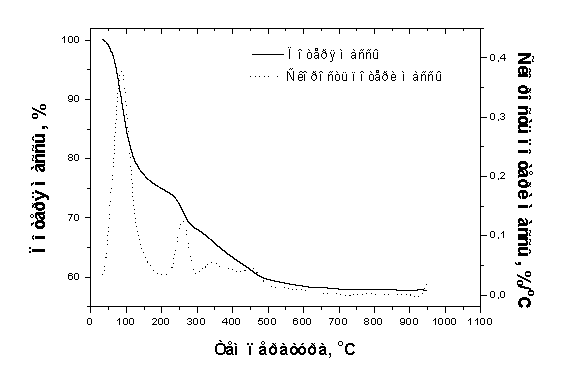
Рис.3.4 Термогравиметрия образца, полученного через 2мин после начала
синтеза.
3
(М/Z = 17, 16), N2 (М/Z = 28, 14), NO (М/Z = 30), O2 (М/Z
= 32, 16), и NO2 (М/Z = 46, 30).
Таким образом, основная потеря массы в интервале от комнатной
температуры до 200єC связана с выделением воды. При этом максимум выделения H2O
как в термогравиметрических, так и в масс-спектрометрических экспериментах
приходится на 80єC. Этот пик связан с выделением воды, адсорбированной на
частицах порошка ГА [106, 141]. Высота этого пика уменьшается с увеличением
времени сушки образцов [134]. Газовыделение в интервале 200-300єC также связано
с молекулами воды, однако расположенными между частицами AФК [107]. В этом
температурном интервале выделяется также аммиак, NH3, который
появляется в результате разложения NH4OH. Это соединение
используется для регулирования рН в маточном растворе. Однако потеря массы в
этом интервале связана не только с выделением воды и небольшого количества
аммиака. На кривой скорости потери массы при 260єC фиксируется интенсивный пик,
связанный с разложением NH4NO3, который образуется во
время синтеза как побочный продукт реакции [141].

Рис.3.5 Выделение газов из образца, полученного через 2мин после
начала синтеза, в процессе отжига.
Потеря массы 7 % в интервале 300-600єC связана с двумя причинами.
Первая обусловлена наличием групп HPO42-, адсорбированных
на частицах AФК, которые разлагаются при нагревании согласно реакции 2HPO42
- = P2O74-+ H2O [108].
Доказательством присутствия ионов HPO42 - в образцах,
извлеченных из раствора через 2 мин - 6 ч после начала синтеза, является слабый
пик на ИК-спектрах при 1150 см-1 (рис.3.3) и масс-спектры выделения
воды (рис.3.5). Второй причиной потери массы в данном интервале температур
является разложение Са (NO3) 2. Эта соль образовалась в
процессе получения образца из оставшейся части ионов Ca2+, которые в
течение первых 2 минут еще не вступили в реакцию синтеза, и групп NO3
- в маточном растворе [141]. Разложение Са (NO3) 2
в интервале 500-600єC (рис.3.5) дает потерю массы около 1,5% (рис.3.4). Далее это
количество постепенно уменьшается с увеличением времени синтеза. Потеря массы в
интервале 600-930єC не превышает 0.5%. Потеря массы в этом интервале связана с
термическим разложением CO32 - групп и (или) их реакциями
с другими примесными ионами [60, 141]. Такая малая потеря массы вместе с очень
слабыми признаками десорбции CO2 (М/Z = 44,12) в масс-спектре
(рис.3.5) показывает, что образцы AФК содержат малое количество ионов CO32
- в течение первых минут синтеза.
Дополнительную информацию о составе образцов дали результаты
рентгенографического исследования образцов после масс-спектрометрии (отжиг
проводили в интервале температур от комнатной до 1000оС в вакууме).
Рентгенограммы всех образцов показали однофазный ГА (рис.3.6). Это подтвердили
также данные энерго-дисперсионного анализа (рис.3.7), согласно которым
отношение Са/Р порошка АФК, полученного из осадка спустя 2 минуты после начала
синтеза составляет около 1,66.
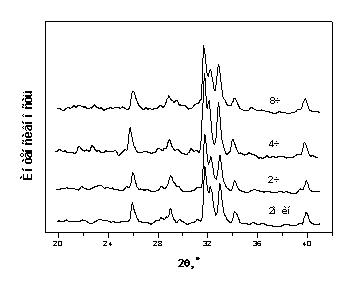
Рис.3.6 Рентгенограммы образцов, полученных после различного
времени синтеза и отжига в вакууме.
Из этих данных можно сделать следующие выводы. Во-первых,
отношение Ca/P в образцах после любого времени синтеза было между 1,5 и 1,67,
т.к. в противном случае сформировалась бы другая кальций-фосфатная фаза (например
β-TКФ, α-TКФ, и т.д. [109]).ФК представляет собой
скопление частиц размерами в несколько сотен нм. Предполагается, что каждая из
таких частиц состоит из сферических нанокластеров*, диаметры которых порядка 1нм, известных
как кластеры Познера [25-27]. Отношение Ca/P для кластера Познера составляет
1,5 [18]. Поскольку отношение Ca/P во всех образцах AФК в данном исследовании
было выше чем 1,5, то избыток кальция, по всей видимости, осадился на
поверхности частиц и образовал слой кальция. Это заключение хорошо согласуется
с недавними исследованиями образцов АФК методами высокоразрешающей электронной
микроскопии [101]. В данной работе был обнаружен тонкий слой CaO на частицах
АФК. Его происхождение, на наш взгляд, связано с окислением слоя кальция на
воздухе. Во-вторых, наблюдаемое на рентгенограммах формирование ГА начинается в
вакууме в районе 600єC. При этой температуре адсорбированная вода уже удалена
из образцов, поскольку парциальное давление воды в вакуумной камере при
температурах отжига образцов более 600єC сравнимо с таковым в начале
масс-спектрометрического анализа (рис.3.5). Следовательно, ОН - ионы,
участвующие в кристаллизации ГА, в основном формируются из воды находящейся
между частицами АФК [107, 110].
В подтверждение решающей роли воды в переходе AФК в ГА приведем
результаты двух контрольных экспериментов. В то время как кристаллизацию AФК в
растворе наблюдали на рентгенограммах образцов через 8 ч после начала синтеза
(рис.3.1), первые признаки кристаллизации (в обоих случаях - в виде
дифракционных линий ГА на рентгенограммах) образцов, высушенных вымораживанием
были обнаружены только через 7 дней после хранения на воздухе. Кроме того,
такие образцы после дополнительной вакуумной дегазации показывали диффузный
максимум в течение 2 месяцев хранения на воздухе (рис.3.8).
3.2 Природа
смещения диффузного максимума
По данным ИК-спектроскопии (рис.3.3), состав исходных частиц
практически не изменяется в течение первых 6ч синтеза. Однако, хотя
рентгенограммы образцов показывают диффузное гало, положение диффузного
максимума смещается в сторону больших углов дифракции (рис.3.2a). Мы
предположили, что этот сдвиг связан с постепенным превращением кластеров,
формирующих частицы AФК, в ячейки ГА. Чем больше ячеек в частице, тем выше
вероятность их соединения, что приводит к образованию нанокристаллов ГА. Таким
образом, преобразование кластеров в ячейки ГА происходит во время первых 6 ч
синтеза и проявляется в виде смещения диффузного максимума в область больших
углов дифракции, а рост нанокристаллов путем объединения ячеек происходит на
дальнейших стадиях синтеза, что отражается в появлении дифракционных максимумов
и увеличении их интенсивностей.
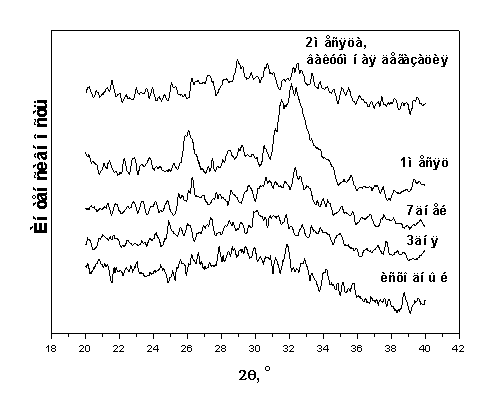
Рис.3.8 Рентгенограммы образцов, высушенных методом вымораживания и
дополнительно обезгаженных в вакууме после хранения в воздухе в течение
различного времени.
Приведенные ниже результаты моделирования картины дифракции таких
частиц свидетельствуют в пользу этой гипотезы. Предположим, что наименьший
нанокристалл (нанокристалл) ГА состоит из 2 гексагональных (или 6 примитивных)
элементарных ячеек, поскольку образование в одну элементарную ячейку не имеет
трансляционной симметрии. Будем считать, что этот нанокристалл является
сферическим, т.е. пренебрежем зависимостью его размера от направления и,
поэтому, имеет диаметр, D=2,3 нм (рис.3.9a). Периоды кристаллической
решетки стехиометрического ГА составляют а = 9,418 Ǻ, с
= 6,884 Ǻ [93]. Согласно формуле Селякова-Шеррера [96], уширение (на
половине высоты) основного максимума ГА при таких размерах нанокристаллов в Cu
Kα
излучении составляет Δq≈3,5є.
Инструментальное уширение дифракционного максимума от плоскости (211) было
измерено для высококристаллического образца, в качестве которого использовали
керамику ГА. Эта величина составила 2% от значения Δq. Поэтому инструментальным уширением
пренебрегли в дальнейшем рассмотрении. Используя межплоскостные расстояния, по
которым можно рассчитать углы дифракции 2θi, и соответствующие относительные интенсивности
дифракционных отражений ГА [93], был вычислен полный дифракционный профиль для
нанокристаллического ГА как сумма профилей отдельных рентгеновских отражений
[103]. Для моделирования профиля дифракционной линии использовали функцию
Pearson VII. Это одна из лучших функций, которые используются для описания
формы профиля дифракционных максимумов в структурном анализе [111]: функция
Pearson VII, а Imax, i, 2θi, ω, m - интенсивность, угловое положение
максимума, ширина на половине высоты и форм-фактор дифракционного максимума,
соответственно.
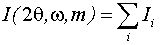 , где
, где
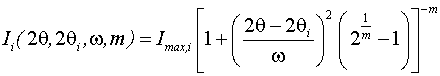 ,
,
Диффузный максимум образца, извлеченного из раствора через 2мин
после начала синтеза, расположен в районе 29,5є (рис.3.10a; Эта кривая -
результат наилучшего приближения к соответствующему экспериментальному профилю
на рис.3.1). В тоже время, расчетный профиль для нанокристаллического ГА с
размерами частиц D показывает основной максимум при 32,2є (рис.3.10б).
Если образец представляет собой смесь AФК и кристаллического ГА, то
рентгенограмма такого образца представляет собой наложение индивидуальных
рентгеновских профилей этих фаз. В случае смеси из исходного AФК, т.е.
полученного через 2 мин после начала синтеза, и 60 вес. % нанокристаллов ГА
размерами 2,3 нм, наложение рентгенограмм дает кривую на рис.3.10в.
Видно, что основной максимум, немного смещается в область больших углов
дифракции и появляется слабый вторичный максимум в интервале 47-50є.
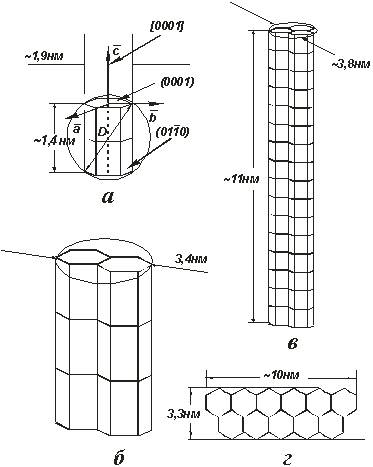
Рис.3.9 Схематическое представление модели нанокристалла ГА (a)
и объединений нанокристаллов, которые показывают процесс образования
кристаллов игольчатой (б, в) и пластинчатой (г) форм, удлиненных
в направлении [0001]. г - направление [0001] перпендикулярно рисунку.
Присутствие вторичного максимума в этом интервале наблюдали в
нескольких работах, посвященных исследованию AФК [6,28,105]. С увеличением
концентрации ГA в AФК основной максимум смещается в область больших углов
дифракции, а вторичный максимум становится более интенсивным.
Наконец, для 100% нанокристаллического ГА, положение максимума
соответствует 32,2о (рис.3.10б). Таким образом, угловое
смещение тем выше, чем выше отношение ГA/AФК, что хорошо согласуется с
экспериментальными данными об изменении положения диффузного максимума для
образцов, извлеченных из раствора после различного времени синтеза (рис.3.2a,
б).
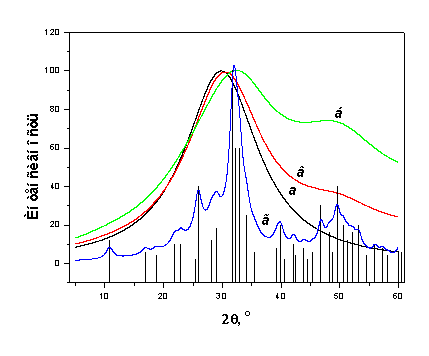
Рис.3.10. Дифракционные профили от АФК (a), ГА с размерами
частиц 2.3 нм (б), смеси 60вес. % ГА/40вес. % AФК (в) и ГА с
размерами частиц 10нм (г).
В работе [102] с использованием высокоразрешающей, просвечивающей
электронной микроскопии было показано, что образцы, показывающие на
рентгенограммах диффузный максимум, состоят из нанокристаллов ГА размерами
около 10-30 нм и некоторого количества истинно аморфного вещества. Для
нанокристалла размером 10 нм, уширение максимумов, уменьшается более чем 5 раз,
и моделируемый дифракционный профиль имеет вид, показанный на рис.3.10г
(при этом существенное возрастание интенсивности отражений [112] не
рассматривается). Этот профиль подобен тем, которые обычно появлялся "на”
диффузном максимуме. Например, они фиксируются на дифрактограммах образцов
после 8-12 ч синтеза (рис.3.1), что обычно рассматривают как начало
кристаллизации AФК [6,10,11,15]. Следовательно, появление нанокристаллов ГА и
увеличение их количества приводит к смещению главного максимума, а последующий
рост нанокристаллов ГА приводит к появлению рентгенограммы, характерной для
нанокристаллического гидроксилапатита. Появление нанокристаллов на начальном
этапе синтеза происходит быстрее, чем их рост.
3.3 О
механизме перехода и кристаллизации AФК в ГА
На основании результатов, изложенных выше, предлагается схема
перехода AФК в ГА в условиях быстрого осаждения. Исходя из данных пункта 3.1,
структуру частиц в образцах, дающих на рентгенограммах аморфное гало на
начальном этапе синтеза, можно представить следующим образом (рис.3.11a).
Частица состоит из сферических кластеров, разделенных захваченными молекулами
воды. Частица покрыта слоем кальция. Этот слой включает небольшое количество
ионов HPO42 - и практически не содержит ионов CO32-,
хотя специальные меры для уменьшения концентрации CO32 - (HCO3-)
в растворе не применялись. В высушенном образце слой кальция в некоторой
степени гидратирован в зависимости от условий сушки. Кроме того, на этом слое
адсорбировано небольшое количество Ca (NO3) 2 и намного
большее NH4NO3, по всей вероятности, в виде молекул.
Поскольку отношение Ca/P во внутренних кластерах ниже 1,67 (кальций
присутствует также в виде поверхностного слоя на частицах), дополнительные
количества ионов кальция и гидроксильных групп должны проникнуть в кластеры,
чтобы образовались ячейки ГА. Ионы Са2+ и ОН - могут
диффундировать из поверхностного слоя частиц АФК и окружающего раствора,
соответственно. Кроме того, ионы ОН - могут образоваться вследствие
диссоциации воды, захваченной между кластерами. Как известно, оба процесса,
диффузия и диссоциация, сильно зависят от температуры.
