Контроль содержания металлов электрохимическими методами в сточных водах гальванических цехов машиностроительных предприятий
ВВЕДЕНИЕ
металл токсикологический сточный
гальванический
В Республике Беларусь гальваническое
производство функционирует более чем на 140 предприятиях. Наибольшее
распространение получили цинковые, никелевые, фосфатные, хромовые, медные и
другие покрытия. В общем объеме производства гальванических покрытий более 80 %
занимает цинкование как наиболее дешевый и надежный способ защиты от
коррозионного разрушения металлических конструкций.
Гальванические (электрохимические) покрытия
являются одним из самых распространенных методов защиты изделий от коррозии,
придания им необходимых эксплуатационных характеристик и декоративных свойств.
Гальванические технологии широко применяются в машиностроении, приборостроении,
авиационной, электронной и радиотехнической промышленности, в других областях.
Выбор способа нанесения гальванопокрытия определяется его видом и толщиной,
требованиями к свойствам, размерами и формой обрабатываемых деталей, а также
применяемыми подготовительными и заключительными операциями обработки.
В процессах обработки поверхности с
цельюпридания ей антикоррозионных и декоративныхсвойств используются
разнообразные реагенты,содержащие тяжелые металлы. Они входят в состав побочных
продуктов этого производства -твердых и жидких отходов, сточных вод, выбросов в
атмосферу. Также все гальванотехнические процессы сопряжены с большим
водопотреблением, а следовательно, и с большим количеством образующихся сточных
вод. В результате предприятия, в составе которых функционируют гальванические
цехи (участки), являются основными источниками поступления токсичных тяжелых
металлов в водные объекты. При этому становленные нормативы допустимых
концентраций по ионам тяжелых металлов для сточных вод, отводимых в
канализационные сети, часто не выполняются, что затрудняет работу городских
очистных сооружений. При очистке сточных вод гальванических производств на
локальных очистных сооружениях образуются осадки,которые относятся к отходам
3-4 классов опасности. В процессе применения технологических растворов
образуются шламы, которые практически не используются и хранятся на площадках
предприятий. Поэтому контроль содержания металлов в сточных водах
гальванических цехов является актуальным на сегодняшний день.
1. МЕТАЛЛЫ В СТОЧНЫХ ВОДАХ ГАЛЬВАНИЧЕСКИХ ЦЕХОВ
МАШИНОСТРОИТЕЛЬНЫХ ПРЕДПРИЯТИЙ
При производстве машиностроительной продукции
многие предприятия для повышения коррозионной стойкости и улучшения внешнего
вида металлических деталей наносят на них гальванические покрытия из водных
растворов или расплавов солей металлов.
Крупнейшие предприятия Республики Беларусь, где
функционирует гальваническое производство: РУП «Белорусский металлургический
завод» (Жлобин), РУП«Гомельский завод литья и нормалей», ОАО «Минский
подшипниковый завод», РУП «Гомсельмаш», РУП «БелАЗ» (Жодино), ПРУП «Минский
автомобильный завод», РУП «Минский тракторный завод», ЗАО «Атлант»(Минск),
РУПП «Витязь» (Витебск) и др. Наибольшее распространение получили цинковые,
хромовые, никелевые, медные и кадмиевые покрытия.
Воздействие на окружающую среду гальванического
производства в значительной степени зависит от организации водного хозяйства,
эффективности работы очистных сооружений и использования образующихся в
процессепроизводства осадков и шламов.В гальваническом производстве неизбежно
образуются токсичные сточные воды, которые необходимо обезвреживать.
Сточные воды гальванических производств содержат
такие металлы, как хром, никель, свинец, медь, кадмий, цинк, олово и др.
Длительное их поступление в организм с водой или пищей даже в незначительных
дозах приводит в результате накопления в органах и тканях к нарушению
функционирования центральной нервной системы, внутренних органов, эндокринной
системы и других жизненно важных систем организма.
По данным Государственного водного кадастра в
водные объекты Беларуси в 2013 г. отведено 974 млн м3 сточных вод, среди
которых, как и ранее, количественно преобладали нормативно-очищенные воды.
Вторую позицию занимали сточные воды, сбрасываемые без очистки(табл.1), объем
которых из года в год увеличивался. Однако в 2013 г. их количество по сравнению
с предыдущим годом уменьшилось на 28 млн м3 (рис. 1).
Таблица 1 - Отведение различных категорий
сточных вод в водные объекты в областях Беларуси и г.Минске в 2013 г., млн м3
|
Область,
|
Всего
|
|
из
них
|
|
|
|
недостаточно
|
не
требующих
|
нормативно-
|
|
|
очищенных
|
очистки
|
очищенных
|
|
1
|
2
|
3
|
4
|
5
|
|
Всего
|
974
|
2,92
|
317
|
654
|
|
Брестская
|
176
|
0,09
|
104
|
72
|
|
Витебская
|
128
|
0,08
|
40
|
88
|
|
Гомельская
|
124
|
0,09
|
27
|
98
|
|
Гродненская
|
89
|
0,05
|
7
|
82
|
|
Минская
|
183
|
2,11
|
118
|
62
|
|
Могилевская
|
99
|
9,49
|
12
|
87
|
|
г.Минск
|
174
|
0,00
|
9
|
165
|
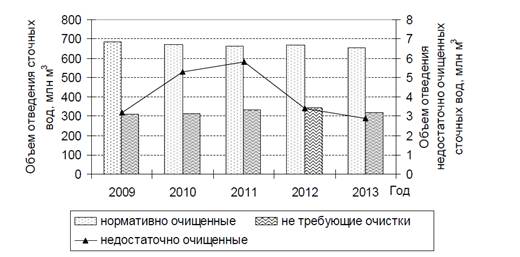 Рисунок 1 -
Динамика отведения сточных вод в водные объекты
Рисунок 1 -
Динамика отведения сточных вод в водные объекты
В 2013 г. в целом для страны количество сточных
вод,содержащих загрязняющие вещества, составило 890 млн м3 и уменьшилось по
сравнению с 2012 г. на 59 млн м3.
Количество загрязняющих веществ, сброшенных в
составе сточных вод в водные объекты, представлено в таблице 2. Следует
отметить, что в последние годы отмечается тенденция к уменьшению объемов сброса
соединений азота.Так, по сравнению с 2012 г. их суммарное поступление в водные
объекты страны сократилось на 0,5 тыс.т.
Общее количество сброшенных в водные объекты
в2013 г. металлов оказалось наименьшим за пятилетний период,а по сравнению с
2012 г. сократилось на 128 т. При этом основное количество (381,7 т) приходится
на железо, цинк составляет 24,75 т, никель - 5,66 и хром - 3,32 т.
Свинец содержится в сточных водах Гомеля (0,35
т), Минска (0,82), Могилева (0,22), Минской (0,1) и Витебской (0,1 т) областей.
При этом по сравнению с2012 г. его сброс со
сточными водами Минска увеличился в2,6 раза, Могилева и Гомеля напротив
уменьшился на 0,18 и 0,05 т соответственно, в Минской и Витебской области
остался на прежнем уровне.
Таблица 2 - Сброс загрязняющих веществ в составе
сточных вод в Беларуси в 2009-2013 гг.
