|
№
|
Источник и термическая обработка
|
Частота поглощения, см−№
|
спектр
|
|
1
|
Парацианоген (оксамидный)
|
1570 v. s.
|
A
|
|
2
|
Парацианоген (цианид ртути)
|
1570 v. s.
|
A′
|
|
3
|
1. нагрев до 450° 24 часа в вакууме
|
1570 v. s.
|
as A
|
|
4
|
1. нагрев до 450° 16 часов в воздухе
|
1100 m., 640 v. s.
|
B
|
|
5
|
2. нагрев до 450° 16 часов в воздухе
|
1570v. s.,810 w.,640 w.
|
B′
|
|
6
|
1. дисоциация в H2SO4
(конц.) по H2O
|
2070 v. v. s., 1570 v. s.
|
C
|
|
7
|
2. диссоциация в H2SO4
(конц.) по H2O
|
2070v. w., 1570 v. s.
|
C′
|
|
8
|
6. нагрев при 250° 6 часов в вакууме
|
2070 w., 1570 v. s.
|
C′
|
|
9
|
6. нагрев при 450° 16 часов в воздухе
|
1100 m., 640 v. s.
|
as B
|
|
10
|
7. нагрев при 450° 16 часов в воздухе
|
1570 v. s., 810 w., 640 w.
|
as B′
|
|
11
|
Парацианоген (цианид серебра)
|
1570 v. s.
|
as A′
|
Окисление парацианогена в растворе концентрированной серной
кислоты, добавляемой по каплям к кипящей азотной кислоте, дает раствор
бледно-соломенного цвета. С добавлением ртути раствор сульфата приобрел
коричневый хлопьевидный осадок, который постепенно формировался в
янтарно-игольчатые кристаллы цианида ртути. Оба раствора парацианогена в
кислотах и их производных, со ртутью имеют небесно-голубую флуоресценцию [16].
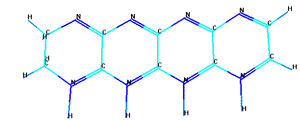
Согласно ранним воззрениям [17] парацианоген обладает
двумерной или квазитрехмерной структурой. Предположительные структуры
парацианогена по данным работы приведены на рисунке 4.
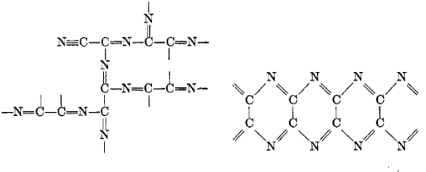
Рисунок 4 - Предположительные структуры парацианогена
Расчеты расширенным методом Хюккеля четырех модельных
структур парацианогена (рисунок 5), выполненные в работе [18] показали, что
наибольшей энергетической устойчивостью характеризуется нециклическая структура
1. Ароматическая структура 4 является энергетически наименее устойчивой.
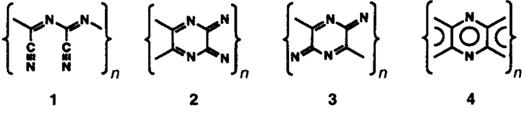
Рисунок 5 - Модельные структуры парацианогена, рассчитанные в
работе
Расчеты методом эффективного валентного гамильтониана двух
модельных структур парацианогена (рисунок 6), выполненные в работе [19], дали в
качестве результата металлический тип проводимости для парацианогена, что
отличается от экспериментальных результатов, свидетельствующих о
полупроводниковом типе проводимости парацианогена.
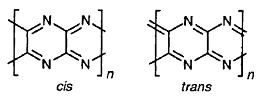
Рисунок 6 - Циклические структуры парацианогена,
рассмотренные в работе
Изучение удельной электропроводности двух образцов
парацианогена, приготовленных из цианада ртути и оксамида проведено в работе
[5]. Температурная зависимость удельной электропроводности двух образцов
парацианогена приведена на рисунке 7. Рассчитанное значение ширины запрещенной
зоны составило 1,12 эВ для парацианогена из цианида ртути и 1,14 эВ для
парацианогена из оксамида.
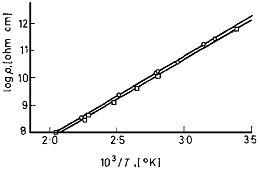
□ - парацианоген из цианада ртути, ○ -
парацианоген из оксамида
Образцы парацианогена были изучены в работе [20] методами
ИК-спектроскопии, твердотельного 13C-MAS-ЯМР и масс-спектроскопии. Изучались
образцы парацианогена, приготовленные методом пиролиза цианида серебра,
УФ-полимеризацией дициана и распылением графитового электрода в азотной плазме.
Все три изученных образца парацианогена имеют похожие
спектральные и термальные характеристики. Методы ИК - и ЯМР-спектроскопии
показали присутствие во всех изученных образцах присутствие атомов углерода в
состоянии sp2 гибридизации в форме структурного звена - C=N-.
На основании этого заключения автором работы [20] был сделан
вывод, что парацианоген представляет собой двумерный полимер с сопряженными
связями (рисунок 8).
