Получение бифазной системы ГФ/В-ТКФ из аморфного фосфата кальция
Содержание
Введение
.
Литературный обзор
.1
Аморфный фосфат кальция (АФК)
.2
Кристаллическая структура гидроксилапатита
.3
β-трикальций
фосфат
.4
Методы синтеза
.5
Метод осаждения из водных растворов
.6
Качественный и количественный фазовый анализ
.6.1
Качественный фазовый анализ
.6.2
Количественный фазовый анализ
.6.3
Определение фазы в многофазном образце методом подмешивания внутреннего
стандарта
.
Экспериментальная часть
.1
Материалы и методы
.2
Результаты и обсуждения
Выводы
Литература
Введение
Материалы для костных имплантатов в настоящее
время вызывают большой интерес вследствие растущих запросов ортопедии. Наиболее
известным и востребованным представителем этого класса материалов является
гидроксилапатит Са10(РО4)6(ОН)2 (ГА). Предшественником образования
гидроксилапатита является аморфный фосфат кальция (АФК) [1]. Получают ГА и АФК
методом осаждения из водных растворов [2]. Однако получаемый при этом осадок
содержит также нитратные примеси, которые являются токсичными и вредными для
организма человека. Процесс очистки АФК от примесей приводит к удалению ионов
кальция из раствора, необходимых для формирования стехиометрического ГА на
более поздних стадиях синтеза [3], что является причиной изменения фазового
состава образцов.
В медицинской практике применение ГА в чистом
виде в качестве материала для имплантатов часто невозможно из-за его низких
прочностных свойств. На основании многочисленных исследований было показано,
что наиболее оптимальным для медицинских применений является состав 60% ГА/40% β-ТКФ.
Ввиду изменения фазового состава образцов при очистке от примесей
представляется интересным исследование оптимального состава кальций-фосфатных
материалов с различным стехиометрическим отношением Са/P для получения бифазной
системы ГА/β-ТКФ, а также исследование
стабильности АФК в различных средах.
Целью дипломной работы является изучение
возможности получения бифазной системы ГА/β-ТКФ
из аморфного фосфата кальция, а также исследование его стабильности в различных
средах (водные растворы, воздух).
1. Литературный обзор
.1 Аморфный фосфат кальция (АФК)
В процессе получения ГА из водных растворов на
ранних стадиях осаждения образуется промежуточная фаза [1,4-6], аморфный фосфат
кальция (АФК). Это вещество дает широкий диффузный максимум на рентгенограммах
в интервале углов дифракции 2q 29-30,5о (рис. 1), характерный
для аморфных соединений. АФК является метастабильной фазой и наблюдается в
течение нескольких часов после начала синтеза [7-9]. Первые следы
нанокристаллического ГА можно обнаружить на рентгенограммах через 5-7 часов
после начала синтеза при температурах 20-25оС (рис. 2). При этом интенсивность
диффузного максимума уменьшается, а его положение смещается в сторону больших
углов 2θ
примерно
на 1,5о.
Химический анализ, а также структурные исследования
осадков ГА с различным исходным отношением Ca/P реагентов, показали [10], что
АФК имеет отношение Ca/P около 1,5 (табл. 1), при этом диффузные максимумы
практически совпадают (рис. 3).
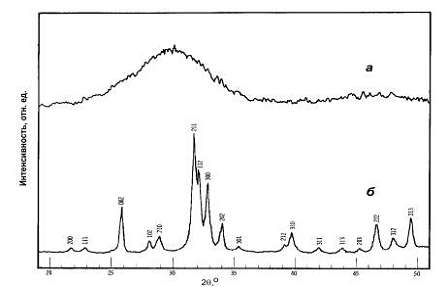
Рис. 1. Рентгенограммы АФК (а) и ГА (б) [7].
Познер и др. [11] на основании анализа функции
радиального распределения атомов предложил кластерную модель строения частиц
АФК. Согласно модели Познера, АФК состоит из сферических кластеров Ca9(PO4)6
диаметром около 9.5Е (рис.4)[12-14]. Образование ГА связывается с упорядочением
этих кластеров в пространстве и образованием дальнего порядка в расположении
атомов (ионов), характерном для кристаллических тел.