|
Показатель
|
Раз-
|
|
|
Год
|
|
|
|
мерность
|
2009
|
2010
|
2011
|
2012
|
2013
|
|
Органические
|
|
8
|
8
|
8
|
9
|
8
|
|
вещества
(БПК5)
|
тыс.т
|
|
|
|
|
|
|
Нефть
и нефтепро-
|
|
|
|
|
|
|
|
дукты
в растворен-
|
тыс.т
|
0,13
|
0,12
|
0,11
|
0,12
|
0,10
|
|
ном
и эмульгирован-
|
|
|
|
|
|
|
|
ном
состоянии
|
|
|
|
|
|
|
|
Взвешенные
|
тыс.т
|
13
|
13
|
13
|
12
|
14
|
|
Сульфаты
|
тыс.т
|
63
|
56
|
60
|
61
|
58
|
|
Хлориды
|
тыс.т
|
73
|
65
|
71
|
75
|
72
|
|
Аммонийный
азот
|
тыс.т
|
5,40
|
5,50
|
5,90
|
5,70
|
5,30
|
|
Нитритний
азот
|
тыс.т
|
0,19
|
0,16
|
0,20
|
0,18
|
0,15
|
|
Нитратный
азот
|
тыс.т
|
3,70
|
3,50
|
3,40
|
3,16
|
|
Медь
|
т
|
6,70
|
5,00
|
6,20
|
7,00
|
5,80
|
|
Другие
металлы (же-
|
|
|
|
|
|
|
|
лезо
общее, цинк,
|
т
|
421
|
494
|
516
|
543
|
415
|
|
никель,
хром общий)
|
|
|
|
|
|
|
Кобальт (0,17 т) поступал в реки со сточными
водами Гомеля, молибден (3,76) - Витебской области, фториды (10,47)
сбрасывались со сточными водами Гомеля, фенолы - со сточными водами Гомельской
области (1,62), а также городов Гродно (0,26) и Могилева (0,06 т). Среди указанных
загрязняющих веществ, следует обратить внимание на рост в 2013 г в сравнении с
прошлым годом сброса фторидов на 2,21 т[1]
. ХИМИЧЕСКАЯ И ТОКСИКОЛОГИЧЕСКАЯ ХАРАКТЕРИСТИКА
МЕДИ, НИКЕЛЯ, КОБАЛЬТА, ЦИНКА
.1 Химическая и токсикологическая характеристика
кобальта
В соединениях кобальт проявляет переменную
валентность. В простых соединениях наиболее устойчив Со(П), в комплексных -
Со(Ш). Для Со(I) и Co(IV) получены только немногочисленные комплексные
соединения. При обыкновенной температуре компактный кобальт стоек против
действия воды и воздуха. Мелко раздробленный кобальт, полученный
восстановлением его оксида водородом при 250 °С ,на воздухе самовоспламеняется,
превращаясь в СоО. Компактный кобальт начинает окисляться на воздухе выше 300
°С; при красном калении он разлагает водяной пар. С галогенами кобальт легко
соединяется при нагревании, образуя галогениды СоХ2. При нагревании кобальт
взаимодействует с S, Se, P, As, Sb, С, Si, В, причем состав получающихся
соединений иногда не удовлетворяет указанным выше валентным состояниям
(например, Со2Р, Co2As, CoSb2, Со3С, CoSi3). В разбавленных соляной и серной
кислотах кобальт медленно растворяется с выделением водорода и образованием
соответственно хлорида СоCо2 и сульфата CoSO4. Разбавленная азотная кислота растворяет
кобальт с выделением оксидов азота и образованием нитрата Co(NO3)2. Едкие
щелочи осаждают из растворов солей Со2+ синий гидроксид Со(ОН)2, которая
постепенно буреет вследствие окисления кислородом воздуха до Со(ОН)3.
Из простых соединений Со(IП) известны лишь
немногие. При действии фтора на порошок Со или СоCl2 при 300-400 °С образуется
коричневый фторид CoF3. Комплексные соединения Со(Ш) весьма устойчивы и
получаются легко. Например, KNO2 осаждает из растворов солей Со (П), содержащих
СН3СООН, желтый труднорастворимый гексанитрокобальтат (III) калия K3[Co(NO2)6].
[11]
В природные воды соединения кобальта попадают в
результате процессов выщелачивания их из медноколчедановых и других руд, из
почв при разложении организмов и растений, а также со сточными водами
металлургических, металлообрабатывающих и химических заводов. Некоторые
количества кобальта поступают из почв в результате разложения растительных и
животных организмов.
Соединения кобальта в природных водах находятся
в растворенном и взвешенном состоянии, количественное соотношение между
которыми определяется химическим составом воды, температурой и значениями рН.
Растворенные формы представлены в основном комплексными соединениями, в т.ч. с
органическими веществами природных вод. Соединения двухвалентного кобальта
наиболее характерны для поверхностных вод. В присутствии окислителей возможно
существование в заметных концентрациях трехвалентного кобальта.
Кобальт относится к числу биологически активных
элементов и всегда содержится в организме животных и в растениях. С
недостаточным содержанием его в почвах связано недостаточное содержание
кобальта в растениях, что способствует развитию малокровия у животных
(таежно-лесная нечерноземная зона). Входя в состав витамина В12, кобальт весьма
активно влияет на поступление азотистых веществ, увеличение содержания
хлорофилла и аскорбиновой кислоты, активизирует биосинтез и повышает содержание
белкового азота в растениях. Вместе с тем повышенные концентрации соединений
кобальта являются токсичными.
В речных незагрязненных и слабозагрязненных
водах его содержание колеблется от десятых до тысячных долей миллиграмма в 1
дм3, среднее содержание в морской воде 0.5 мкг/дм3.
ПДКв составляет 0.1 мг/дм3, ПДКвр 0.01 мг/дм3 .
[10]
2.2 Химическая и токсикологическая характеристика
меди
Образуя химические соединения, атом меди может
отдавать один, два или три электрона, проявляя степень окисления соответственно
+1, +2 и +3. При этом наиболее устойчивыми являются соединения меди (II), а
наименее устойчивыми - соединения меди (III).
Медь относится к малоактивным металлам.
Стандартный электродныйпотенциал меди равен +0,34 В, что определяет ее место в
ряду стандартных электродных потенциалов: оно находится правее водорода. При
обычных условиях она не взаимодействует с водой, растворами щелочей, соляной и
разбавленной серной кислотой.
Однако в кислотах-сильных окислителях (например,
азотной и концентрированной серной)-медь растворяется.
Как малоактивный металл медь обладает достаточно
высокой стойкостью к коррозии, влажной атмосфере, содержащей углекислый газ,
медь покрывается зеленоватым налетом карбоната меди.В большинстве известных
соединений медь проявляет степень окисления + 2.
Соединения меди (II) - оксид СuО и гидроксид
Сu(ОН)2-довольно устойчивы. Этот гидроксид амфотерен,хорошо растворяется в
кислотахи в концентрированных щелочах.
Гидроксид меди (II) - труднорасворимое в воде
вещество голубого цвета. Принагреванииразлагается,образуя оксид меди (II)
черного цвета.
Темный цвет окисленных медных изделий обусловлен
наличием на их поверхности этого оксида. Для ионов меди (II) Сu2+ характерно
образование комплексных соединений, например K2[Cu(CN)4]-тетрацианокупрат (II)
калия.
Из других комплексных соединений меди (II)
отметим соединение с аммиаком. Если к раствору хлорида меди (II) прилить
небольшое количество раствора аммиака, то выпадет осадок гидроксида меди (II).
Если добавить избыток аммиака, то гидроксид
растворится с образованием комплексного соединения темно-синей окраски,
характерной для аммиачного комплекса меди. Эта реакция является качественной на
ион меди (II).
Соединения меди (III), например Cu203 или KCu02,
встречаются редко, они малоустойчивы. Устойчивость соединений меди (I) выше,
однако и они в водных растворах легко подвергаются диспропорционированию (реакции
самоокисления-самовосстановления). [12]
Медь - один из важнейших микроэлементов.
Физиологическая активность меди связана главным образом с включением ее в
состав активных центров окислительно-восстановительных ферментов. Недостаточное
содержание меди в почвах отрицательно влияет на синтез белков, жиров и
витаминов и способствует бесплодию растительных организмов. Медь участвует в
процессе фотосинтеза и влияет на усвоение азота растениями. Вместе с тем,
избыточные концентрации меди оказывают неблагоприятное воздействие на
растительные и животные организмы.
Содержание меди в природных пресных водах
колеблется от 2 до 30 мкг/дм3, в морских водах - от 0.5 до 3.5 мкг/дм3.
Повышенные концентрации меди (до нескольких граммов в литре) характерны для
кислых рудничных вод.
В природных водах наиболее часто встречаются
соединения Cu(II). Из соединений Cu(I) наиболее распространены
труднорастворимые в воде Cu2O, Cu2S, CuCl. При наличии в водной среде лигандов
наряду с равновесием диссоциации гидроксида необходимо учитывать образование
различных комплексных форм, находящихся в равновесии с акваионами металла.
Основным источником поступления меди в природные
воды являются сточные воды предприятий химической, металлургической
промышленности, шахтные воды, альдегидные реагенты, используемые для
уничтожения водорослей. Медь может появляться в результате коррозии медных
трубопроводов и других сооружений, используемых в системах водоснабжения. В
подземных водах содержание меди обусловлено взаимодействием воды с горными породами,
содержащими ее (халькопирит, халькозин, ковеллин, борнит, малахит, азурит,
хризаколла, бротантин).
Предельно допустимая концентрация меди в воде
водоемов санитарно-бытового водопользования составляет 0.1 мг/дм3 (лимитирующий
признак вредности - общесанитарный), в воде рыбохозяйственных водоемов - 0.001
мг/дм3. [10]
.3 Химическая и токсикологическая характеристика
никеля
Химические свойства никеля. В химические
отношении Ni сходен с Fe и Со, но также и с Cu и благородными металлами. В
соединениях проявляет переменную валентность (чаще всего 2-валентен). Никель -
металл средней активности. Поглощает (особенно в мелкораздробленном состоянии)
большие количества газов (H2, СО и других); насыщение никеля газами ухудшает
его механические свойства. Взаимодействие с кислородом начинается при 500 °C.