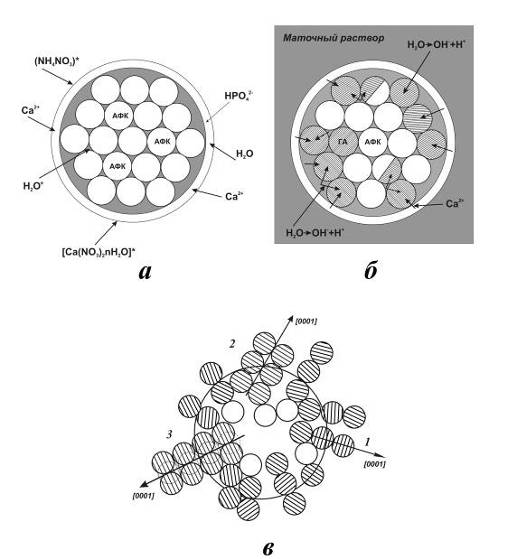
Рис.3.11. Схематическое представление частицы AФК (a), начало
(б), развитие (в) кристаллизации. H2O - адсорбированная и гидратная
вода; * - соли, сформировавшиеся в высушенной частице.
Весьма вероятно, что этим объясняется значительное ускорение
перехода АФК в ГА при увеличении температуры синтеза [12, 113]. Поскольку
диффузионный поток ионов кальция идет из поверхности в объем, то первые ячейки
ГА формируются в поверхностных слоях частиц АФК (рис.3.11б).
По-видимому, ячейка ГА является элементарной по размеру, потому что такое
образование является самым устойчивым, т.е. наименее растворимым среди других
возможных начальных конфигураций фосфатов кальция при данных условиях синтеза
[27, 109, 114]. Чем больше образуется ячеек ГА, тем выше вероятность их
соединения плоскостями (0001) по механизму автоэпитаксии (рис.3.11в
нанокристалл 1 и рис.3.9a). Соединение другими плоскостями также
возможно, но менее вероятно, так как эти плоскости более рыхлые (например
плоскости  ; нанокристалл 2 на рис.3.11в и
рис.3.9б). Вследствие преимущественного соединения ячеек ГА их базисными
плоскостями, первоначально округлые нанокристаллы постепенно трансформируются в
кристаллиты ГА, удлиненные в направлении [0001] (нанокристалл 3 на рис.3.11в
и рис.3.9в).
; нанокристалл 2 на рис.3.11в и
рис.3.9б). Вследствие преимущественного соединения ячеек ГА их базисными
плоскостями, первоначально округлые нанокристаллы постепенно трансформируются в
кристаллиты ГА, удлиненные в направлении [0001] (нанокристалл 3 на рис.3.11в
и рис.3.9в).
Эта картина хорошо согласуется с экспериментальными данными.
Формирование нанокристаллов ГА, имеющих размеры в несколько элементарных ячеек,
и увеличение их числа и размера с увеличением времени синтеза приводит к сдвигу
главного максимума и, далее, к появлению острых максимумов на рентгенограммах.
Исследования при помощи высокоразрешающей электронной микроскопии
[16,29,95,115,116] показали, что нанокристаллы ГА формируют оболочку из
игольчатых нанокристаллов в поверхностных слоях исходной частицы AФК в конце
перехода АФК в ГА. Кроме того, форм-фактор больших (вызревших) кристаллитов ГА
субмикронных размеров находится в интервале 5-10, при этом они имеют форму
пластинок [28,102]. Нанокристаллы ГА были обнаружены в округлых
рентгено-аморфных частицах размерами 10-50нм в диаметре [29,95].
Следовательно, даже при самом низком форм-факторе, нанокристал
имеет размеры 25нм в длину, 5нм в ширину и при пластинчатой геометрии - толщину
в 1 или 2 элементарных ячеек.
Эта оценка хорошо согласуется с предложенной моделью, согласно
которой можно получить кристаллиты ГА различных размеров и форм путем
соединения ячеек ГА, включая кристаллиты в виде пластинок толщиной в несколько
размеров ячейки (рис.3.9г), которые характерны для минералов апатита
[117].
Выводы к
разделу 3
1. Установлены особенности строения
дифракционно-аморфного вещества, известного как аморфный фосфат кальция, АФК,
на начальной стадии процесса кристаллизации ГА из раствора. Показано, что это
вещество, считавшееся ранее аморфным, представляет собой смесь аморфного
вещества и нанокристаллического ГА. Нанокластеры, которые составляют частицы
АФК непосредственно после формирования, в процессе синтеза ГА переходят в
элементарные ячейки ГА, затем - в нанокристаллы ГА размером в несколько
параметров решетки. Содержание нанокристаллов ГА в частицах АФК увеличивается с
течением синтеза.
2. Предложены схема перехода АФК→ГА и механизм
роста ГА путем автоэпитаксии нанокристаллов ГА, т.е. объединением элементарных
ячеек ГА в определенных кристаллографических направлениях. Они качественно
объясняют как результаты данной работы, так и последние сведения о формах
нанокристаллов ГА и их образований, полученные с помощью техники атомного
разрешения.
. Показано, что ограничение или, тем более,
блокирование диффузии кальция и (или) гидроксильных групп в нанокластеры путем
уменьшения содержания молекул воды в гидратированном слое тормозит их конверсию
в ГА и позволяет сохранять кальций-фосфатный осадок (или порошок) в аморфном
состоянии длительное время. Этот результат свидетельствует в пользу
предложенной схемы фазового перехода.
Основные результаты исследований, представленные в разделе 3,
опубликованы в [141-144].
Раздел 4.
Изменения морфологии, фазового состава и структуры в высокодисперсных порошках
га в процессе термического отжига
Многочисленные применения ГА в виде порошков, гранул, плотных
и пористых керамик требуют предварительной термообработки и (или) отжига
(спекания) образцов при определенной температуре. При этом функциональные
свойства конечных изделий во многом определяются структурой и свойствами
исходного порошка ГА, а также теми процессами, которые происходят при его
нагреве. Это особенно касается порошков ГА, содержащих примеси в виде отдельных
фаз, соединений или примесных частиц (ионов) в кристаллической решетке. Поэтому
одной из наиболее важных задач является исследование изменений структуры и
примесного состава ГА, происходящих в процессе нагрева и отжига.
В данном разделе изложены результаты исследования морфологии,
фазового состава и структуры ГА в процессе отжига порошковых образцов, причем
основное внимание уделяется вопросам изменения структурных параметров,
связанных с наличием и термическими реакциями решеточных примесей. Несмотря на
значительное количество работ [42,118-126], имеющиеся сведения по этой проблеме
неоднозначны и противоречивы. Они рассматриваются далее при обсуждении
полученных результатов.
Проблему исследовали для порошков ГА, синтезированного при
низкой (комнатной) и высокой (96оС) температурах.
.1
Морфологические, фазовые и структурные изменения в нанокристаллических порошках
ГА в процессе отжига
Процедура синтеза и получения порошков изложена в разделе 2.
Порошки отжигали в интервале температур от комнатной до 1200оС в
воздушной атмосфере в течение часа.
На рис.4.1.1 приведены дифрактограммы порошков ГА,
отжигавшихся при различных температурах в течение часа. Как отмечалось ранее
(раздел 3), исходный порошок ГА дает дифрактограмму, характерную для
нанокристаллического вещества. Дифракционные линии значительно уширены. В связи
с перекрытием линий из-за уширения в интервале углов дифракции 30-34о,
где расположены наиболее интенсивные отражения ГА, затруднительно определить
положение пиков с необходимой степенью точности. Это обстоятельство осложняет
проведение структурных исследований. Такая картина наблюдается до температур
около 800оС (рис.4.1.1), при которых активизируются диффузионные
процессы, и начинается рост частиц [127]. Эти процессы сопровождаются
образованием совершенных кристалликов ГА (или их совершенствованием), о чем
свидетельствует уменьшение ширины дифракционных линий, их лучшее разрешение.
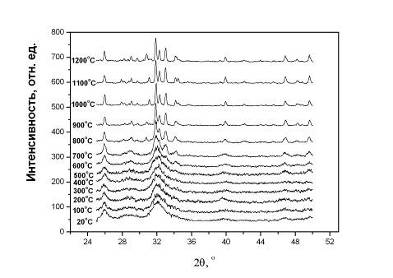
Рис.4.1.1 Дифрактограммы порошков ГА после отжига при
различных температурах.
Интенсивные рекристаллизационные процессы практически
заканчиваются при температурах 1100-1300оС, и, как будет показано
далее (раздел 5), наиболее совершенный по структуре и функциональным
характеристикам ГА может быть получен в результате отжига вблизи 1100оС.
Тем не менее, дифракционная линия в районе 25.9о
от плоскости (002) проявляется отчетливо на всех рентгенограммах. Кроме того,
для порошков из некоторых хорошо вызревших осадков, удается выделить положение
наиболее интенсивного отражения (211) кристаллического ГА. Эти данные позволили
оценить параметры решетки исходного порошка как а=9,38±0,01Е, с=6,853±0,007Е.
Близкие значения параметров получены путем электронографирования образцов. В
силу специфики метода [96], большинство линий на электронограммах хорошо
разрешается (рис.4.1.2). Используя метод съемки со стандартом (Al), были
найдены значения параметров: а=9,39±0,02Е, с=6,98±0,01Е. Таким образом,
постоянные решетки для исходного порошка отличаются от стехиометрических
значений ГА (а=9,418Е, с=6,884Е, PDF № 9-432 [93]) и больше
соответствуют параметрам карбонат-замещенного ГА (а=9,386 Е, с=6,901 Е
[118]). Действительно, ИК-спектры исходного порошка (рис.4.1.3) обнаруживают
присутствие карбонатных групп в структуре ГА по характерному пику при 875 см-1,
а также - по широкой полосе в интервале 1380-1500 см-1 [54]. Увеличение
температуры отжига в интервале 800-1200оС приводит не только к
увеличению кристалличности, но и к процессам десорбции продуктов разложения
карбонатных групп из кристаллической решетки ГА. На рис.4.1.4 показана
температурная зависимость размеров областей когерентного рассеяния
рентгеновских лучей (ОКР). Как видно, размеры ОКР практически не меняются
вплоть до 800оС, когда активизируются диффузионные процессы. Размеры
ОКР исходного порошка при низких температурах отжига лежат в тех же пределах,
что и размеры первичных частиц ГА. Частицы порошка имеют широкий интервал
субмикронных размеров, что хорошо видно на рис.4.1.5 (образцы после отжига при
300оС или 500оС). Однако каждая из таких частиц
образована из первичных наночастиц, размер которых находится в интервале 10-50
нм и составляет обычно 20 нм (см., например, строение частицы А на рис.4.1.5).
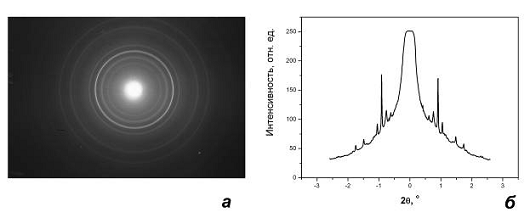
Рис.4.1.2 Электронограмма исходного порошка, нанесенного на
стандартную пленку Al (а), и результат ее микрофотометрирования (б); наиболее
интенсивные линии соответствуют отражениям от Al.
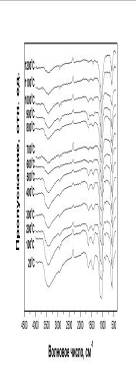
Рис.4.1.3 ИК - спектры порошка ГА после отжига при различных
температурах.
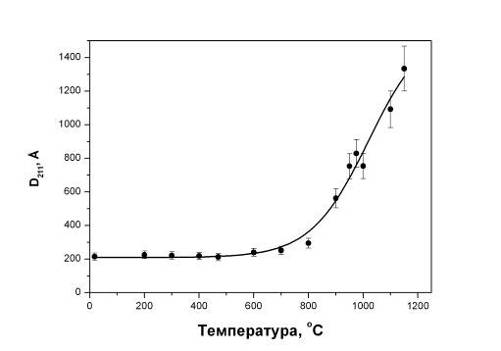
Рис.4.1.4 Зависимость размеров ОКР в порошках от температуры
отжига.
При 800оС начинают формироваться различия между
отдельными частицами, а при 900оС фиксируются отдельные
(нередко-ограненные) зерна, характерные для поликристаллического образца с их
последующим ростом при более высоких температурах. Увеличение степени
кристалличности порошка при высоких температурах позволяет определить параметры
решетки (рис.4.1.6). Они отличаются от стехиометрических значений
(горизонтальные прямые) наличием примесей в кристаллической решетке. Это
приводит к образованию нестехиометрии в спекающихся исходных порошках.
Кроме результатов масс-спектрометрии (раздел 3) и ИК-спектров
(раздел 3; рис.4.1.3), об этом свидетельствуют дифрактограммы порошков после
отжига при температурах выше 700оС: на них фиксируется образование a - и b-Ca3 (PO4) 2 (рис.4.1.1, 4.1.7),
что характерно для ГА с отношением 1,5<Ca/P<1,67. Данный результат
представляет практический интерес, поскольку указывает на пути получения
полифазных керамик фосфата кальция с заданным фазовым составом из порошков
низкотемпературного синтеза.
4.2
Термические структурные изменения и примесные реакции в порошках ГА,
синтезированных при высокой температуре
Как отмечалось в разделах 3 и 4, наноразмеры кристалликов
исходного порошка ГА, получаенного при низкой температуре синтеза, были
основной причиной плохого разрешения дифракционных максимумов, что затрудняло
проведение структурных исследований вплоть до 800оС. Однако вблизи
этой и при более высоких температурах отжига порошок ГА постепенно теряет
свойства, характерные для наноразмерных объектов. Кроме того, такой порошок
часто является нестехиометрическим по составу, что приводит к образованию
вторичных фаз при высоких температурах.
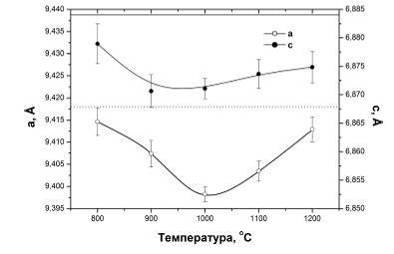
Рис.4.1.6. Параметры решетки нестехиометрического ГА после
отжига при различных температурах.
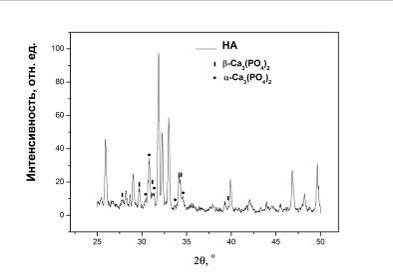
Рис.4.1.7 Качественный рентгеновский фазовый анализ порошка
после отжига при 1200оС.
Однако многие применения в промышленности, медицине и
биологии требуют нанопорошков ГА. Для выяснения возможных путей улучшения
стехиометрии и кристалличности частиц при одновременном сохранении их
наноразмерности структурное и примесное состояния порошков, полученных при
высокотемпературном синтезе (96оС), исследовали в широком интервале
температур 20-1100оС. Процедура синтеза и получение порошка подробно
описана в разделе 2.
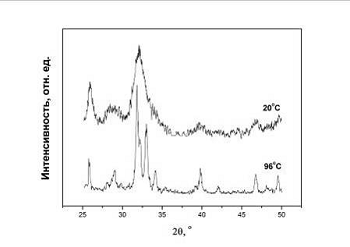
Рис.4.2.1 Дифрактограммы порошков ГА, синтезированного при 20
и 96оС.