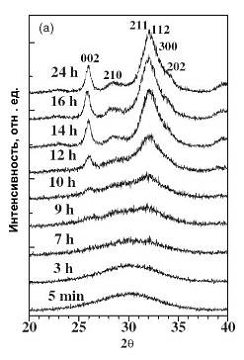
Рис. 2. Рентгенограммы осадка ГА после различного
времени кристаллизации в маточном растворе [7].
Таблица 1.
Атомное отношение Ca/P в начале синтеза и в АФК
[10]
|
Исходное
отношение Са/Р в растворе
|
1,25
|
1,33
|
1,50
|
2,00
|
|
Отношение
Са/Р в АФК
|
1,40±0,01
|
1,47±0,01
|
1,49±0,01
|
1,49±0,01
|
Кластерная модель Познера получила широкое
распространение. Однако в последнее время появилась точка зрения, что АФК
состоит из нанокристаллических частиц. Данное заключение было сделано на основе
данных [15-17], просвечивающей электронной микроскопии высокого разрешения и
микродифракции на образцах АФК.
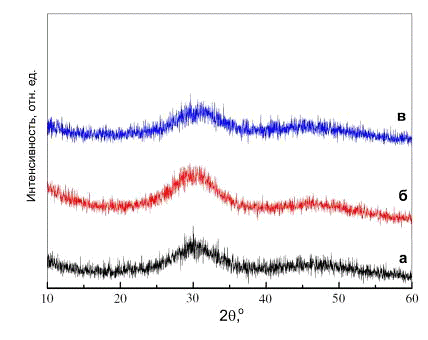
Рис. 3. Рентгенограммы АФК с исходным атомным
отношением Ca/P в маточном растворе 1,25 (а), 1,5(б) и 2,0 (в) [10].

Рис. 4. Кластерная модель строения ГА. В белом
круге выделен “кластер Познера” [13].
1.2 Кристаллическая структура гидроксилапатита
Гидроксилапатит кристаллизуется [18] в
гексагональной сингонии с пространственной группой Р63/т и параметрами
элементарной ячейки а=b=9.432Е, c=6.881Е (таблица 2). ГА представляет собой
слоистый кристалл, содержащий более 100 атомов в элементарной ячейке (рис. 5).
Кристалл ГА имеет две структурных
подсистемы (рис. 5): первую создают Са- каналы с группами ОН- внутри них, а
вторая - это остовый каркас, в который ионы X-F-,  ,
,  могут
внедряться с малой вероятностью, а такие ионы, как СО32-, могут изоморфно
замещать РО4- - группы. Ионы ОН-, расположенные в кальциевых каналах, могут
изоморфно замещаться на ионы Cl- и F- .
могут
внедряться с малой вероятностью, а такие ионы, как СО32-, могут изоморфно
замещать РО4- - группы. Ионы ОН-, расположенные в кальциевых каналах, могут
изоморфно замещаться на ионы Cl- и F- .
Таблица 2.
Кристаллохимические параметры
некоторых фосфатов кальция.
|
Название
|
Химическая
формула
|
Пространственная
группа
|
Параметры
решетки
|
|
|
|
нм
|
угол,
о
|
|
Октакальций
фосфат
|
Ca8H2(PO4)6∙5H2O
|
P1
|
a=0,9529;
b=1,899; c=0,6855;
|
α=90,14;
β=92,52; γ=108,67
|
|
a-Трикальций
фосфат
|
Са3(РО4)2
|
Р21/а
|
а=1,2887;
b = 2,728; c =1,5219
|
b = 126.2
|
|
b-Трикальций
фосфат
|
Са3(РО4)2
|
а=
1,0428; c = 3,7378
|
|
|
Гидроксилапатит
|
Са5(РО4)3ОН
|
Р63/m
|
а
= 0,9432; c = 0,6880
|
|
|
Кальций
- дефицитный гидроксилапатит
|
Ca9(HPO4)(PO4)5OH
|
Р63/m
|
а
=0,944; c=0,6881
|
|
|
Фторапатит
|
Са5(РО4)3F
|
Р63/m
|
а
= 0,9377; c = 0,6880
|
|
|
Хлорапатит
|
Са5(РО4)3С1
|
Р63/m
|
а
= 0,9641; c = 0,6771
|
|
|
Карбонат-апатит
|
Са10(РО4)6CO3
|
Рa
|
a
= 0,9529; b=1,910; с = 0,686
|
b = 87.97
|
На рис.6 показан фрагмент структуры
гидроксилапатита. Группы [РО4 ] образуют тетраэдры со средним расстоянием
Р-О=1.53±0.02Е. Атомы Са занимают в структуре ГА два кристаллографически
независимых положения. Находящийся в положении 2 атом Са окружён шестью атомами
кислорода, принадлежащих группам РО43- и ОН- , в то время как атом Са,
занимающий положение 1, имеет окружение атомами кислорода близкое к
октаэдрическому. Атомы Ca в положении 2 образуют треугольник в плоскости,
перпендикулярной оси С. Треугольники повёрнуты друг относительно друга на 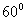 вдоль этой
оси. В структуре фторапатита атомы F расположены в центре таких треугольников.