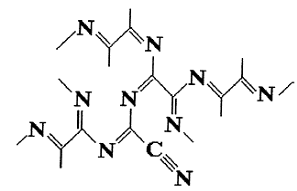
Рисунок 8 - Возможная структура парацианогена на основании
вывода работы
Парацианоген, полученный методом термолиза цианида ртути, был
изучен в работе [21] методами DRIFT, ЭПР, UV-VIS-NIR и рентгенофотоэлектронной
спектроскопии.
Рассчитанная из данных оптической спектроскопии (рисунок 9)
ширина запрещенной зоны в парацианогене составляет Eg = 0,85 эВ.
Это значение Eg характерное для полупроводника
хорошо согласуется с удельной электропроводностью 2,9∙10-5 См∙см-1
парацианогена.
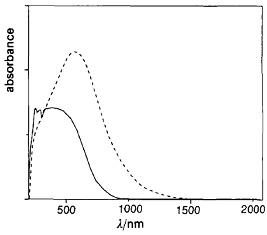
Рисунок 9 - Спектр поглощения порошкообразного парацианогена
(жирная линия) и продукта его термической обработки при 713 К в течение 40
минут (прерывистая линия)
Особенности фотополимеризации цианогена УФ-излучением и
солнечным светом в различных растворителях изучены в работе [22]. Возможные
структуры парацианогена, предположенные в этой работе приведены на рисунке 10.
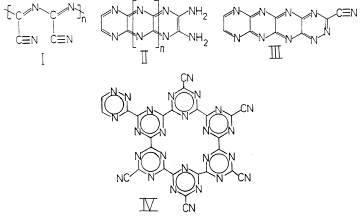
Рисунок 10 - Возможные структуры парацианогена, рассмотренные
в работе
Спектры оптического поглощения фотополимеров цианогена в УФ и
видимой области, полученные в работе [22], приведены на рисунках 11 и 12.
Согласно анализу изученных электронных спектров парацианогена, автором работы
[22] сделан вывод, что фотополимеры цианогена в растворителях представляют
собой структуры типа IV на 1,3,5-триазина с терминальными циано-группировками.
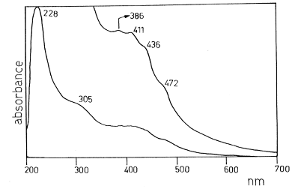
Рисунок 11 - Электронный спектр фотополимеризованного цианогена
в растворе тетрагидрофурана
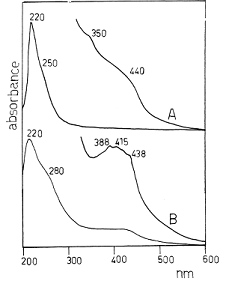
Рисунок 12 - Электронный спектр фотополимеризованного
цианогена: (A) (С2N2) x фотополимер,
полученный в метаноле с помощью УФ-облучения лампой, (B) (С2N2)
x фотополимер, полученный в этаноле под воздействием солнечного
света
2. Методы
молекулярного моделирования сопряженных полимеров
Органические сопряженные полимеры являются типичными
органическими соединениями с чередующимися двойными связями. В связи с этим для
моделирования их свойств требуется производить адекватный учет поведения
электронов π-связях [23-24].
Для моделирования структуры и свойств сопряженных полимеров
имеются достаточно эффективные и точные полуэмпирические и неэмпирические
варианты метода молекулярных орбиталей (МО) [24]. Таким образом,
квантовохимические расчеты являются в настоящее время одним из наиболее важных
инструментов физико-химических исследований структуры и свойств сопряженных
полимеров.
В основе современной квантовой химии лежит уравнение
Шредингера для стационарных состояний. Обычно используют адиабатическое
приближение, т.е. предполагают, что ядра неподвижны и можно решать уравнения
только для движения электронов [24]. Однако даже в этом случае точно решить
уравнение Шредингера можно только для одноэлектронных систем. Поэтому в
квантовохимических расчетах используют различные приближенные методы, наиболее
распространенным из которых является метод самосогласованного поля (ССП) или
метод Хартри-Фока. В этом методе полагается, что каждый электрон движется в
поле атомных ядер и эффективном усредненном поле других электронов.
Многоэлектронную волновую функцию ищут в виде антисимметричного произведения
спин-орбиталей, т.е. одноэлектронных МО ф i (m), умноженных на
спиновые волновые функции a (альфа) или b (бэтта) соответствующего электрона
(1) [24].
Ф = ||ф1a ф1b ф2a ф2b..
фNa фNb||. (1)
Для расчетов молекулярных систем с закрытыми оболочками (где
нет неспаренных электронов) используют обычно ограниченный метод Хартри-Фока
(RHF), когда каждая одноэлектронная МО фi (m) занята двумя
электронами с противоположными спинами. Тогда уравнение Шредингера
преобразуется в систему интегрально-дифференциальных уравнений для движения
каждого отдельного электрона (2) [23-24]:
F ф i = e iф i,
(2)
где F - фокиан (гамильтониан специального вида в
приближении ССП);i - энергия i-й МО.
Молекулярные орбитали ф i (m), как правило, ищут в
виде линейных комбинаций атомных орбиталей x i (m) (приближение
ЛКАО) (3):
ф i (m) = Sumj [Cmj (m) x j
(m)], (3)
где Cmj - искомые коэффициенты.