На рис.4.2.1 приведены дифрактограммы порошков ГА,
полученного при 20 и 96оС. В отличие от синтеза при низкой
температуре, дифракционные линии порошка ГА, синтезированного при 96оС,
вблизи 31,8 (211), 32,2 (112) и 32,9о (300) хорошо разрешаются. Это
обстоятельство говорит о более высокой степени кристалличности порошков ГА из
высокотемпературного синтеза, что позволяет провести структурные исследования
(рис.4.2.2). С этой целью была получена серия образцов путем отжига порошков ГА
от комнатной температуры до 1100оС (время отжига при определенной
температуре 1 ч). По дифрактограммам (рис.4.2.3) были определены параметры
решетки ГА в соответствии с соотношением (2.2). Средняя абсолютная погрешность
определения параметров решетки Dа, Dс, составляла ±0.003Е. На
рис.4.2.4 показана зависимость параметров решетки ГА от температуры отжига. Ход
зависимостей имеет немонотонный характер, причем амплитуда изменения параметра
решетки а почти в полтора раза больше чем у параметра с.
Полученную зависимость можно разделить на несколько температурных интервалов:
комнатная температура - 500оС, 500-600оС, 600-700оС,
700-800оС, 800-1050оС и 1050-1100оС.
Рассмотрим каждый из них.
Первый участок кривой характеризуется уменьшением параметров
решетки a и с от значений 9,433±0,003 Е и 6,904±0,003 Е при
комнатной температуре до 9,407±0,003 Е и 6,898±0,003 Е при 200оС.
При этом более значительным является изменение параметра a. Исходные
значения параметров существенно отличаются от соответствующих значений для
стехиометрического ГА (a=9,418 Е, c=6,884 Е) [93]. ИК-спектры исходного
порошка (рис.4.2.5) обнаруживают наличие примесей, из которых лучше всего
проявляются карбонатные группы CO32-. Следовательно этот
факт, не позволяет объяснить увеличение параметра решетки а относительно
стехиометрического значения. С другой стороны, маточный раствор, в котором идет
кристаллизация ГА, содержит ионы NH4+, а также H3O+.
Масс-спектры газообразных продуктов, выделяющихся из порошков ГА в процессе
отжига (рис.4.2.6), показывают, что наиболее интенсивные пики десорбции при 120оС
принадлежат массовым числам (массам) 17, 18, которые могут давать NH3,
ОН - и NH4+, H2O
соответственно. Хорошо известно, что замещение карбонатными группами ОН - групп
в кристаллической решетке ГА ведет к увеличению параметра решетки а
[42].

Рис.4.2.3 Дифрактограммы порошков ГА после отжига при
различных температурах.
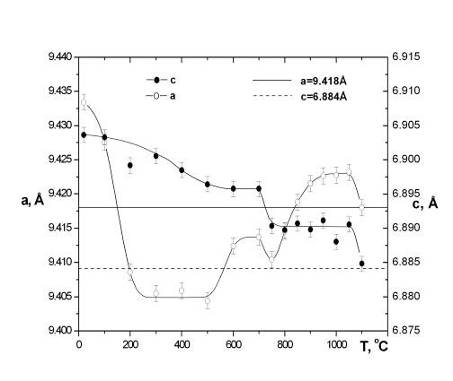
Рис.4.2.4 Параметры решетки ГА после отжига при различных
температурах.
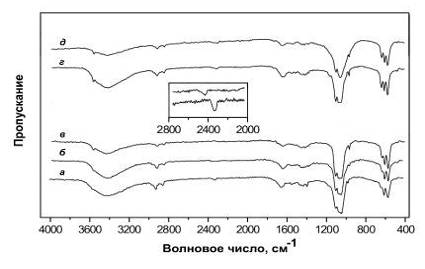
Рис.4.2.5 ИК-спектры порошков ГА при различных температурах: a
- комнатная температура; отжиг при б - 400, в - 600, г -
800, д - 1100оС. На вставке показаны фрагменты вблизи полос
поглощения СО2 в образцах, отжигавшихся при: нижний - 500, верхний -
800оС. Масштаб фрагментов увеличен.
Данные масс-спектрометрии хорошо коррелируют с результатами
термогравиметрии (рис.4.2.7). Скорость потери массы (пунктирная кривая) имеет
максимум при 120oC, причем ход кривой почти совпадает с таковым для
массового числа 18. Небольшие различия обусловлены тем, что потеря массы
является интегральным эффектом, который определяется выделением всех
газообразных продуктов, образовавшихся в результате нагрева образцов.
Возможность ионного замещения NH4+®Ca2+
рассматривалась ранее в карбонат-замещенных апатитах в присутствии NH4+
[119]. Образование ионов гидроксония H3O+ возможно в
соответствии с реакцией [129]:
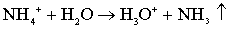 . (5.1)
. (5.1)
Ионный радиус NH4+ (1,43Е) больше ионного радиуса
H3O+ (1,09Е), который в свою очередь больше ионного
радиуса Ca2+ (0,99Е) [42]. Оба типа ионных замещений возможны и
приводят к увеличению параметра решетки а.

Рис.4.2.6 Масс-спектры газообразных продуктов, выделяющихся из
порошков ГА в процессе отжига.
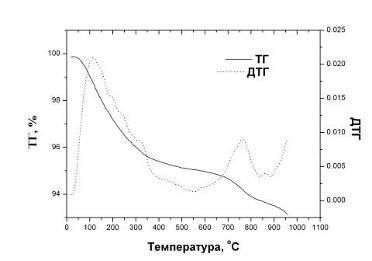
Рис.4.2.7 Потеря массы (ТГ) и скорость потери массы (ДТГ) порошков
ГА.
В процессе нагрева образцов ГА при температурах, превышающих 100оС,
оба иона разлагаются в соответствии с реакциями:
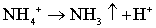 , (5.2)
, (5.2)
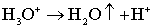 . (5.3)
. (5.3)
При этом на масс-спектрах должны фиксироваться выделения летучих продуктов
NH3 и H2O в интервале температур от комнатной до 200оС,
когда
параметры решетки уменьшаются. Однако появление 17 массы в
масс-спектрах может быть связано не только с выделением летучего NH3,
но с образованием ионов ОН-, происхождение которых связано с
процессом ионизации молекул воды в масс-спектрометре:
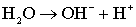 ; (5.4)
; (5.4)
при этом отношение массовых чисел 17/18 лежит в пределах 0,23-0,32
[130]. На рис.4.2.8 показано отношение масс 17/18 в зависимости от температуры
отжига. Как видно, это отношение является постоянным и приблизительно равным
0,3, что близко к значениям, приведенным в литературе. Таким образом, появление
17 массы является результатом образования ОН - групп, а не выделения
аммиака. Следовательно, уменьшение параметра решетки а является
результатом разложения ионов H3O+, так называемой
"решеточной воды”, занимающих места ионов Ca2+ в
кристаллической решетке ГА.
Вместе с тем необходимо отметить, что возможность существования
иона гидроксония вне жидкости не является общепризнанной [129, 131].
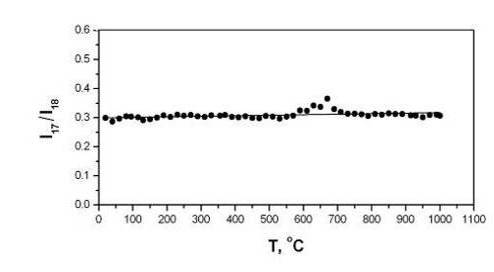
Рис.4.2.8 Отношение интенсивностей выделения газообразных
продуктов с массами 17 и 18 в зависимости от температуры отжига порошков ГА.
Поэтому вопрос об электронном состоянии молекул воды в катионных
позициях ГА нуждается в отдельном исследовании, вероятно - резонансными
методами.
500-600оС
Итак, процесс отжига ГА до 500оС сопровождается
десорбцией воды, адсорбированной поверхностями частиц, и решеточной воды, а
также небольшого количества СО2. Однако распределение ионов СО32
- в позициях А и В кристаллической решетки ГА, согласно данным
ИК-спектроскопии (рис.4.2.5 и табл.5.1), меняется незначительно. Следовательно,
выделение углерод - содержащих групп из образцов, нагретых до 500оС,
происходит в основном с поверхности частиц, а большая часть карбонатных групп
остается внедренной в решетку ГА. При этом параметры решетки при 500оС
имеют значения а=9,404 Е, с=6,901 Е, соответствующие значениям для
карбонат-замещенного ГА (КГА) [120].
Важные сведения о примесном состоянии ГА в данном температурном
интервале дают ИК-спектры (рис.4.2.5). Во-первых, полосы поглощения,
соответствующие колебаниям OH - групп при 625 и 3560 см-1,
становятся отчетливо разрешаемыми и увеличиваются по интенсивности с увеличением
температуры отжига (рис.4.2.5в и рис.4.2.9). Кроме того, ГА со смешанным
АВ типом карбонатного замещения становится ГА В типа карбонатного замещения
поскольку исчезают полосы поглощения, характерные для А типа карбонатного
замещения (табл.5.1). Во-вторых, плохо разрешаемая ранее полоса поглощения при
875-880 см-1 смещается к значению 873 см-1. Появляется
полоса поглощения СО2 в районе 2330 см-1 при 500оС
и исчезает при 600оС. Параметры решетки показывают небольшое
уменьшение параметра с и значительное увеличение параметра а до
600оС. Масс-спектры (рис.4.2.6) в данном температурном интервале
обнаруживают десорбцию газообразных продуктов с массовыми числами 28 и 44,
которые соответствуют СО2, а также 18, связанной с выделением воды.
Эти результаты можно объяснить, исходя из таких процессов разложения СО32-
групп в А позиции кристаллической решетки ГА [121,122]:
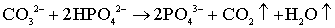 , (5.5)
, (5.5)
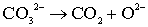 , (5.6) и
, (5.6) и
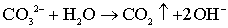 . (5.7)
. (5.7)
Реакция (5.5) не дает вклада в увеличение числа OH - групп.
Более детальный анализ кривых - других массовых чисел подтверждает
предположение о том, что в данном интервале температур происходит несколько
процессов разложения. Изменения весьма слабого пика М=12 и довольно сильного
М=16, которые являются побочными пиками 44 массы, происходят в целом
согласованно. Однако пик М=12 имеет два максимума при 500оС и 570оС
(рис.4.2.10).
Таблица 5.1
Полосы поглощения в ИК-спектрах КГА ионов n3 CO32-(1400-1600см-1).
Значения волновых чисел указаны в см-1. Условные обозначения: с-сильная,
ср-средняя, сл-слабая, оч-очень, п- плечо.
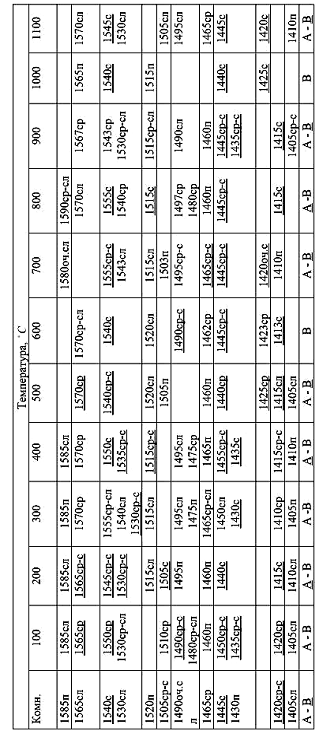

Рис.4.2.10. Масс - спектры изменений 12 и 44 массовых чисел в
зависимости от температуры отжига порошков.
Это свидетельствует о том, что процесс выделения СО2
идет в две стадии. При этом основной пик 44 массы (рис.4.2.10) и побочный при
28 (рис.4.2.6) не разделяются из-за большой интенсивности. Учитывая, что
разложение СО32 - групп сопровождается появлением OH
- групп в решетке (рис.4.2.9), вода не выделяется, а содержание НРО42
- групп в исходном порошке низкое, можно предложить следующую реакцию при
500оС:
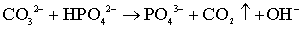 . (5.8)
. (5.8)
Число OH - групп при 600оС изменяется слабо
по сравнению с 500оС (рис.4.2.9). В свою очередь, ИК-спектры
(рис.4.2.5) также показывают уменьшение интенсивности полос поглощения в
интервале 1650-1300 см-1, характерных для колебаний СО32
- групп, при 600оС. При этом интенсивность полосы при 873 см-1,
наоборот, усиливается, что, по-видимому, связано с наложением полос n3 СО32 - и Са2+-О2 - (875
см-1) [44] в случае, если разложение карбонатных групп СО32
- происходит с образованием ионов О2 - в А позиции при 600оС
(табл.5.1). Следовательно, разложение карбонатных групп СО32 - при
570оС происходит в соответствии с реакцией (5.6).
Появление пика 18 массы на масс-спектрах при 540оС
между пиками выделения углерод-содержащих газов (12, 16 и 44 массы) может быть
объяснено как результат реакции разложения ионов НРО42 - групп
[123], присутствующих в ГА:
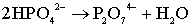 . (5.9)
. (5.9)
Данная реакция происходит при отжиге кальций - дефицитного ГА
(КДГА) в интервале температур 350-650оС с максимумом при 400оС.
Аналогичную тенденцию имеет изменение 18 массы при температурах более 400оС
(рис.4.2.6), когда заканчиваются процессы выделения воды. Кроме того, в
интервале температур 400-700оС появляется слабая полоса поглощения
около 720 см-1 (рис.4.2.5в), связанная с колебаниями ионов P2O74
- (эта характеристическая полоса свидетельствует о присутствии НPO42
- ионов и их термическом разложении в образце согласно реакции (5.9)
[123,132]. Следовательно, вблизи 540оС, когда замедляется разложение
карбонатных групп по реакции (5.8), большинство ионов НРО42 - претерпевают
разложение в соответствии с реакцией (5.9), что в конечном итоге приводит к
небольшому возрастанию интенсивности 18 массы в интервале 500-600оС.
Данный процесс позволяет объяснить увеличение параметра решетки а в этом
температурном интервале, поскольку ион P2O74 - имеет
больший радиус, чем ион НPO42 - [124].
600-700оС
В данном температурном интервале резко возрастает интенсивность
полосы поглощения ОН - групп (рис.4.2.9). Данная тенденция
связывается с диссоциативной адсорбцией (разложением) поверхностью частиц ГА
водяного пара из атмосферы, в которой производится отжиг, и последующей
диффузией ионов ОН - и Н+ в кристаллическую решетку ГА
[125]. При 700оС интенсивность полосы поглощения ОН - групп
достигает 90% максимального значения без
дополнительного образования ионов ОН - посредством решеточных
реакций. При 650оС обнаруживаются интенсивные пики газовыделения
(рис.4.2.6). Однако их происхождение не связано с разложением СО32
- групп кристаллической решетки, поскольку в масс-спектрах (рис.4.2.10)
отсутствуют выделения газов с массовым числом 12 в этом температурном
интервале. Более того, отсутствует выделение воды (17 и 18 массы). Параметры
решетки (рис.4.2.4) при этих температурах практически не изменяются. Этот факт
находится в хорошем согласии с тем, что при 700оС наблюдается
максимальная концентрация ионов Р2О74 - в КГА
[123]. Основные пики газовыделения в масс-спектрах (рис.4.2.6) соответствуют 28
и 32 массам. Можно предположить, что их появление связано с разложением ионов
NO3-. Однако деструкция ионов NO3 - происходит
в интервале 500-600оС, поскольку Са (NO3) 2
начинает разлагаться при 500оС и затем плавится при 560оС
[129]. Отношение интенсивностей выделения газообразных продуктов с массами
14/28 и 16/32 не соответствуют значениям, характерным для выделения N2
и O2 (0,078 и 0,036 вместо 0,137 и 0,218 соответственно) [130].