вдоль этой
оси. В структуре фторапатита атомы F расположены в центре таких треугольников.
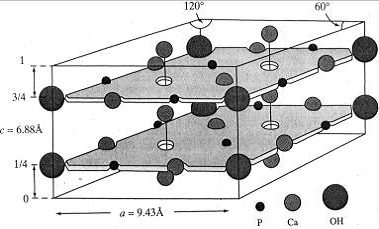
Рис.5 Упрощенный вид элементарной
ячейки гидроксилапатита.
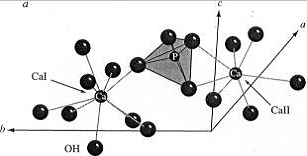

Рис. 6. Фрагмент структуры
гидроксилапатита:  -
координационные полиэдры кальция; б - анионные столбики ионов
-
координационные полиэдры кальция; б - анионные столбики ионов 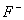 , ,
, ,  .
.
В случае ГА группы располагаются
немного выше или ниже центра. Атомы 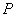 окружены четырьмя атомами
окружены четырьмя атомами 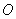 и образуют
тетраэдр практически правильной формы, лишь с небольшим искажением. При
довольно сложной координации атомов
и образуют
тетраэдр практически правильной формы, лишь с небольшим искажением. При
довольно сложной координации атомов 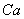 в ячейке ГА образуются 75 связей - (без учета
связей с кислородом иона гидроксила), 24 связи -, 6 связей -
в ячейке ГА образуются 75 связей - (без учета
связей с кислородом иона гидроксила), 24 связи -, 6 связей - 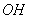 и 2 связи .
и 2 связи .
Стехиометрический ГА может быть
также описан в моноклинной сингонии (пространственная группа Р21/b с
параметрами решетки, а=9.4214(8)Е, b=2a, c=6.8814(7)Е, g=120°), причем такое описание
не связано с ограничениями, обусловленными зеркальной симметрией. Снижение
симметрии до моноклинной является результатом упорядоченного расположения ОН- в
кальциевых каналах, а также взаимного упорядочения этих каналов таких образом,
что происходит двукратное увеличение параметра b элементарной ячейки. ГА
моноклинной сингонии может быть получен только гидротермальным синтезом или
термообработкой ГА в атмосфере водной пары.
.3 β-трикальций
фосфат
β-ТКФ (β-трикальций
фосфат) является ортофосфатом кальция со стехиометрическим составом Са3(РО4)2.
Он не может выпадать в осадок из водного раствора, а получается только через
кальцинирование при температуре выше 800°С.
(HPO4)(PO4)5 (OH)® Са3(РО4)2 + Н2О
Выше 1125°С он превращается в a-ТКФ. На температуру
превращения фазы сильно влияют примеси. При комнатной температуре, β-ТКФ имеет
меньшую растворимость, чем a-ТКФ.
В медицине β-ТКФ находит
применение как в чистой фазе, так и в смеси с гидроксилапатитом (так называемый
« бифазный фосфат кальция») в качестве материала для замены кости.
Кристаллографические данные β-ТКФ
следующие (рис.7): ромбоэдрическая система 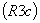 ,
, 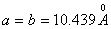 ;
; 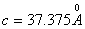 (гексагональная ось); число атомов
в ячейке
(гексагональная ось); число атомов
в ячейке 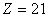 ; плотность
3.067
; плотность
3.067  .
.