Таким образом, совокупность атомных орбиталей (АО) xj
является базисом для построения молекулярной волновой функции фi.
Количество базисных АО как правило недостаточно велико, т.е. базис является
неполным. Тем не менее, известно, что для адекватного описания диффузного
поведения электронов необходимо чтобы базисный набор АО мог достаточно точно
передавать распределение электронной плотности в молекуле. Выполнить это
требование на практике бывает достаточно трудно и выбор подходящего базисного
набора является серьезной методологической задачей [23-24].
Для определения величин Cmj используется
вариационная процедура минимизации полной электронной энергии молекулы Е. Для
этого решается система линейных уравнений специального вида (4) [23-24]:
j [ (Fij - e mSij)
Cmj] = 0 (4)
где соответствующие матричные элементы гамильтониана имеют
вид (5)
ij = Fij'+ Sumkl [Pkl
(<ij|kl> - 1/2<ik| jl>] (5)
где Sij - интеграл перекрывания АО xi и
xj;ij' - матричный элемент одноэлектронного
гамильтониана, включающий кинетическую энергию электронов и энергию
взаимодействия электронов и атомных ядер;kl - матрица зарядов и
порядков связей;
<ij|kl> - кулоновский многоцентровый двухэлектронный
интеграл вида (6):
<ij|kl> = Integral [xi* (1) xj*
(1) ·1/r12 · xk (2) xl (2) dt1 dt 2]
(6)
Решение системы (4) проводят методом самосогласования. Для
этого в качестве нулевого приближения берется достаточно произвольная
совокупность коэффициентов Cmj, по ним строится матрица Fij
и решение (4) дает новый набор Cmj, которые дают новую матрицу Fij
и т.д., пока коэффициенты Cmj не перестанут изменяться [23-24].
3.
Полуэмпирические методы квантовой химии
На практике при машинных расчетах структуры и свойств
химических обычно пользуются как неэмпирическими (ab initio), так и
полуэмпирическими методами. Они отличаются методикой вычисления матричных
элементов, описывающих электрон-электронные и электрон-ядерные взаимодействия в
системе (4). В полуэмпирических методах для этой цели используют приближенные
эмпирические формулы и известные из экспериментов параметры атомов. В
неэмпирических методах проводится непосредственный аналитический расчет
матричных элементов [25].
В прошедшие годы ранее на практике исследователи чаще
пользовались полуэмпирическими методами квантовой химии, чем более сложными и
требующими гораздо больше (на несколько порядков) машинного времени
неэмпирическими методами [24]. В последние годы с ростом вычислительных
возможностей компьютерной техники неэмпирические методы квантовой химии заняли
прочное место в моделировании сопряженных полимеров [26,38].
Тем не менее, многие практические задачи в области
моделирования сопряженных проводящих органических полимеров пока не поддаются
решению неэмпирическими методами даже после предельного их упрощения. Следует
особо отметить, что из-за ограниченности машинного времени большинство
неэмпирических расчетов возможны лишь в базисах небольшого и среднего размеров.
Но даже если расчет будет возможен и в достаточно большом базисе, будет найдено
не точное решение уравнения Шредингера, а лишь его решение в приближении
Хартри-Фока. Таким образом, существует авторитетное мнение, что по сути
неэмпирические квантово-химические методы все равно являются приближенными
[23-24].
Полуэмпирические расчеты в настоящее время проводят в
валентных приближениях CNDO, INDO и NDDO [24]. В этих приближениях расчет
проводится только для валентных электронов, а электроны внутренних оболочек
включают в остов молекулы; используют минимальный базис; пренебрегают
значительной частью кулоновских интегралов (6). Последнее упрощение является
наиболее существенным и позволяет значительно упростить расчет. Неточность
расчета при этом можно частично компенсировать за счет удачного подбора
параметров. В приближении CNDO (Complete Neglect of Differential Overlap)
учитываются только одноцентровые интегралы типа <ii|ii> и двухцентровые
интегралы типа <ii|kk>. В приближении INDO (Intermediate Neglect of
Differential Overlap) дополнительно учитываются кулоновские интегралы, у
которых все четыре орбитали xi, xj, xk,xl
принадлежат одному атому. В приближении NDDO (Neglect of Diatomic
Differential Overlap), кроме интегралов, которые учитываются в приближениях
CNDO и INDO, в расчет дополнительно включают интегралы <ij |kl>, у
которых орбитали xi и xj принадлежат одному атому, а xk
и xl - другому. Так как выбор параметров и эмпирических формул
неоднозначен, существуют различные модификации всех этих методов.
В последние годы наиболее широко из полуэмпирических
используют MNDO-подобные методы (приближение NDDO), к которым относят MNDO
[27], AM1 [28] и PM3 [29]. Популярности MNDO-подобных методов в немалой степени
способствовало некоммерческое распространение программ AMPAC и MOPAC,
включающих эти методы [30]. Все три метода незначительно отличаются друг от
друга и дают примерно одинаковые (вполне удовлетворительные для большинства
практических ситуаций) результаты. Подробный обзор применения MNDO-подобных
методов к различным задачам приводятся в работе [23-24]. Особенностью метода
AM1 является несколько лучшее описание межмолекулярных взаимодействий [23],
тогда как в методе PM3 разработаны параметры для большего числа элементов, в т.
