Взаимосвязь парциальных процессов при знакопеременной поляризации медного электрода в растворах с ионами
МИНОБРНАУКИ
РОССИИ
ФЕДЕРАЛЬНОЕ
ГОСУДАРСТВЕННОЕ БЮДЖЕТНОЕ ОБРАЗОВАТЕЛЬНОЕ
УЧРЕЖДЕНИЕ
ВЫСШЕГО
ПРОФЕССИОНАЛЬНОГО ОБРАЗОВАНИЯ
«ВОРОНЕЖСКИЙ
ГОСУДАРСТВЕННЫЙ УНИВЕРСИТЕТ»
Химический
факультет
Кафедра
физической химии
Выпускная
работа бакалавра
по
направлению 020100 Химия
ВЗАИМОСВЯЗЬ
ПАРЦИАЛЬНЫХ ПРОЦЕССОВ ПРИ ЗНАКОПЕРЕМЕННОЙ ПОЛЯРИЗАЦИИ МЕДНОГО ЭЛЕКТРОДА В
РАСТВОРАХ С ИОНАМИ Fe3+
Обучающийся
Свиридова
В.Е.
Руководитель
к.х.н.,
доц. Кондрашин В.Ю.
Воронеж 2015
Реферат
Взаимосвязь парциальных процессов при
знакопеременной поляризации медного электрода в растворах с ионами Fe3+:
Бакалаврская работа / Свиридова В.Е. руководитель к.х.н., доц. Кондрашин В.Ю. -
Воронеж. ун-т. Химический ф-т. Кафедра физич. химии. 2015. 60 с.
Ключевые слова: Медь. Знакопеременная
поляризация. Взаимосвязь парциальных процессов.
Цель работы состояла в изучении взаимной связи
парциальных катодных и анодных реакций на медном электроде в растворах с
бескислородным окислителем при знакопеременной поляризации.
Исследовалось анодное окисление меди после
предварительной катодной поляризации в растворах:
0,2 M Na2SO4,
,2 M Na2SO4 +
0,01 M H2SO4,
,2 M Na2SO4 +
0,001 M H2SO4,
,2 M Na2SO4 +
0,001 M H2SO4 + 0,001 M Fe3+,
,2 M Na2SO4 +
0,01 M H2SO4 + 0,01 M Fe3+,
,2 M Na2SO4 +
0,001 M H2SO4 + 0,005 M Fe3+.
Методы исследования: хронопотенциометрический,
поляризационных кривых.
Илл.18. Библ.44. Прил.7.
Оглавление
Введение
Глава
1. Обзор литературы
.1
Анодное растворение меди
.1.1
Анодное растворение меди в хлоридных и сульфатных средах
.1.2
Анодное растворение меди в щелочном растворе
.2
Специфика растворения металлов под действием переменного тока
.3.Взаимосвязь
электродных процессов на сложных электродах
Глава
2. Объекты и методы исследования
.1
Объект исследования
.2
Электрохимическая ячейка. Подготовка растворов для эксперимента
.3
Хронопотенциометрический метод
.4
Метод поляризационных кривых
.5
Обработка экспериментальных данных
Глава
3. Экспериментальные данные и их обсуждение
.1
Постановка задачи и исследования
.2
Анодное окисление меди после предварительной катодной поляризации в нейтральном
растворе Na2SO4
.3
Растворение в подкисленных сульфатных средах
.4
Растворение медного электрода в растворах, содержащих ионы Fе3+…..40
.5
Обсуждение результатов
Выводы
Список
литературы
Приложение
Введение
Набор парциальных электрохимических процессов,
вызванных прохождением переменного тока большой амплитуды через границу металла
и раствора электролита, обычно намного шире набора процессов, возникающих под
действием постоянного тока. Это объясняется, в частности, тем, что
нестационарность поляризации и особенно смена ее знака ведут к значительному
изменению химического состава поверхностного слоя металла и приэлектродной
области раствора.
Понятно, что переменно-токовые процессы идут на
электроде не одновременно. Напротив, их протекание разделено во времени, а сама
их реализация может иметь как строго периодический характер, так и обладать
очевидными признаками непериодичности [1, 2]. Но в любом случае возможность
свободного варьирования скоростей процессов в катодном и анодном полупериодах
(практически в неограниченных пределах) позволяет заметить такие их
взаимосвязи, которые в иных ситуациях остаются замаскированными. В результате
переменный ток больших амплитуд оказывается привлекательным инструментом при
изучении и моделировании взаимной связи, взаимного влияния парциальных
электродных процессов на различных сложных электродах.
В предыдущих работах, выполненных на кафедре
физической химии, установлено четкое влияние продуктов катодной реакции на
последующее окисление меди и медно-цинковых фаз в анодном полупериоде при
переменно-токовой поляризации электродов [1-4].
Это влияние обеспечивают гидроксидные ионы в приэлектродной области раствора
электролита, которые возникают в результате катодного восстановления молекул
воды:
2Н2О + 2е- → Н2
+ 2ОН-.
Адсорбируясь на поверхности электрода и частично
диффундируя в глубь раствора, эти ионы способны изменить природу электродного
процесса в следующем анодом полупериоде [1]. Однако в работе [3] было обнаружено,
что влияние катодного процесса на анодный имеет место даже тогда, когда pH
приэлектродной области раствора после прохождения катодного импульса тока
изменяется незначительно, оставаясь заведомо меньше 7.
В растворах, содержащих окислители H2O2,
NO3¯,
NO2¯,
и др., протекание катодного тока вызывает образование гидроксидных ионов
приэлектродной области, в результате чего увеличивается pHs.
Все это оказывает влияние, часто весьма сильное, на природу последующего
анодного процесса [5]. Данная проблема решалась в ИФХ РАН и на кафедре
физической химии, и здесь уже ясность достигнута. Как продолжение этих работ
представляет интерес установить природу взаимного влияния парциальных
электродных реакций, вызванных знакопеременной поляризацией в растворах с бескислородными
окислителями.
Если же в рабочий раствор ввести окислитель, не
содержащий кислорода, то его катодное восстановление не должно сопровождаться
ростом pHs
и генерацией OН--ионов;
например,
Fe3+
+ e-
→ Fe2+.
В такой ситуации ожидается, что влияние катодной
реакции на анодное окисление меди будет сведено до минимума.
Цель работы состояла в изучении взаимной связи
парциальных катодных и анодных реакция на медном электроде в растворах с
бескислородным окислителем при знакопеременной поляризации.
В задачи работы входило:
. Изучение влияния продуктов катодных
реакций на последующие анодное окисление меди в нейтральных и подкисленных
сульфатных средах при знакопеременной поляризации.
. Изучение роли бескислородного
окислителя Fe3+
во взаимной связи парциальных электродных реакций на медном электроде при
знакопеременной поляризации.
Глава 1. Обзор литературы
.1 Анодное
растворение меди
Анодное растворение меди - сложный процесс,
который включает в себя различные стадии: адсорбции-десорбции компонентов раствора,
перенос электрона. Кроме того, присутствуют стадии диффузии, адсорбции
продуктов окисления меди и их десорбции. В этом процессе возможно образование
растворимых продуктов окисления или труднорастворимых соединений в виде оксидов
и гидроксидов меди, солей или основных солей. Скорость растворения металла
зависит от потенциала, а также и от природы и концентрации анионов раствора,
которые оказывают значительное влияние на прочность и состав образующихся
комплексных соединений. Так, в случае хлоридных растворов образуется комплексы
типа [CuCln](n-1)-,
где п =1- 4[1, 3, 4]. В сульфатных средах комплексы непрочные. При высоких
анодных потенциалах возможно окисление воды с выделением кислорода [3, 6].
Медь способна ионизироваться с образованием
одно- и двухзарядных ионов по схемам [3, 6]:
Cu → Cu+ + e-,
(1.1)→ Cu2+ + 2e-. (1.2)
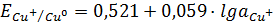 (1.3)
(1.3)
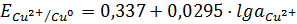 . (1.4)
. (1.4)
Прямые (E-lga),
отвечающие равновесным электродным потенциалам металлической меди с одно- и
двухзарядными ионами (1.3) и (1.4), благодаря разным предлогарифмическим
коэффициентам, имеют разные наклоны. Прямые пересекаются в точке с координатами
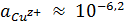 М и E = 0,153 В
(рис. 1.1) [8, 9], что отвечает значению стандартного электродного потенциала
М и E = 0,153 В
(рис. 1.1) [8, 9], что отвечает значению стандартного электродного потенциала 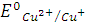 = 0,153 В.
При
= 0,153 В.
При 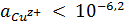 М и Е < 0,153 В ионизация меди
термодинамически более вероятна до однозарядного состояния, а при
М и Е < 0,153 В ионизация меди
термодинамически более вероятна до однозарядного состояния, а при 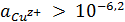 М и при Е > 0,153 В - до
двухзарядного. В точке пересечения медь находится в равновесии как с ионами Сu+, так и с
ионами Сu2+. Равновесие
меди с собственными ионами в хлоридной, цианидной, бромидной среде и в
растворах других подобных комплексообразователей отвечает первому из
рассмотренных случаев. Константы устойчивости таких комплексов достаточно
велики [1], поэтому в результате образования комплексов концентрация свободных
ионов меди редко превышает 10-6,2 М. В сульфатных, нитратных,
перхлоратных и других средах, анионы которых не образуют с ионами меди прочных
комплексов, активность потенциалопределяющих ионов, обычно выше, чем 10-6,2
М, поэтому термодинамически обусловлена ионизация меди до Сu2+-состояния.
В гидрокарбонатных средах область электродных потенциалов меди находится вблизи
пересечения прямых, описываемых уравнениями (1.3) и (1.4) [9].
М и при Е > 0,153 В - до
двухзарядного. В точке пересечения медь находится в равновесии как с ионами Сu+, так и с
ионами Сu2+. Равновесие
меди с собственными ионами в хлоридной, цианидной, бромидной среде и в
растворах других подобных комплексообразователей отвечает первому из
рассмотренных случаев. Константы устойчивости таких комплексов достаточно
велики [1], поэтому в результате образования комплексов концентрация свободных
ионов меди редко превышает 10-6,2 М. В сульфатных, нитратных,
перхлоратных и других средах, анионы которых не образуют с ионами меди прочных
комплексов, активность потенциалопределяющих ионов, обычно выше, чем 10-6,2
М, поэтому термодинамически обусловлена ионизация меди до Сu2+-состояния.