Из оксидов наиболее важен NiO - зеленоватые кристаллы, практически
нерастворимые в воде (минерал бунзенит). Гидрооксид выпадает из растворов
никелевых солей при прибавлении щелочей в виде объемистого осадка яблочно-зеленого
цвета. При нагревании никель соединяется с галогенами, образуя NiX2. Сгорая в
парах серы, дает сульфид, близкий по составу к Ni3S2. МоносульфидNiS может быть
получен нагреванием NiO с серой.
В ряду напряжений Ni стоит правее Fe (их
нормальные потенциалы соответственно -0,44 в и -0,24 в) и поэтому медленнее,
чем Fe, растворяется в разбавленных кислотах. По отношению к воде никель
устойчив. Органические кислоты действуют на никель лишь после длительного
соприкосновения с ним. Серная и соляная кислоты медленно растворяют никель;
разбавленная азотная - очень легко.
При взаимодействии с кислотами образуются соли
2-валентного Ni. Почти все соли Ni (II) и сильных кислот хорошо растворимы в
воде, растворы их вследствие гидролиза имеют кислую реакцию. Труднорастворимы
соли таких сравнительно слабых кислот, как угольная и фосфорная. Большинство
солей никеля разлагается при прокаливании (600- 800 °C). Одна из наиболее
употребительных солей - сульфат NiSO4 кристаллизуется из растворов в виде
изумрудно-зеленых кристаллов NiSO4·7H2O - никелевого купороса. Сильные щелочи
на никель не действуют, но он растворяется в аммиачных растворах в присутствии
(NH4)2CO3 с образованием растворимых аммиакатов.
При повышенных температурах никель
взаимодействует с оксидами азота, SO2 и NH3. При действии СО на его
тонкоизмельченный порошок при нагревании образуется карбонилNi(CO)4.
Термической диссоциацией карбонила получают наиболее чистый никель. [11]
Присутствие никеля в природных водах обусловлено
составом пород, через которые проходит вода: он обнаруживается в местах
месторождений сульфидных медно-никелевых руд и железо-никелевых руд. В воду
попадает из почв и из растительных и животных организмов при их распаде.
Повышенное по сравнению с другими типами водорослей содержание никеля
обнаружено в сине-зеленых водорослях. Соединения никеля в водные объекты
поступают также со сточными водами цехов никелирования, заводов синтетического
каучука, никелевых обогатительных фабрик. Огромные выбросы никеля сопровождают
сжигание ископаемого топлива.
Концентрация его может понижаться в результате
выпадения в осадок таких соединений, как цианиды, сульфиды, карбонаты или
гидроксиды (при повышении значений рН), за счет потребления его водными
организмами и процессов адсорбции.
В поверхностных водах соединения никеля
находятся в растворенном, взвешенном и коллоидном состоянии, количественное
соотношение между которыми зависит от состава воды, температуры и значений рН.
Сорбентами соединений никеля могут быть гидроксид железа, органические вещества,
высокодисперсный карбонат кальция, глины. Растворенные формы представляют собой
главным образом комплексные ионы, наиболее часто с аминокислотами, гуминовыми и
фульвокислотами, а также в виде прочного цианидного комплекса. Наиболее
распространены в природных водах соединения никеля, в которых он находится в
степени окисления +2. Соединения Ni3+ образуются обычно в щелочной среде.
Соединения никеля играют важную роль в
кроветворных процессах, являясь катализаторами. Повышенное его содержание
оказывает специфическое действие на сердечно-сосудистую систему. Никель
принадлежит к числу канцерогенных элементов. Он способен вызывать респираторные
заболевания. Считается, что свободные ионы никеля (Ni2+) примерно в 2 раза
более токсичны, чем его комплексные соединения.
В речных незагрязненных и слабозагрязненных
водах концентрация никеля колеблется обычно от 0.8 до 10 мкг/дм3; в
загрязненных она составляет несколько десятков микрограммов в 1 дм3. Средняя
концентрация никеля в морской воде 2 мкг/дм3, в подземных водах - n.103
мкг/дм3. В подземных водах, омывающих никельсодержащие горные породы,
концентрация никеля иногда возрастает до 20 мг/дм3.
Содержание никеля в водных объектах
лимитируется: ПДКв составляет 0.1 мг/дм3 (лимитирующий признак вредности -
общесанитарный), ПДКвр - 0.01 мг/дм3 (лимитирующий признак вредности -
токсикологический).[10]
.4 Химическая и токсикологическая характеристика
цинка
Степень окисления цинка в соединениях равна +2.
Нормальный окислительно-восстановительный потенциал, равный 0,76В,
характеризует цинк как активный металл и энергичный восстановитель. На воздухе
при температуре до 100 °Сцинк быстро тускнеет, покрываясь поверхностной пленкой
основных карбонатов. Во влажном воздухе, особенно в присутствии СО2, происходит
разрушение металла даже при обычных температурах. При сильном нагревании на
воздухе или в кислороде цинк интенсивно сгорает голубоватым пламенем с
образованием белого дыма оксида цинка ZnO. Сухие фтор, хлор и бром не
взаимодействуют с цинком на холоде, но в присутствии паров воды металл может
воспламениться, образуя, например, ZnCl2. Нагретая смесь порошка цинка с серой
дает сульфид цинкZnS. Сульфид цинк выпадает в осадок при действии сероводорода
на слабокислые или аммиачные водные растворы солей Zn. Гидрид ZnH2 получается
при взаимодействии LiАlН4 с Zn(CH3)2 и другими соединениями цинка;
металлоподобное вещество, разлагающееся при нагревании на элементы. Карбид
цинка ZnC2 получен при нагревании цинка в токе ацетилена. Сильные минеральные
кислоты энергично растворяют цинк, особенно при нагревании, с образованием
соответствующих солей. При взаимодействии с разбавленной НCl и H2SO4 выделяется
Н2, а с НNО3 - кроме того, NO, NO2, NH3. С концентрированной НCl, H2SO4 и
HNO3цинк реагирует, выделяя соответственно Н2, SO2, NO и NO2. Растворы и
расплавы щелочей окисляют цинк с выделением Н2 и образованием растворимых в
воде цинкитов. Интенсивность действия кислот и щелочей на цинк зависит от
наличия в нем примесей. Чистый цинк менее реакционноспособен по отношению к
этим реагентам из-за высокого перенапряжения на нем водорода. В воде соли цинка
при нагревании гидролизуются, выделяя белый осадок гидрооксидаZn(OH)2. Известны
комплексные соединения, содержащие цинк, например [Zn(NH3)4]SО4 и другие. [11]
Попадаетцинк в природные воды в результате
протекающих в природе процессов разрушения и растворения горных пород и
минералов (сфалерит, цинкит, госларит, смитсонит, каламин), а также со сточными
водами рудообогатительных фабрик и гальванических цехов, производств
пергаментной бумаги, минеральных красок, вискозного волокна и др.
В воде существует главным образом в ионной форме
или в форме его минеральных и органических комплексов. Иногда встречается в
нерастворимых формах: в виде гидроксида, карбоната, сульфида и др.
В речных водах концентрация цинка обычно
колеблется от 3 до 120 мкг/дм3, в морских - от 1.5 до 10 мкг/дм3. Содержание в
рудных и особенно в шахтных водах с низкими значениями рН может быть
значительным.
Цинк относится к числу активных микроэлементов,
влияющих на рост и нормальное развитие организмов. В то же время многие
соединения цинка токсичны, прежде всего его сульфат и хлорид.
ПДКв Zn2+ составляет 1 мг/дм3 (лимитирующий
показатель вредности - органолептический), ПДКвр Zn2+ - 0.01 мг/дм3
(лимитирующий признак вредности - токсикологический). [10]
. ЭЛЕКТРОХИМИЧЕСКИЕ МЕТОДЫ ОПРЕДЕЛЕНИЯ МЕТАЛЛОВ
Электрохимические методы анализа (ЭХМА) основаны
на использовании процессов, протекающих на поверхности электрода или в
приэлектродном пространстве, и измерении электрического параметра системы
(разности потенциалов, силы тока, количества электричества, омического
сопротивления, электропроводности и др.), значения которого функционально
связаны с составом и концентрацией (специфическими свойствами) раствора, т.е.
пропорциональны количеству определяемого вещества в анализируемом растворе. Эти
зависимости используют для количественного и качественного определения веществ.
ЭХМА классифицируют по измеряемому параметру (табл. 3).
Таблица 3 - Классификация ЭХМА по измеряемому
параметру
|
Измеряемый
параметр
|
Метод
|
|
|
1.