ч. и металлов [31].
Следует отметить, что из всех MNDO-подобных методов метод AM1
пожалуй наиболее подходящий к изучению структурных, электронных и
фотофизических свойств органических электропроводящих полимеров с сопряженными
связями [32,35-37].
При расчетах систем с открытыми оболочками (т.е. с
неспаренными электронами) необходимо учитывать электронную корреляцию [33]. В
приближении Хартри-Фока движение электронов не коррелированно, т.е. вероятность
найти электрон в некоторой точке пространства не зависит от местонахождения
других электронов, распределение в пространстве которых задано одноэлектронными
функциями. В результате двум электронам с одинаковым спином не запрещено
занимать одну и ту же точку пространства, что противоречит принципу Паули и
ведет к ошибкам. Одним из методов учета электронной корреляции является
неограниченный метод Хартри-Фока (UHF), когда электроны с разными направлениями
спинов должны занимать, в отличие от RHF, разные МО. Неограниченный метод
Хартри-Фока имеет один серьезный недостаток, особенно существенный при расчетах
спиновых: плотностей [34]: волновая функция UHF не соответствует чистому
спиновому состоянию, т.е. содержит примеси состояний более высоких
мультиплотностей. Степень чистоты спинового состояния можно оценить, сравнивая
расчетную величину <S2> с номинальной, равной S (S+1).
Допустимым считается завышение порядка 10% [34]. Другой способ учета
корреляционных эффектов состоит в использовании в рамках RHF метода
конфигурационного взаимодействия (CI). В этом случае многоэлектронную волновую
функцию ищут в виде линейной комбинации большого числа детерминантов (1). При
расчетах MNDO-подобными методами систем с открытыми оболочками используют как
UHF, так и CI.
4.
Экспериментальная часть
На основании выполненного литературного обзора были выдвинуты
следующие практические задачи работы:
. На основании изучения литературных данных относительно
возможных структур парацианогена построить модельные структуры различных
химических форм парацианогена и с помощью метода молекулярной механики
определить их относительную устойчивость;
. Квантовохимическим методом MNDO-AM1 рассчитать наиболее
устойчивые структурные модели парацианогена двух топологических рядов;
. Провести сравнение рассчитываемых спектроскопических
характеристик модельных форм парацианогена с известными экспериментальными
данными оптической и ИК-спектроскопии.
4.1
Использованные подходы и программное обеспечение
Для расчета свойств и характеристик модельных структур
парацианогена был использован следующий подход. Предварительно оптимизированные
методом молекулярной механики с силовым полем ММ+ модельные структуры
парацианогена рассчитывались полуэмпирическим квантовохимическим методом AM1 в
ограниченном по спину приближении MNDO. В ходе расчетов производилась
оптимизация молекулярной геометрии градиентным методом Поллака-Рибера изучаемых
модельных структур парацианогена без каких-либо ограничений по симметрии или
структурных характеристик по достижению оптимизационного предела в 0,02
ккал/моль. Для оптимизированных структур определялись термодинамические
(энергии связывания, энтальпии образования) и электронные (энергии граничных
молекулярных орбиталей) характеристики. Фотофизические характеристики
рассчитывались с использованием полуэмпирического метода ZINDO/S в приближении
подхода конфигурационного взаимодействия (КВ) с однократными возбуждениями CIS.
Использовалось два подхода определения параметра предельного возбуждения электрона
с высшей занятой молекулярной орбитали (ВЗМО) модельной структуры для метода
конфигурационного взаимодействия. В первом методе в качестве параметра
предельного возбуждения бралось значение на 0,01 эВ меньше рассчитываемого
методом MNDO/AM1 значения потенциала ионизации модельной структуры. Во втором
методе в качестве параметра предельного возбуждения бралось значение на 0,01 эВ
меньше рассчитываемого методом ZINDO/S значения потенциала ионизации модельной
структуры, полученной методом MNDO/AM1.
Все проведенные расчеты выполнены с помощью лицензионной
программы HyperChem версии 7.52.
4.2
Полученные результаты
Модельные структуры парацианогена, оптимизированные методом
молекулярной механики в приближении силового поля ММ+ приведены на рисунках.
Установлено, что наиболее устойчивыми структурами модельных форм парацианогена
являются следующие топологические структуры:
) ациклическая форма (рисунок 13 а)),
) циклическая форма с боковыми NH2-группировками
(рисунок 13 в)).
На основании этих двух топологических форм были построены и
рассчитаны модельные структуры парацианогена с различным прогрессивно
увеличивающимся числом структурных группировок.
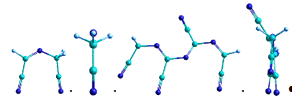
На рисунке 13 приведены оптимизированные методом MNDO/AM1
модельные структуры ациклической формы парацианогена с различным числом
структурных группировок. Можно видеть, что с ростом числа структурных
группировок сохраняется линейный структурный мотив с чередованием двойных
связей.