В гидрокарбонатных средах область электродных потенциалов меди находится вблизи
пересечения прямых, описываемых уравнениями (1.3) и (1.4) [9].
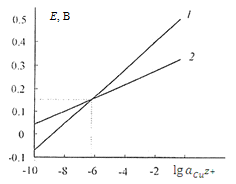
Рис.1.1. Зависимость
равновесных электродных потенциалов 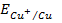 (1) и
(1) и 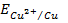 (2) от логарифма активности
потенциалопределяющих ионов [8, 9]
(2) от логарифма активности
потенциалопределяющих ионов [8, 9]
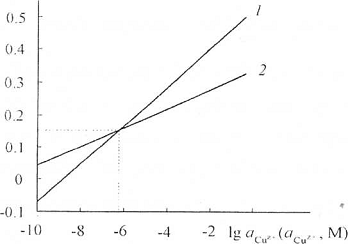
Термодинамика равновесных реакций
металлической меди с продуктами ее окисления в зависимости от pH дана на
диаграмме Пурбе (рис. 1.2).
Рис.1.2. Диаграмма
Е - pH для системы медь - вода при 25°С [10].
Рассматриваемая область pH ограничена значениями
0 и 14. Цифры, расположенные у линий, отвечают номеру соответствующей
равновесной реакции, а показатель степени - логарифму активности растворимого
продукта (Cu+,
Cu2+,
НСuO2¯
). Пунктирные линии на диаграмме выделяют область термодинамической
устойчивости воды:
2H3O+
+ 2e-
↔ H2
+ 2H2O,
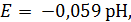
 O2 + 2H2O + 4e- ↔ 4OH-, (1.5)
O2 + 2H2O + 4e- ↔ 4OH-, (1.5)
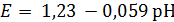 .
.
В кислых растворах (до pH 2,5…5)
возможно образование лишь растворимых частиц - Cu+ или Cu2+ в
соответствии с (1.1) и (1.2); равновесия с медным электродом описываются
уравнениями (1.3) и (1.4).
В нейтральных и щелочных растворах
протекают процессы образования Cu2O и затем CuO (или
гидратированной формы Cu(OH)2):
Cu2O + 2H+ + 2e- ↔ 2Cu + H2O, (1.6)
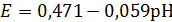 ,
,
CuO + 2H+ + 2e- ↔ Cu2O + H2O,
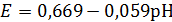 ,
,
Cu(OH)2
+ 2H+ + 2e- ↔ Cu2O + 3H2O,
(1.7)
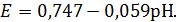
Линии, характеризующие равновесие
между Cu2+, Cu2O и CuO,
соответствуют реакциям и описываются уравнениями:
2Cu2+
+ H2O
+ 2e-
↔ Cu2O
+ 2H+,
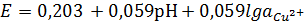 ,
,
Cu2+ + H2O ↔ CuO + 2H+, (1.15)
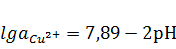 ,
,
Cu2+ + 2H2O ↔ Cu(OН)2
+ 2H+,
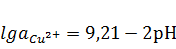 .
.
При высоких pH возможно образование растворимых
продуктов НСuO2¯,
равновесия которых с Cu2O,
CuO и Cu(OH)2
и соответствующие уравнения электродных потенциалов даны ниже:
2HCuO2¯
+ 4H++2e↔Cu2O+3H2O,
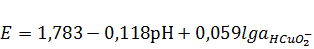 ,+ H2O ↔ HCuO2¯
+ 2H+,
,+ H2O ↔ HCuO2¯
+ 2H+,
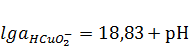 ,(OH)2 ↔ HCuO2¯
+ H+, (1.23)
,(OH)2 ↔ HCuO2¯
+ H+, (1.23)
1.1.1 Анодное растворение в
хлоридныхи сульфатных средах
Характерный вид анодной поляризационной кривой
медного электрода в хлоридном растворе имеет два ярко выраженных участка (рис. 1.3)
[1, 11]. Первый из них (ab)
в
полулогарифмических координатах Е - lgi
линеен
с угловым коэффициентом 0,059 В и отвечает активному растворению меди с
образованием однозарядных ионов Cu+.
В последующих реакциях, протекающих на поверхности электрода или в
приэлектродном слое, образуются прочные комплексы типа [CuCln](n-1)-.
Константы нестойкости Кн комплексов [CuCln](n-1)-
малы: для CuCl
-
1,2·10-4, для [CuCl4]3-
- 8,9·10-7 [1], поэтому в результате образования комплексов
концентрация свободных ионов меди не превышает 10-6,2 М.
Потенциалопределяющей стадией активного растворения меди в кислой и нейтральной
хлоридной среде является ее ионизация по уравнению [1, 11-13]:
Cu
↔
Cu+
+ e-.
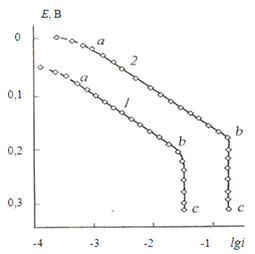
Рис.
1.3. Анодные поляризационные кривые
вращающегося медного электрода (1245 об/мин) при концентрациях HCl: 1М (1) и 2М (2) [13].
Энергия активации этой электродной реакции мала,
поэтому скорость велика. Анодный потенциал электрода определяется в основном
концентрационной поляризацией. Потенциал мало отличается от равновесного
потенциала, и его значение определяется активностью ионов Cu2+
в приэлектродном слое раствора:
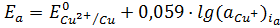 , (1.8)
, (1.8)
где 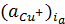 - активность ионов меди в
приэлектродном слое при плотности анодного тока ia.
С
учетом константы нестойкости комплексов уравнение (1.8) можно записать как [12,
13]:
- активность ионов меди в
приэлектродном слое при плотности анодного тока ia.
С
учетом константы нестойкости комплексов уравнение (1.8) можно записать как [12,
13]:
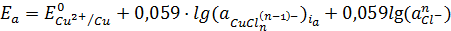 , (1.9)
, (1.9)
где 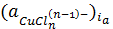 и
и 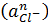 - активности комплексов и хлорид-ионов
в приэлектродном слое.
- активности комплексов и хлорид-ионов
в приэлектродном слое.
Активность комплексов [CuCln](n-1)- в
приэлектродном слое находится в прямой зависимости от анодного тока ia. Согласно
[11, 14], эта зависимость в условиях стационарной диффузии, когда концентрация
комплексов в объеме раствора может быть принята равной нулю, имеет вид:
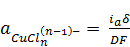 (1.10)
(1.10)
где δ - толщина
диффузионной зоны, D -
коэффициент диффузии, F
- постоянная Фарадея. Подставив (1.10) в (1.9), получим:
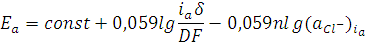 . (1.11)
. (1.11)
Из (1.11) следует, что при
постоянном потенциале 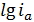 является линейной функцией
является линейной функцией 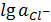 и зависит от условий перемешивания
раствора. Увеличение концентрации хлорид-иона в растворе и скорости вращения
электрода облегчают анодную реакцию, смещая активные участки поляризационных
кривых Е - сторону меньших значений
потенциалов (рис. 1.3, кривые 1, 2) [12, 15, 16]. При этом область активного
растворения меди расширяется. В работах [12, 17] сделано заключение о том, что
хлорид-ионы принимают непосредственное участие в электрохимическом процессе
растворения меди, и с уменьшением их концентрации в растворе координационное
число комплекса [CuCln](n-1)- понижается.
Так, при концентрации Cl--ионов 1М в
растворе преимущественно образуются комплексы состава [СuС12]-.
и зависит от условий перемешивания
раствора. Увеличение концентрации хлорид-иона в растворе и скорости вращения
электрода облегчают анодную реакцию, смещая активные участки поляризационных
кривых Е - сторону меньших значений
потенциалов (рис. 1.3, кривые 1, 2) [12, 15, 16]. При этом область активного
растворения меди расширяется. В работах [12, 17] сделано заключение о том, что
хлорид-ионы принимают непосредственное участие в электрохимическом процессе
растворения меди, и с уменьшением их концентрации в растворе координационное
число комплекса [CuCln](n-1)- понижается.
Так, при концентрации Cl--ионов 1М в
растворе преимущественно образуются комплексы состава [СuС12]-.
В [12, 14, 16, 18] было установлено,
что скорость анодного процесса контролируется диффузией продуктов окисления
меди от поверхности электрода в объем раствора. Это заключение подтверждается
линейной зависимостью 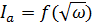 (где ?? - скорость вращения электрода), которая
экстраполируется в начало координат (рис. 1.4).
(где ?? - скорость вращения электрода), которая
экстраполируется в начало координат (рис. 1.4).
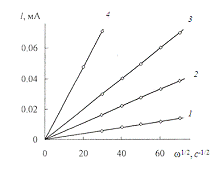
Рис.1.4. Зависимость
тока медного диска от числа его оборотов в 0,1 М растворе НСl при
потенциалах: -0,01 (1), 0,01 (2), 0,03 (3) и 0,05 В (4) (по н.к.э.) [18].
В соответствии с вышесказанным
активное растворение меди в хлоридных растворах можно представить следующим
образом. В начале происходит адсорбция хлорид-ионов на поверхности меди с
образованием заряженного комплекса с последующим отрывом электрона [1, 12, 14,
19]:
Clv¯ ↔
Cls¯ ↔ Clads¯,+
Clads¯ ↔
[CuCl]ads¯,
[CuCl]ads¯
↔ [CuCl]ads + е-.
(1.12)
Поверхностныекомплексы [CuCl]ads
затем могут переходить в приэлектродный слой и диффундировать в объем раствора
или гидратироваться и распадаться на гидратированные ионы Cl-
и Cu+.
[CuCl]ads ↔ [CuCl]s,
(1.13)
[CuCl]s
↔ [CuCl]v, (1.14)
[CuCl]s · aq ↔ Cus+·aq
+ Cls¯·aq.