Потенциал, Е, В
|
Потенциометрия
|
-прямая
потенциометрия (ионометрия) -потенциометрическое титрование
|
|
2.
Ток, I, мкА
|
-классическая
полярография (прямая вольтамперометрия) -инверсионная вольтамперометрия
-амперометрическое титрование
|
|
3.
Количество электричества, Q, Кл
|
Кулонометрия
|
-прямая
кулонометрия -косвенная кулонометрия
|
|
4.
Удельная электропроводность
|
Кондуктометрия
|
|
|
5.
Масса, m, г
|
Электрогравиметрия
|
|
Электрохимические методы довольно селективны
(кроме кондуктометрии), поэтому с их помощью количественно определяют одни
элементы в присутствии других, раздельно определяют разные формы одного
элемента, делят сложные смеси и идентифицируют их компоненты, а также
концентрируют некоторые микропримеси. Электрохимические методы широко применяют
для контроля состава природных и сточных вод, почв и пищевых продуктов,
технологических растворов и биологических жидкостей. Соответствующие методики
не требуют сложного оборудования, в них не используются высокие температуры и
давления. Разные электрохимические методы различаются по чувствительности,
точности, экспрессности и другим показателям, а потому хорошо дополняют друг
друга.
.1 Потенциометрия
Потенциометрические методы анализа подразделяют
на прямую потенциометрию (ионометрию) и потенциометрическое титрование. Методы
прямой потенциометрии основаны на прямом применении уравнения Нернста для
нахождения активности или концентрации участника электродной реакции по
экспериментально измеренной ЭДС цепи или потенциалу соответствующего электрода.
При потенциометрическом титровании точку эквивалентности определяют по резкому
изменению (скачку) потенциала вблизи точки эквивалентности.
Ионометрия - потенциометрический метод
исследования состава раствора с использованием ионоселективных электродов
(ИСЭ), при помощи которых можно определять концентрацию более 50 катионов,
анионов и молекулярных соединений. Наиболее широко применяются электроды,
селективные к ионам F-,C1-, CN-, S2-, NO3-, Н+, NH4+, Na+, Cu2+, Са2+, и Mg2+,
ИСЭ для определения содержания газов (СО2, NН3, H2S, NO) и органических
соединений (ацетилхолина, мочевины и т.д.).
Ионометрия обладает некоторыми принципиальными
преимуществами перед другими методами:
. Позволяет определять активную
концентрацию иона или молекулы на фоне его общей концентрации.
. Измерения можно проводить в
непрозрачных, мутных и окрашенных средах и вязких пастах.
. Время установления равновесного
потенциала ИСЭ чаще всего составляет секунды, что позволяет автоматизировать
проведение анализа.
. Относится к группе неразрушающих
методов контроля.
. Характерен широкий диапазон измерений,
который находится в пределах 1-10-6 моль/дм3, а в некоторых случаях и до 10-8
моль/дм3. Погрешность определения при прямой потенциометрии 2-10%, при
проведении потенциометрического титрования - 0,5-1,0%.
. Унифицированность аппаратуры,
возможность создания стационарных и переносных приборов.
К недостаткам следует отнести следующие:
. Селективность основной части электродов
не так велика, чтобы производить непосредственное измерение активной
концентрации анализируемого иона в любой среде.
. Возможность создания электродов,
селективных к многозарядным ионам, ограничена точностью измерения ЭДС.
. Для всех электродов характерен дрейф
стандартного потенциала, что требует периодической проверки градуировки
прибора.
Основой ионоселективных электродов является
полупроницаемая мембрана (ИСМ), обладающая селективной ионной проводимостью.
Если такая мембрана, например, проницаемая только для иона М, разделяет два
раствора MX различной концентрации (a1>a2), то в результате возникает
разность потенциалов между растворами, разделенными ИСМ, которую называют
мембранным потенциалом. Величина потенциала является функцией активной
концентрации иона М, что и используется в данном методе.
Для измерения разности потенциалов а,
следовательно, и активной концентрации иона М, необходимо обеспечить
электрический контакт между внутренним раствором (стандартный раствор с
известной концентрацией электролита) и исследуемым раствором. Для этой цели,
как правило, используют хлорсеребряные электроды сравнения.
Величина электродвижущей силы (ЭДС, Е) такой
системы связана с активностью определяемого иона следующим уравнением:
=E0 + 0,059 lg a/z (3.1)
где константа Е0 определяется выбором вспомогательных
электродов сравнения и их электродных потенциалов,- заряд определяемого иона.
Существует три способа количественного
определения концентрации вещества в потенциометрии.
. Метод концентрационного элемента
основан на измерении разности потенциалов между двумя идентичными ИСЭ,
находящимися в растворе известного состава и в анализируемом растворе с
неизвестным содержанием определяемого иона. Вычисления активной концентрации
ведутся с использованием вышеприведенных уравнений. Метод эффективен в автоматических
методах анализа.
. Метод градуировочнного графика основан
на измерении потенциала индикаторного электрода в растворе с неизвестной
концентрацией определяемого иона и расчете этой концентрации по уравнению
регрессии, найденному по серии градуировочных растворов с известной
концентрацией этого же иона. Для снижения погрешности анализа градуировочный
график строят по серии растворов, состав которых (концентрация инертного
электролита и рН) максимально приближен к составу анализируемого раствора. Как
правило, для этого во все измеряемые растворы вводят специальные фоновые
растворы, обеспечивающие постоянство ионной силы, рН и устраняющие мешающее
влияние ионов, сопутствующих определяемому. В настоящее время выпускают
иономеры, в которых отсчет активности (обычно p-функция) любого иона можно
производить непосредственно по шкале измерительного прибора после его настройки
по стандартным растворам. При работе с такими приборами нет необходимости в
построении калибровочного графика.
. Метод стандартных добавок основан на
введении в анализируемый раствор объемом Vx, содержащий неизвестную
концентрацию потенциалопределяющего иона Cх, точного объема Vдоб раствора этого
же иона с известной концентрацией Cдоб и вычислении Cх по измеренному значению
изменения потенциала индикаторного электрода E по формуле:
х=( Cдоб*Vдоб)/( Vx+ Vдоб)(10 -E/S- Vx/(Vx+
Vдоб)) -1 (3.2)
Основное достоинство этого метода заключается в
том, что все измерения проводятся в присутствии всех компонентов пробы. Поэтому
хорошей точности определения можно достичь, даже если существенная часть
определяемого иона находится в комплексной форме.
Ионоселективные электроды обычно классифицируют
по агрегатному состоянию электродноактивного материала[2].
Стеклянные электроды. Мембрана изготавливается
из стекол различного состава. Стеклянный электрод представляет собой трубку
специального сорта стекла с выдутым на ее конце шариком с тонкой стенкой.
Внутрь электрода заливают раствор электролита с известной величиной активности
ионов и помещают электрод сравнения - хлорсеребряный электрод.
Твердофазные электроды. Мембраны этих электродов
созданы на основе моно- и поликристаллов труднорастворимых в воде солей.
Электроды этого типа применяются для определения галогенид-ионов (Е-, С1-, Вг-,
1-), ионов S2-,Сu2+ ,Рb2- Существенным достоинством этих электродов является
длительный срок работы. Однако число твердых ионных кристаллических соединений,
обладающих селективной ионной проводимостью, ограничено.
Электроды с жидкой мембраной. В этих электродах
используется диафрагма, поры которой заполнены раствором электродноактивного
вещества в органическом растворителе. Ассортимент жидкостных ИСЭ постоянно
увеличивается. В настоящее время изготавливаются электроды селективные к ионам
К+, Ca2+, Cu2+, Mg2+, Pb2+, Mn2+, NH4+, МO3-, СО3-, SO42- а также к ионогенным
органическим соединениям (поверхностно-активные вещества, ацетилхолин и др.).
Газовые электроды позволяют определять активную
концентрацию следующих газов: CO2, NН3, NO2, H2S и т. д. В основе лежит реакция
этих газов с водой, в результате которой образуются ионы Н или ОН, которые, в
свою очередь, определяются рН-стеклянным электродом.
Ферментные, бактериальные и иммуноэлектроды
(биологические сенсоры) существенно расширяют рамки ионометрии, позволяя
определять концентрацию органических соединений в водных растворах (глюкозы,
мочевины, антибиотиков, ряда аминокислот и др.), и находят применение в
медицинской практике. В случае ферментных электродов на поверхности мембран
нанесен слой иммобилизированного фермента, который способен катализировать
превращения только одного субстрата. В результате такого превращения образуется
ион, к которому чувствителен данный ИСЭ. [3]
.2 Вольтамперометрия
Вольтамерометрическими называют методы анализа,
основанные на регистрации и изучении зависимости тока, протекающего через
электролитическую ячейку, от внешнего наложенного напряжения. Графическое
изображение этой зависимости называют вольтамперограммой. Анализ
вольтамперограммы дает информацию о качественном и количественном составах анализируемого
раствора.