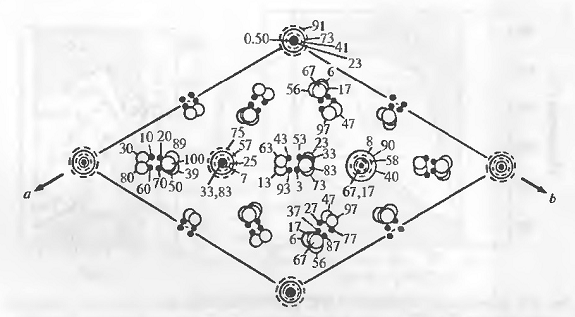
Рис.7. Структура элементарной ячейки
β-ТКФ.
.4 Методы синтеза
Существует три основных метода
синтеза фосфатов кальция: осаждением из растворов (“мокрый метод”),
твердофазный синтез (“сухой метод”) и гидротермальный синтез. На практике
преобладает синтез фосфатов кальция из водных растворов. Данный способ зависит
от большого количества изменяющихся факторов, которые не всегда дают
возможность достичь хорошую воспроизводимость, сохранить стехиометрическое
соотношение Са/Р в процессе синтеза, т.е. получить порошок с заданными и
физико-химическими свойствами. Среди множества факторов наиболее важными
являются - рН раствора, температура реакции и продолжительность синтеза.
Твердофазный синтез ГА является более длительным и энергоемким. Кроме того,
этим методом тяжело достичь гомогенности конечного продукта.
В зависимости от метода синтеза,
можно получить порошки с различной морфологией, удельной поверхностью,
стехиометрией и степенью кристалличности.
В результате смешения водных
растворов соединений, содержащих ионы Са2+ и РО43-, при сохранении рН≥7
по методу осаждения, образуется осадок ГА. В качестве источника Са2+
используются СаС12, Ca(NO3)2, Са(ОН)2, СаСО3, CaSO4 • 2Н2О, (СН3СОО)2Са и др.
Как источник фосфатов- ионов могут быть использованы Н3РО4, NH4H2PO4,
(NH4)2HPO4 или К3РО4. Для регулирования рН и введения ОН- групп применяют водные
растворы аммиака или гидроксидов щелочных металлов.
.5 Метод осаждения из водных
растворов
Для мокрых методов [18] является
характерным образование на начальной стадии осадка, который является
нестехиометрическим ГА.
Среди множества мокрых методов можно
выделить несколько "классических", которые с теми или иными
изменениями приводятся в литературе. Одним из них является метод, который
основан на использовании нитрата кальция:
10Ca(NO3)2+
6(NH4)2HPO4+ 8NH4OH => Са10(РО4)6(OН)2
+ 20NH4NO3 +6H2O
Также в качестве источника Са можно
использовать Ca(NO3)2:
10Ca(NO3)2 +
6KH2PO4 + 14NaOH =>Са10(РО4)б(OН)2
+ 6KNO3 + 14NaNO3 + 12Н2О
Рост кристаллов и их выделение из
раствора происходит постепенно; отфильтрованный осадок ГА промывают водой и
спиртом, затем сушат при температуре 40-50°С.
Также был предложен метод, в котором
ГА получается по реакции:
СаС12 + 6(NH4)2HPO4 + 8NH4OH
=>Са10(РО4)6(ОН)2 + 20NH4C1 + 6H2O
После отстаивания осадок промывают
декантацией до рН=7, фильтруют на воронке Бюхнера и сушат на воздухе при
температуре 100°С. Затем осадок прокаливают в муфельной печи при температуре
1000°С в течение 2-х часов.
Другая группа классических методов
получения ГА кальция мокрым способом основана на реакции :
Са(ОН)2 + 6Н3РО4 Þ
Са10(РО4)6(ОН)2 +18Н2О
Указанная выше базовая реакция между
Са(ОН)2 и Н3РО4 может быть трансформирована таким образом, что одни реагенты
будут заменены на другие (иногда частично) того же назначение. Например,
описано получение ГА на основе реакции:
Са(ОН)2 + 6(NH4)2HPO4 => Са10(РО4)6(ОН)2
+ 12NH4OH + 6Н2О
В результате смешивания
эквимолекулярных количеств реагентов в течении 1часа и последующего высушивания
осадка при 85°С получали порошок белого цвета, который прокаливали при 1000°С
на воздухе в муфельной печи.