(1.15)
При относительно невысоких концентрациях
хлорид-ионов (~0,01..0,05М) преимущественно образуются комплексы с одним
лигандом, диффузия которых (1.14) определяет скорость процесса в стационарных
условиях. В случае большей концентрации С1--ионов (или
незначительной поляризации) возможно образование комплексов с большим
координационным числом как на поверхности электрода (вслед за (1.12)), так и в
приэлектродном слое:
[CuCl]ads
+ (n-1)Clads¯
↔ ([CuCln](n-1)-)ads,
([CuCln](n-1)-)ads
↔ ([CuCln](n-1)-)s, [CuCl]s +
(n-1)Cls¯
↔ ([CuCln](n-1)-)s,s+·aq
+ Cls¯·aq
↔ ([CuCln](n-1)-)s.
Далее происходит диффузия продуктов окисления
меди из приэлектродного слоя в объем раствора, которая контролирует скорость
анодного процесса в стационарных условиях:
([CuCln](n-1)-)s
↔ ([CuCln](n-1)-)v.
Таким образом, процесс окисления меди
осуществляется при участии анионов хлора с образованием на поверхности элктрода
комплексных соединений [CuCl]ads,
являющихся исходным компонентом для всех последующих реакций.
В отличие от хлоридных сред, растворение меди в
сульфатных средах протекает по двум стадиям [19-20]:
Cu ↔ Cuads+
+ e-,
Cuads+ ↔
Cuads2+ + e-, (1.16)ads2+
→ Cus2+,
Cus2+
→ Cuv2+.
Исследования, проведенные на
вращающемся дисковом электроде, показали, что величина тока зависит от числа
оборотов диска, и зависимость линейна и экстраполируется в начало
координат [9, 12]. Это указывает на то, что контролирующей стадией при
растворении металла является диффузия продуктов окисления меди. При этом
поляризационная кривая описывается уравнением [11, 14]:
Снятие диффузионных ограничений с
ростом числа оборотов диска приводит к тому, что наклон с 0,03 В увеличивается
до 0,04 В. Причиной тому является смена лимитирующей стадии, вызванная ростом
скорости диффузионного переноса. На участке анодной поляризационной кривой с
наклоном 0,04 В контроль принадлежит стадии (1.16) [11, 14].
Форма расчётных поляризационных
кривых такая же, как и в случае хлоридных сред, однако, кривые смещены в
положительную область, поскольку в сульфатных средах не происходит образования
прочных комплексов. В случае диффузионной кинетики наклон поляризационных
кривых в области больших плотностей тока составляет 0,0295 В.
1.1.2 Анодное растворение меди в
щелочном растворе
В щелочном растворе естественно
предположить адсорбцию гидроксидионов на медном электроде. Вместе с тем, долгое
время существовало сомнение в возможности адсорбции воды на поверхности меди.
Считалось, что в нейтральных растворах потенциалы адсорбции воды на меди равны
-1,1… -1,2 В [22]. В настоящее время новейшими методами исследования
установлено, что адсорбция воды на меди имеет место даже в крепких растворах
кислот и в широком интервале потенциалов. Наряду с адсорбцией воды на металлах,
в том числе и на меди, имеется достаточно высокая концентрация гидроксид-ионов,
которые играют решающую роль в анодных процессах.
В работах [22] новым методом
контактного электросопротивления доказана специфическая адсорбция
гидроксид-ионов на некоторых металлах, в том числе и на меди. Поэтому в
настоящее время можно определенно считать, что активное анодное растворение
меди в щелочном растворе протекает через стадию взаимодействия поверхности
металла с ионами OHads. Последние образуются
благодаря адсорбции OH- из объема
раствора, и при диссоциативной адсорбции воды [22]:
H2Oads ↔
H+ads + OH¯ads.
Участие в анодном процессе OH¯ads
приводит к образованию поверхностных комплексов CuOH¯ads
[1, 23]:
Cu + OH¯ads
↔ [CuOH¯]ads
+ e-. (1.17) (1.42)
Возникновение [CuOH¯]ads
вызвано частичной, а при значительной анодной поляризации - полной потерей
электрона с его переходом в металлическую фазу (1.17).
Далее возможны два варианта развития процесса. В
первом случае поверхностные комплексы [CuOH¯]ads
могут гидратироваться переходить в приэлектродный слой раствора (активное
растворение):
CuOHads
↔
CuOHs.
Затем CuOHs
как
основание средней силы [1, 2] можетчастично диссоциировать на гидратированные
ионы OH-
и ионы Сu+,
которые диффундируют в объем раствора:
CuOHs ↔ Cus+·aq
+ OHs¯·aq.
При этом контроль принадлежит стадии (1.17) [1].
Этот механизм получил название гидроксидного [14].
Второй вариант, реализующийся при значительной
анодной поляризации,
состоит в смещении отрицательного заряда от
кислорода к меди в поверхностном адсорбционном комплексе:
CuOHads → Cu(O-H)ads.
Образовавшиеся адсорбционные соединения Cu(O-H)ads
труднорастворимы, блокируют активные участки поверхности и вызывают пассивацию
электрода. При этом потенциал электрода смещается в положительную сторону [11].
Одновременно эти соединения могут преобразовываться в фазу СuОН
или, дегидратируясь, - в фазу Сu2О:
Cu(O-H)ads → CuOH,
2Cu(O-H)ads
→
Cu2O
+ Н2О.
Анодная поляризационная кривая медного электрода
в щелочном растворе чаще всего имеет два характерных пика: А1 и А2 (рис. 1.5)
[1]. После этапа активного растворения, возникает пассивация, связанная с
блокированием активной поверхности электрода труднорастворимыми продуктами
анодной реакции. В работах [12, 14, 17] установлено, что первая ступень
окисления связана с электрохимическим процессом образования оксида Cu(I)
по
реакции (1.7) (пик А1). Считается, что образование труднорастворимого слоя Сu2O
на поверхности медного электрода осуществляется в два основных этапа [1]. На
первом поверхность покрывается двумерными зародышами, которые формируются на
энергетически активных центрах, образовавшихся в начале процесса растворения
меди. Продолжительность первого этапа, по данным [1], не превышает 5 минут.
Второй этап - разрастание зародышей до полного заполнения поверхности.
Вторая стадия растворения меди заключается в
формировании смеси оксидов и гидроксидов Сu(II)
(пик А2). При этом потенциал сдвигается в положительную сторону, а ток резко
уменьшается. Согласно [12, 14, 15, 17] сначала оксид Cu2O
окисляется до гидроксида Cu(OH)2,
который с течением времени (особенно при повышенной температуре) разлагается и
переходит в CuO, что и
является конечным продуктом второй ступени окисления меди.
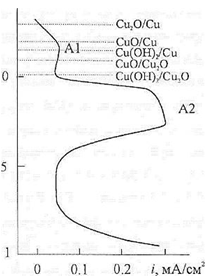
Рис.1.5. Анодная
потенциодинамическая поляризационная кривая на меди в 0,1 М KOH
при скорости развертки потенциала 10 мВ/с [22].
Согласно [1], при достаточно высоких потенциалах
(но не превышающих потенциал выделения кислорода) в концентрированных щелочных
средах возможна и третья ступень окисления меди с образованием купрат-ионов HCuO2¯
и CuO22¯.
В результате многократно повторяющихся циклов гальваностатической поляризации
эти ионы способны образовывать неустойчивый высший оксид Cu2O3.
При дальнейшей анодной поляризации, когда
потенциал превосходит потенциал кислородного электрода, происходит разложение
воды по реакции (1.5). При катодной поляризации в щелочных растворах происходит
восстановление труднорастворимых продуктов в обратном порядке: сначала
восстанавливается CuO
и/или Cu(OH)2
до Cu2O,
а затем Cu2O
до металлической меди [11, 12, 17, 22].
Таким образом, кинетика анодного растворения
меди в стационарном режиме в кислых и щелочных средах весьма сильно
различается. В первом случае контролирующей стадией является диффузионный отвод
продуктов окисления из приэлектродного слоя в объем раствора, а собственно
электрохимическая стадия - быстрая, то есть квазиобратимая. По этой причине
анодные потенциалы меди в области активного растворения описываются уравнением
Нернста. Насыщение приэлектродного слоя относительно CuCl
образует
на поверхности медного электрода слой мелкокристаллической соли. Слой резко
уменьшает электрохимически активную поверхность электрода, что обусловливает
сдвиг электродного потенциала в положительную сторону («солевая пассивность»).
В случае щелочного раствора область активного
растворения меди невелика и находится при более отрицательных потенциалах, чем
активное растворение меди в кислых средах. Переход в пассивное состояние
сопровождается резким уменьшением анодного тока и последующим образованием
оксидов и гидроксидов меди на поверхности электрода. В универсальной буферной
смеси переход от механизма растворения меди в кислой среде к гидроксидному
механизму начинается при pH 4,1 (рис. 1.6), [12].
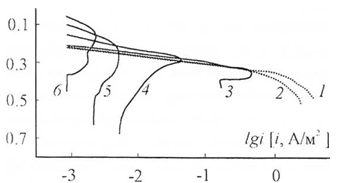
Рис.1.6. Анодные
поляризационные кривые растворения меди в растворах УБС с pH:
1 - 1,80, 2 - 2,09, 3 - 4,10, 4 - 6,80, 5 - 9,15, 6 - 11,90 [12].
При увеличении pH раствора участки активного
растворения меди смещаются в сторону отрицательных потенциалов, указывая на то,
что растворение меди облегчается, причем протяженность этих участков
уменьшается. При этом потенциал пассивации смещается в сторону отрицательных
потенциалов, и ток пассивации снижается. Зависимость скорости активного
растворения от щелочности раствора говорит о влиянии гидроксид-ионов на анодный
процесс.
1.2 Специфика растворения металлов
под действием переменного тока
катодный анодный медный электрод
Влияние переменного тока на электрохимические
процессы в значительной степени зависит от его формы и основных параметров:
плотности тока в катодный и анодный полупериоды, частоты, амплитуды,
длительности каждого полупериода. Большое значение при этом имеет состав
электролита, температура и др. [6, 24].
Переменный ток различной частоты используется
при электрохимическом полировании металлов, растворении благородных металлов,
анодировании. Он позволяет получать высококачественные корозионностойкие и
декоративные покрытия [6, 24-26]. Распределение продуктов растворения различных
металлов в приэлектродной зоне раствора при переменно-токовой поляризации
существенно отличается от такового при наложении на электрод постоянного тока
[27, 28].