Для регистрации вольтамперограмм нужна
электролитическая ячейка, состоящая из индикаторного электрода (иногда его
называют рабочим электродом) и электрода сравнения. Электродом сравнения обычно
служит насыщенный каломельный электрод или слой ртути на дне электролизера
(донная ртуть). В качестве индикаторного электрода используют ртутный капающий
электрод, микродисковые платиновые или графитовые электроды (вращающиеся или
стационарные).
Первый вольтамперометрический метод -
полярография. Рабочим электродом в нем служит капающий ртутный электрод. Эту
методику обычно применяют для определения ионов металлов.
Электролитическая ячейка состоит из ртутного
капающего электрода и электрода сравнения, погруженных в анализируемый раствор.
Для изготовления ртутного капающего электрода используют тонкий (d=0,05 мм)
стеклянный капилляр длиной 5-10 см. Один конец капилляра с помощью
полиэтиленовой трубки присоединяют к резервуару со ртутью. Если резервуар
поднять на достаточную высоту и укрепить в фиксированном положении на штативе,
то под давлением из капилляра через равные промежутки времени будут вытекать
капли ртути. Величина поверхности ртутной капли в стационарных условиях хорошо
воспроизводится, и это позволяет получать воспроизводимые результаты измерений.
В основе качественного полярографического
анализа лежит величина полуволны, характеризующая природу вещества. Его
числовое значение показывает, насколько легко восстанавливается на электроде
данное вещество. Потенциал полуволны непосредственно связан со стандартным
потенциалом данной окислительно-восстановительной системы, поэтому он для
одного и того же вещества будет зависеть от состава фонового электролита.
Потенциалы полуволн для различных ионов величины известные.
Прямая пропорциональная зависимость между
диффузионным током (или высотой волны) и концентрацией определяемого вещества
позволяет использовать полярографию для количественного определения
электроактивных веществ. Существует три способа количественного определения
концентрации вещетва: метод градуировочного графика, метод стандартов и метод
добавок.
Метод градуировочного графика. Для построения
градуировочного графика готовят серию из 4-5 растворов определяемого вещества с
известными концентрациями разбавлением более концентрированного исходного
стандартного раствора. Для определения используют наиболее подходящий для
определяемого элемента фоновый электролит. Для каждого из приготовленных
растворов регистрируют полярограмму в одинаковых, строго контролируемых
условиях эксперимента. По полученным полярограммам строят градуировочный график
в координатах высота волны (или диффузионный ток) -концентрация. В идентичных
условиях регистрируют полярограмму анализируемого раствора и, измерив высоту
волны, по градуировочному графику находят искомую концентрацию.
Метод стандартов. При наличии пропорциональной
зависимости между высотой волны и концентрацией определяемого вещества можно
значительно сократить время анализа, не строя градуировочных графиков, а
сравнивая высоты волн на полярограммах анализируемого и стандартного растворов,
полученные в идентичных условиях. Тогда искомую концентрацию рассчитывают по
следующей формуле:
Сх=Сстhх/hст (3.3)
где Сх - концентрация вещества в анализируемом
растворе;
Сст - концентрация стандартного раствора;х -
высота волны на полярограмме анализируемого раствора;ст - высота волны на
полярограмме стандартного раствора.
Метод добавок. После того как полярограмма
анализируемого раствора записана, в ячейку добавляют известное количество
определяемого вещества и записывают полярограмму раствора с добавкой. Тогда
искомую концентрацию рассчитывают по следующей формуле:
Сх=Сдоб h1/ (h1- h2 ) (3.4)
где Сх - концентрация вещества в анализируемом
растворе;
Сдоб - концентрация добавочного раствора;-
высота волны анализируемого раствора;- высота волны раствора с добавкой.[5]
Второй метод - вольтамперометрия. Различают:
амперометрическое титрование и инверсионную вольтамперометрию.
Индикаторным электродом обычно служит
вращающийся платиновый или графитовый микроэлектрод, который отличается от РКЭ
тем, что:
имеет другой интервал поляризации от -0,50…-1 В
до +1,4…1,6 В (область поляризации любого электрода определяется потенциалами
участков электрохимической реакции фонового материала и материала электродов);
поверхность электродов во время регистрации
вольтамперограммы не возобновляется, т. е. легко загрязняется продуктами
реакции, поэтому для получения достоверных результатов необходимо производить
очистку электродов перед каждой регистрацией вольтамперограммы.
Амперометрическое титрование - используется для
установления точки стехиометричности в окислительно-восстановительном
титровании. Он основан на регистрации изменения тока какого-либо из участков
химической реакции, протекающей в процессе титрования.
А(определямоеве-во) + Т(титрант) = Р(продукт)
(3.5)
Собирают электролитическую ячейку, на
индикаторном электроде устанавливают потенциал предельного тока окисления
(восстановления) электроактивного участка реакции (А, Т или Р). В качестве
электрода сравнения используют хлорсеребрянный электрод.
В электролизер с анализируемым веществом и
фоновым раствором прибавляют 0,1 мл раствора титранта из бюретки, через минуту
фиксируют значение тока, после прибавляют очередную порцию титранта (0,1 мл),
через минуту фиксируют значение тока и т.д. Величина тока будет сначала
постоянной и очень маленькой, затем начнет увеличиваться или наоборот сначала
увеличится, а потом будет оставаться постоянным.
Для построения кривой титрования необходимо
иметь три точки до начала изменения величины тока и три после. Кривую
титрования строят в координатах: I - VT.
Кривая титрования состоит из двух линейных
участков и по положению точки их пересечения находят объем титранта, пошедший
на реакцию с определяемым веществом.
Инверсионная вольтамперометрия (ИВА). Этот метод
детально разработан советскими электрохимиками-аналитиками, в частности
А.Г.Стромбергом и его учениками. Так как метод ИВА не требует применения
больших количеств ртути, он сравнительно безопасен. ИВА включает операцию
предварительного концентрирования определяемого элемента (как правило,
металла). Поэтому нижняя граница определяемых содержаний в этом методе
составляет 10-9 - 10-10 М, что на несколько порядков ниже, чем в классической
полярографии.
Чтобы провести анализ, пробу переводят в
раствор, а затем при постоянном перемешивании ведут электролиз этого раствора.
В качестве катода при этом используют неподвижную каплю ртути. В простейшем
случае продолжительность электролиза выбирают так, чтобы все катионы
определяемого металла успели восстановиться на катоде и накопиться там в виде
амальгамы. Затем полярность электродов меняют, капля ртути становится анодом,
начинается быстрый процесс анодного растворения накопившегося металла.
Возникает ток растворения, который и является аналитическим сигналом. Его
величина пропорциональна концентрации металла в капле. Так как объем капли во
много раз меньше объема исходного раствора, концентрация определяемого металла
в капле во много раз больше начальной. Во столько же раз усиливается
аналитический сигнал. Кроме высокой чувствительности, метод ИВА обладает и
другими достоинствами. Можно одновременно накопить на катоде (в капле ртути)
сразу несколько металлов, а затем раздельно регистрировать токи их анодного
растворения (на полярограмме наблюдают несколько пиков при разных потенциалах).
По положению пиков можно опознать соответствующие металлы, а по высоте или
площади пиков - рассчитать содержание каждого металла в исходной пробе. Такой
способ анализа обычно применяют для селективного определения следовых количеств
токсичных тяжелых металлов (свинец, кадмий и др.) в воде и пищевых продуктах, а
также для контроля микропримесей в химических реактивах и полупроводниковых
материалах. Точность инверсионной вольтамперометрии немного уступает точности
классической полярографии, но вполне достаточна для решения многих
химико-аналитических задач. [6]
Также, в качестве индикаторного электрода могут
использоваться твердые электроды.В инверсионном анализе с твердыми электродами
используют прежде всего две группы электродов:
электроды из благородных металлов;
Индикаторные электроды из платины и графита
отличаются от капающего ртутного электрода, во-первых, тем, что они имеют
другой интервал поляризации, и, во-вторых, что их поверхность во время
регистрации вольтамперограммы не возобновляется. На этих электродах разряд
ионов водорода протекает значительно легче, поэтому область поляризации
ограничена значительно более низкими отрицательными потенциалами. Таким
образом, твёрдые электроды используются в основном для измерений в области
положительных потенциалов, где ртуть уже растворяется .