Можно получать высококачественный ГА
из смеси Са(ОН)2, Са3(РО4)2, (NH4)3PO4, и Н3РО4, которая соответствует составу
конечного продукта, с последующей ультразвуковой обработкой при частоте 10-30
кГц.
Образование ГА также возможно при
добавлении Са(ОН)2 к водной суспензии Са3(РО4)2 при перемешивании в интервале
температур от 5 до 100°С в атмосфере инертного газа; рН смеси поддерживают
равным 10, регулируя количество подаваемого Са(ОН)2 до тех пор, пока Са/Р не
составит 1.6. Далее взаимодействие проводят до момента, когда Са/Р достигает
1.67, поддерживая рН = 7-11.
ГА получают при быстром смешивании
растворов Ca(NO3)2 и К3РО4. Исходное отношение Са/Р составляет 1.58, а через 6
часов - 1.67. Осаждением из растворов могут быть полученные Mg-, Y-, Zn-, Cd-
замещенные аппатиты.
.6 Рентгеновский фазовый анализ
.6.1 Качественный фазовый анализ
Определение фазового состава образца
[19] является наиболее распространенной задачей рентгеноструктурного анализа.
Каждая фаза имеет свою кристаллическую решетку и характеризуется определенным набором
межплоскостных расстояний. Поэтому для решения задачи о том, какая фаза
присутствует в образце достаточно, рассчитав рентгенограмму или дифрактограмму,
снятую по методу поликристалла (порошка), сравнить полученный ряд
межплоскостных расстояний с табличными значениями. Совпадение опытных и
табличных значений d/n и относительной интенсивности линий позволяет однозначно
идентифицировать присутствующую в образце фазу.
Сравнение с табличными результатами
начинают с наиболее интенсивных линий. Если 3-4 наиболее интенсивных линии
предполагаемой фазы отсутствуют, то полученные значения d/n следует сравнивать
с табличными для другой фазы.
Если в анализируемом образце
присутствуют несколько фаз, то на рентгенограмме наложены дифракционные картины
от всех этих фаз, причем интенсивность линий каждой фазы зависит от ее объемной
доли. В этом случае пользование таблицами затруднительно, поскольку наиболее
сильные линии рентгенограммы могут принадлежать разным фазам, и возникает
необходимость проверки большого числа их возможных комбинаций. В этих случаях
нужно знать химический состав образца и (или) провести его металлографический
анализ. Комплексное применение методов необходимо как для ускорения фазового
анализа, так и для однозначности его.
Важным вопросом является
чувствительность качестственного фазового анализа, т. е. минимальное количество
фазы, которое можно определить в многофазных композициях. Чувствительность
определяется соотношением интенсивности наиболее сильной линии на
рентгенограмме фазы и интенсивности фона.
Если линии определяемой фазы
размыты, чувствительность качественного фазового анализа резко понижается.
При проведении фазового анализа
желательно применение селективно поглощающих фильтров. Фильтр ослабляет
сплошной спектр и позволяет избавиться от b-линий,
которые существенно затрудняют идентификацию фаз в многофазных композициях.
Крупнокристаллические образцы
следует во время съемки вращать, чтобы увеличить количество вещества, которое
участвует в создании рентгеновской картины. Однако, если крупнозерниста только
одна из фаз, ее линии можно сразу выделить, сопоставив рентгенограммы, снятые с
вращением и без вращения образца.
Чувствительность качественного
фазового анализа можно повышать очищением образцов от загрязнений.
.6.2 Количественный фазовый анализ
Количественный фазовый анализ - это
определение количества какой-либо одной фазы или ряда фаз в многофазных
композициях.
Существует много разновидностей
методов рентгеновского количественного фазового анализа. Но в каждом из них
происходит сравнение интенсивности так называемых «аналитических» линий каждой
фазы - это линии наибольшей интенсивности, свободные от наложения других линий
анализируемой фазы или остальных фаз многофазного образца.
Ясно, что для выделения
аналитических линий необходимо знать качественный фазовый состав или
предварительно провести качественный фазовый анализ образца.