При переменно-токовой поляризации следует
различать ток, который идет на заряжение двойного электрического слоя
(емкостная составляющая) и на электрохимические процессы (фарадеевская
составляющая) [6]. Величина емкостной составляющей ic
при
заданной амплитуде колебания потенциала электрода ΔЕ
пропорциональна емкости двойного электрического слоя на металле с и частоте
поляризующего тока ƒ:
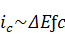 ,
,
а фарадеевская составляющая
поляризующего тока iф является
только функцией потенциала и не зависит от частоты (для электрохимических
реакций с контролирующей стадией ионизации или разряда ионов). Учет емкостной
составляющей переменного тока важен для правильной интерпретации
электрохимических явлений. Увеличение плотности переменного тока приводит к
уменьшению доли ic [29] этой
составляющей переменного тока.
Специфика анодного растворения
металлов при переменно-токовой поляризации состоит, прежде всего, в том, что
продукты анодного растворения металла в катодный полупериод могут
восстанавливаться или не восстанавливаться из-за низких значений потенциала, а
также из-за высокого перенапряжения процесса. В зависимости от этого выделяют
несколько характеров анодного растворения металлов под действием переменного
тока.
В наиболее простом случае
переменно-токовой поляризации образующиеся в анодный полупериод ионы металла в
последующий катодный полупериод не восстанавливаются, поскольку в этот
полупериод протекают реакции восстановления окислителей. Это обычно
восстановление растворенного кислорода и выделение водорода. К таким металлам
относятся, например, алюминий, магний, цирконий в непассивирующих средах
[29-31]. В пассивирующих средах продуктами окисления металлов являются
труднорастворимые оксиды или гидроксиды. Если в катодный полупериод они не
восстанавливаются, то может происходить оксидирование металла. Возникающий в
анодный полупериод пассивирующий слой будет включать как адсорбционную, так и
оксидную компоненты [29]. В отличие от оксидной, адсорбционная компонента легко
восстанавливается в катодный полупериод и переводит металл в активное
состояние. Поэтому при соответствующем подборе частоты переменного тока, когда
оксидная компонента еще не образуется, металл будет в активном состоянии.
Установлено, что при поляризации алюминия, магния и циркония в сульфатных и
нитратных средах образуются пассивирующие слои с небольшим содержанием
адсорбционной компоненты. Следовательно, эти металлы при переменно-токовой
поляризации не образуют растворимых продуктов окисления, но в анодный
полупериод оксидируются.
В общем случае пассивирующий слой,
возникающий на поверхности металлов в анодный полупериод тока, всегда частично
восстанавливается в катодный полупериод. Степень его восстановления зависит от
того, насколько сильно электрод был запассивирован в предшествующий анодный
полупериод, то есть от его анодного потенциала. Поскольку при любом потенциале
и плотности поляризующего тока необходимо некоторое время для образования
пассивирующего слоя, то при высоких частотах характерна заметная скорость
растворения. При низких же частотах образуются фазовые оксиды, восстановление
которых в катодный полупериод затруднено [32].
Таким образом, растворение металлов
в непассивирующих средах полностью определяется кинетикой реакции в анодный
полупериод и всегда эквивалентно фарадеевской составляющей переменного тока.
Скорость растворения в пассивирующих средах оказывается равной скорости
разрушения образующегося оксидного слоя. При этом металлы периодически
переходят из активной области в область пассивного состояния. Подобный эффект
может прослеживаться на благородных металлах, например, Pt,
Rh,
Ir и др. и их
сплавах [33]. В пассивирующих средах растворение благородных металлов
лимитируется скоростью разрушения пассивирующего слоя.
Такие металлы, как железо, свинец,
олово, медь, цинк и др. в анодный полупериод окисляются в активном состоянии, а
в катодный полупериод их собственные ионы или комплексы частично
восстанавливаются [34]. При соблюдении равенства суммы плотностей фарадеевского
тока в анодный и катодный полупериоды должно выполняться равенство:
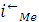 =
= 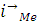 +
+ 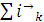 ,
,
где и 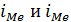 - скорости анодно-катодных реакций
на электродах с участием собственных ионов метала, " суммарная скорость
других катодных реакций. Материальный эффект растворения металла тем самым
будет определяться разностью скоростей реакции ионизации и восстановления
собственных ионов металла, или, другими словами, суммарной скорости катодных
реакций, не связанных с разрядом собственных ионов:
- скорости анодно-катодных реакций
на электродах с участием собственных ионов метала, " суммарная скорость
других катодных реакций. Материальный эффект растворения металла тем самым
будет определяться разностью скоростей реакции ионизации и восстановления
собственных ионов металла, или, другими словами, суммарной скорости катодных
реакций, не связанных с разрядом собственных ионов:
- = .
Для повышения скорости растворения активных
металлов необходимо облегчить протекание реакции восстановления окислителей.
Это достигается увеличением концентрации последних. Для ускорения растворения
металлов с большим перенапряжением водорода и высокой обратимостью
катодноанодных реакций относительно собственных ионов, таких как свинец, олово,
цинк, необходимо облегчить диффузионный подвод окислителей за счет изменения
гидродинамических условий [35].
Следует заметить, что в определенных средах на
рассматриваемых металлах, так же, как и на легко пассивирующихся, могут
образовываться адсорбционные или фазовые слои, тормозящие растворение металлов
[36].
.3 Взаимосвязь
электродных процессов на сложных электродах
В электрохимической теории сложных электродов
принято считать, что процессы на таких электродах протекают независимо друг от
друга. Это положение возведено в ранг принципа, получившего название принципа
независимого протекания электродных процессов [34]. Однако тщательное изучение
процессов на сложных электродах в ряде случаев четко указывало на их очевидную
взаимосвязь. Так, при потенциостатическом восстановлении кислородсодержащих
окислителей облегчается анодное растворение некоторых металлов (Fe,
Cr, Zn,
Mg, Sn
и
др.) [35, 36]. Причиной ускоряющего действия окислителя является взаимодействие
продуктов его восстановления (а не частиц окислителя) с поверхностью металла.
Такими анодно активными промежуточными продуктами выступают ОН--ионы,
которые образуются в катодной реакции, адсорбируются на поверхности металла и
влияют на кинетику окисления металла.
Схематично растворение металла в присутствии
кислородсодержащих окислителей представляется следующим образом. При
восстановлении окислителя происходит генерация OH-
частиц
(помимо диссоциативной адсорбции воды) по общей схеме:
Ох + nН3O+
+ mе- →
Red +
nОНads,
(1.47)
и при диссоциативной адсорбции воды:
H2Oads →
H+ads + OH-ads.
ЕслиионыOH-ads
- непосредственно взаимодействуют с металлом:
Me + nOH-ads →
[Me(OH)]ads + ne-,
[Me(OH)]ads + nН3O+
→ Men+ + 2nH2O,
то увеличение концентрации гидроксид-ионов
облегчает растворение металла. Оно способно идти при более отрицательном потенциале,
чем потенциал анодной реакции в отсутствие восстановления кислородсодержащего
окислителя [37-40].
Увеличение концентрации OH--ионов
на поверхности металла может происходить не только благодаря восстановлению
кислородсодержащих окислителей, но и катодного выделения водорода. Поэтому
увеличение скорости растворения кобальта, хрома, железа можно наблюдать при
введении в электролит солей металлов с более низким перенапряжением водорода (Pt,
Cu и др.) [34, 35].
Ускорение реакции ионизации указанных металлов также объясняется участием в
анодном процессе поверхностных OH--ионов.
Они образуются на диспергированных локальных микроэлектродах Pt,
Rh, Pd,
Cu и затем
диффундируют к активным участкам поверхности основного металла.
Пониженная концентрация ионов Н3O+ на границе
металл/раствор автоматически ведет к росту концентрации ионов OH-. В режиме
предельного диффузионного тока по Н3O+ (у
поверхности металла 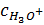 ~ 10-7М).
Превышение концентрации кислородсодержащего окислителя выше некоторого
критического значения приводит к новой катодной реакции его восстановления, где
донором протонов уже становится молекула воды. При этом процесс (1.47)
заменяется на:
~ 10-7М).
Превышение концентрации кислородсодержащего окислителя выше некоторого
критического значения приводит к новой катодной реакции его восстановления, где
донором протонов уже становится молекула воды. При этом процесс (1.47)
заменяется на:
Ох + рН3O+ + mе-
→ Red + nОНads.
В этих условиях продукты растворения
металлов могут образовывать труднорастворимые слои и тормозить анодную реакцию
[37].
Таким образом, ускорение реакции
катодного выделения водорода влияет на анодное окисление многих металлов и
может в одних случаях приводить к активации растворения металла за счет
повышения концентрации хемосорбированных на поверхности электрода ионов, а в
других к торможению растворения вследствие защелачивания приэлектродного
пространства и образования фазовых оксидов и гидроксидов на поверхности
металла.
В случае переменно-токовой
поляризации анодные и катодные процессы, как и при коррозии металлов, протекают
на одной и той же поверхности. Однако они разделены во времени. Возможность же
изменять скорости катодных и анодных реакций практически в неограниченных
пределах позволяет увидеть такие взаимосвязи между этими реакциями, которые при
коррозионных исследованиях могут оставаться незамеченными.
Главным и фактически единственным
путем влияния катодной реакции на анодное окисление меди до последнего времени
считался путь с участием ионов ОН- Обнаруженная же в работе [3,4]
взаимосвязь парциальных процессов в условиях увеличения pHs подробному
изучению не подвергалась. Этим были определены задачи настоящего исследования.
Глава 2. Объекты и методы
исследования
2.1 Объекты
исследования
Объектом исследования служил стационарный
электрод из меди (99,99%), армированной с трех сторон эпоксидной смолой.
Поверхность электрода зачищали на шлифовальной бумаге с последовательным
уменьшением размера зерна абразива до марки Р1000, полировали на фетре или
фильтровальной бумаге, обезжиривали этиловым спиртом и промывали
дистиллированной водой.
Рабочие растворы были выбраны таким образом,
чтобы анодное окисление металлов было кинетически простым и проходило в активном
состоянии. В таком случае специфика переменно-токового окисления металла и, в
частности, влияние катодных продуктов на последующее окисление в анодном
полупериоде выступали наиболее ярко.
Для исследования электродных процессов
использовали сульфатные среды состава:
,2 M Na2SO4 +
0,01 M H2SO4,
,2 M Na2SO4 +
0,001 M H2SO4,
,2 M Na2SO4 +
0,001 M H2SO4 + 0,001 M Fe3+,
,2 M Na2SO4 +
0,01 M H2SO4 + 0,01 M Fe3+,
,2 M Na2SO4 +
0,001 M H2SO4 + 0,005 M Fe3+.