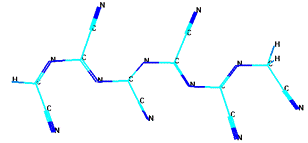
а) ациклическая форма шестичленного парацианогена
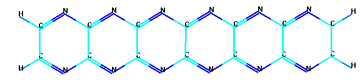
б) циклическая форма шестичленного парацианогена
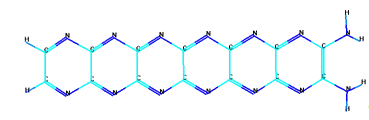
в) циклическая форма шестичленного парацианогена с боковыми
NH2-группировками
г) циклическая форма четырехчленного парацианогена
Рисунок 13 - Модельные формы парацианогена
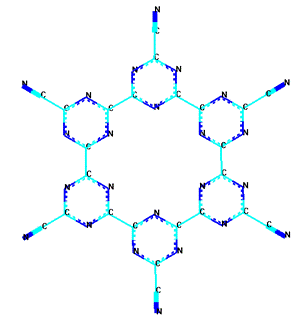
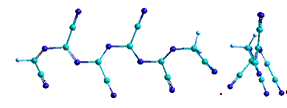
Рисунок 14 - Модельная полициклическая форма парацианогена
 = 2 N
= 4
= 2 N
= 4
Рисунок 15 - Оптимизированные структуры модельной
ациклической формы парацианогена (в двух проекциях) с различным количеством
циклов
 = 6
= 6
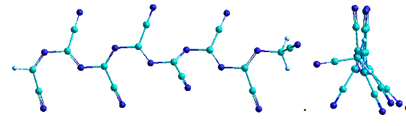 = 8
= 8
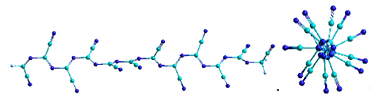 = 12
= 12
Рисунок 15 (продолжение) − Оптимизированные структуры
модельной ациклической формы парацианогена (в двух проекциях) с различным
количеством циклов
Зависимость нормированной энергии связи для изученных
модельных структур парацианогена от числа атомов в структуре представлена на
рисунке 16. Согласно представленным данным, можно видеть, что линейная
ациклическая форма парацианогена обладает большей энергетической устойчивостью,
чем циклическая.
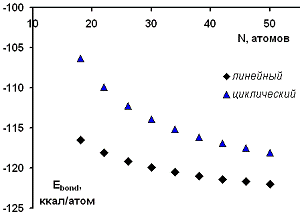
Рисунок 16 - Зависимость нормированной энергии связи для
изученных модельных структур парацианогена от числа атомов в структуре
Зависимость рассчитанной энтальпии образования для изученных
ациклических модельных структур парацианогена от числа структурных группировок
в молекуле представлена на рисунке 17. Согласно представленным данным, можно
видеть, что для линейной ациклической формы парацианогена каждая последующая
группировка вносит добавочный энергетический вклад в энтальпию образования равный
52,13 ккал/моль.
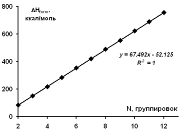
Рисунок 17 - Зависимость энтальпии образования для изученных
ациклических модельных структур парацианогена от числа структурных группировок
в молекуле
Рассчитанные значения ширины энергетической щели для
изученных ациклических модельных структур парацианогена приведены на рисунке
18.
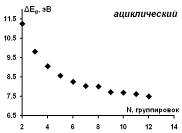
Рисунок 18 - Зависимость рассчитанных значений ширины
энергетической щели для изученных ациклических модельных структур парацианогена
от числа структурных группировок в молекуле
Можно видеть систематическое снижение ширины энергетической
щели (аналог запрещенной зоны) с ростом размера ациклических модельных структур
парацианогена.
Можно оценить асимптотически достигаемое предельное значение
ширины энергетической щели в 6,7 эВ.
Зависимость рассчитанного значения дипольного момента для
изученных ациклических модельных структур парацианогена от числа структурных
группировок в молекуле представлена на рисунке 19.
Согласно представленным данным, можно видеть, что для
линейной ациклической форма парацианогена начиная с 3-х структурных группировок
каждая последующая группировка приводит к монотонному росту значения дипольного
момента.
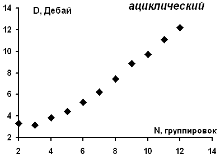
Рисунок 19 - Зависимость рассчитанных значений дипольного
момента для изученных ациклических модельных структур парацианогена от числа
структурных группировок в молекуле
На рисунке 20 приведены рассчитанные значения для частоты
наиболее длинноволнового электронного перехода в оптических спектрах изученных
ациклических модельных структур парацианогена с помощью двух вышеописанных
подходов.
Можно видеть, что с ростом числа структурных группировок в
молекулах ациклических модельных структур парацианогена пик длинноволнового
поглощения в оптических спектрах смещается область дальнего ультрафиолета.
Для изученных ациклических модельных структур парацианогена
пиков поглощения в видимой области спектра предсказано не было.
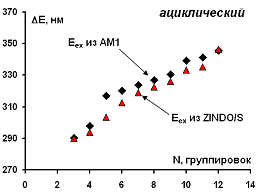
Рисунок 20 - Зависимость рассчитанных значений
длинноволнового электронного перехода для изученных ациклических модельных
структур парацианогена от числа структурных группировок в молекуле
Вид спектров поглощения изученных ациклических модельных
структур парацианогена представлен на рисунке 21. Вид спектров поглощения
изученных ациклических модельных структур парацианогена свидетельствует, что
наибольшее поглощение наблюдается в коротковолновой области УФ-диапазона.