.6.3 Определение фазы в многофазном
образце методом подмешивания внутреннего стандарта
Метод применим только для порошковых
образцов. Для этого, к анализируемому порошку добавляют известное количество
порошка-эталона. В качестве стандарта используют порошки химически стабильных
веществ с кубической решеткой, дающие на рентгенограммах интенсивные и и узкие
линии. Нужно, чтобы линии стандарта не накладывались на линии анализируемой
смеси, а аналитические линии эталона и фазы были обязательно свободны от
наложений. Затем тщательно перемешивают и определяют отношение интенсивности
аналитических линий анализируемой фазы и стандарта.
Недостатком метода внутреннего
стандарта является то, что добавление вещества-эталона приводит к разбавлению
определяемых фаз, а следовательно, к снижению чувствительности и точности
анализа количества фазы при ее малом содержании.
2. Экспериментальная часть
.1 Материалы и методы
Синтез АФК проводили методом
осаждения из водных растворов [2]. Соотношение реагентов выбирали из такого
расчёта, чтобы в результате получить АФК с молярным отношением Са/P=1,5ч2,0.
Синтез проводили путём вливания раствора гидрофосфата аммония в раствор нитрата
кальция для образования суспензии и тщательно перемешивали в течение минуты. Отделение
АФК от раствора проводили при помощи центрифугирования. Для очистки АФК от
примесей использовали промывку осадка дистиллированной водой. Полученный осадок
помещали в морозильную камеру для удаления воды путём возгонки льда при
отрицательной температуре (-23ч25°С) в течение месяца. Полученный сухой порошок
АФК хранили при отрицательной температуре. Для определения фазового состава
образцов после очистки от примесей АФК отжигали при 1000°С в течение часа. От
полученных образцов снимали рентгенограммы при помощи рентгеновского
дифрактометра ДРОН-2.0 (U=30 кВ, I=10 mA) в медном излучении. Определение
фазового состава образцов осуществлялось путём сравнения рентгенограмм
исследуемых образцов со стандартами ГА и β-ТКФ [20]. Для исследования
стабильности АФК в различных средах, исходные образцы откачивали в вакууме до
давления 3·10-2 мм.рт.ст. Затем образцы помещали в дистиллированную воду.
Другая серия образцов хранилась на воздухе. Эксперименты проводились при
комнатной температуре. Стехиометрическое отношение Са/P определялось исходя из
известного фазового состава образцов по формуле:
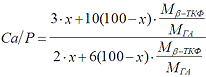
где М β-ТКФ и МГА -
молярная масса β-ТКФ и ГА
соответственно, х- весовая доля β-ТКФ в %.
.2 Результаты и обсуждения
На рис.1 приведены ИК-спектры
исходного АФК со стехиометрическим отношением Са/P=1,67. Видно, что на
ИК-спектрах присутствуют полосы поглощения при 3180 см-1 соответствующие
колебаниям ионов NH4, а также 1385 см -1, принадлежащая колебаниям ионов НNО3-.
Наличие этих полос поглощения на ИК-спектрах образцов АФК связано с
присутствием в последних нитратных примесей (нитрат кальция, нитрат аммония).
Удаление этих примесей приводит к изменению фазового состава образцов, что
хорошо видно на рентгенограммах образцов содержащих примеси и очищенных от
примесей (рис.2).
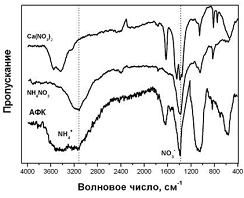
Рис.1. ИК-спектры образцов аморфного
фосфата кальция, NH4NO3 и Ca(NO3)2.
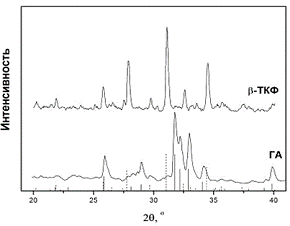
Рис.2 . Фазовый состав образцов АФК,
отожженных при 10000С, без предварительной очистки от примесей (ГА) и после
очистки (β-ТКФ).
Как видно, фазовый состав образцов
заметно изменяется при очистке от примесей. Видно, что очищенные от примесей
образцы содержат одну фазу - β-ТКФ, а не очищенные от примесей -
ГА.
На рис.3 приведена зависимость
отношения Са/P образцов АФК до и после очистки от примесей для исходных
образцов АФК с различным стехиометрическим отношением Са/P.