Деаэрацию растворов проводили продуванием аргона
(ГОСТ 10157-79) в течении 50-60 мин. Катодное восстановление кислорода не
применяли, оно могло вызвать нежелательное изменение pH раствора.
Все
растворы готовили на дистиллированной воде из реактивов марки «химически
чистый».
2.2. Электрохимическая ячейка
Подготовка рабочих растворов для
эксперимента.
Электрохимическая ячейка, изготовленная из
стекла, с объемом рабочего пространства 0,10 л имела разделенные пространства
для рабочего и вспомогательного электрода. Ее верхняя часть содержала несколько
отверстий, позволяющих опускать в рабочее пространство электрод, капилляр
Луггина, индикаторный медный электрод и трубку для подачи аргона. Удаление
растворенного кислорода проводили продуванием через рабочий раствор аргона
(скорость около 0,5 мл/с) в течение 50-60 минут при непрерывном размешивании,
магнитной мешалкой.
2.3
Хронопотенциометрический метод
Для изучения взаимосвязи парциальных электродных
процессов использовали реверсирование тока с электронной стабилизацией.
Регулировка силы тока катодного и анодного направления осуществлялась
независимо друг от друга. Использовали режим поляризации: первое включение
анодного тока на 3 с, затем катодный ток на 3 с, после чего - снова анодный ток
прежнего значения на 5-10 с. Отклик потенциала электрода регистрировали
самопишущим потенциометром ЛКД 4-003, получая зависимость «потенциал-время» (E-t).
Для
увеличения входного сопротивления и уменьшения входной емкости потенциометра
был изготовлен специальный усилитель мощности исследуемого сигнала. Он представлял
собой операционный усилитель (микросхему) К284УД1А с входами на полевых
транзисторах, который работал в режиме повторения напряжения. Эти меры
позволили добиться входного сопротивления потенциометра (по постоянному току)
более 109 Ом, а входной емкости не более 10-11 Ф. Такие
параметры измерительного устройства фактически исключали шунтирование пары
«исследуемый электрод - электрод сравнения».
Выбор упомянутого режима поляризации создавал
ряд удобств при проведении эксперимента. Во-первых, запись хронопотенциограмм
могла проводиться электромеханическим прибором, точность которого заметно
превышает точность осциллографа. Во-вторых, продолжительная анодная и катодная
поляризация [1], не сопряжены с появлением ощутимых емкостных токов, что
значительно упрощает интерпретацию экспериментальных данных. Всё это дает
возможность выделить на E,t
-
зависимости определенные фазы развития электродных процессов: ионизацию
металла, его осаждение на электрод, выделение водорода и пр.
В качестве электрода сравнения применяли
насыщенный хлоридсеребряный электрод, собственный потенциал которого равен
0,194 В. В работе все потенциалы указаны по шкале стандартного водородного
электрода.
2.4 Метод поляризационных кривых
Методом поляризационных кривых
измеряли предельный катодный ток восстановления ионов Fe3+.
Поляризационные кривые получали в потенциодинамическом режиме, используя
потенциостат П5848. После подготовки поверхности электрода и обескислороживания
рабочего раствора на электрод подавали потенциал, равный стационарному (ок.
0,05 В). Скорость изменения потенциала задавали равной (-2) мВ/c. Протекающий
ток регистрировали по миллиамперметру М2020. Регистрацию тока заканчивали,
когда наступало его быстрое возрастание, вызванное катодным выделением
водорода.
В остальном методика не
отличалась от той, что использовалась при получении хронопотенциограмм.
2.5
Обработка
экспериментальных данных
Преимущественно анализировались
хронопотенциограммы, полученные при втором включении анодного тока. Как
правило, все опыты повторяли от 2 до 5 раз.
При заданных гидродинамических условиях участки
анодного растворения меди в большинстве случаев показывали расхождения величин
потенциалов в пределах ± 5 мВ. На приведенных в главе 3 рисунках такой разброс
значений не может быть изображен. Амплитудные значения катодных потенциалов
попадали в коридор случайных ошибок шириной до ± 0,1 В, что отмечено далее
общепринятым способом.
Глава 3.
Экспериментальные результаты и их обсуждение
3.1 Постановка задачи исследования
Влияние продуктов катодного процесса на анодное
окисление меди особенно легко наблюдать в слегка подкисленном или нейтральном
растворе «индифферентной» соли: KClO4, Na2SO4,
NaCl (рис.3.1.).
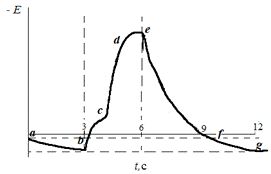
Рис.3.1. Хронопотенциограмма
реверсивной поляризации медного электрода в растворе «индифферентной» соли
(схематически).
В перхлоратном или сульфатном
растворе первое включение анодного тока сопровождается стадийным окислением
металла в активном состоянии с контролирующей второй стадией [40] (участок аb):
Cu → Cu+ + e-
(быстро), (3.1)+ → Cu2+ + e-
(медленно). (3.2)
В концентрированной хлоридной
среде продуктом окисления меди будут комплексные ионы меди (I):
Cu
+
nCl- →
СuCln(n-1)+
+
e- (быстро),
(3.3)
где n = 2, 3 или 4 в
зависимости от концентрации ионов Cl-
в растворе [40].
Процессы (3.1)-(3.3)
-
тривиальны, они ведут к накоплению ионов Cu2+ или СuCln(n-1)+
в приэлектродной области раствора.
Последующее включение катодного
тока вызывает частичное восстановление образовавшихся ионов меди, не продиффундировавших
в глубь раствора:
Сus2+
+
2e-
→
Cu (3.4)
или
СuCln(n-1)+
+
e-
→ Cu
+ nCl-.
(3.5)
На хронопотенциограмме это
восстановление отмечено более или менее продолжительной задержкой потенциала
(участок bc). При возникновении концентрационных ограничений по ионам Cu2+
или СuCln(n-1)+
процесс (3.4) или (3.5) постепенно заменяется восстановлением молекул Н2О.
При этом потенциал смещается в отрицательную область (участок сd)
и появляется новая катодная реакция (участок de):
2Н2О + 2e-
→ Н2 + 2ОН-.
Образующиеся гидроксидные ионы
способны частично адсорбироваться на поверхности медного электрода [41-43]:
2H2O + 2e-
→ H2 + 2OH-ads. (3.6)
Другая часть ионов остается в
приэлектродной зоне или диффундирует в глубь раствора:
2H2O + 2e-
→ H2 + 2OH-s. (3.7)
При протекании процесса (3.7)
pH приэлектродной зоны раствора может увеличиваеться на несколько единиц.
Подщелачивание околоэлектродного пространства зависит от способа подготовки
поверхности электрода, плотности и продолжительности протекания катодного тока,
гидродинамического режима опыта и др.
Следовательно, процессами (3.6)
и (3.7) создаются условия, при которых электрод, находящийся в нейтральной или
подкисленной среде, фактически оказывается погруженным в щелочной раствор.
Развивающиеся в такой ситуации электродные реакции представляют собой объект
нашего исследования.
При повторном включении
анодного тока поведение металла будет определяться составом как возникнувшего
адсорбционного слоя, так и того раствора, который граничит с поверхностью
электрода. Очевидно, в первую очередь станут потребляться OH-ads-ионы,
вызывая образование соединения адсорбционного типа [43] (участок ef):
Cu + OH-ads
→ (СuOH)ads
+ e-. (3.8)
По мере исчерпания OH-ads
в анодное окисление будут вовлекаться ионы приэлектродной области раствора OH-s,
не продиффундировавшие в глубь раствора:
Cu + OH-s
→ (СuOH)ads
+ e-. (3.9)
Если же предшествующая катодная
поляризация электрода отличается значительной величиной и продолжительностью,
то не исключено анодное образование трехмерной фазы оксида меди (I) [44]:
2Cu + 2OH-s
→ Cu2O + H2O + 2e-.
Однако в нашем исследовании
последний процесс интереса не представлял, и его возникновение, по возможности,
исключалось.
Следовательно, ионы ОН-ads
и ОН-s, генерируемые реакцией восстановления молекул
воды, выступают в роли интермедиата. Они создают термодинамические предпосылки
для возникновения на металле анодных процессов, не характерных для данной
среды. Реакции образования (СuOH)ads замедляют достижение потенциала
первого включения анодного тока, в чем и состоит влияние продуктов катодного
процесса на последующее анодное окисление металлической фазы. По мере
приближения к потенциалу первого включения анодного тока адсорбционный комплекс
(СuOH)ads способен переходить в раствор в виде продукта Cu2+
или СuCln(n-1)+
(участок
fg). После протекания процессов (3.8) и (3.9) создаются условия для
возобновления реакций растворения металла в активном состоянии (3.1) -
(3.3).
Эффективность влияния продуктов
катодной реакции на последующее анодное растворение металла можно
характеризовать безразмерным отношением электрических зарядов:
ε = Qef
/ Qbe (3.10)
где Qef
= iAtef
-
часть анодного заряда, пройденного через электрод на участке ef,
а Qbe = iKtbe
-
катодный заряд, пройденный через электрод на участке хронопотенциограммы be
(см. рис. 3.1). При исключении влияния процесса на процесс Qef
<< Qbe,
и тогда e
»
0. В противоположном случае e » 1.
Представленная выше очередность
электродных превращений подсказывает ряд простых приемов снижения их
взаимосвязи. Подавление или устранение влияния процесса на процесс может быть
достигнуто такими способами, которые ограничивают или исключают участие
интермедиатов ОН-ads и ОН-s в
электродных процессах. Один из таких способов нам видится во введении в рабочий
раствор окислителей, катодное восстановление которых не вызывает изменения pH
приэлектродной зоны раствора. К таким окислителям относятся, в частности, ионы Fe3+.
3.2 Анодное
окисление меди после предварительной катодной поляризации в нейтральном
растворе Na2SO4
Как было указанно выше, основой
взаимного влияния парциальных электродных процессов, вызванных протеканием
переменного тока через границу медной фазы и водного раствора, выступают
гидроксидные ионы. Для подтверждения данной взаимосвязи объектами исследования
служили стационарный медный электрод и неподвижный сульфатный раствор.