Можно предполагать, что с ростом длины ациклических модельных
структур парацианогена в районе 17-18 структурных группировок будет достигнута
синяя область видимого спектра, следовательно, этот структурный тип
молекулярных форм будет иметь черно-фиолетовую окраску.
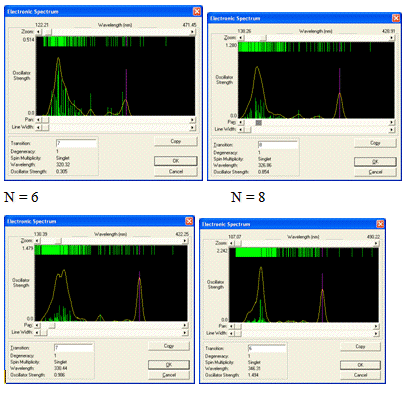
Рисунок 21 - Спектры поглощения изученных ациклических
модельных структур парацианогена
На рисунке 22 приведены оптимизированные методом MNDO/AM1
модельные структуры циклической формы парацианогена с различным числом
структурных группировок. Можно видеть, что с ростом числа структурных
группировок сохраняется линейный структурный мотив с чередованием двойных
связей в сопряженной π-системе.
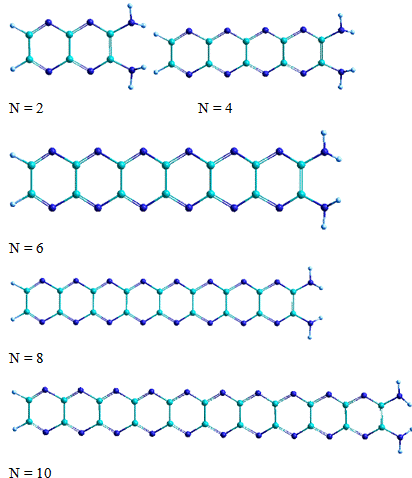
Рисунок 22 - Оптимизированные структуры модельной циклическая
формы парацианогена
Зависимость рассчитанной энтальпии образования для изученных
циклических модельных структур парацианогена от числа структурных группировок в
молекуле представлена на рисунке 23. Согласно представленным данным, можно
видеть, что для изученной циклической формы парацианогена каждая последующая
группировка вносят добавочный энергетический вклад в энтальпию образования
равный 43,18 ккал/моль.
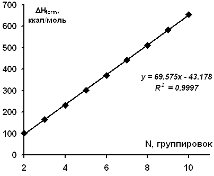
Рисунок 23 - Зависимость энтальпии образования для изученных
циклических модельных структур парацианогена от числа структурных группировок в
молекуле
Рассчитанные значения ширины энергетической щели для
изученных ациклических модельных структур парацианогена приведены на рисунке
11. Можно видеть систематическое снижение ширины энергетической щели (аналог
запрещенной зоны) с ростом размера циклических модельных структур
парацианогена. Можно оценить асимптотически достигаемое предельное значение
ширины энергетической щели в 5,4 эВ.
Зависимость рассчитанного значения дипольного момента для
изученных ациклических модельных структур парацианогена от числа структурных
группировок в молекуле представлена на рисунке 24. Согласно представленным
данным, можно видеть, что для изученной циклической формы парацианогена каждая
последующая группировка приводит к монотонному росту значения дипольного
момента.
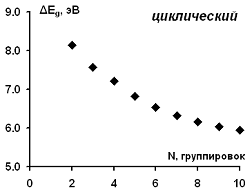
Рисунок 24 - Зависимость рассчитанных значений ширины
энергетической щели для изученных циклических модельных структур парацианогена
от числа структурных группировок в молекуле
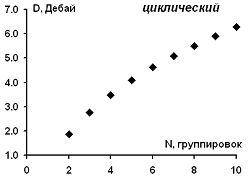
Рисунок 25 - Зависимость рассчитанных значений дипольного
момента для изученных циклических модельных структур парацианогена от числа
структурных группировок в молекуле
На рисунке 26 приведены рассчитанные значения для частоты
наиболее длинноволнового электронного перехода в оптических спектрах изученных
циклических модельных структур парацианогена с помощью двух вышеописанных
подходов. Можно видеть, что с ростом числа структурных группировок в молекулах
ациклических модельных структур парацианогена пик длинноволнового поглощения в
оптических спектрах смещается из область дальнего ультрафиолета в красную
область видимого спектра.