Кроме того, при 650оС фиксируются пики выделения газов с массами 2
(Н2+), 15 (NH+) и 16 (NH2+)
(рис.4.2.11). В этом интервале также отмечается отклонение отношения
интенсивностей выделения газообразных продуктов с массами 17/18 от постоянного
значения (рис.4.2.8). По-видимому, при этих температурах имеют место частичное
разложение адсорбированных примесей, а также их возможные реакции на
поверхности частиц КГА. Этим обстоятельством объясняется малое изменение
параметров решетки при этих температурах. Предполагается, что такие примеси
могут давать полосы поглощения на ИК - спектрах (рис.4.2.5) при 2910 и 2840 см-1.
Эти полосы присутствуют на спектрах вплоть до 1250оС и, вероятно,
связаны с образованием NxHy соединений.
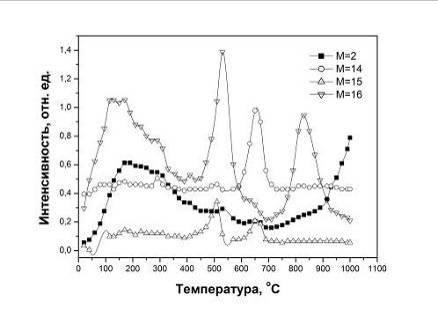
Рис.4.2.11. Масс-спектры выделения газообразных продуктов с
массовыми числами 2, 14, 15 и 16 в зависимости от температуры отжига порошков.
700-800оС
При 800оС наблюдается небольшое уменьшение
интенсивности полосы поглощения, связанной с колебаниями ОН - групп
(рис.4.2.9). Кроме того, на ИК-спектрах появляются полосы поглощения СО2
(рис.4.2.5), а также полосы поглощения карбонатных групп СО32-,
характерные для АВ типа КГА (табл.5.1). Параметры решетки (рис.4.2.4) сперва уменьшаются
в интервале 700-750оС, затем параметр а увеличивается в то
время как параметр с практически не изменяется. Масс-спектры (рис.4.2.6)
фиксируют появление пиков 18 и 44 масс, связанных с выделением Н2О и
СО2.
Резкое падение параметров решетки можно считать последствием
разложения ионов P2O74 - [123]:
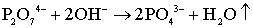 . (5.10)
. (5.10)
В результате реакции выделяется вода и наблюдается уменьшение
числа OH - групп. Оба эффекта наблюдаются на практике. Выделение
воды показывает (рис.4.2.6), что реакция фактически начинается при 650оС
и усиливается с увеличением температуры. Развитие реакции не позволяет достичь
максимального числа ОН - групп в кристаллической решетке при 700оС
благодаря внешнему гидролизу и является причиной уменьшения числа ОН - групп
при высоких температурах (на рис.4.2.9 пунктирная кривая иллюстрирует
предполагаемое изменение числа ОН - групп в интервале 600-800оС).
Поскольку ионный радиус P2O74 - намного больше
ионного радиуса PO43-, то разложение P2O74
- должно вызвать уменьшение параметров решетки. Этот эффект наблюдается
при 700-750оС. По литературным данным, разложение P2O74
- заканчивается при температурах около 900оС [123]. Однако при
700оС начинается процесс разложения карбонатных групп СО32-,
который налагается на описанный выше процесс, усиливается с повышением
температуры и, в целом, приводит к увеличению параметра решетки а.
800-1050оС
ИК-спектры (рис.4.2.5 и табл.5.1) показывают, что КГА АВ типа
преобразуется в КГА В типа при 1000оС. Масс-спектры (рис.4.2.6) фиксируют
выделение СО2 в интервале температур 750-950оС с
максимумом при 830оС. Параметр решетки а постепенно
увеличивается и выходит на плато, в то время как параметр с осциллирует
около среднего значения с экспериментальной погрешностью. Параметры решетки определяются
количеством карбоната в КГА В типа: чем больше карбоната, тем меньше параметр а-прямая
пропорция, в то время, как параметр с изменяется незначительно [57]. В
данном исследовании была обнаружена ожидаемая обратная корреляция, т.е.
параметр решетки а (в интервале 750-950оС) увеличивается с
уменьшением количества карбоната в решетке, а параметр решетки с
меняется незначительно. Однако эта корреляция при повышении температуры отжига
становится нелинейной, что может быть связано с изменением типа карбонатного
замещения в КГА.
Отжиг при 800оС приводит к образованию преимущественно
А типа КГА вместо В типа КГА при 700оС (табл.5.1). Это следует из
значительного уменьшения площади пика при 1300-1555 см-1, где
локализованы основные полосы поглощения В типа КГА, и изменения характеристик
полосы n2 CO32-. Последняя уменьшается в
интенсивности почти в 2 раза и образует дублет вместо полосы при 875 см-1,
которая выглядела как один пик. Пики в дублете, хотя и плохо разрешаются, четко
локализованы при 873 и 878 см-1 и имеют отношение интенсивностей 6:
10. Также появляется полоса поглощения СО2 при 2340 см-1
- такая же, как после отжига при 500оС (рис.4.2.5в, г и
вставка). Эти данные являются подтверждением того факта, что СО32
- группы в В позициях КГА разлагаются. По всей видимости, продукты
разложения выделяются из решетки через октаэдрические каналы с промежуточным
образованием карбонатных групп в А позиции КГА [121]. Это должно приводить к
увеличению интенсивности основной полосы СО32 - групп в А
позиции при 1500-1600см-1 и уменьшению полос поглощения СО32
- групп в В позиции КГА, что и наблюдали (табл.5.1).
Разложение СО32 - групп возможно в
соответствии с реакцией [121]:
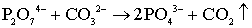 . (5.11)
. (5.11)
Из двух ионов PO43-, один остается в анионной
подрешетке вместо иона P2O74-, второй -
занимает В позицию иона СО32-. Однако разложение СО32
- групп может также происходить в два этапа. Первый этап идет в
соответствии с реакцией (5.6). Образовавшийся ион О2-, как известно,
является весьма реактивным [125] и на втором этапе взаимодействует с ионом P2O74
- согласно уравнению реакции:
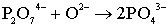 . (5.12)
. (5.12)
с образованием двух ионов PO43-. В любом
случае, эти реакции усиливают результаты реакции (5.7), которая все еще имеет
место при данных температурах. Этим обстоятельством объясняется плохая
корреляция выделения воды и СО2 на масс-спектрах (рис.4.2.6).
Реакции (5.8) и (или) (5.9) позволяют объяснить ход изменения
параметров решетки. Заполнение двух анионных позиций, занятых ранее P2O74
- и СО32-, ионами PO43 - ведет
к улучшению степени совершенства кристаллической решетки и, следовательно, к
приближению постоянных решетки к стехиометрическим значениям ГА.
Несмотря на то, что существуют две области выделения СО2
(450-600 и 750-950оС), СО32 - ионы не
полностью изымаются из КГА до 1000оС. Кроме того, на соответствующих
ИК-спектрах фиксируются следы NO3-, CO2 и
(вероятно) NхHу, чем объясняются несколько
завышенные значения параметров решетки (рис.4.2.4).
1050-1100oC
Интенсивности полос поглощения OH - групп на
ИК-спектрах (рис.4.2.9) уменьшаются вследствие процесса дегидроксилирования,
который начинается в КГА [126]. Параметры решетки резко уменьшаются до
стехиометрических значений ГА (горизонтальные линии на рис.4.2.4). На ИК-спектрах
при 1100оС появляются полосы поглощения, характерные для b-трикальций фосфата (b-ТКФ) при 950 и 1020 см-1.
Кроме того, полосы n3, n4 PO43
- значительно уширены, что свидетельствует о мелкодисперсности
образовавшейся фазы (рис.4.2.5д). Однако, содержание этой фазы в порошке
КГА было слишком малым для ее обнаружения при помощи рентгеновской
дифрактометрии, поскольку чувствительность данного метода составляет 0,5 вес. %
по b-ТКФ [133].
Потерю массы в КГА в интервале температур 500-900оС
обычно связывают с разложением карбонатных групп СО32 - [120,
121]. Измеренная потеря массы составила 1,6 вес. % в интервале температур
500-900оС (рис.4.2.7), что соответствует разложению около 2,2 вес. %
ионов СО32-. В соответствии с реакцией (5.6), это
приводит к образованию около 0,6 вес. % ионов О2 - в кристаллической
решетки ГА. Даже предполагая, что все О2 - ионы сосредоточены в А
позициях и все позиции ионов Ca2+ и PO43 - заняты,
это количество является слишком малой частью в процессах гидролиза КГА, в
котором происходит разложение карбонатных групп (для сравнения: в
стехиометрическом ГА содержится около 3,3 вес. % ионов ОH-. А из 0,6
вес. % ионов О2 - путем соединения с ионами Н+ может
образоваться немногим большая масса - 0,64 вес. % ОН - групп). Кроме
того, процесс потери массы связан также с десорбцией воды, которая по давлению
в камере масс-спектрометра (рис.4.2.6) сравнима с десорбцией СО2, а
в интервале 500-600оС обусловлен поверхностными реакциями. К тому
же, процесс разложения карбонатных групп СО32 - в кристаллической
решетке начинается к 800оС, когда позиции OH - групп
почти полностью заполнены. Эти соображения показывают, что при общей потере
массы в 2,2 вес. % доля потери массы, связанной с образованием ионов О2-,
должна быть еще меньше 0,64 вес. %. Из этих оценок можно сделать два важных
вывода:
) ионы О2-, образующиеся в процессе разложения
карбонатных групп СО32-, играют вторичную роль в
процессах гидролиза КГА;
) анализ данных потери массы КГА, содержащего примеси, может
привести к ошибочным результатам без параллельных масс-спектрометрических
исследований.
Выводы к
разделу 4
1.1 Порошки ГА, полученные в результате низкотемпературного
синтеза, состоят из частиц, размеры которых находятся в субмикронном интервале
со средним размером около 20нм, что совпадает с размером ОКР, определенным по
рентгенограммам порошков. Нанокристаллическое состояние сохраняется в этих
порошках после отжига в воздухе до примерно 800оС.
.2 После отжига в интервале температур 800-1200оС
в порошках наблюдается объединение частиц и их последующее укрупнение.
Одновременно происходит увеличение размеров ОКР, полученных по рентгенограммам
порошков. С увеличением температуры отжига средние размеры частиц становятся
больше, чем соответствующие значения ОКР. Таким образом, изначально нанокристаллический
порошок превращается в кристаллическое образование из спеченных частиц,
обладающих дефектной структурой.
.3 Показано, что исходный нанокристаллический порошок ГА
является нестехиометрическим по составу, т.е. отношение Са/Р<1,67.
.1 Показано, что синтез ГА при высокой температуре (96оС)
позволяет получить ГА со значительно меньшим количеством примесей (примерно 6-7
вес. % против 10-12 вес. % у низкотемпературного порошка). Основную часть среди
них занимают адсорбированные и решеточные молекулы воды и карбонатные группы в
анионных позициях. В связи с примесными замещениями в обеих подрешетках
удаление примесей в результате высокотемпературного отжига приводит к
формированию ГА стехиометрического состава.
.2 Впервые обнаружен немонотонный ход зависимости параметров
решетки образцов ГА после отжига в интервале температур 20-1200оС.
Он связан с присутствием в решетке ГА примесей NH4+, H2O,
HPO42-, P2O74-, CO32
- и их реакциями между собой, а также ионами, образующими решетку ГА. Эти
реакции сопровождаются выделением газообразных продуктов.
Основные результаты исследований, представленные в разделе 4,
опубликованы в [145-152].
Раздел 5.
Структурные изменения при спекании карбонизированных нанопорошков га в связи с
газовыделением
Рассмотренные выше (разделы 3,4) особенности строения
порошков ГА, связанные с наличием примесей, должны найти свое отражение в
зависимостях физических свойств порошковых материалов от температуры отжига.
Такие зависимости представляют интерес как с точки зрения получения новой
информации о процессе спекания твердых тел, так и разработки новых керамических
кальций-фосфатных материалов.
Данная глава посвящена исследованию особенностей спекания
порошковых прессовок ГА, связанных с газовыделением, при температурах 600-1200оС,
когда происходят решеточные реакции и развиваются диффузионные процессы. В
таком аспекте эта проблема практически не изучена.
5.1
Морфология и структура порошков
Для сравнительного анализа были задействованы образцы
порошков трех типов - №1, №2 и №3, которые отличались способом получения и,
следовательно, морфологией, структурой и примесным состоянием соответствующих
порошковых прессовок.
Порошок №1 был получен в результате низкотемпературного
синтеза. Полная характеристика такого порошка изложена в подразделе 4.1 Он
состоял из частиц с размерами в очень широком интервале (рис.5.1а, а').
Вместе с тем, в подразделе 4.1 отмечалось, что подобные частицы являются
агломератами из наночастиц. Дифрактограграммы порошка №1 характеризуются
значительным уширением максимумов (рис.5.2а). Оценка размеров
кристалликов (точнее, ОКР - областей когерентного рассеяния) по уширению
дифракционных максимумов дает значение около 20 нм (раздел 4.1).
Порошок №2 был получен в результате такого же
низкотемпературного синтеза, однако для увеличения степени дисперсности осадка
(и, предположительно, порошка) в реакционный раствор вводили ПАВ.
Микрофотографии порошка №2 показывают, что введение ПАВ действительно приводит
к уменьшению агломерации, поскольку размеры крупных частиц редко превышали 5мкм
(рис.5.1б, б'), а область субмикронных частиц начиналась с меньших
размеров, чем у порошка №1. Вместе с тем, уширение дифракционных максимумов на
дифрактограммах от порошка №2 (рис.5.2б) проявлялось в несколько меньшей
степени, чем для порошка №1. Средний размер нанокристалликов (ОКР), оцененный
по уширению дифракционных максимумов, составил здесь около 30 нм.
Следовательно, порошок №2 характеризовался несколько большей дисперсностью и
степенью кристалличности.
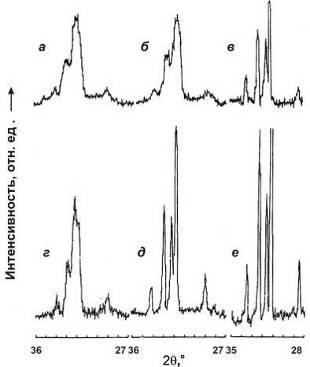
Рис.5.2 Дифрактограммы порошков №1 (а), №2 (б),
№3 (в) и прессовки из порошка №1 после отжига в течение 1 часа при 700°С (г), 900°С (д) и 1150°С (е).
Порошок №3 был получен твердофазным методом при температуре
800оС [134] и состоял из частиц, интервал размеров которых был
близок к таковому у порошка №2 (Рис.5.1в, в'). Однако наличие острых
дифракционных максимумов на дифрактограмме (Рис.5.2в) показало, что
частицы порошка №3 содержали более крупные кристаллики, чем порошки №1 и 2.
Таким образом, порошок №3 имел наибольшую степень кристалличности. Порошки №1 и
№2 предварительно отжигали в воздухе при 600оС в течение 1ч для
снятия возможных искажений решетки и избавления от примесей и остатков побочных
продуктов синтеза (которые разлагаются и выделяются при температурах до 600оС).
Степень кристалличности отожженных порошков, определяемая по уширению
дифракционных максимумов, изменялась слабо (разделы 3,4). Затем из всех трех
порошков готовили прессовки (раздел 2).