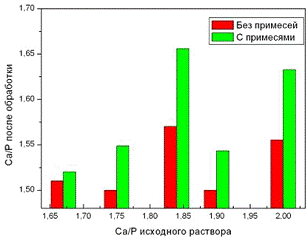
Рис. 3. Изменение отношения Са/P в
образцах АФК после очистки от примесей.
Видно, что наибольшее значение
отношения Са/P после очистки от примесей имеют образцы АФК с исходным
отношением Са/P =1,84 и 2,0 , что соответствует содержанию β-ТКФ в
образцах АФК 55% и 65% соответственно. Последние два образца обладают фазовым
составом близким к двухфазной системе, которая используется в медицинских
применениях (60%ГА/ 40%β-ТКФ).
Следующим этапом работы стало исследование стабильности АФК на воздухе и в
воде. Как показывают результаты (рис.4), оба образца кристаллизуются в воде в
течение 2-3 дней, о чём свидетельствует смещение диффузного максимума в область
больших углов (от 30 до 32°), а также появление уширенных линий, характерных
для нанокристаллического ГА в районе 25 и 30ч33°. В то же время хранение
образцов АФК на воздухе (рис. 5) не приводит к изменению дифракционного профиля
в течение недели.
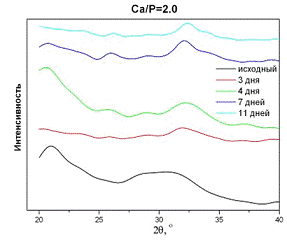
Рис.4. Фазовый состав образцов АФК
после хранения в воде с молярным отношением Са/P=2,0.
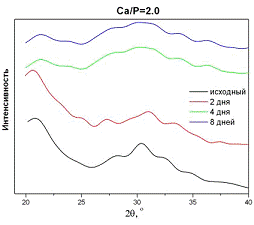
Рис.5. Фазовый состав образцов АФК
после хранения на воздухе с молярным отношением Са/P=2,0.
Было обнаружено, что содержание β-ТКФ в
образцах после хранения в воде изменяется (рис. 6). При этом с начала
количество β-ТКФ
возрастает (при малых временах хранения), а затем постепенно уменьшается. Этот
результат, по-видимому, связан с тем обстоятельством, что ионы Са, находящиеся
в гидратированной оболочке вокруг кластеров, при помещении образцов АФК в воду
переходят в раствор. В результате фазовый состав образцов соответствует β-ТКФ. При
больших временах хранения АФК в воде, когда в растворе устанавливается
равновесная концентрация ионов Са2+, начинается обратный процесс: ионы Са2+
проникают в кластеры Познера и образуют элементарные ячейки ГА. В результате
чего количество β-ТКФ
уменьшается.
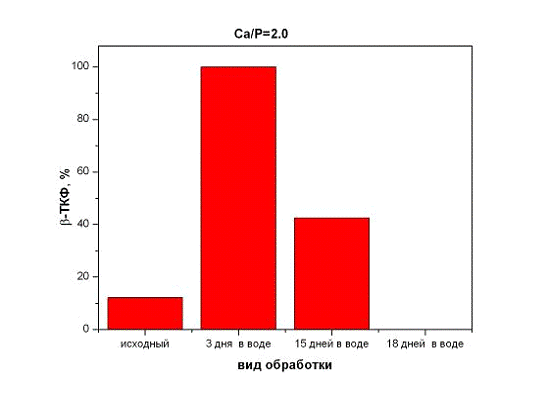
Рис.6. Содержание β-ТКФ в
образцах после хранения в воде с молярным отношением Са/P=2,0.
Выводы
Оптимальные условия получения АФК
без примесей такие, при которых исходный АФК имеет отношение Са/Р=1,84 и 2,0.
При этом образцы после отжига при 10000С имеют фазовый состав 55% ГА/45% β-ТКФ. Такие
отношения являются оптимальными для получения двухфазной системы, которая
используется для медицинских применений. Данная двухфазная система может быть
использована для получения кальций-фосфатных цементов.
Обнаружено изменение фазового
состава образцов АФК, помещенных в жидкость. Оно связано с уходом ионов Са2+ в
раствор при малых временах хранения осадка в жидкости и диффузией ионов Са2+ в
кластеры Познера, что приводит к образованию ГА при больших временах хранения
осадка в жидкости
Вакуумная откачка образцов АФК
позволяет сохранить кальций-фосфатный осадок в аморфном состоянии в течение
недели.