Поверхностные свойства Pt, Au, прежде всего это
касается относительно лёгкого образования на поверхности оксидов, являются
причиной осложнения при электроосаждении и растворении плёнок металлов или их
малорастворимых соединений.
Пористые угольные или графитовые материалы
принципиально нельзя использовать в электрохимическом инверсионном анализе без
предварительной подготовки (диффузия раствора в поры, восстановление кислорода
в порах, большой емкостной ток). Поэтому используют импрегнированные электроды
(электроды из спектрального графита или углерода, пропитанные подходящим
наполнителем, в качестве которого чаще всего используют парафин, полиэтилен,
подходящие смолы). Также используются угольные пастовые электроды, главное
преимущество которых - легкость воспроизведения поверхности. Электроды из
пиролитического графита и стеклоуглерода не требуют никакой подготовки
(пропитки). Наиболее часто они используются в виде стационарного или
вращающегося диска .
В электрохимическом инверсионном анализе для
определения As, Hg, Se, Cu, Sb, относящихся к группе электроположительных
элементов, наиболее часто используют электроды из золота. Мышьяк относится к
числу элементов, для концентрирования которых на поверхности твердого электрода
необходимо наличие вспомогательного элемента, например золота. Оно облегчает
выделение мышьяка на поверхности электрода за счет образования
интерметаллических соединений (ИМС). Широкое применение нашли золото-пленочный
и золотой вращающийся дисковый электрод, последний очень прост в обращении и
обладает лучшей воспроизводимостью, но немногим уступает в пределах обнаружения
золото-пленочным электродам [13].
В настоящее время научно-производственные
предприятия предлагают различные комплексы на базе вольтамперометрического
анализатора для определения микроколичеств тяжелых металлов в различных
объектах анализа. Напримервольтамперометрические комплексы
"Экотест-ВА" для анализа тяжелых металлов.
Модульная конструкция вольтамперометрического
анализатора «Экотест-ВА» обеспечивает возможность работы как с
раздельнымиэлектродами (стационарный рабочий электрод, модули с вращающимся
дисковым рабочим электродом, так и с комбинированным электродом «3 в 1». Кроме
этого, анализаторы могут управляться внешним устройством типа «Автосамплер»,
обеспечивающим автоматизацию процесса замены анализируемой среды.
Каждая комплектация допускает дальнейшее
расширение другими рабочими электродами, электрохимическими ячейками и
подключение автосамплера.[14]
.3 Кулонометрия
Метод прямой кулонометрии пригоден для
определения только электроактивных веществ, поскольку, в его основе лежит
непосредственноеэлектропревращение вещества на электроде. Прямые кулонометрические
измерения можно проводить, поддерживая постоянной либо силу тока (необходимо
иметь гальваностат), либо потенциал рабочего электрода (необходимо иметь
потенциостат).
Погрешность измерения Q зависит от точности
измерения времени, поскольку современные приборы позволяют очень точно измерять
даже небольшие токи. Прямая кулонометрия при постоянной силе тока является
более простым, но менее селективным способом, поскольку в определенный момент
времени может пойти реакция с участием мешающих веществ, фонового электролита
или растворителя, и выход по току начинает уменьшаться по экспоненциальному
закону.
Чаще применяют прямуюкулонометрию при постоянном
потенциале рабочего электрода. Потенциал электрода выбирают в области
предельного тока; в этом случае ток, протекающий через ячейку, будет
уменьшаться по экспоненциальному закону в соответствии с уменьшением
концентрации электроактивного вещества.
Однако в аналитической практике этот способ
измерения количества электричества применяют редко. Чаще измеряют ток, а не
количество электричества.
Электролиз ведут до достижения остаточного тока
It, величина которого определяется требуемой точностью. Так, если допустима
погрешность порядка 0.1%, то электролиз можно считать завершенным при It ~
0.001•I0.
Прямая кулонометрия - высокочувствительный и
точный метод анализа, легко поддающийся автоматизации. Общая погрешность метода
может составлять 0.5%. При проведении электролиза в течение 103 спри силе тока
1 мкА принципиально возможно определить до 10‾9 г вещества.
Наиболее распространенный метод
кулонометрического анализа - кулонометрическое тирование в гальваностатическом
режиме. Он отличается простотой аппаратурного оформления, высокой точностью и
надежностью.
Кулонометрическое титрование в значительной
степени сохраняет аналогию с другими титриметрическими методами. Основное
различие состоит в том, что в обычных титриметрических методах титрант заранее
готовится по точной навеске или стандартизируется по специальным установочным веществам.
В методе кулонометрического титрования титрант генерируется электрохимическим
методом. Это значительно сокращает время анализа и позволяет использовать в
качестве титранта малоустойчивые, летучие растворы, например, CI2; Вr2; SnCI2 и
др. Такой титрант называется электрогенерированным кулонометрическим титрантом,
аэлектрод, на котором его получают, генераторным.
Титрант в косвенной кулонометрии может быть
электрогенерирован из материала электрода (серебро, медь, хром), из
растворителя ( Н+ и ОН- при электролизе воды) или из дополнительно вводимого
вспомогательного вещества.
Количество электричества при кулонометрическом
титровании, когда сила тока поддерживается постоянной, легко рассчитывается по
результатам измерения времени, затраченного на электролиз:
= I • t. (3.6)
Поскольку время и ток можно измерять с очень
высокой точностью, главным фактором, который определяет точность всего метода,
становится фиксация точки эквивалентности.
Точка эквивалентности (т. е. появление малейшего
избытка реагента)может быть зафиксирована различными методами:
а) с помощью химических индикаторов (крахмал в
случае с йодом);
б) электрохимическими методами (амперометрия или
потенциометрия);
в) спектрофотометрическими методами.
Следует подчеркнуть, что в кулонометрическом
титровании в отличие от классического титриметрического анализа при индикация
точки эквивалентности измеряется не эквивалентный объем титранта, а время,
затраченное на электрогенерацию титранта к моменту достижения точки
эквивалентности в реакции взаимодействия определяемого вещества с электрохимически
получаемым титрантом .На основании полученного результата по закону Фарадея
рассчитывается масса титранта, по величине которой находится количество
анализируемого вещества.
В кулонометрическом титровании используют
химические реакции различного типа: кислотно-основного взаимодействия,
окисления-восстановления, комплексообразования, осаждения. Можно определять
неорганические и органические вещества.
В общем случае установка для кулонометрического
титрования при постоянной силе тока содержит следующие узлы:
) источник постоянного тока;
) устройство для определения количества
электричества;
) электролитическую ячейку с генераторным
электродом;
) индикаторную систему для определения
конца титрования;
) хронометр для определения
продолжительности электролиза.
Кулонометрическое титрование имеет значительные
преимущества не только перед прямой кулонометрией (быстрота выполнения,
нетрудоемкость анализа, отсутствие дорогостоящей аппаратуры, возможность
анализа неэлектроактивных веществ), но и перед объемным титрованием, так как в
амперостатическом титровании не надо заранее готовить рабочие растворы и
устанавливать их точную концентрацию.
При кулонометрическом титровании в качестве
электрогененерированных титрантов можно применять вещества, малоустойчивые в
обычных условиях и поэтому непригодные для приготовления рабочих растворов,
например, Cu2SO4, SnCl2, Cl2, Br2 и др.
Это единственный из косвенных методов анализа
(методов титрования), который не требует приготовления стандартных растворов.
Кулонометрический метод можно автоматизировать и
управлять им дистанционно, что имеет большое значение для определения
радиоактивных элементов.
Кулонометрия при постоянной силе тока пригодна
для определения малых количеств препарата (до 10-6 моль/дм3) с небольшой погрешностью.
Регулируя силу тока, можно легко и с высокой
точностью вводить в раствор небольшие порции реагента, тогда как в классическом
титриметрическом анализе дозировка малых объемов даже сильно разбавленных
растворов приводит к значительным ошибкам.
Так как современные приборы позволяют определить
силу тока и время с погрешностью не более 0,01 %, точность кулонометрического
титрования лимитируется только величиной индикаторной погрешности и составляет,
как правило, от 0,05 до 0,1 %.
Метод характеризуется высокой селективностью,
позволяя определять вещества в растворе без предварительного химического
разделения.
По совокупности характеристик амперостатическая
кулонометрия является точным, чувствительным, простым и надежным методом.[4]
.4 Кондуктометрия
Кондуктометрия - основана на измерении
электропроводности раствора и применяется для определения концентрации солей,
кислот, оснований и т.д. При кондуктометрических определениях обычно используют
электроды из одинаковых материалов, а условия их проведения подбирают таким
образом, чтобы свести к минимуму вклад скачков потенциала на обеих границах
раздела электрод/электролит (например, используют переменный ток высокой
частоты). В этом случае основной вклад в измеряемый потенциал ячейки вносит
омическое падение напряжения IR, где R - сопротивление раствора.