5.2 Усадка
порошковых прессовок
Кривые линейной усадки образцов прессовок (нумерация образцов
соответствует нумерации порошков) показывают, что труднее всего процесс
спекания идет в образце №3 (рис.5.3). Этот результат находится в хорошем
согласии с известными [127], следуя которым, чем крупнее кристаллики ГА в
частицах, тем при более высокой температуре выходит на плато зависимость
плотности от температуры отжига прессовок (кривая спекания). Образец №1,
степень кристалличности которого была наименьшей, уплотнялся быстрее других.
Интенсивная линейная усадка образца №1 практически заканчивалась к 1100оС.
В образце №2 при повышении температуры отжига выше 1100оС
происходила дальнейшая усадка. Это же, но в большей степени, относится к
образцам №3.
Данные о линейной усадке не дают однозначного ответа, почему
итоговая линейная усадка образцов №2 больше, чем у образов №1, которые в
сравнении с первыми имели даже несколько большую исходную пористость (образцы:
№1-60%, №2-57%, №3-51%; раздел 2), к тому же были изготовлены из порошка с
меньшей степенью кристалличности, что обычно приводит к большей итоговой
усадке. Для выяснения этого вопроса были получены кривые объемной усадки
(рис.5.4).
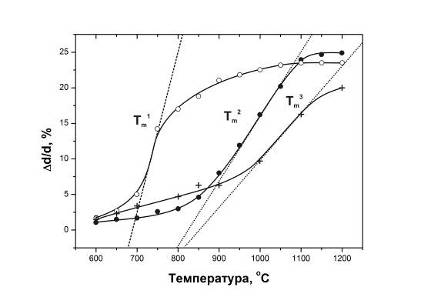
Рис.5.3 Линейная усадка образцов №1 (o), №2 (l) и №3 (+). Температуры
наибольшей скорости усадки образцов, найденные из наибольших значений тангенса
угла наклона касательных, Тm1 (№1) - 725оС, Тm2
(№2) - 980оС, Тm3 (№3) - 1050оС.
В отличие от кривых линейной усадки, для образцов №№1,2 они
имеют немонотонный характер. Причем, оба образца вплоть до 1100оС
обнаруживают качественно подобные немонотонные участки на этих кривых. Наиболее
ярко они проявляются для образца №1, где можно выделить три таких участка: абв,
вгд, деж. Практически каждый из них отражает один и тот же
процесс: начало, ускорение, достижение максимальной скорости, замедление и -
практически - остановка (квазиостановка) усадки. На следующем участке процесс
повторяется. Кривые скорости объемной усадки, полученные дифференцированием
зависимостей на рис.5.4, представлены на рис.5.5 Они также показывают, что
между ходом усадки образцов №1 и 2 принципиальной разницы нет: каждый образец
имеет три пика скорости усадки, которые лишь несколько сдвинуты друг
относительно друга на шкале температур.
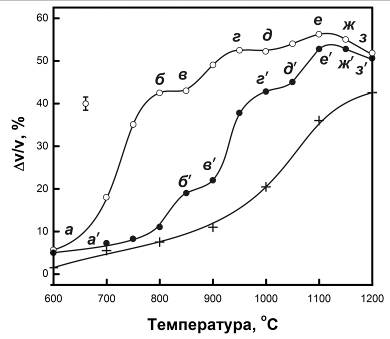
Рис.5.4 Объемная усадка образцов №1 (o), №2 (l) и №3 (+). Погрешность
измерения ±2,5%.
Ранее было обнаружено [135], что при отжиге прессовок из
порошков типа №1 и №2 наблюдаются пики газовыделения в интервале температур
600-1100оС. Предполагая, что подобный эффект имел место в данном
случае, и что при газовыделении терялась некоторая масса, было определено
изменение массы образцов в процессе усадки при различных температурах в
изучаемом интервале. Оказалось, что образцы №1 и 2 действительно теряли массу в
процессе отжига. Кроме того, были обнаружены три области температур, в которых
потеря массы была особенно значительной. Такие области наблюдали для обоих
типов образцов (рис.5.6). Сопоставим экспериментальные кривые для образцов №1,
на которых отмеченные особенности проявились наиболее заметно (Рис.5.4-5.6).
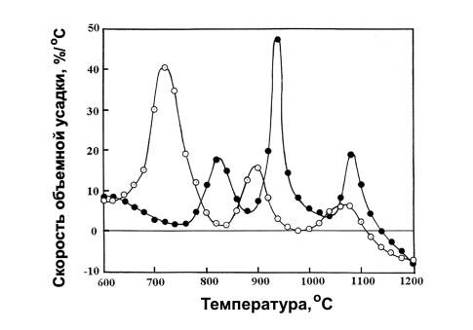
Рис.5.5 Скорость объемной усадки в образцах №1 (o) и №2 (l).
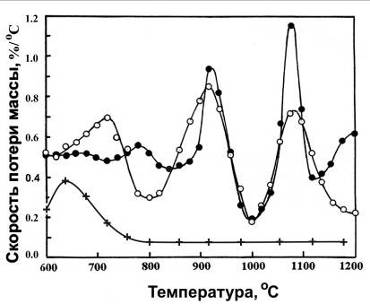
Рис.5.6 Скорость потери массы в образцах №1 (o) и №2 (l) и №3 (+).
Исследованный интервал температур отжига прессовок можно
разбить на более узкие интервалы а) 600-800, 850-950 и 1000-1100°С, а также б) 800-850,
950-1000 и 1100-1200°С. В интервалах типа а) происходила объемная
усадка образцов и расположены пики скоростей усадки, отражающие кинетику этого
процесса. В этих же интервалах температур наблюдались и пики скоростей потери
массы (газовыделения). В интервалах типа б) расположены минимумы скоростей
усадки и скоростей газовыделения, а также квазиплато на кривых объемной усадки,
т.е. в них имели место почти остановки усадки. Подобные корреляции можно
установить и при сопоставлении соответствующих кривых образца №2. Однако здесь
они выражены не так четко. В частности, на кривой объемной усадки отсутствуют
платообразные участки, и вместо квазиостановки можно говорить лишь о
существенном замедлении усадки. Таким образом, оказалось, что процессы газовыделения
и усадки тесно связаны между собой.
5.3 Плотность
и микротвердость образцов
Усадка приводила к уплотнению и изменению свойств образца, в
особенности тех, которые чувствительны к плотности, например, - микротвердости.
Рис.5.7 показывает, что основное увеличение плотности образца действительно
происходило в температурных интервалах типа а). Особенно наглядно процесс
уплотнения отражался на кривых изменения микротвердости образцов (рис.5.8).
Кривые образцов №1 и №2 на рис.5.8 подобны соответствующим кривым объемной
усадки на рис.5.4 Например, для образца №1 кривая микротвердости состоит из
участков а’’б’’в’’, г’’д’’е’’
и е’’ж’’з’’, которые
подобны участкам абв, где и ежз на кривой объемной усадки.
Весьма убедительно проявлялась также квазиостановка усадки: когда на участках бв
и гд в интервалах температур типа б) усадка в пределах погрешности
измерения не обнаруживалась, микротвердость образцов также увеличивалась весьма
незначительно - кривые имели почти горизонтальные участки б’’в’’
и г’’д’’.
Таким образом, для образцов №1 и №2 потеря массы вследствие
газовыделения стимулировала усадку, что в итоге приводило к увеличению
плотности и микротвердости. Такие зависимости имели место в интервале
температур 600-1100°С, пока пористость в уплотняющемся образце была
открытой (рис.5.9). При температурах вблизи 1100°С достигалась наибольшая
объемная усадка, при этом плотности образцов соответствовала пористость менее
8% (для образцов №2 - около 3%). Это значило, что полученная в этих условиях
керамика отличалась закрытой микропористостью.

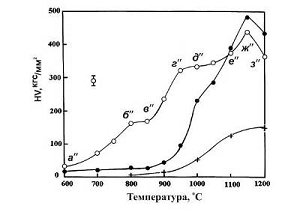
Рис.5.8 Изменение микротвердости в процессе уплотнения
образцов №1 (o), №2 (l) и №3 (+).
Дальнейшее повышение температуры отжига приводило к
отрицательным результатам. При температурах 1100-1150°С изменялся знак объемной
усадки (равно как и скорости объемной усадки), т.е. наблюдалось не уменьшение,
а увеличение объема образцов относительно спеченных при 1100°С (участок ежз на
рис.5.4). При 1150°С плотность и микротвердость достигали наибольших
значений. Однако при более высоких температурах отжига плотность не
увеличивалась, а микротвердость образцов резко падала (участок ж’'з’’
на рис.5.8).
5.4 Сущность
и механизм процессов
Основные пики десорбции воды были расположены в интервале
температур 100-500°С. Разложение побочных продуктов синтеза
заканчивалось к 650оС (разделы 3,4). Таким образом, из прессовок,
нагретых до температур выше 600°С, практически вся вода и побочные продукты были
удалены.
Образовавшиеся из раствора частицы ГА состояли из
нанокристаллических ядер, окруженных оболочкой из гидратированного
аморфоподобного вещества. Благодаря этому веществу, частицы в порошках №1 и №2
были "склеены" в агломераты очень широкого интервала размеров.
Другими словами, нанокристаллические ядра в агломерированных частицах были
отделены дрг от друга прослойками из гидратированного аморфоподобного вещества.
Кроме адсорбированной воды, такая прослойка содержала остатки загустевшего
маточного раствора из гидратированных ионов реагентов и побочных продуктов
синтеза. (Осадки перед сушкой очищали путем промывки и центрифугирования.
Однако даже после 4-х кратной процедуры очистки в высушенных осадках, т.е.
порошках, обнаруживали следы частиц раствора; разделы 3,4). Отжиг порошков
перед компактированием при 600оС (2ч) позволил избавиться от летучих
компонентов и побочных продуктов синтеза. При этом степень кристалличности
порошка изменилась очень слабо (разделы 3,4).
Эффективный рост кристалликов в частицах происходит при
температурах выше 600°С. Это показали дифрактограммы, на которых
фиксировалось увеличение интенсивности и сужение отражений (увеличение
кристалличности) одновременно с уменьшением интегральной интенсивности
(площади) диффузного максимума под пиками (уменьшение количества
нанокристаллического вещества) в процессе отжига (усадки) образцов (рис.5.2г-е
и разделы 3,4). В интервале температур 600-1200оС наблюдали два
пика газовыделения - между 680 и 800оС (720оС - образец
№1 и 780оС - образец №2). Они являются следствием термического
разложения примесных ионов CO32-. Эти ионы замещают
преимущественно PO43 - группы в решетке ГА (В-тип
замещения) (рис.5.10). В результате разложения карбонатных групп и выделения
продуктов разложения (преимущественно в виде СО2) в решетке
образуются анионные вакансии, которые вследствие их происхождения назовем примесными.

Рис.5.10. Масс-спектр выделения газообразных продуктов с
массовым числом 44 в зависимости от температуры отжига порошков.
Скорость образования примесных вакансий зависит от скорости
реакции разложения. Согласно кинетике газовыделения, скорость реакции
значительно меняется с повышением температуры (рис.5.6).
Усадка связана с потоком вакансий от пор к местам стока
вакансий. Судя по имеющимся данным сверхразрешающей электронной микроскопии
[101] и по слабому изменению кристалличности после предварительного отжига
порошков (рис.5.2а и рис.5.2г), нанокристаллы ГА слабодефектны.
Поэтому основным стоком вакансий является поверхность нанокристаллов, что ведет
к усадке образца. Скорость усадки зависит от коэффициента диффузии и
концентрации вакансий [136]. "Впрыскивание” в имеющийся поток примесных
вакансий не изменяет его градиента, поскольку разложение ионов CO32
- равновероятно в любой точке кристаллика. Однако увеличивается общая
концентрация вакансий, что ведет к увеличению скорости диффузии, а значит и
скорости залечивания пор, т.е. скорости усадки.
Мера влияния примесных вакансий на процесс спекания зависит
от величины отношения концентраций примесных вакансий и вакансий,
существовавших в образце до начала процесса разложения карбонатных ионов.
По-видимому, это отношение значительно, поскольку кинетика усадки очень хорошо
следует кинетике газовыделения (рис.5.5 и 5.6). Для температурных интервалов,
где реакция разложения не идет или ее скорость малая - усадка определяется
концентрацией ”собственных” вакансий (равновесных при данной температуре и
образовавшихся вследствие растворения пустоты в кристалл [136]). Величина и
скорость такой усадки весьма мала (платообразные участки типа бв, гд,
б'в' и г'д' на рис.5.4 и 5.5). Убедительным аргументом
для такого заключения является сравнение со значительно меньшей величиной
усадки при одинаковых температурах в образцах №3, которые - при сравнимой
пористости - состояли из более совершенных и беспримесных кристалликов
(рис.5.4).
После спекания при 900оС образовалась керамика ГА
со сквозной пористостью около 20% (рис.5.7 и 5.9а). Из такого тела газообразные
продукты разложения карбонатных ионов свободно выделялись. Повышение
температуры отжига до 1000оС имело результатом уменьшение пористости
до 12%, однако поры все еще соединялись между собой и - вероятнее всего - с
поверхностью (рис.5.7 и 5.9б). Наименьшая пористость наблюдалась в
керамиках, полученных при температурах отжига 1000-1150оС. Как
отмечалось, в образцах №1 пористость была менее 8%, а в образцах №2 - около 3%.
Ее формировали изолированные микропоры размером менее 2мкм (рис.5.7 и 5.9в).
Пористость такого уровня обычно относят к замкнутой [137].
В интервале температур отжига 1000-1120оС
наблюдали последний, третий, выброс газообразных продуктов из спекающихся
образцов (пик газовыделения при 1080оС; рис.5.6). При весьма малой
закрытой и закрывающейся микропористости выделение газа через систему пор было
весьма затруднительным, если вообще возможно. Давление газа, накапливающегося в
порах внутри керамики, могло оказаться столь высоким, что привело бы к
разрушению керамики.
Другой причиной, которая могла бы привести к (или
содействовать) разрушению, был переход части нестехиометрического
слабокарбонизированного ГА - из которого состояли образцы №1 и №2 - в β-ТКФ, что обычно имеет место при температурах отжига выше 1000оС
[138]. Этот переход действительно наблюдался при отжиге порошков №1 и №2 при
высоких температурах, причем после отжига при 1200оС в образцах №1
фиксировали около 15 вес. % β-ТКФ (рис.4.1.7). По мере
развития этого высокотемпературного фазового перехода в кристаллической решетке
накапливались напряжения, поскольку объем октаэдрической фазы β-ТКФ в 6,6 раза больше порождающего объема ГПУ фазы ГА (объем
элементарной ячейки β-ТКФ равен 3521 Е3,
а ГА - 529 Е3).
Следовательно, газовыделение в интервале температур отжига
прессовок 600-1200оС можно преимущественно связать с десорбцией
продуктов распада карбонатных групп CO32-, а выше 1000оС
- еще и с выделением молекул воды, образовавшихся из гидроксильных ионов
кристаллической решетки в результате перехода части ГА в β-ТКФ (раздел 3,4).
Одна или обе из указанных причин приводили к растрескиванию
керамики и освобождению газа. На микрофотографиях скола керамики, полученной в
результате спекания при 1200оС, хорошо видны трещины, пронизывающие
ее объем (рис.5.9г). Растрескивание и, вследствие, разбухание керамики
приводили к увеличению ее объема, что хорошо видно на участках типа ежз
е'ж'з', где усадка и скорость усадки меняли знак (рис.5.4 и 5.5), а
микротвердость резко уменьшалась (рис.5.8).