Отсутствие конвективного потока в данном случае позволяет утверждать, что
основная часть ОН--ионов остается в приэлектродной зоне раствора. Их
массоперенос в глубь раствора совершается только путем нестационарной
молекулярной диффузии. При смене направления тока на анодное электрод некоторое
время оказывается в среде измененного химического состава. Понятно, что природа
анодных процессов на металле в таких условиях может кардинально измениться.
На рис.3.2. представлена хронопотенциограмма
реверсивной поляризации медного электрода в нейтральном растворе сульфата
натрия.
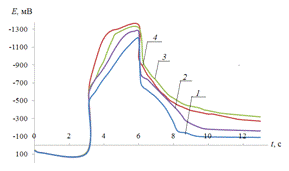
Рис.3.2.
Хронопотенциограмма реверсивной поляризации медного электрода в неподвижном
растворе 0,2 М Na2SO4: iA = 0,2 мА/см2,
iK
= 1(1), 2(2), 3(3), 4(4) мА/см2..
В неподвижном растворе, где
массоперенос ограничен, потенциал анодного растворения в активном состоянии
достигается через 10-20 с и более, в зависимости от тока предшествующей катодной
поляризации. Величина анодного заряда, необходимого для достижения потенциала
активного растворения металла (участок ab - см. рис. 3.1) первого анодного
включения тока, имеет значение от 2 до 5 мКл/cм2. Оценка по закону
Фарадея показывает, что такой заряд должен создавать от 5 до 15 монослоев
(CuOH)ads. В данном случае речь может идти об образовании слоя
оксида Cu2O [1].
Как сказано выше, от
таких условий эксперимента мы отказались. Эксперимент проводили в размешиваемых
с постоянной скоростью растворах, что позволило добиться удовлетворительной
воспроизводимости результатов. В частности, движущийся раствор устранял
искажающее влияние выделяющихся пузырьков водорода на нестационарную диффузию.
Рис. 3.3.
Хронопотенциограмма реверсивной поляризации медного электрода в размешиваемом
растворе 0,2 М Na2SO4: iA = 0,2 мА/см2,
iK
= 1(1), 2(2), 3(3), 4(4) мА/см2.
При размешивании раствора, с
ускорением массопереноса, эффект влияния процесса на процесс становится не
столь внушительным. Теперь время достижения потенциала анодного растворения
меди в активном состоянии уменьшается до 3-5 с, а анодные заряды сокращаются до
0,6 - 1 мКл/cм2. Такие заряды способны создавать не более одного
монослоя адсорбционного соединения CuOHads.
В неподвижном нейтральном
растворе Na2SO4, где условия для влияния процесса на
процесс оптимальны, величина (3.10) приближается к 1(на рисунке не показано).
Однако размешивание раствора уменьшает e примерно в 5 раз.
Описанная в п. 3.1
последовательность электродных превращений подсказывает пути целенаправленного
влияния на эффективность взаимосвязи парциальных электродных реакций. Помимо
ужесточения гидродинамических условий, эффект влияния процесса на процесс может
быть уменьшен при изменении состава рабочего раствора, исключающего образование
гидроксидных ионов при катодной поляризации электрода. В частности, подкисление
рабочего раствора, резко повышающего его буферную емкость, исключает
образование значительного числа гидроксидных ионов. Их образование может быть,
если только достигнуты диффузионные ограничения по ионам водорода.
3.3 Растворение в
подкисленных сульфатных средах
Подкисление раствора приводит к
тому, что влияние продуктов катодной реакции на последующее анодное окисление
металла в значительной степени подавляется. Тот путь влияния процесса на
процесс, который описан для нейтрального раствора (см. п. 3.2), остается в силе
и для кислых сред. Однако добавка сильной кислоты, резко увеличивая буферную
емкость раствора, способна, до определенных значений катодного тока iK,
стабилизировать pHs. Очевидно, для перехода pHs в
щелочную область теперь потребуются более высокие iK, чем в
нейтральном растворе Na2SO4.
В подкисленных растворах
возникновение соединения адсорбционного типа (CuOH)ads по (3.8) и
(3.9) возможно при условии превосходства iK над предельным катодным
током восстановления ионов водорода iKd. В противном
случае катодный ток будет преимущественно восстанавливать только ионы водорода
(см. процесс (3.6)), ионы OH-ads и OH-s
в такой ситуации практически не возникают. Это иллюстрирует рис. 3.4, из
которого следует, что заметный рост времени достижения потенциала активного
растворения металла происходит при iK > 5…6 мА/см2.
Действительно, после этого порога наблюдается быстрое удлинение «хвоста»
хронопотенциограммы. Соответственно возрастает и число монослоев адсорбционного
соединения (СuOH)ads, возникающего на участке ef. Например, при iK
= 3 мА/см2 оно равно всего лишь 0,22, а при iK = 20 мА/см2
- уже 2,37. Для порогового тока iK = 5…6 мА/см2
эта величина равна 1,48.
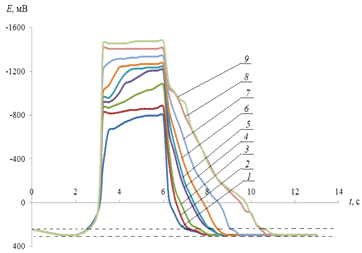
В полном согласии с
предложенной трактовкой, уменьшение концентрации H2SO4
удлиняет период достижения потенциала активного растворения (рис.3.5). В данном
случае можно говорить о возрастании эффективности влияния продуктов катодной
реакции на анодное окисление. Напротив, хронопотенциограммы, полученные в
растворе 0,2 М H2SO4, показывают очевидное подавление
связи парциальных электродных процессов (иллюстрацию не приводим).
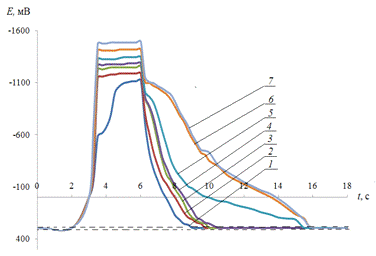
Рис.3.5.
Хронопотенциограмма реверсивной поляризации медного электрода в размешиваемом
растворе 0,2 М Na2SO4 + 0,001 М H2SO4:
iA = 0,2 мА/см2, iК = 2(1), 3(2), 5(3), 7(4),
10(5), 15(6), 20(7) мА/см2.
На рис. 3.6 приведены
зависимости параметра e от величины тока катодной
поляризации iK для растворов с разным содержанием H2SO4.
С ростом плотности катодного тока эффект влияния продуктов катодной реакции на
последующее анодное растворение металла уменьшается (кривые 1,2 и 3). Самый
сильный эффект влияния - в нейтральном растворе 0,2 М Na2SO4
(кривая
6).
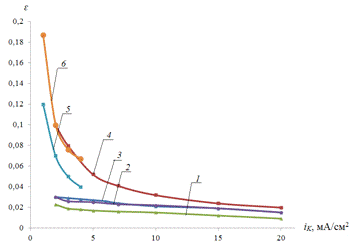
Рис.3.6. Зависимость
параметра e от величины тока катодной поляризации iK
для растворов с различным содержанием H2SO4: 1 - 0,15 М Na2SO4
+
0,05 M H2SO4;
2 - 0,19 М Na2SO4
+
0,01 M H2SO4;
3 - 0,2 М Na2SO4
+
0,05 M H2SO4;
4 - 0,2 М Na2SO4
+
0,001 M H2SO4;
5 - 0,2 M H2SO4;
6 - 0,2 М Na2SO4
iA
= 0,2 мА/см2 (размешиваемый раствор).
3.4 Окисление медного электрода в
растворах, содержащих ионы Fe3+
Стандартный потенциал
окислительно-востановительной пары Fe3+/Fe2+
составляет + 0,771 В. Поскольку в уравнение восстановления данных ионов
Fe3+
+ e-
↔ Fe2+
водородные ионы не входят,
значение равновесного электродного потенциала определяется лишь соотношением
концентраций Fe3+
и Fe2+,
но не зависит от pH.
В наших экспериментах потенциалы
восстановления ионов водорода и молекул воды достигают значений -1,0…-1,2 В,
что на 1,7…1,9 В отрицательнее стандартного электродного потенциала
окислительно-восстановительной пары Fe3+/Fe2+.
Есть основания полагать, что поляризация медного электрода (анодная и катодная)
происходит при одновременном катодном восстановлении ионов Fe3+:
Fe3+
+ e-
→ Fe2+.
Вероятно, оно протекает в
условиях, близких предельному катодному диффузионному току. В этом состоит одно
из главных отличий окисления меди в сульфатных средах (при реверсировании тока)
от её окисления в неагрессивных растворах: H2SO4, Na2SO4,
HCl и т. д.
На рис. 3.7 приведены катодные
поляризационные кривые восстановления ионов Fe3+
в подкисленных растворах сульфата натрия. Выход на предельный катодный
диффузионный ток четко выражен. При концентрации Fe3+,
равной 1 мМ, предельный ток составляет 0,09±0,01 мА/см2, а при
концентрации 0,01 - около 0, 90 мА/см2.
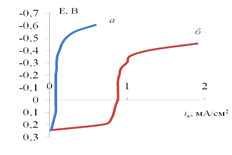
Рис. 3.7.
Катодные потенциодинамические (2 мВ/с) поляризационные кривые медного электрода
в растворе 0,2 М Na2SO4
+ 1 мM
Fe3+
+ 1мM
H2SO4
(а), 0,2 М Na2SO4
+ 0,01 M
Fe3+
+ 1мM
H2SO4
(б).
Главный момент влияния ионов Fe3+
- после включения катодного тока. Восстановление этого окислителя создает
конкуренцию двум катодным реакциям:
2Н+ + 2e-
®
Н2,
Н2О + 2e-
®
Н2 + 2ОН-.
В результате образование ОН--ионов
частично или полностью подавляется, и переход к реакции образования ионов меди
ускоряется. Таким образом, реакция восстановления бескислородного
деполяризатора должна способствовать подавлению влияния процесса на процесс.
Следует ожидать сокращение анодного участка ef хронопотенциограммы, на котором
образуется соединение адсорбционного типа (CuOH)ads, а также более
быстрый переход к активному растворению металла на участке fg.
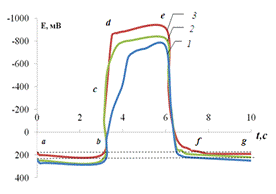
Рис. 3.8.