Электропроводность однокомпонентного раствора можно связать с его
концентрацией, а измерение электропроводности электролитов сложного состава
позволяет оценить общее содержание ионов в растворе и применяется, например,
при контроле качества дистиллированной или деионизованной воды. В другой
разновидности кондуктометрии - кондуктометрическом титровании - к
анализируемому раствору порциями добавляют известный реагент и следят за
изменением электропроводности. Точка эквивалентности, в которой отмечается
резкое изменение электропроводности, определяется из графика зависимости этой
величины от объема добавленного реагента. [6]
3.5 Электрогравиметрия
Электрогравиметрия электрохимический метод
количественного анализа, основанный на определении увеличения массы рабочего
электрода вследствие выделения на нем определяемого компонента в результате
электролиза. Как правило, определяемое вещество осаждают в виде металла (или
оксида) на предварительно взвешенном платиновом катоде (или аноде). Момент
завершения электролиза устанавливают с помощью специфической чувствительной
качественной реакции на определяемый ион. Рабочий электрод промывают,
высушивают и взвешивают. По разности масс электрода до и после электролиза определяют
массу выделившегося металла или оксида.[7]
. ОПРЕДЕЛЕНИЕ КОНЦЕНТРАЦИЙ МЕДИ, КОБАЛЬТА,
СВИНЦА В ВОДЕ ПОЛЯРОГРАФИЧЕСКИМ МЕТОДОМ
.1 Перечень методик определения концентраций
меди, кобальта, свинца в воде
Контроль качества объектов окружающей среды производится
по нормативно-технической документации с грифом «разрешено к применению в РБ».
Существует «Перечень методик выполнения измерения,допущенных к применению в
деятельности лабораторий экологического контроля предприятий и организаций РБ»,
согласно которому медь, кобальт и свинец рекомендуется определять по следующим
МВИ (таблица.4).[8]
Таблица 4 - Перечень методик выполнения
измерения концентрации меди и кобальта в сточных водах
|
Наименование
методики выполнения измерений в данном перечне
|
Д
- диапазон измерений; нижний, верхний пределы измерений; ХП - характеристика
погрешности методики выполнения измерений
|
|
1
|
2
|
|
МВИ
концентрации кобальта фотометрическим методом с β-нитрозо-α-нафтолом
|
Д
- 1,0 - 12 мкг/пробе Д - 0,25 - 15 мкг/пробе Д - 1,0 - 40 мкг/дм3; ХП - 8 -
12%
|
|
МВИ
концентрации кобальта полярографическим методом
|
Д
- 25 - 125 мг/пробе Д - св. 0,1 мг/дм3
|
|
МВИ
концентрации кобальта методом рентгенофлюорисценции
|
Д
- 0,005 - св.1,0 мг/дм3; ХП - 15%
|
|
МВИ
концентрации кобальта методом ААС
|
Д
- 0,2 - 1,6 мкг/дм3; ХП - 10%
|
|
МВИ
концентрации кобальта фотометрическим методом с нитрозо-R-солью
|
Д
- 0,028 - 2,5 мг/дм3; ХП - 25%
|
|
МВИ
концентрации кобальта методом флуориметрии
|
Д
- 0,0001 - 0,0005 мг/дм3; ХП - 65% Д - 0,0005 - 0,002 мг/дм3; ХП - 40% Д -
0,002 - 0,005 мг/дм3; ХП - 25% Д - 0,005 - 0,01 мг/дм3; ХП - 20%
|
|
МВИ
концентрации кобальта методом рентгенофлюорисценции
|
Д
- 0,011 - 0,1 мг/дм3; ХП - 5%
|
|
МВИ
концентрации меди фотометрическим методом с диэтилдитиокарбаматом натрия
|
Д
- 0,005-0,1 мг/пробе; Д - 0,01-0,15 мг/пробе; Д - 1,0-30 мкг/пробе; Д -
0,01-5 мг/дм3;
|
|
МВИ
концентрации меди фотометрическим методом с дикупралом
|
|
МВИ
концентрации меди полярографическим методом
|
Д
- 0,02-0,5 мг/дм3;ХП-5-1%;
|
|
МВИ
концентрации меди фотометрическим методом с диэтилдитиокарбаматом свинца
|
Д
- 0,01-0,08 мг/дм3;ХП-25-50%; Д - 0,005-0,1 мг/дм3;ХП-9,3%;
|
|
МВИ
концентрации меди фотометрическим методом с пикрамин-эпсилон
|
Д
- 0,01-0,1 мг/дм3;ХП-5%; Д - 0,5-40 мкг/пробе;
|
|
МВИ
концентрации меди методом флуометрии
|
Д
- 0,05-0,1 мг/дм3;ХП-50%; Д - 0,1-0,2 мг/дм3;ХП-25%;
|
|
МВИ
концентрации меди методом рентгенофлюоресценции
|
Д
- 0,011-1,0 мг/дм3;ХП-25%; Д-0,001-св1,0мг/дм3;ХП-10-50%
|
|
МВИ
концентрации меди фотометрическим методом с неокупроином
|
Д
- 2-200 мкг/пробе;
|
|
МВИ
концентрации меди методом ААС
|
Д
- 0,2-6,0 мг/дм3;
|
|
МВИ.
МН 3369-2010. Методика выполнения измерений содержания металлов в жидких и
твердых матрицах методом атомной абсорбционной спектрометрии
|
Д
- 0,005-0,01 мг/дм3 Д - 0,01-10 мг/дм3
|
|
МВИ
концентрации свинца фотометрическим методом с дитизоном.
|
Д
- 2 - 30 мкг/дм3
|
.2 Методика № 2.2.41.1 МВИ концентрации свинца
полярографическим методом
Рассмотрим определение концентрации свинца на
примере МВИ концентрации свинца полярографическим методом. Метод основан на
электрохимическом концентрировании свинца (II) на ртутно-графитовом электроде
при потенциале предельного диффузионного тока (-1,2 В) с последующей
регистрацией величины максимального анодного тока, электрорастворения осадка (
Еп = -0,6 В). Рабочий электрод формируется путем концентрирования ртути из
разбавленных растворов и соли на поверхности графитового дискового электрода.
Величина регистрируемого аналитического сигнала соответствует концентрации
свинца в анализируемом растворе.
Определению свинца не мешают: медь, кадмий,
хром, железо, алюминий, если количество их и свинца соизмеримы.
Продолжительность одного определения, включая
УФ-облучение и приготовление реактивов, годных сутки; 4,5 часа, серии из 10
проб - 8 часов.
.3 Отбор проб
Пробы воды отбираются согласно НВН
3.3-5.3.01-85.
Объем пробы воды должен быть не менее 200 см3.
Пробу можно не консервировать.
.4 Средства измерений, вспомогательные
устройства, реактивы и материалы
Полярограф универсальный ПУ - 1.
Графитовый электрод. А. с. 998590 (СССР).
Полярографическая ячейка с донной ртутью.
Ртутно-кварцевая лампа типа ПРК -2 по ТУ 16-316.
Баллон со сжатым газом аргоном ГОСТ 10157-79.
Весы аналитические, отвечающие требованиям ТУ
25-06-1131-75
Колбы исполнения 2,1-го класса точности, вместимостью
25,50, 100, 500 и 1000 см3 по ГОСТ 1770-74Е.
Пипетки исполнения 4, 6, 1-го класса точности,
вместимостью 1, 2, 5, 10 см3 по ГОСТ 20922-74.
Калий хлористый, KCl, ТУ 6-09-3895-79, «осч(м)
23-3».
Свинец металлический ТУ 6-09-1490-75.
Ртуть (II) азотнокислая 1-водная, ГОСТ 4520-78,
Hg(NO3)*H2O, х.ч.
Перикись водорода 30%, H2O2, х.ч., ГОСТ
10929-76.
Кислота соляная, HCl, х.ч., ГОСТ 3118-77.
.5 Требования безопасности
При выполнении работ необходимо соблюдать
правила техники безопасности для химических лабораторий, а также правила
техники безопасности по работе со ртутью.
.6 Подготовка к выполнению измерений
Включение и подготовка полярографа ПУ-1 к работе
проводится в соответствии с правилами эксплуатации полярографа, которые
изложены в инструкции к прибору.
Приготовление реактивов.
.1 Стандартный раствор свинца (II). Основной
раствор. 0,1 г металлического свинца растворяют в 50 см3HNO3 (1:1) при
нагревании, приливают 50 см3 бидистиллированной воды, кипятят для удаления
окиси азота. После этого раствор переводят в мерную колбу и разбавляют водой до
1000 см3, 1 см3 раствора содержит 0,1 мг свинца (II).