В результате спекания в интервале 950-1000оС получалась
однофазная керамика ГА, которая при примерно 20% -й пористости имела
микротвердость 320кгс/мм2. Это значение близко к микротвердости
трабекулярной кости (около 300 кгс/мм2 [139]). А керамика, спеченная
в интервале 1100-1150оС, была двухфазной (ГА+β-ТКФ) и плотной (керамику с пористостью менее 5% относят к
”сверхплотной" [140]). При этом значения микроствердости были близки к
таковой у эмали (около 450 кгс/мм2; эмаль является самой твердой
костной тканью скелета [140]). Эти характеристики позиционировали полученные
керамики как весьма перспективные биоматериалы для костных замещений.
Выводы к
разделу 5
1. Обнаружен новый примесный
эффект активизации спекания в образцах ГА. Механизм эффекта предположительно
связан с увеличением концентрации вакансий в объеме кристалликов ГА за счет
появления примесных вакансий, образующихся в результате термического разложения
карбонатных ионов и выделения продуктов разложения из решетки. Оценки
показывают, что концентрация примесных вакансий на несколько порядков превышает
концентрацию вакансий, которая обуславливает процессы уплотнения и спекания в
образцах ГА, не содержащих карбонатных ионов в решетке ГА.
2. Результаты работы были
опорными в разработке новых биоактивных материалов на основе ГА. В частности,
используя слабокарбонизированные высокодисперсные порошки ГА, были получены
керамические материалы двух типов: однофазные пористые (пористость около 20%) и
бифазные (ГА+β-ТКФ) плотные (пористость менее 8%).
Благодаря эффекту активизации спекания, пористую керамику получали при
температурах отжига 900-1000оС, т.е. на 200-300оС
меньших, чем по известным способам. Функциональные характеристики керамических
материалов удовлетворяли таковым для эффективных заменителей костной ткани.
Основные результаты исследований, представленные в разделе 5,
опубликованы в [153-156].
Выводы
В работе системно исследованы структурные изменения в
фосфатах кальция на основе гидроксилапатита, связанные с влиянием примесей, на
стадиях осаждения из раствора, получения порошка, а также, его отжига и
спекания в широком интервале температур. Получены такие основные научные и
практические результаты:
. Решена задача по установлению особенности строения
дифракционно-аморфного вещества, известного как аморфный фосфат кальция (АФК)
на начальной стадии процесса кристаллизации ГА из раствора. Показано, что
нанокластеры, которые составляют частицы АФК сразу после формирования, в
процессе синтеза ГА переходят в элементарные ячейки ГА, затем - в нанокристаллы
ГА размером в несколько параметров решетки. Отношение ГА/АФК в образовавшейся
двухфазной смеси увеличивается с течением процесса. К концу фазового перехода
начинается рост нанокристаллов ГА, что приводит к появлению дифракционных
максимумов.
2. Предложены схема перехода АФК→ГА и
возможный механизм роста ГА путем автоэпитаксии нанокристаллов ГА. Они
качественно объясняют как результаты данной работы, так и последние сведения о
формах нанокристаллов ГА и их образований, полученные с помощью техники
атомного разрешения.
. Показано, что ограничение или, тем более,
блокирование диффузии кальция и (или) гидроксильных групп в нанокластеры путем
уменьшения содержания молекул воды в гидратированном слое тормозит их конверсию
в ГА и позволяет сохранять кальций-фосфатный осадок (или порошок) в аморфном
состоянии длительное время. Этот результат свидетельствует в пользу
предложенной схемы фазового перехода.
. Исследованы морфологические и структурные
особенности, а также примесный состав порошков ГА, полученных в результате
синтеза при низкой (20оС) и высокой (96оС) температурах.
Частицы в порошках ГА характеризовались произвольной формой и
широким интервалом размеров. Вместе с тем, все они состояли из наночастиц:
округлых, размерами около 20 нм - в низкотемпературном порошке, и несколько
удлиненных, размерами около 40 нм - в высокотемпературном. Размеры наночастиц
совпадали со значениями ОКР в этих порошках. Таким образом, оба порошка были
нанокристаллическими.
Установлена природа и происхождение примесей в порошках.
Ключевой интерес представили решеточные примеси. В обоих порошках ими
преимущественно оказались молекулы воды, захваченные в катионные позиции
("решеточная” вода) и карбонатные ионы CO32-,
которые замещали фосфатые группы PO43 - (В-тип
замещения).
. Изучены изменения параметров решетки и фазовый
состав в порошках ГА после отжига в интервале температур 20-1200оС.
Параметры решетки ГА в образцах низкотемпературного порошка,
отожженных при 800-1200оС, были ниже их табличных значений. К тому
же, при высоких температурах отжига часть ГА переходила в β-ТКФ. Отсюда следовало, что порошок был нестехиометрическим и имел
отношение Са/Р<1,67.
Впервые получена зависимость параметров решетки ГА от
решеточных примесей после отжига в интервале температур 20-1200оС.
Обнаружен немонотонный ход зависимости, который связан с определенными
реакциями примесных ионов и выделением образующихся газообразных продуктов из
решетки. В связи с примесными замещениями в обеих подрешетках отношение Са/Р в
порошке оказалось близким к 1,67, и в результате высокотемпературного отжига
(термического удаления примесей) образовался однофазный продукт из
стехиометрического ГА.
. Обнаружен новый примесный эффект активизации
спекания в твердом теле. Эффект заключается в ускорении спекания порошковых
прессовок карбонизированного ГА в случае одновременного выделения газообразных
продуктов из кристаллической решетки этого вещества. Механизм эффекта
предположительно связан с увеличением концентрации вакансий в объеме
кристалликов ГА за счет "примесных" вакансий, которые образуются в
результате термического разложения карбонатных ионов и выделения продуктов
разложения из решетки. Увеличение концентрации вакансий обусловливает
интенсификацию диффузионных процессов, а значит - и спекания в прессовке.
7. Результаты работы были опорными в разработке новых
биоактивных материалов на основе ГА. В частности, используя
слабокарбонизированные высокодисперсные порошки ГА, были получены керамические
материалы двух типов: однофазные пористые (пористость около 20%) и бифазные
(ГА+β-ТКФ) плотные (пористость менее 8%). Благодаря эффекту активизации
спекания, пористую керамику получали при температурах отжига 900-1000оС,
т.е. на 200-300оС меньших, чем по известным способам. Функциональные
характеристики керамических материалов удовлетворяли таковым для эффективных
заменителей костной ткани.
Список
использованных источников
1. Баринов
С.М. Биокерамика на основе фосфатов кальция [текст] / С.М. Баринов, В.С.
Комлев. - М.: Наука, 2005. - 204 с.
2. Posner
A. S. Refinement of the hydroxyapatite structure [текст] / A. S. Posner, A.
Perloff, A. F. Diorio // Acta Cryst. - 1958. - Vol.11. - P.308.
. Robinson
R. A. An electron-microscopic study of the crystalline inorganic component of
bone and its relationship to the organic matrix [текст] / R. A. Robinson // J.
Bone Joint Surg. Am. - 1952. - Vol.34. - P.389.
. Kay
M.I. Crystal structure of hydroxyapatite [текст] / M.I. Kay, R. A. Young, A. S.
Posner // Nature. - 1964. - Vol. 204. - P.1050.
. Mackie
P. E. Monoclinic structure of synthetic Ca5 (PO4) 3Cl,
chlorapatite [текст] / P. E. Mackie, J. C. Elliott, R. A. Young // Acta Cryst.
- 1972. - Vol. B28. - P.1840.
. Eanes
E. D. Intermediate states in the precipitation of hydroxyapatite [текст] / E.
D. Eanes, I. H. Gillessen, A. S. Posner // Nature. - 1965. - Vol. 208. -
P.365-367.
. Tung
M. S. An intermediate state in hydrolysis of amorphous calcium phosphate
[текст] / M. S. Tung, W. E. Brown // Calcif. Tissue Int. - 1983. - Vol.35. -
P.783-790.
. Boskey
A. L. Formation of hydroxyapatite at low supersaturation [текст] / A. L.
Boskey, A. S. Posner // J. Phys. Chem. - 1976. - Vol.80. - P.40-45.
. Posner
A. S. Synthetic amorphous calcium phosphate and its relation to bone mineral
structure [текст] / A. S. Posner, F. Betts // Acc. Chem. Res. - 1975. - Vol.8.
- P.273-281.
. Holmes
J. M. Surface Areas by Gas Adsorption on Amorphous Calcium Phosphate and
Crystalline Hydroxyapatite [текст] / J. M. Holmes, R. A. Beebe // Calcif.
Tissue Int. - 1971. - Vol.7. - P.163-174.
. Gadaleta
S. J. Fourier Transform Infrared Spectroscopy of the Solution-Mediated
Conversion of Amorphous Calcium Phosphate to Hydroxyapatite: New Correlations
Between X-Ray Diffraction and Infrared Data [текст] / S. J. Gadaleta, E. P.
Paschalis, F. Betts, R. Mendelsohn, A. L. Boskey // Calcif. Tissue. Int. - 1996.
- Vol.58. - P.9-16.
. Liu
C. Kinetics of hydroxyapatite precipitation at pH 10 to 11 [текст] / C. Liu, Y.
Huang, W. Shen, J. Cui // Biomaterials. - 2001. - Vol.22. - P.301-306.
. Termine
J. D. Calcium phosphate formation in vitro: II. Effects of environment on
amorphous-crystalline transformation [текст] / J. D. Termine, R. A. Peckauskas,
A. S. Posner // Arch. Biochem. Biophys. - 1970. - Vol.140. - P.318-325.
. Termine
J. D. Calcium phosphate formation in vitro: I. Factors affecting initial phase
separation [текст] / J. D. Termine, A. S. Posner // Arch. Biochem. Biophys. -
1970. - Vol.140. - P.307-317.
. Kim
S. Direct observation of hydroxyapatite nucleation from amorphous phase in a
stoichiometric calcium/phosphate aqueous solution [текст] / S. Kim, H. S. Ryu,
H. Shin, H. S. Jung, K. S. Hong // Chemistry Letters. - 2004. - Vol.33. - № 10.
- P.1292-1293.
. Eanes
E. D. An Electron Microscopic Study of the Formation of Amorphous Calcium
Phosphate and Its Transformation to Crystalline Apatite [текст] / E. D. Eanes,
J. D. Termine, М. U. Nylen // Calc. Tissue. Res. - 1973. - Vol.12. - P.143-158.
. Posner
A. S. Calculation of the X-Ray intensities from arrays of small crystallites of
hydroxyapatite [текст] / A. S. Posner // Arch. Biochem. Biophys. - 1968. -
Vol.124. - P.604-615.
. Li
Y. In vitro synthesis and characterization of amorphous calcium phosphates with
various Ca/P atomic ratios [текст] / Y. Li, W. Weng // J Mater Sci: Mater Med.
- 2007. - Vol.18. - P.2303-2308.
. Nelson
L. S. Amorphous calcium phosphates of different composition give very similar
EXAFS spectra [текст] / L. S. Nelson, C. Holt, J. E. Harries, D. W. L. Hukins
// Physica B. - 1989. - Vol.158. - P.105-106.
. Betts
F. An x-ray radial distribution study of amorphous calcium phosphate [текст] /
F. Betts, A. S. Posner // Mat. Res. Bull. - 1974. - Vol.9. - P.353-360.
. Fawcett
R. W. A Radial Distribution Function Analysis of an Amorphous Calcium Phosphate
with Calcium to Phosphorus Molar Ratio of 1.42 [текст] / R. W. Fawcett // Calc.
Tissue Res. - 1973. - Vol.13. - P.319-325.
. Glimcher
M. J. Recent studies of bone mineral: is the amorphous calcium phosphate theory
valid? [текст] / M. J. Glimcher, L. C. Bonar, M. D. Grynpas, W. J. Landis, A.
H. Roufosse // J. Cryst. Growth. - 1981. - Vol.53. - P.100-119.
. Harries
J. E. Conversion of amorphous calcium phosphate into hydroxyapatite
investigated by EXAFS spectroscopy [текст] / J. E. Harries, D. W. L. Hukins, C.
Holt, S. S. Hasnain // J. Cryst. Growth. - 1987. - Vol.84. - P.563-570.
. Eanes
E. D. Extended X-ray absorption fine structure (EXAFS) studies on calcium in
crystalline and amorphous solids of biological interest [текст] / E. D. Eanes,
L. Powers, J. L. Costa // Cell Calcium. - 1981. - Vol.2. - P.251-262.
. Betts
F. A structural model for amorphous. calcium phosphate [текст] / F. Betts, A.
S. Posner // Trans. Amer. Cryst. Ass. - 1974. - Vol.10. - P.73-84.
. Betts
F. Atomic Structure of Intracellular Amorphous Calcium Phosphate Deposits
[текст] / F. Betts, N. C. Blumenthal, A. S. Posner, G. L. Becker, A. L.
Lehninger // Proc. Nat. Acad. Sci. - 1975. - Vol.72. - P. 2088-2090.
. Onuma
K. Cluster Growth model for hydroxyapatite [текст] / K. Onuma, A. Ito // Chem.
Mater. - 1998. - Vol.10. - P.3346-3351.
. Suvorova
E.I. Size Effect in X-ray and Electron Diffraction Patterns from Hydroxyapatite
Particles [текст] / E.I. Suvorova, P. A. Buffat // Cryst. Reports. - 2001. -
Vol.46, № 5. - P.722-729.
. Suvorova
E.I. Electron diffraction and high-resolution transmission electron microscopy
in characterization of calcium phosphate precipitation from aqueous solutions
under biomineralization conditions [текст] / E.I. Suvorova, P. A. Buffat //
European Cells and Materials. - 2001. - Vol.1. - P.27-42.
. Suvorova
E.I. Electron Diffraction from Micro - and Nanoparticles of Hydroxyapatite
[текст] / E.I. Suvorova, P. A. Buffat // Journal of Microscopy. - 1999. - Vol.
196. - № 1. - P.46-58.
. Skrtic
D. Silica - and zirconia-hybridized amorphous calcium phosphate: Effect on
transformation to hydroxyapatite [текст] / D. Skrtic, J. M. Antonucci, E. D.
Eanes, R. T. Brunworth // J Biomed Mater Res. - 2002. - Vol.59. - P.597-604.
. Big
A. Magnesium inftuence on hydroxyapatite crystallization [текст] / A. Big, G.
Falini, E. Foresti, M, Gazzano, A, Ripamonti, N. Roveri, Journal of Inorganic
Biochemistry. - 1993. - Vol.49. - P.69-78.
. Abbona
F. A XRD and TEM study on the transformation of amorphous calcium phosphate in
the presence of magnesium [текст] / F. Abbona, A. Baronnet // Journal of
Crystal Growth. - 1996. - Vol.165. - P.98-105.
. Posner
A. S. Role of ATP and Mg in the Stabilization of Biological and Synthetic
Amorphous Calcium Phosphates [текст] / A. S. Posner, F. Betts, N. C. Blumenthal
// Calcified Tissue International. - 1976. - Vol.22. - P. 208-212.
. Blumenthal
N. C. Stabilization of Amorphous Calcium Phosphate by Mg and ATP [текст] / N.
C. Blumenthal, F. Betts, A. S. Posner // Calcif. Tiss. Res. - 1977. - Vol.23. -
P.245-250.
. Lundager
H. E. Effects of cadmium on crystallization of calcium phosphates / H. E.
Lundager Madsen, F. Abbona, E. Barrese // Cryst. Res. Technol. - 2004. Vol.39.
- №.3. - P.235 - 239.
. Hidaka
S. Effects of indium and iron ions on in vitro calcium phosphate precipitation
and crystallinity [текст] / S. Hidaka, Y. Okamoto, K. Abe, K. Miyazaki //
Journal of Biomedical Materials Research. - 1996. - Vol.31. - P.11-18.