Литература
синтез фосфат кальций
E.D.
Eanes, I.H.
Gillesen, A.S.
Posner. Nature,
1965, 208, 365-367.. Jarcho, C.H. Bolen, M.B. Thomas, J. Bobick, J.F. Kay, R.H.
Doremus, Hydroxylapatite synthesis and characterization in dense
polycrystalline form. J. Mater. Sci., 1976, 11, 2027-2035.. Zyman, D.
Rokhmistrov, V. Glushko. Structural and compositional features of amorphous
calcium phosphate at the early stage of precipitation. J. Mater. Sci: Mater
Med., 2010, 21, 123-130..S. Tung, W.E. Brown, An intermediate state in
hydrolysis of amorphous calcium phosphate, Calcif. Tissue Int., 1983, 35,
783-790..S. Posner, F. Betts, Synthetic amorphous calcium phosphate and its
relation to bone mineral structure, Acc. Chem. Res., 1975, 8, 273-281.. Liu, Y.
Huang, W. Shen, J. Cui, Kinetics of hydroxyapatite precipitation at pH 10 to
11, Biomaterials, 2001, 22, 301-306.. Kim, H.S. Ryu, H. Shin, H.S. Jung, K.S.
Hong, Direct observation of hydroxyapatite nucleation from amorphous phase in a
stoichiometric calcium/phosphate aqueous solution, Chemistry Letters, 2004,
33(10), 1292-1293..D. Eanes, J.D. Termine, М.U.
Nylen, An Electron Microscopic Study of the Formation of Amorphous Calcium
Phosphate and Its Transformation to Crystalline Apatite, Calc. Tissue. Res.,
1973, 12, 143-158..S. Posner Calculation of the X-Ray intensities from arrays
of small crystallites of hydroxyapatite, Arch. Biochem. Biophys., 1968, 124,
604-615. . Li, W. Weng, In vitro synthesis and characterization of amorphous
calcium phosphates with various Ca/P atomic ratios, J Mater Sci: Mater Med.,
2007, 18, 2303-2308.. Betts, A.S. Posner, An x-ray radial distribution study of
amorphous calcium phosphate, Mat. Res. Bull., 1974, 9, 353-360. .W. Fawcett, A
Radial Distribution Function Analysis of an Amorphous Calcium Phosphate with
Calcium to Phosphorus Molar Ratio of 1.42, Calc. Tissue Res., 1973, 13,
319-325. . Betts, A.S. Posner, A structural model for amorphous calcium
phosphate, Trans. Amer. Cryst. Ass., 1974, 10, 73-84.. Onuma, A. Ito, Cluster
Growth model for hydroxyapatite, Chem. Mater., 1998, 10, 3346-3351..I.
Suvorova, P.A. Buffat, Size Effect in X-ray and Electron Diffraction Patterns
from Hydroxyapatite Particles, Cryst. Reports, 2001,46, № 5, 722-729. .I.
Suvorova, P.A. Buffat, Electron diffraction and high-resolution transmission
electron microscopy in characterization of calcium phosphate precipitation from
aqueous solutions under biomineralization conditions, European Cells and
Materials, 2001, 1, 27-42..I. Suvorova, P.A. Buffat, Electron Diffraction from
Micro- and Nanoparticles of Hydroxyapatite, Journal of Microscopy, 1999, 196, №
1, P. 46-58.
С.
М. Баринов, В. С. Комлев, Биокерамика на основе фосфатов кальция. М., Наука,
2005.
Я.
С. Уманский. Рентгенография металлов и полупроводников, М.: Металлургия,
1969.Data Base (Sets 1-46) PDF-2 46 6A JUN96 [Электронный ресурс]: / Copyright
(C) JCPDS-ICDD 1996 JCPDS - International Centre for Diffraction Data 12 Campus
Boulevard Newtown Square, PA 19073-3273 U.S.A. - 1 электрон. опт. диск (CD-ROM)
: цветн. ; 12 см. - (PDF-2, 1996). - Систем. требования: Pentium-486 ; 32 Mb
RAM ; CD-ROM Windows 98/2000/NT/XP. - Название с титул. экрана.
1.