Рабочий раствор. 1 см3 основного раствора
разбавляют до 100 см3 дистиллированной водой. 1 см3 раствора содержит 0,001 мг
свинца (II). Раствор готовят перед определением.
.2 Раствор ртути (II) 2*10-3 М. Растворяют 0,34
г Hg (NO3)2*H2Oв 50 см3HNO3 (1:1) и разбавляют водой до 1000 см3. Раствор
хранят в склянках из темного стекла.
.3 Раствор хлористого калия, 2 М. Растворяют
149,11 г хлористого калия в мерной колбе на 1000 см3.
4.7 Построение калибровочного графика
В мерные колбы емкостью 100 см3 приливают
рабочий раствор свинца (II) в количествах 0, 1, 3, 5, 7, 10 см3; 5 см3 2 М
раствора хлористого калия; 0,2 см3 концентрированной соляной кислоты; 0,2 см3 перекиси
водорода. Доводят до метки бидистиллированной водой, перемешивают. Концентрации
полученных растворов соответственно равны: 0; 0,01; 0,03; 0,05; 0,07; 0,10
мг/дм3. В полярографическую ячейку помещают 20 см3 раствора, 0,2 см3 раствора
ртути (II) и удаляют кислород. Накопление проводят в течении 2 минут при
напряжении -1,1 V, снимают переменнотоковуюполярограмму при амплитуде
переменного напряжения 30 mV, скорости развертки напряжения 6 mV/сек.
Графитовый электрод перед каждым измерением зачищают калькой.
Строят градуировочный график, откладывая на оси
абсцисс концентрацию свинца (II) в мг/дм3 по оси ординат - высоту пика в мм по
средним значениям из 5 параллельных измерений.
.8 Выполнение измерений
В мерную колбу емкостью 100 см3 помещают 5 см3 2
М раствора хлористого калия, 0,2 см3 концентрированной соляной кислоты, 2 см3
30 %-ного раствора перекиси водорода и доводит до метки анализируемой водой.
Тщательно перемешивают и раствор помещают в колбу емкостью 200 см3 из
кварцевого стекла. Данный раствор подвергают УФ- облучению в течению 4-х часов.
20 см3 обработанной таким образом воды с добавкой 0,2 см3 ртути (II)
полярографируется.
4.9 Вычисление результатов измерения
Концентрацию свинца (II) в анализируемой воде
рассчитывают по градуировочному графику или по методу стандартных добавок.
Содержание свинца (II) с использованием
градуировочного графика находят
=C1* (V0+∆V)/V0 (4.1)
где С1 - концентрация свинца (II) по
градуировочному графику, мг/дм3;- объем воды, взятой для анализа, см3;
∆V - объем добавок растворов, см3.
Содержание свинца (II) в воде по методу
стандартных добавок находят по формуле:
С= (Ccт*H0*∆V)/((H*(V0+∆V)/V0-H0)*Vob)
(4.2)
гдеCcт- концентрация стандартного раствора
определяемого компонента, мг/дм3;- первоначальная высота пика, мм;
∆V - объем введенной добавки стандартного
раствора определяемого компонента, см3;- высота пика определяемого компонента
после введения добавки, мм;- объем пробы воды, введенной в ячейку, см3;общий
объем раствора в ячейке, см3.
=Vnp+VHg2++ ∑∆V (4.3)
где ∑∆V - общий объем стандартного
раствора свинца.
За результат анализа принимают среднее
арифметическое двух параллельных измерений, расхождение между которыми не
больше значений оперативного контроля Д, приведенных в таблице 5 для данного
поддиапазона.
.10 Контроль точности результата
Контроль точности результатов измерений
содержания свинца (II) проводится с помощью нормативов оперативного контроля
воспроизводимости (Д) и точности (К), приведенных для поддиапазона содержания
свинца (II) в таблице 5.
Оперативный контроль воспроизводимости и
точности результатов измерений содержания свинца (II) проводят в соответствии с
Д1.01.9987.3-88. «Методика лабораторного контроля качества измерений состава
сточных вод», по результатам анализа рабочей пробы сточной воды и
соответствующей ей контрольной пробы, полученной методом добавок СО или
растворов свинца (II) с известным содержанием, выполняемого одновременно двумя
аналитиками на двух комплектах посуды, реактивов, средств измерения.
Решение об удовлетворительнойвоспроизводимости
результатов измерений принимают при условии:
Решение об удовлетворительной точности
результатов измерений принимается при условии:
|C - C|≤ К (4.5)
где С1, С2 - результаты измерения содержания
добавки двумя аналитиками;
С - среднее содержание добавки =(С1+С2)/2;
С - введенная добавка;
Д - норматив оперативного контроля
воспроизводимости;
К - норматив оперативного контроля точности.[9]
Таблица 5 - Нормативы оперативного контроля
воспроизводимости и точности результатов измерения содержания свинца (II)
|
Поддиапазон
содержания свинца, мкг/дм3
|
Д
мкг/дм3
|
К
мкг/дм3
|
|
15-100
|
22,3
|
11,2
|
ЗАКЛЮЧЕНИЕ
Целью данной работы является изучение контроля
содержания металлов электрохимическими методами в сточных водах гальванических
цехов машиностроительных предприятий.
Гальваническое производство - один из наиболее
крупных источников образования сточных вод в машиностроении. Основными
загрязнителями сточных вод в гальванических производствах являются ионы тяжелых
металлов, неорганические кислоты и щелочи, цианиды, поверхносто-активные
вещества. Наиболее опасными загрязняющими веществами являются тяжелые металлы -
медь, цинк, кобальт, никель, хром, железо, кадмий и другие. Опасность этих
веществ определяется их высокой токсичностью для живых организмов в
относительно низких концентрациях, а также способностью к биоаккумуляции и
биомагнификации.
Основными электрохимическими методами
определения металлов являются потенциометрия, кулонометрия, вольтамперометрия.
Выбор того или иного метода зависит от определяемого металла, его ожидаемой
концентрации, наличия мешающих компонентов, а также от допустимой погрешности
для данного измерения. Определения производят согласно «Перечня методик
выполнения измерения, допущенным к применению в деятельности лабораторий
экологического контроля предприятий и организаций РБ».
Поскольку концентрации тяжелых металлов в
сточных водах гальванических цехов велики, то чувствительность и точность
электрохимических методов анализа позволяют получить достоверные результаты по
их содержанию. А простота конструкции и относительно невысокая стоимость
приборов для электрохимических определений обусловили их широкое применение в
аналитической практике.
За последние годы 2010 - 2015 разработалась
только одна новая МВИ, с использованием электрохимического метода анализа:
МВИ.МН 4488-2012 Методика выполнения измерений концентрации хлорид-ионов в
сточных и поверхностных водах потенциометрическим методом с солями ртути на
автоматическом титраторе.
СПИСОК ИСПОЛЬЗОВАННОЙ ЛИТЕРАТУРЫ
1. Состояние
природной среды Беларуси: ежегодный экологический бюллетень 2013 год / НАН
Беларуси, М-во природных ресурсов и охраны окружающей среды; ред. В. Ф.
Логинов. - Минск: Минсктиппроект, 2014. - 361 с.
.Ионометрия
в неорганическом анализе / Л. А. Демина, Н.Б. Краснова, Б.С. Юрищева, М.
С.Чупахин . - Москва : Химия, 1991 . - 192 с.
.Ионоселективные
электроды /И.В. Корыта,К.М.Штулик, М.: Мир, 1989. - 272 с.
.Физико-химические
методы анализа / Дмитрович И.Н., Пругло Г.Ф., Федорова О.В., Комиссаренков А.А.
- Санкт-Петербург, 2014.- Ч. 1. - С. 1 - 78.
.Дорохова,
Е.Н. Аналитическая химия. Физико-химические методы анализе / Е.Н. Дорохова,
Г.В.Прохорова. М.: Высшая школа. 1991. 256 с.
.Электрохимические
методы анализа. [Электронный документ] - Режим доступа:
http://crus55.narod.ru/10-Дата доступа: 23.11.2015.
.Электрогравиметрия.
[Электронный документ] - Режим доступа:
http://www.xumuk.ru/encyklopedia/2/5293-Дата доступа: 23.11.2015.
.Перечень
методик выполнения измерений, допущенных к применению в деятельности
лабораторий экологического контроля предприятий и организаций РБ.- Минск 1996. −
Ч.1.
.Сборник
методик выполнения измерений, допущенных к применению в деятельности
лабораторий экологического контроля предприятий и организаций РБ. - Минск 2005.
− Ч.2. - С. 265 - 270.