. Hasvold
O. The Effect of Fluoride on the Transformation of Amorphous Calcium Phosphate
into Crystalline Apatite [текст] / O. Hasvold, S. Dahm // Calcified Tissue
International. - 1976. - Vol.22. - P.425-427
. Termine
J. D. Calcium Phosphate Formation in Vitro II. Effects of Environment on
Amorphous-Crystalline Transformation [текст] / J. D. Termine, R. A. Peckauskas,
A. S. Posner // Archives of Biochemistry and Biophysics. - 1970. - Vol.140. -
P.318-325.
. Lopez-Valero
I. Effects of sodium and ammonium ions on occurrence, evolution and
crystallinity of calcium phosphates [текст] / I. Lopez-Valero, C.
Gomez-Lorente, R. Boistelle // Journal of Crystal Growth. - 1992. - Vol.121. -
P.297-304.
. Klement
R. Basische Phosphate zweiwertige Metalle. V. Phosphate and Hydroxylaptits des
Cadmiums [текст] / R. Klement, F. Zureda // Z. Anorg. Allgem. Chem. - 1940. -
Vol.245. - P.229.
. Narasaraju
T. S. B. Preparation and some physico-chemical aspects of solid solutions of
hydroxylapatite and fluorapatite [текст] / T. S. B. Narasaraju // Ind. J. Chem.
- 1972. - Vol.10. - P.309.
. Rai
U. S. Preparation and characterization of solid solutions of hydroxylapatite
and chlorapatite [текст] / U. S. Rai, K. K. Rao, T. S. B. Narasaraju // Ind. J.
Chem. - 1979. - Vol.18A. - № 2. - P.168.
. Narasaraju
T. S. B. Some physico-chemical aspects of hydroxyapatite [текст] / T. S. B.
Narasaraju, D. E. Phebe // J. Mater. Sci. - 1996. - Vol.31. - P.1-21.
. Narasaraju
T. S. B. Physico-chemical aspects of calcium vanadate apatite [текст] / T. S.
B. Narasaraju, K. K. Rao, U. S. Rai, B. K. Kapoor // Ind. J. Chem. - 1977. -
Vol.15A. - P.1014.
. Gupta
S. K. Determination of solubility products of phosphate and vanadate apatites
of calcium and their solid solutions [текст] / S. K. Gupta, P. V. R. Rao, G.
George, T. S. B. Narasaraju // J. Mater. Sci. - 1987. - Vol.22. - P.1286.
. Termine
J. D. Hydroxide and carbonate in rat bone mineral and its synthetic analogues
[текст] / J. D. Termine, D. R. Lundy // Calcif. Tissue Res. - 1973. - Vol.13. -
P.73.
. Suzuki
M. Physiochemical nature of hard tissue X [текст] / M. Suzuki // Hirosaki
Igaku. - 1973. - Vol.24. - P.450.
. Holager
J. Thermogravimetric Examination of Enamel and Dentin [текст] / J. Holager // J
Dent Res. - 1970. - Vol.49. - P.546-548.
. Aoki
H. Science and medical application of hydroxyapatite [текст] / Aoki H. - Tokyo:
JAAS, 1991. - 245 p.
. Klement
R. New investigation of the inorganic matter of bone and teeth and its
synthesis [текст] / R. Klement // Klin. Wochschr. - 1937. - Vol.16. - P.591.
. Bonel
G. Sur une Nouvelle Apatite Carbonate Synthetique [текст] / G. Bonel, G. Montel
// Compt. Rend. - 1964. - Vol.258. - P.923-926.
. Sonju
Clasen A. B. Quantitative determination of type A and type B carbonate in human
deciduous and permanent enamel by means of Fourier transform infrared
spectrometry [текст] / A. B. Sonju Clasen, I. E.ruyter // Adv Dent Res. - 1997.
- Vol.11. - № 4. - P.523-527.
. LeGeros
R. Z. Two Types of Carbonate Substitution in the Apatite Structure [текст] / R.
Z. LeGeros, O. R. Trautz, E. Klein, J. P. LeGeros // Specialia Experimentia. -
1969. - Vol.15. - № 1. - P.5-7.
. Trautz
O. R. Crystallographic studies of calcium carbonate phosphate [текст] / O. R.
Trautz // Ann. NY. Acad. Sci. - 1960. - Vol.85. - P.145.
. Young
R. A. Reversible high temperature exchange of carbonate and hydroxyl ions in
tooth enamel and synthetic hydroxyapatite [текст] / R. A. Young, M. L.
Bartlett, S. Spooner, P. E. Mackie, G. Bonel // J. Biol. Phys. - 1981. - Vol.9.
- P.1-26.
. LeGeros
R. Z. Effect of carbonate on the lattice parameters of apatite [текст] / R. Z.
LeGeros // Nature. - 1965. - Vol. 206. - P.403-404.
. Driessens
F. C. M. Mechanism of Substitution in Carbonated Apatites [текст] / F. C. M.
Driessens, R. M. H. Veerbeck // Z. Anorg. Allg. Chemie. - 1983. - Vol.504. - P.
195-200.
. Bonel
G. Contribution a 1'etude de la carbonation des apatites.I. Synthese et etude
des proprietes physicochimiques des apatites carbonatees du type A [текст] / G.
Bonel // Ann. Chim. - 1972. - Vol.7. - P.65-88.
. Doi
Y. Pyrolysis-gas chromatography of carbonate apatites used for sintering
[текст] / Y. Doi, T. Koda, M. Adachi, N. Wakamatsu, T. Goto, H. Kamemizu, Y.
Moriwaki, Y. Suwa // J. Biomed. Mater. Res. - 1995. - Vol.29. - P.1451-1457.
. Elliott
J. C. Space group and lattice constants of Ca10 (PO4) 6CO3
[текст] / J. C. Elliott // J. Appl. Cryst. - 1980. - Vol.13. - P.618-621.
. LeGeros
R. Z. Apatite crystallites: Effects of carbonate on morphology [текст] / R. Z.
LeGeros // Science. - 1965. - Vol.155. - P.1409-1411.
. Gross
K. A. Sintered hydroxyfluorapatites. Part I: Sintering ability of precipitated
solid solution powders [текст] / K. A. Gross, L. M. Rodrнguez-Lorenzo //
Biomaterials. - 2004. - Vol.25. - P.1375-1384.
. Zhang
H. G. Preparation of fluoride-substituted hydroxyapatite by a molten salt
synthesis route [текст] / H. G. Zhang, Q. Zhu // J Mater Sci: Mater Med.
- 2006. - Vol.17. - P.691-695.
. Ergun
C. Hydroxylapatite with substituted magnesium, zinc, cadmium, and yttrium.I.
Structure and microstructure [текст] / C. Ergun, T. J. Webster, R. Bizios, R.
H. Doremus // J Biomed Mater Res. - 2002. - Vol.59. - P.305-311.
. Legeros
R. Z. The Effect of some Trace Elements on the Lattice Parameters of Human and
Synthetic Apatites [текст] / R. Z. Legeros, M. A. Miravite, G. B. Quirolgico,
M. E. J. Curzon // Calcified Tissue International. - 1976. - Vol.22. -
P.362-367.
. Gibson
I. R. Preparation and characterization of magnesium/carbonate co-substituted
hydroxyapatites [текст] / I. R. Gibson, W. Bonfield // Journal of Materials
Science: Materials in Medicine. - 2002. - Vol.13. - P.7 685-693.
. Putlayev
V. Silicon-substituted hydroxyapatite ceramics (Si-HAp): densification and
grain growth through the prism of sintering theories [текст] / V. Putlayev, A.
Veresov, M. Pulkin, A. Soin, V. Kuznetsov // Mat. - wiss. u. Werkstofftech. -
2006. - Vol.37. - №.6. - P.416 - 421.
. LeGeros
R. Z. Types of "H2O" in Human Enamel and in Precipitated
Apatites [текст] / R. Z. LeGeros, G. Bonel, R. Legros // Calcif. Tiss. Res. -
1978. - Vol.26. - P.111-118.
. Гегузин
Я.Е. Физика спекания [текст] / Я.Е. Гегузин. - М.: Наука, 1971. - 360 c.
. Балкевич
В.Л. Физика спекания [текст] / В.Л. Балкевич. - М.: Стройиздат, 1984. - 230 c.
. Кингери
У.Д. Введение в керамику [текст] / У.Д. Кингери. - М.: Стройиздат, 1964. - 536
c.
. Doremus
R. H. Bioceramics [текст] / R. H. Doremus // J. Mater. Sci. - 1992. - Vol.27. -
P.285-297.
. Bernache-Assollant
D., Ababou A., Champion E., Heughebaert M. Sintering of calcium phosphate
hydroxyapatite Ca10 (PO4) 6 (OH) 2.I.
Calcination and particle growth [текст] / D. Bernache-Assollant, A. Ababou, E.
Champion, M. Heughebaert // J. Europ. Ceram. Soc. - 2003. - Vol.23. -
P.229-241.
. Raynaud
S., Champion E., Bernache-Assollant D. Calcium phosphate apatites with variable
Ca/P atomic ratio II. Calcination and sintering [текст] / S. Raynaud, E.
Champion, D. Bernache-Assollant // Biomaterials. - 2002. - Vol.23. -
P.1073-1080.
. Орловский
В.П., Суханова Г.Е., Ежова Ж.А., Родичева Г.В. Гидроксилапатитная биокерамика
[текст] / В.П. Орловский, Г.Е. Суханова, Ж.А. Ежова, Г.В. Родичева // ЖВХО. -
1991. - 36. - С.683-690.
. Kim
S. Effect of calcinations of starting powder on mechanical properties of
hydroxyapatite-alumina bioceramic composite [текст] / S. Kim, Y. M. Kong, I. S.
Lee H. L. Kim // Journal of Materials Science: Materials in Medicine. - 2002. -
Vol.13. - №.3. - P.307-310.
. Gautier
S. Toughening characterization in alumina platelet-hydroxyapatite matrix
composites [текст] / S. Gautier, E. Champion, D. Bernache-Assollant // Journal
of Materials Science: Materials in Medicine. - 1999. - Vol.10. - P.533-540.
. Champion
E. Characterization of hot pressed AI203-platelet
reinforced hydroxyapatite composites [текст] / E. Champion, S. Gautier, D.
Bernache-Assollant // Journal of Materials Science: Materials in Medicine. -
1996. - Vol.7. - 125-130.
. Zhang
J. Fabrication of Hydroxyapatite-Zirconia Composites for Orthopedic
Applications [текст] / J. Zhang, M. Iwasa, N. Kotobuki, T. Tanaka, M. Hirose,
H. Ohgushi D. Jiang // J. Am. Ceram. Soc. - 2006. - Vol.89. - №.11. -
P.3348-3355.
. Chaki
T. K. Densification and strengthening of silver-reinforced
hydroxyapatite-matrix composite prepared by sintering [текст] / T. K. Chaki, P.
E. Wang // Journal of Materials Science: Materials in Medicine. - 1994. -
Vol.5. - P.533-542.
. Zhang
X. Toughening of calcium hydroxyapatite with silver particles [текст] / X.
Zhang, G. H. M. Gubbels, R. A. Terpstra, R. Metselaar // Journal of Materials
Science. - 1997. - Vol.32. - P.235-243.
. Gibson
I. R. Effect of Silicon Substitution on the Sintering and Microstructure of
Hydroxyapatite [текст] / I. R. Gibson, S. M. Best, W. Bonfield // J. Am. Ceram.
Soc. - 2002. - Vol.85. - №.11. - P.2771-2777.
. Barinov
S. M. Influence of fluorapatite minor additions on behavior of hydroxyapatite
ceramics [текст] / S. M. Barinov, F.rustichelli, V. P. Orlovskii, A. Lodini, S.
Oscarsson, S. A. Firstov, S. V. Tumanov, P.millet A. Rosengren // Journal of
Materials Science: Materials in Medicine. - 2004. - Vol.15. - №.3. - P.291-296.
. Gross
K. A. Sintered hydroxyfluorapatites. Part III: Sintering and resultant
mechanical properties of sintered blends of hydroxyapatite and fluorapatite
[текст] / K. A. Gross, K. A. Bhadang // Biomaterials. - 2004. - Vol.25. -
P.1395-1405.
. Gross
K. A. Sintered hydroxyfluorapatites. Part II: Sintering and resultant
mechanical properties of sintered blends of hydroxyapatite and fluorapatite
[текст] / K. A. Gross, K. A. Bhadang // Biomaterials. - 2004. - Vol.25. -
P.1385-1395.
. Jarcho
M. Hydroxylapatite synthesis and characterization in dense polycrystalline form
[текст] / M. Jarcho, C. H. Bolen, M. B. Thomas, J. Bobick, J. F. Kay, R. H.
Doremus // J. Mater. Sci. - 1976. - Vol.11. - P. 2027-2035.
. Ткаченко
М.В. Синтез гідроксиапатиту поєднанням хімічної та твердофазної реакцій [текст]
/ М.В. Ткаченко, І.Г. Іванов, З.З. Зиман // Вісник ХДУ, серія "Фізика”. -
1999. - Вип.3, № 440. - С.67-69.
. Fowler
B. O. Infrared studies of apatites.I. Vibrational assignments for calcium,
strontium, and barium hydroxyapatites utilizing isotopic substitution / B. O.
Fowler // Inorg. Chem. - 1974. - Vol.13, № 1. - P. 194-207.
. Fowler
B. O. Infrared studies of apatites. II. Preparation of normal and isotopically
substituted calcium, strontium and barium hydroxyapatites and
spectra-structure-composition correlations [текст] / B. O. Fowler // Inorg.
Chem. - 1974. - Vol.13, № 1. - P. 207-214.
. Blackeslee
K. C. Vibrational Spectra of Hydrothermally Prepared Hydroxyapatites [текст] /
K. C. Blackeslee, R. A. Condrate, Sr. // J. Am. Ceram. Soc. - 1971. - Vol.54, №
11. - P.559-563.
. Coblentz
Society, Inc., "Evaluated Infrared Reference Spectra" in NIST
Chemistry WebBook, NIST Standard Reference Database Number 69, Eds. P. J.
Linstrom and W. G. Mallard, National Institute of Standards and Technology,
Gaithersburg MD, 20899, http://webbook. nist.gov, (retrieved October 19, 2008).
. PDF-2
Data Base (Sets 1-46) PDF-2 46 6A JUN96 [Электронный ресурс]: / Copyright (C)
JCPDS-ICDD 1996 JCPDS - International Centre for Diffraction Data 12 Campus
Boulevard Newtown Square, PA 19073-3273 U. S. A. - 1 электрон. опт. диск
(CD-ROM): цветн.; 12 см. - (PDF-2, 1996). - Систем. требования: Pentium-486; 32
Mb RAM; CD-ROM Windows 98/2000/NT/XP. - Название с титул. экрана.
. Boultif
A. Powder pattern indexing with the dichotomy method [текст] / A. Boultif, D.
Louer // J. Appl. Cryst. - 2004. - Vol.37. - P.724-731.
. Уманский
Я.С. Рентгенография металлов и полупроводников [текст] / Уманский Я.С. - М.:
Металлургия, 1969. - 496 с.
. Горелик
С.С. Рентгенографический и электронооптический анализ [текст] / Горелик С.С.,
Расторгуев Л.Н. - М.: Металлургия, 1970. - 366 с.
97. Fityk [электронный
ресурс]: http://www.unipress. waw. pl/fityk/
<http://www.unipress.waw.pl/fityk/>
. Termine J. D.
Vibrational spectra of some phosphate salts amorphous to X-ray diffraction
[текст] / J. D. Termine, D. R. Lundy // Calc. Tiss. Res. - 1974. - Vol.15. -
P.55-70.
. Boskey A. L.