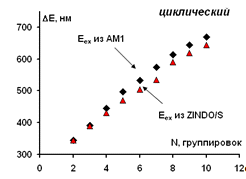
Рисунок 26 - Зависимость рассчитанных значений длинноволнового
электронного перехода для изученных циклических модельных структур
парацианогена от числа структурных группировок в молекуле
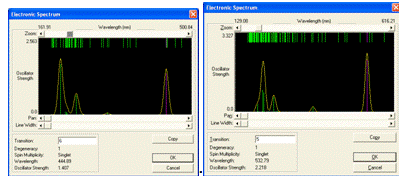
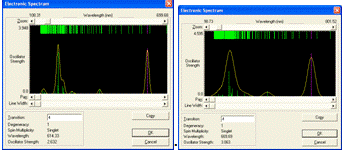
Вид спектров поглощения изученных циклических модельных
структур парацианогена представлен на рисунке 27. Вид спектров поглощения
изученных циклических модельных структур парацианогена свидетельствует, что
наибольшее поглощение наблюдается в как в коротковолновой области УФ-диапазона
так и в видимой части спектра. Можно предполагать, что для этого структурного
тип молекулярной формы парацианогена будет наблюдаться интенсивное поглощение
во всех частях видимого спектра в зависимости от степени полимеризации.
 = 4 N
= 6
= 4 N
= 6
 = 8 N = 10
= 8 N = 10
Рисунок 27 - Спектры поглощения изученных ациклических
модельных структур парацианогена
Сравнение предсказываемые спектрофизических характеристики
для изученных линейных ациклических и циклических молекул парацианогена в с
известными экспериментальными спектроскопическими литературными данными
позволяет сделать предположение, что при водной полимеризации дициана в
основном образуются линейные полимеры парацианогена. Доля же циклических форм
должна быть небольшой.
Заключение
Полуэмпирическим квантовохимическим методом MNDO/AM1 были
изучены модельные структуры парацианогена с различным типом топологии и числом
структурных группировок. Согласно выполненным расчетам наибольшая
энергетическая устойчивость характерна для линейных ациклических молекул
парацианогена. Предсказываемая ширина запрещенной зоны для этого типа молекул с
ростом длины асимптотически приближается к 6,7 эВ. Предсказываемая ширина
запрещенной зоны для циклических молекул парацианогена с ростом длины
асимптотически приближается к 5,4 эВ.
Предсказываемые спектрофизические характеристики для
изученных линейных ациклических и циклических молекул парацианогена в сравнении
с известными экспериментальными спектроскопическими данными позволяют сделать
вывод, что при водной полимеризации дициана в основном образуются линейные
полимеры парацианогена.
Список
использованных источников
1.
Electrochemically synthesised conducting polymeric materials for applications
towards technology in electronics, optoelectronics and energy storage devices /
K. Gurunathan, A. V. Murugan, R. Marimuthu, U. P. Mulik, D. P. Amalnerkar //
Materials Chemistry and Physics. - 1999. - № 61. - P.173-191.
.
Блайт Э.Р. Электрические свойства полимеров / Э.Р. Блайт, Д. Блур - М.:
Физматлит, 2008. - 376 с.
.
Salaneck W. R. Electronic structure of conjugated polymers: consequences of
electron-lattice coupling / W. R. Salaneck, R. H. Friend, J. L. Bredas //
Physics Reports. - 1999. - № 319. - P.231-251.
.
Salamone J. C. Concise Polymeric Materials Encyclopedia / J. C. Salamone - CRC
Press, Florida, 1998. - 1760 p.
.
Graham J. The electrical conductivity of some nitrogen containing heterocyclic
polymers. / J. Graham, D. I Packham. // Polymer. - 1969. - Vol.10. - P.645-651.
.
Salamone. J. C. Polymeric Materials Encyclopedia / J. C. Salamone - CRC Press,
Florida, 1996. - 960 p.
.
Пат.3020246 США, Carbon-nitrogen polymers and method of preparing same / W. L.
Fierce, W. J. Sandner (США). - заявл.28.05.59; опубл.06.02.62. - 2 с.
.
Kryszewski M. Semiconducting Polymers / M. Kryszewski - PWN-Polish Scientific
Publishers, Warszawa, 1980. - 710 p.
.
Schmidt C. L. New directions in carbonitride research: synthesis of resin-like
dense-packed C3N4 using a hydrogen-free precursor / C. L.
Schmidt, M. Jansen // J. Mater. Chem. - 2010. - № 20. - P.183-192.
.
Coll P.organic chemistry in Titan’s atmosphere: new data from laboratory
simulations at low temperature / P. Coll, D. Coscia, M. C. Gazeau // Adv. Space
Res. - 1995. - № 16. - P.93-103.
.
Coustenis A. Modeling Titan’s thermal infrared spectrum for high-resolution
space observations / A. Coustenis, T. Encrenaz, B. Bezard // Icarus. - 1993. №
102. - P.240-260.
.
Hudson R. L. Reactions of nitriles in ices relevant to Titan, comets, and the
interstellar medium: formation of cyanate ion, ketenimines, and isonitriles /
R. L. Hudson, M. H. Moore // Icarus - 2004. - № 172. - P.466-478.
.
Brotherton T. K. The Synthesis and Chemistry of Cyanogen / T. K. Brotherton, J.
W. Lynn // Chemical Review - 1959. - Vol.59. - № 5. - P.843-883.
.
Bowden F. P. Initiation and propagation of explosion in azides and fulminates /
F. P. Bowden, H. T. Williams // Proc. R. Soc. Lond. A. - 1951. - Vol. 208. -
P.176-188.
.