Хронопотенциограмма реверсивной поляризации медного электрода в размешиваемом
растворе 0,2 М Na2SO4 + 0,01 M Fe3+
+ 1 мМ Н2SO4:
iA = 0,2 мА/см2, iK
= 1(1), 1,5(2), 2(3) мА/см2.
В полном соответствии с
предположением, из рис. 3.7, наблюдается резкое сокращение времени перехода от
процесса (3.8) к процессу (3.2). Рис. 3.7 имеет еще одно характерное отличие от
аналогичных иллюстраций, приведенных ранее. Потенциал растворения меди в
активном состоянии ab примерно на 30 мВ положительнее потенциала растворения в
отсутствие окислителя. Это смещение вызвано одновременным саморастворением
электрода с Fe3+-деполяризацией,
в результате чего общая скорость перехода металла в раствор по уравнению (3.2)
возрастает. Понятно, что саморастворение - тоже причина сокращения участка de.
В какой степени ток восстановления деполяризатора отличен от предельного
катодного тока - сказать пока невозможно.
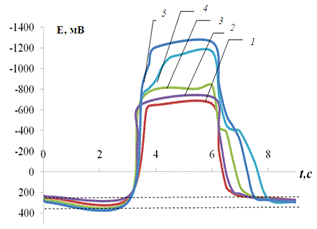
Рис.3.8.
Хронопотенциограмма реверсивной поляризации медного электрода в размешиваемом
растворе 0,2 М Na2SO4 + 1 мM Fe3+
+ 1 мМ Н2SO4:
iA = 0,2 мА/см2, iK
= 1,8(1), 2,8(2), 3,8(3), 5(4), 7(5) мА/см2.
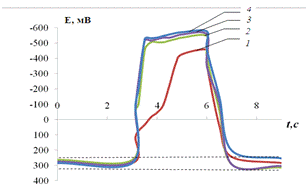
Рис.3.9.
Хронопотенциограмма реверсивной поляризации медного электрода в размешиваемом
растворе 0,2 М Na2SO4 + 5 мM Fe3+
+ 0,001 М Н2SO4:
iA = 0,2 мА/см2, iK
= 1(1), 2(2), 3(3), 4(4) мА/см2.
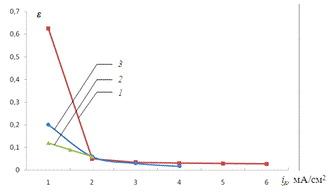
Рис.3.10. Зависимость
параметра e от величины тока катодной поляризации
iK для растворов с различным содержанием Fe3+
- ионов: 1 - 0,2 М Na2SO4
+
0,001 M
H2SO4
+ 0,001 М Fe3+;
2 - 0,2 М Na2SO4
+
0,001 M
H2SO4
+ 0,005 М Fe3+;
3 - 0,2 М Na2SO4
+
0,001 M
H2SO4
+ 0,01 М Fe3+.
Как и следовало ожидать, при
снижении концентрации окислителя переход к потенциалу активного растворения
меди происходит медленнее (рис. 3.8, 3.9). Напротив, подкисление рабочего
раствора, а значит, увеличение его буферной емкости способствует подавлению
процесса генерации гидроксидных ионов.
На рис. 3.10 показана
зависимость ε от iK.
С ростом плотности катодного iK
тока эффект влияния продуктов катодной реакции на последующее анодное
растворение металла в растворах с добавкой Fe3+-ионов
уменьшается, что выражается снижением ε. Здесь
следует учесть, что величина ε
= Qef / Qbe
- относительная. С ростом iK,
как это и следует и всех предыдущих хронопотенциограмм, абсолютное влияние
процесса на процесс усиливается, однако относительное - снижается.
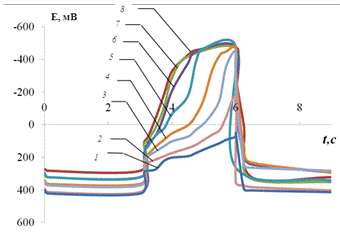
Рис. 3.11.
Хронопотенциограмма реверсивной поляризации медного электрода в размешиваемом
растворе 0,2 М Na2SO4 + 0,001 M H2SO4
+ 0,001 M Fe3+:
iK = 0,05 мА/см2, iА = 5(1), 3(2), 1(3),
0,5(4), 0,2(5), 0,1(6), 0,05(7), 0,01(8) мА/см2.
На рис. 3.11 изображены
хронопотенциограммы, полученные при изменении тока анодной поляризации, но при
постоянстве катодного тока. Примечательно, что с ростом iA
особенно
четко выделяются участки bc (см. рис. 3.1.). Напомним, что здесь
восстанавливаются ионы Cu2+, образовавшиеся в предшествующем анодном
процессе. Относительно быстрый переход от катодной реакции к анодному
потенциалу Cu2+/Cu-электрода
(примерно за 1 с) обязано саморастворению электрода. С включением внешней
анодной поляризации скорость перехода возрастает.
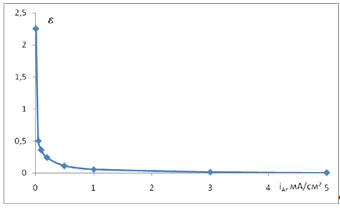
Рис.3.12. Зависимость
параметра e от величины анодного тока iA в
растворе 0,2 М Na2SO4 + 0,001 M H2SO4
+ 0,001 M Fe3+.
На рис. 3.12 дана зависимость e
- iA,
построенная на основе данных рис. 3.11. С увеличением плотности анодного тока
относительное влияние процесса на процесс уменьшается.
3.5 Обсуждение
результатов
На основе полученных данных,
предлагается следующая трактовка процессов, протекающих на медном стационарном
электроде при его знакопеременной поляризации.
Окисление медного электрода
начинается при его погружении в сульфатный водный раствор, содержащй ионы Fe3+.
Парциальный анодный процесс в сульфатной среде (0,2 М Na2SO4)
представляет собой окисление до растворимых форм меди по двухстадийному
механизму[19-20]:
Cu ↔ Cuads+
+ e-,ads+ ↔ Cuads2+
+ e-.
Контролирующая стадия - отрыв
второго электрона. Сопряженный катодный процесс - это восстановление Fe3+
до
Fe2+:
Fe3+
+ e-
↔ Fe2+.
Первое включение анодного тока
вызывает смещение потенциала от стационарного значения в положительную область.
Это нарушает баланс скоростей, характерный для саморастворения. Скорость
растворения меди равна iA0
= iK + iA,
где iA
- внешний анодный ток, а iK
- скорость восстановления ионов Fe3+.
За исключением последнего обстоятельства, анодный процесс на медном электроде
не отличается от процесса, идущего в растворе без добавок окислителя.
Не успевшие продиффундировать в
глубь раствора ионы металла будут восстанавливаться при включении катодного
импульса:
Cu2+
+ 2e-
→ Cu.
Восстановление идет
одновременно с восстановлением Fe3+.
Пока катодный ток не превосходит
предельную скорость восстановления ионов Fe3+, т. е. iK < iKd,
восстановление молекул воды невозможно. При этом генерация ОН-
исключена. В том случае, когда iK  iKd и особенно,
когда iK
iKd и особенно,
когда iK  iKd, неизбежно
восстановление воды,
iKd, неизбежно
восстановление воды,
2H2O + 2e- → H2 + 2 OH-,
со всеми возникающими осложнениями,
связанными с повышением pH приэлектродной области
раствора.
Таким образом, в режиме мгновенного
перехода от катодного процесса к анодному, при iK 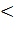 iKd,
бескислородный деполяризатор практически исключают влияние процесса на процесс.
По мере увеличения катодного тока влияние добавок Fe3+-ионов постепенно
сходит на нет, а при iK iKd оно вообще
исчезает. Можно предположить, что скорость образования ионов OH- здесь равна
iK
iKd,
бескислородный деполяризатор практически исключают влияние процесса на процесс.
По мере увеличения катодного тока влияние добавок Fe3+-ионов постепенно
сходит на нет, а при iK iKd оно вообще
исчезает. Можно предположить, что скорость образования ионов OH- здесь равна
iK  iKd, т.е. вклад iKd оказывается
очень малым:
iKd, т.е. вклад iKd оказывается
очень малым:
Cu + OH-ads
→ (CuOH)ads + e-,
Cu + 2OH-s
→ Cu2O +H2O + 2e-.
В подкисленных растворах Na2SO4, обладающих
значительно большей буферной емкостью, чем нейтральные растворы, образование
соединения адсорбционного типа (CuOH)ads маловероятно. Оно возможно
при условии, что iК превосходит предельный катодный ток
восстановления ионов водорода iKd (Н+). Если
данное условие не выполняется, и i < iKd (Н+),
то преимущественно будут восстанавливаться только ионы водорода, а ионы OH-ads
и OH-s в такой ситуации практически не возникают.
Понижение рН рабочего раствора
приводит к тому, что влияние продуктов катодной реакции на последующее анодное
окисление металла подавляется. При этом взаимосвязь парциальных электродных
процессов, которая описана для нейтрального раствора, остается в силе и для
кислых. Однако добавка сильной кислоты резко увеличивает буферную емкость
раствора, а значит, способна до определенных значений катодного тока iK,
стабилизировать pHs. Исходя из выше упомянутого, для перехода pHs
в щелочную область теперь потребуются более высокие величины iK, чем
в растворе Na2SO4, без добавки кислоты.
Расчет параметра ε показал,
что в неподвижном нейтральном растворе Na2SO4, где условия
для влияния процесса на процесс оптимальны, его величина приближается к 1. При
размешивании раствора параметр уменьшается примерно в 5 раз; соответственно,
возрастает доля анодного тока, расходуемого на окисление металла до Cu2+-ионов.
В подкисленных размешиваемых средах ε еще меньше. Общая тенденция
в изменении параметра такова, что ε уменьшается с ростом
катодного тока. В подкисленных растворах ε приобретает минимальные
значения и тоже уменьшается с ростом iK.
Из экспериментальных данных следует,
что ионы Fe3+,
восстанавливаясь в катодном полупериоде, создают конкуренцию реакции
образования ионов ОН-. В результате влияние парциальных катодных
процессов на последующие анодное окисление меди в анодном полупериоде
подавляется или вообще исчезает.