Conversion of amorphous calcium phosphate to microcrystalline hydroxyapatite. A
pH-dependent, solution-mediated, solid-solid conversion [текст] / A. L. Boskey,
A. Posner // J. Phys. Chem. - 1973. - Vol.77. - №19. - P.2313-2317.
. Kim S. Influence of
Ca/P ratios of starting solutions on the crystallization of amorphous calcium
phosphate to hydroxyapatite [текст] / S. Kim, H. - S. Ryu, H. S. Jung, K. S.
Hong // Met. Mater. Int. - 2004. - Vol.10. - №2. - P.171-175.
. Urch H. Calcium
phosphate nanoparticles with adjustable dispersability and crystallinity
[текст] / H. Urch, M. Vallet-Regi, L.ruiz, J. M. Gonzalez-Calbet, M. Epple //
J. Mater. Chem. - 2009. - Vol. 19. - P.2166-2171.
. Tao J. Evolution of
amorphous calcium phosphate to hydroxyapatite probed by gold nanoparticles
[текст] / J. Tao, H. Pan, J. Wang, J. Wu, B. Wang, X. Xu, R. Tang // J. Phys.
Chem. - C 2008. - Vol.112. - P.14929-14933.
. Rietveld H. M. A
profile refinement method for nuclear and magnetic structures [текст] / H. M.
Rietveld // J. Appl. Cryst. - 1969. - Vol.2. - P.65-71.
. Bienenstock A.
Calculation of the X-ray intensities from arrays of small crystallites of
hydroxyapatite [текст] / A. Bienenstock, A. S. Posner. // Arch. Biochem.
Biophys. - 1968. - Vol.124. - P.604-615.
. Celotti G.
Crystallinity in apatites: how can a truly disordered fraction be distinguished
from nanosize crystalline domains? [текст] / G. Celotti, A. Tampieri, S. Sprio,
E. Landi, L. Bertinetti, G. Martra, C. Ducati. // J. Mater. Sci.: Mater. Med. -
2006. - Vol.17. - P.1079-1087.
. Anderson C. W.
Programmed temperature dehydration studies of octacalcium phosphate [текст] /
C. W. Anderson, R. A. Beebe, J. S. Kittelberger // J. Phys. Chem. - 1974. -
Vol.78. - P.1631-1635.
. Sedlak J. M.
Temperature programmed dehydration of amorphous calcium phosphate [текст] / J.
M. Sedlak, R. A. Beebe // J. Coll. Interf. Sci. - 1974. - Vol.47. - №2. -
P.483-489.
. Gee A. Pyrophosphate
formation upon ignition of precipitated basic calcium phosphate [текст] / A.
Gee, V. R. Deitz // J. Amer. Chem. Soc. - 1955. - Vol.77. - P.2961-2965.
. Dorozhkin S. V.
Calcium orthophosphates in nature, biology and medicine [текст] / S. V.
Dorozhkin // Materials. - 2009. - Vol.2. - P.399-948.
. Christoffersen J.
The importance of formation of hydroxyl ions by dissociation of trapped water
molecules for growth of calcium hydroxyapatite crystals [текст] / J.
Christoffersen, J. Dohrup, M. R. Christoffersen // J. Cryst. Growth. - 1998. -
Vol.186. - P.275-282.
. Young R. A. Profile
shape functions in Rietveld refinements [текст] / R. A. Young, D. B. Wiles. //
J. Appl. Cryst. - 1982. - Vol.15. - P.430-438.
. Гинье А.
Рентгенография кристаллов. Теория и практика [текст] / Гинье А. - М.:
Физматгиз, 1961. - 604 с.
. Boskey A. L. Conversion
of amorphous calcium phosphate to microcrystalline hydroxyapatite. A
pH-dependent, solution-mediated, solid-solid conversion [текст] / A. L. Boskey,
A. Posner // J. Phys. Chem. - 1973. - Vol.77. - №19. - P.2313-2317.
. Kanzaki N. Calcium
phosphate clusters [текст] / N. Kanzaki, G. Treboux, K. Onuma, S. Tsutsumi, A.
Ito // Biomaterials. - 2001. - Vol.22. - P.2921-2929.
. Nylen M. U.
Molecular and ultrastructural studies of non-crystalline calcium phosphates
[текст] / M. U. Nylen, E. D. Eanes, J. D. Termine // Calc. Tissue. Res. - 1972.
- Vol.9. - P.95-108.
. Lazic S.
Microcrystalline hydroxyapatite formation from alkaline solution [текст] / S.
Lazic // J. Cryst. Growth. - 1995. - Vol.147. - P.147-154.
. Whittaker E. J. W.
Crystallography: An introduction for Earth Science (and other solid-state)
students [текст] / Whittaker E. J. W. - Pergamon Press, Oxford, 1981. - 254 с.
. Feki H. E. Structure
refinements by the Rietveld method of partially substituted hydroxyapatite: Ca9Na0.5
(PO4) 4.5 (CO3) 1.5 (OH) 2
[текст] / H. E. Feki, J. M. Savariault, A. Ben Salah // Journal of Alloys and
Compounds. - 1999. - Vol.287. - P.114-120.
. Vignoles M.
Occurrence of nitrogenous species in precipitated B-type carbonated
hydroxyapatites [текст] / M. Vignoles, G. Bonel // Calcif. Tiss. Int. - 1987. -
Vol.40. - № 2. - P.64-70.
. Gibson I. R. Novel
synthesis and characterization of an AB-type carbonate-substituted
hydroxyapatite [текст] / I. R. Gibson, W. Bonfield // J. Biomed. Mater. Res. -
2002. - Vol.59. - P.697-708.
. Dowker S. E. P.
Infrared study of trapped carbon dioxide in thermally treated apatites [текст]
/ S. E. P. Dowker, J. C. Elliott // J. Solid State Chem. - 1983. - Vol.47. -
P.164-173.
. Apfelbaum F. An FTIR
study of carbonate in synthetic apatites [текст] / F. Apfelbaum, H. Diab, I.
Mayer, J. D. B. Featherstone // J. Inorg. Biochem. - 1992. - Vol.45. -
P.277-282.
. Berry E. E. The
structure and composition of some calcium-deficient apatites [текст] / E. E.
Berry // J. Inorg. Nucl. Chem. - 1967. - Vol.29. - P.317-327.
. MacLennan G. The
crystal structure of dicalcium phosphate, CaHPO4 [текст] / G.
MacLennan, C. A. Beevers // Acta Cryst. - 1955. - Vol.8. - P.579-583.
. Trombe J. C. Some
features of the incorporation of oxygen in different oxidation states in
apatite lattice [текст] / J. C. Trombe, G. Montel // J. Inorg. Nucl. Chem. -
1978. - Vol.40. - P.15-21.
. Kijima T.
Preparation and Thermal Properties of Dense Polycrystalline Oxyhydroxyapatite
[текст] / T. Kijima, M. Tsutsumi // J. Am. Ceram. Soc. - 1979. - Vol.62. -
P.455-460.
. Ruys A. J.
Hydroxyapatite sintering characteristics: correlation with powder morphology by
high-resolution microscopy [текст] / A. J.ruys, C. C. Sorrell, A. Brandwood, B.
K.milthorpe // J. Mater. Sci. Lett. - 1995. - Vol.14. - P.744-747.
. Prevey P. S. X-ray
diffraction characterization of crystallinity and phase composition in
plasma-sprayed hydroxylapatite coatings [текст] / P. S. Prevey // Journal of
Thermal Spray Technology. - 2000. - Vol.9. - №.3. - P.369-376.
. Глинка Н.Л. Общая
химия [текст] / Глинка Н.Л. - Л.: Химия, 1977. - 704 с.
. Cornu A.compilation
of mass spectral data [текст] / Cornu A., Massot R. - London: Heyden and Son
Ltd, 1974. - 1096 p.
. Simpson D. R.
Substitutions in apatite: I potassium-bearing apatite [текст] / D. R. Simpson
// Am Mineral. - 1968. - Vol.53. - P.432-44.
. Fowler B. O.
Infra-red spectra of hydroxyapatite, octacalcium phosphate and pyrolysed
octacalcium phosphate [текст] / B. O. Fowler, E. C. Moreno, W. E. Brown //
Arch. Oral. Biol. - 1966. - Vol.11. - P.477-492.
. Raynaud S.
Determination of Calcium/Phosphorus Atomic Ratio of Calcium Phosphate Apatites
Using X-ray Diffractometry [текст] / S. Raynaud, E. Champion, D. B. Assollant,
J. P. Laval, // J. Am. Ceram. Soc. - 2001. - Vol.84. - №.2. - P.359-66.
. Ткаченко М.В. Синтез
гідроксиапатиту поєднанням хімічної та твердофазної реакцій [текст] / М.В.
Ткаченко, І.Г. Іванов, З.З. Зиман // Вісник ХДУ: серія "Фізика”. - 1999. -
Вип.3. - № 440. - С.67-69.
. Zyman Z. Z.
Possibilities for strengthening hydroxyapatite ceramics [текст] / Z. Z. Zyman,
I. G. Ivanov, V.I. Glushko // J. Biomed. Mater. Res. - 1999. - Vol.46. - P.73 -
79.
. Пинес Б.Я. Спекание,
крип, отдых, рекристаллизация и другие явления, обусловленные самодиффузией в
кристаллических телах [текст] / Б.Я. Пинес // Успехи физических наук. - 1954. -
Т. LII. - Вып.4. - С.501-559.
. Њlуsarczyk A. Porous
hydroxyapatite ceramics [текст] / A. Њlуsarczyk, E. Stobierska, Z. Paszkiewicz
// J. Mater. Sci. lett. - 1999. - Vol.18. - P.1163 - 1165.
. Zhou J. High
temperature characteristics of synthetic hydroxyapatite [текст] / J. Zhou, X.
Zhang, J. Chen, S. Zeng, K. De Groot // J. Mater. Sci: Mater. in Med. - 1993. -
Vol.4. - P.83-85.
. Hench L. L.
Bioceramics [текст] / L. L. Hench // J. Am. Ceram. Soc. - 1998. - Vol.81. - №7.
- P.1705-1728.
. Low I. M.
Depth-Profiling of Crystal Structure, Texture, and Microhardness in a
Functionally Graded Tooth Enamel [текст] / I. M. Low // J. Am. Ceram. Soc. -
2004. - Vol.87. - №.11. - P.2125-2131.
. Zyman Z. On
impurities and the internal structure in precipitates occurring during the
precipitation of nanocrystalline calcium phosphate [текст] / Z. Zyman, M.
Epple, D. Rokhmistrov, V. Glushko // Mat. - wiss. u. Werkstofftech. - 2009. -
Vol.40, № 4. - P.297-301.
. Rokhmistrov D. V. On
the nature of amorphous calcium phosphate at the initial stages of
hydroxyapatite crystallization [текст] / D. V. Rokhmistrov, Z. Z. Zyman, K. A.
Kornieieva, V.I. Glushko // German-Ukrainian Symposium on Nanoscience and
Nanotechnology, 22-25 Sept. 2008. - Essen, Germany, 2008. - P.62.
. Корнеева Е.А.
Особенности картины дифракции рентгеновских лучей коллоидных систем на основе
гидроксилапатита [текст] / Е.А. Корнеева, Д.В. Рохмистров, З.З. Зыман //
Дифракционные методы исследования вещества: от молекул к кристаллам и
наноматериалам: конф. - шк. для молодых ученых, 30 июня - 3июля 2008 г.: тезисы
докл. - Черноголовка, Россия, 2008. - С.24.
. Зыман З.З.
Образование гидроксиапатита из аморфного фосфата кальция [текст] / З.З. Зыман,
Д.В. Рохмистров, Е.А. Корнеева, В.И. Глушко // ХІІ Міжнародна конференція з
фізики і технології тонких плівок та наносистем, 18-23 травня, 2009,
Івано-Франківськ, Україна, 2009. - С.181-182.
. Зыман З.З.
Исследование процессов газовыделения гидроксилапатита при отжиге [текст] / З.З.
Зыман, Д.В. Рохмистров, В.И. Глушко, И.Г. Иванов // Вісник ХНУ, серія
"Фізика". - 2006. - №739. - вип.9. - С.159-163.
. Zyman Z. Evolution
in the impurity state of hydroxyapatite during post-precipitation treatment
[текст] / Z. Zyman, M. Epple, D. Rokhmistrov, V. Glushko, I. Ivanov //
Biomaterialen. - 2006. - Vol.7. - № 3. - P.253.
. Зыман З.З. Получение
карбонат-замещенного гидроксилапатита из водных растворов [текст] / З.З. Зыман,
Д.В. Рохмистров, В.И. Глушко, Е.А. Корнеева // Физические явления в твердых
телах: 8-я междунар. конф., 11-13 дек. 2007 г.: тезисы докл. - Х., 2007. -
С.139.
. Zyman Z. Impurity
effects in the structure of nanocrystalline hydroxyapatite during calcinations
[текст] / Z. Zyman, D. Rokhmistrov, I. Ivanov, V. Glushko, M. Epple //
Fundamental and Clinical Applications: 8th Essen Symposium on
Biomaterials and Biomechanics, 21-23 Sept. 2005. - University Duisburg-Essen,
Essen, Germany, 2005. - P.18.
. Zyman Z.
Investigation of Gas release at heating of hydroxylapatite pressings [текст] /
Z. Zyman, I. Ivanov, D. Rokhmistrov // Crystal Materials 2005 ICMM 2005:
intern. conf., 30 May-2 June, 2005, Kharkov, Ukraine, 2005. - p.36.
. Зыман З.З.
Температурная зависимость параметров решетки гидроксилапатита [текст] / З.З.
Зыман, Д.В. Рохмистров, В.И. Глушко, И.Г. Иванов // Физические явления в
твердых телах: 7-я междунар. научн. конф., 12-16 дек. 2005 г.: тезисы докл. -
Х., 2005. - С.44.
. Zyman Z. The
influence of foreign ions on the crystal lattice of hydroxyapatite upon heating
[текст] / Z. Zyman, D. Rokhmistrov, I. Ivanov, M. Epple // Mat. - wiss. u.
Werkstofftech. - 2006. - Vol.37. - № 6. - P.530-532.
. Zyman Z. Z. Thermal
impurity reactions and structural changes in slightly carbonated hydroxyapatite
[текст] / Z. Z. Zyman, D. V. Rokhmistrov, V.I. Glushko, I. G. Ivanov // J Mater
Sci: Mater Med. - 2009. - Vol. 20. - P.1389-1399.
. Zyman Z. Z.
Sintering peculiarities for hydroxyapatite with different degrees of
crystallinity [текст] / Z. Z. Zyman, I. G. Ivanov, D. V. Roсhmistrov, V.I. Glushko,
N. V. Tkachenko, S. M. Kijko // J. Biomed. Mater. Res. - 2001. - Vol.54. - № 2.
- P.256-263.
. Иванов И.Г.
Особенности кривых распределения частиц порошка и кристаллитов керамики
гидроксилапатита по размерам [текст] / И.Г. Иванов, Д.В. Рохмистров, З.З. Зыман
// Физические явления в твердых телах: 5-я междунар. конф., 25-26 окт. 2001 г.:
тезисы докл. - Х., 2001. - С.77.
. Зыман З.З.
Возможности улучшения функциональных свойств керамики в связи с особенностями
строения и свойств исходного порошка гидроксилапатита [текст] / З.З. Зыман,
Д.В. Рохмистров, И.Г. Иванов // Функционализированные материалы: синтез,
свойства и использование: междунар. конф., 24-29 сент. 2002 г.: тезисы докл. -
К., 2002. - С.31-32.
. Зыман З.З. Улучшение
механических свойств керамики гидроксилапатита смешиванием порошков различного
происхождения [текст] / З.З. Зыман, Д.В. Рохмистров, И.Г. Иванов // Физические
явления в твердых телах: 6-я междунар. конф., 28-29 окт. 2003 г.: тезисы докл.
- Х., 2003. - С.90.