Carley A. F. Polymerisation of cyanogen on graphite and graphite supported
copper films / A. F. Carley, M. Chinn, C. R. Parkinson. // Surface Science. -
2002. - № 17. - P.563-567.
.
Bircumshaw L. L. Paracyanogen: its formation and properties. Part I / L. L.
Bircumshaw, F. M. Tayler, D. H. Whiffen // J. Chem. Soc. - 1954. - № 15. -
P.31-35.
.
Brotherton T. K. The Synthesis and Chemistry of Cyanogen. / T. K. Brotherton,
J. W. Lynn // Chemical Review. - 1959. - Vol.59. - №5. - P.843-883.
.
Whangbo M. H. Conjugated one and two dimensional polymers / M. H. Whangbo, R.
Hoffman, R. B. Woodward // Proclamations of Royal Society London. - 1976. - №
366. - P.23-46.
.
Bredas J. L. Valence effective Hamiltonian technique for nitrogen containing
polymers: Electronic structure of polypyrrole, pyrolized polyacrylonitrile,
paracyanogen, polymethineimine, and derivatives / J. L. Bredas, B. Themans, J.
M. Andre // The Journal of Chemical Physics. - 1983. - №.78. - P.137-148.
.
Leon M. Paracyanogen reexamined / M. Leon // Journal of Polymer Science Part A:
Polymer Chemistry. - 1993. - Vol.31. - № 10. - P.2595-2600.
.
Jenneskens L. W. Structural Studies on Paracyanogen and Paraisocyanogen / L. W.
Jenneskens, J. G. Mahyt, E. J. Vlietstra // Journal of Chemical Society. -
1994. - Vol.90. - № 2. - P.327-332.
.
Cataldo F. On cyanogen photopolymerization / F. Cataldo // European Polymer
Journal. - 1999. - № 35. - P.571-579.
.
Минкин В.И. Теория строения молекул / В.И. Минкин, Б.Я. Симкин, Р.М. Миняев. -
Ростов-на-Дону: Феникс, 1997. - 560 с.
.
Минкин В.И. Молекулярный дизайн таутомерных систем / В.И. Минкин, Л.П.
Олехнович, Ю.А. Жданов. - Ростов-на-Дону: Изд-во Ростов, 1993. - 272 с.
.
Vukmirovic N. Charge Carrier Motion in Disordered Conjugated Polymers: A
Multiscale Ab Initio Study / N. Vukmirovic, L. W. Wang // Nano Lett. - 2009. -
№ 12. - P.396-400.
.
Dewar M. J. MNDO results for molecules containing hydrogen, carbon, nitrogen
and oxygen / M. J. Dewar, W. Thiel // J. Am. Chem. Soc. - 1977. - Vol.99. - №
15. - P.4907-4917.
.
Dewar M. J. AM1: a new general purpose quantum mechanical molecular model / M.
J. Dewar, E. G. Zoebisch, E. F. Healy // J. Am. Chem. Soc. - 1985. - Vol.107. -
№ 15. - P.3902-3909.
.
Stewart J. P. Optimization of parameters for semiempirical methods II.
Applicatios / J. P. Stewart // J.comput. Chem. - 1989. - Vol.10. - № 2. -
P.221-264.
.
Stewart J. P. Mopac: a semiempirical molecular orbital program / Stewart J. P.
// J.comput. Aided Mol. Des. - 1990. - Vol.4. - № 1. - P.1-105.
.
Stewart J. P. Optimization of parameters for semiempirical methods III.
Extensions of PM3 to Be, Mg, Zn, Ga, Ge, As, Se, Cd, In, Sn, Sb, Te, Hg, Tl,
Pb, and Bi / J. P. Stewart // J.comput. Chem. - 1991. - Vol.12. - № 3. -
P.320-341.
.
Birgerson J. Electronic structure of some conjugated polymers for electron
transport / J. Birgerson, N. Johansson, A. Pohl // Synthetic Metals. - 2001. -
№ 122. - P.67-72.
.
Бучаченко А.Л. Стабильные радикалы. Электронное строение, реакционная
способность и применение / А.Л. Бучаченко А.М. Вассерман. - М:. Химия, 1973. -
408 с.
.
Кларк Т. Компьютерная химия / Т. Кларк - М.: Мир, 1990. - 383 с.
.
Bredas J. L. Electronic structure of π-conjugated oligomers and polymers:
a quantum-chemical approach to transport properties / J. L. Bredas, D.
Beljonne, J. Cornil // Synthetic Metals. - 2002. - № 122. - P.107-116.
.
Lawrence T. Spectroscopic Study of Polyaniline Emeraldine Base: Modelling
Approach / T. Lawrence, Jr. Sein, W. Yen // Synt. Met. - 2004. - № 143. -
P.1-12.
.
Medhat I. Spectroscopic Study of Polyaniline Emeraldine Base: Modelling
Approach / I. Medhat, K. Eckhard // Acta Chim. Slov. - 2005. - № 52. -
P.159-163.
.
Ferretti A. Ab initio complex band structure of conjugated polymers: Effects of
hydrid density functional theory and GW schemes / A. Ferretti, G. Mallia, L.
Martin-Samos // Phys. Rev. B: Condens. Matter Mater. Phys., - 2012. - №34 -
P.85-102.