Одновременно ионы Fe3+ обладают
буферным действием из-за высокой склонности к гидролизу:
Fe3+ + ОН-
↔ FeОН2+,
(3.11)
FeОН2+
+ ОН- ↔ Fe(ОН)2+.
(3.12)
Пока в приэлектродной области
раствора реализуются равновесия (3.11) и (3.12), ее рН не возрастатет выше 4-5 единиц
[8]. Следовательно, добавление ионов Fe3+ в рабочий
раствор создается такой же эффект, как и подкисление раствора (см. п. 3.3).
Выводы
1. Условия, при которых особенно велико
влияние продуктов катодного восстановление Н2О на анодное окисление
меди - стационарный электрод в неподвижном нейтральном растворе Na2SO4.
Включение анодного тока после предшествующей катодной поляризации может
создавать поверхностное соединение Cu2Oтолщиной
до 10 монослоев. Параметр ε при
этом приближается к единице.
. Подкисление раствора Na2SO4
с добавкой 1мМ Н2SO4
способствует снижению влияния процесса на процесс. Включение анодного тока
после катодной поляризации образовывает соединение адсорбционного типа (CuOH)ads
толщиной максимум 1 - 2 монослоя. Параметр ε при
этом снижается до 0,02 - 0,03. С увеличением содержания Н2SO4
в растворе связь процессов становится еще ниже.
. Бескислородные ионы Fe3+,
введенные в подкисленный раствор Na2SO4,
создают конкуренцию катодному восстановлению молекул воды и обеспечивают
дополнительный буферный эффект. В результате влияние процесса на процесс
снижается еще сильнее: адсорбционного соединения (CuOH)ads
образуется доля монослоя, а ε = 0,015 - 0,02.
Список литературы
1. Куксина О.Ю. Растворение меди, α-
β-латуней
в хлоридных средах при инфранизкочастотной переменно-токовой поляризации:
Дисс.... канд. хим. наук / О.Ю.Куксина - Воронеж: Воронеж, ун-т, 2006. - 178 с.
2. Куксина О. Ю., Кондрашин В.Ю.,
Маршаков И.К. Парциальные электродные процессы при переменно-токовой
поляризации меди в хлоридных и нитратных средах. // Защита металлов. 2004. Т.
40. №6 С. 646-652.
3. Чекмарева Н.А. Переменно-токовое
окисление меди и серебра в растворах с различным ионным составом. Диплом.
работа. / Руководитель к.х.н., доц. Кондрашин В.Ю. - Воронеж. ун-т. Химический
ф-т. Кафедра физ. химии. 2008. 41 с.
4. Филипцова И.В. Анодное окисление
меди в хлоридных растворах при знакопеременной поляризации. Диплом. работа. /
Руководитель к.х.н., доц. Кондрашин В.Ю. - Воронеж, ун-т. Химический ф-т.
Кафедра физ. химии. 2009. 61 с.
6. Шульгин Л.П. Электрохимические
процессы на переменном токе / Л.П. Шульгин - Л.: Наука, 1974. -70 с.
7. Лошкарев Ю.М. Особенности структуры
и некоторых свойств цинковых покрытий, электроосажденных импульсным током из
щелочных электролитов / Ю.М. Лошкарев [и др.] // Электрохимия - 1994. - Т. 30,
№10. - С. 1287-1290.
8. Грушевская С.Н. Кинетика начального
этапа анодного образования CuCl
на меди и ее низколегированных сплавах с золотом / С.Н. Грушевская, Т.А.
Кузнецова, А.В. Введенский // Защита металлов. - 2001. - Т. 37, №6. -
С.613-623.
9. Крейзер И.В. Растворение меди при
катодной поляризации в кислых средах: дисс....канд. хим. наук / И.В. Крейзер. -
Воронеж, 2002. - 182 с.
10. Справочник химика: в 3-х т. / под.
ред. Б.П. Никольского. - M.
- Л: Химия, 1964. - Т. 3: Химическое равновесие и кинетика. Свойства растворов,
электродные процессы. - 1008 с.
11. Скорчеллетти В. В. Теоретические
основы коррозии металлов / В.В.Скорчеллетти. - Л: Химия, 1973. - 264 с.
12. Алтухов В.К. Механизм анодного
окисления меди в растворах хлоридов / В.К. Алтухов [и др.]; Воронеж, гос. ун-т.
- Воронеж, 1978. - 24 с. -Деп. в ВИНИТИ 26.10.78, № 1357-78.
13. Алтухов В.К. Влияние концентрации
хлоридионов на кинетику диодного растворения меди / В.К. Алтухов [и др.] //
Химия и хим. технология. - 1972. - Т. 15, № 11. - С. 1752-1754.
14. Скорчеллетти В.В. Теоретическая
электрохимия / В.В. Скорчеллетти. - Л. : Химия, 1973. - 608 с.
15. Могиленко В.Ф. Коррозия меди в
растворах хлоридов/ В.Ф. Могиленко, В.Н. Ковтун // Вопросы химии и хим.
технологии : Респ. межвед. на-\ ч.-техн. сб. - 1985. - №. 78. - С. 36-41.
16. Алтухов В.К. Влияние хлорида на
ионизацию и пассивацию меди / В.К. Алтухов, Т.А. Моргунова // Защита металлов.
- 1981. - Т. 17, №5. - С. 557-560.
17. Казанцев А.А. О механизме анодного
растворения меди в растворах галогенидов и роданидов / А.А. Казанцев, В.А.
Кузнецов // Электрохимия. - 1984. - Т. 20, № 7. - С. 934-939.
18. Алтухов В.К. Анодное окисление меди,
серебра и свинца в растворах хлоридов / В.К. Алтухов [и др.] // Защита
металлов. - 1978. - Т. 14, № 4. - С. 474-480.
19. Введенский А.В. Анодное окисление
меди в разбавленных хлоридных растворах / А.В. Введенский, И.К. Маршаков //
Защита металлов. - 1983. - Т.19,№1. - С.79-83.
20. Анодное поведение меди в сульфатные
растворах / В.К. Алтухов, И.К. Маршаков, В.С. Воронцев, Т.К. Клепинина //
Электрохимия. -1976. -Т.12.№1. - С. 88-91.
21. Могиленко В.Ф. Влияние добавки ионов
хлора на анодное растворение меди в серной кислоте / В.Ф. Могиленко, Ю.М.
Лошкарев // Электрохимия. - 1992. - Т.28,№1. - С. 21-26.
22. Маричев В.А. Метод контактного
электросопротивления для исследования in
situ поверхности
металлов в электролитах. Кинетика субмонослойного окисления меди и серебра в
щелочных и хлоридных растворах / В.А. Маричев // Электрохимия. 1999. Т.35. №4.
С. 466-473.
23. Кондрашин В.Ю., Куксина О.Ю.,
Маршаков И.К. О роли парциальных катодных процессов при переменно-токовом
окислении меди в хлоридных средах // Вестн. Воронеж. гос. ун-та. Сер. Химия.
Биология. Фармация. 29005. №1. С. 49-57
24. Костин Н.А. Импульсный электролиз /
Н.А. Костин, B.C.
Кублановский, В.А. Заблудовский. - Киев: Наукова думка, 1989. - 168 с.
25. Лошкарев Ю.М. Особенности структуры
и некоторых свойств цинковых покрытий, электроосажденных импульсным током из
щелочных электролитов / Ю.М. Лошкарев [и др.] // Электрохимия - 1994. - Т. 30,
№10. - С. 1287-1290.
26. РеваО.В.Осаждениенаноструктурированных
композиционномодулированных покрытий Cu-Sn
и Cu-Zn
в условиях периодического изменения плотности тока / О.В. Рева, Т.Н.
Воробьева,
В.В. Свиридов // Электрохимия - 1999. - Т. 35, №9. - С. 1070-1072.
27. Гамбург Ю.Д. Электрохимическая
кристаллизация металлов и сплавов / Ю.Д. Гамбург - М.: Янус - К, 1997. - 384 с.
28. Кости Н.А. Концентрированные
изменения в приэлектродном слое и составляющие поляризации при импульсном
электролизе / Н.А. Костин, В.И.Черненко // Докл. АН УССР Серия Б. - 1982. - №5.
- С. 53-57.
29. Михайловский Ю.Н. Электрохимический
механизм коррозии металлов под действием переменного тока IV. Растворение
алюминия и магния при поляризации переменным током / Ю.Н. Михайловский //
Журнал физической химии. - 1963. - Т. 37. №5. С. 1196-1200
30. Михайловский Ю.Н. Коррозия металлов
под действием переменных токов в жидких электролитах и влажных почвах. Автореф.
дисс.... д-ра хим. наук / Ю.Н. Михайловский. - Москва 1963. - 27с.
31. Томашов Н.Д. Влияние частоты
переменного тока на скорость коррозии алюминия в серной кислоте / Н.Д. Томашов,
Струков // Журн. Физ. Химии. - 1968. Т. 42. №4. С. 931-934
32. Косинцев В. И. Электросинтез оксида
алюминия с развитой поверхностью / В.И. Косинцев, В.В. ВКоробочкин, Е.П.
Ковалевский // Матер. Межрегион. конф. с междунар. участием. Красноярск, 1996.
С.59.
33. Кузнецов О.Г.Использование
переменноготокадля электрохимическогоизвлечения никеля, кобальта, иплатиновых
металлов
из промпродуктов никелевого производства / О.Г. Кузнецов, С.Ф. Белов, А.М.
Левин // Наукоемкие химические технологии 2002. С. 139-140.
34. Михайловский Ю.Н. Электрохимический
механизм коррозии металлов под действием переменного тока. I. Коррозионное и
электрохимическое поведение железа в электролитахпри поляризации переменным
током / Ю.Н. Михайловский // Журн. физ. химии. 1963. Т. 37. №3. С. 553-558.
35. Михайловский Ю.Н. Михайловский Ю.Н.
Электрохимический механизм коррозии металлов под действием переменного тока.
III. Коррозионное и электрохимическое поведение железа в электролитах при
поляризации переменным током / Ю.Н. Михайловский // Журн. физ. химии. 1963. Т.
37. №3. С. 553-558.
36. Сметанина Е.И. Разработка материалов
описания электросинтезов оксида меди на переменном токе / Е.И. Сметанина,
В.И.Коробочкин // Перспект. хим. технологии и матер. Пермь 1997 С. 254
37. Маршаков А.И. Особенности активного
растворения хрома в сернокислых растворах в присутствии кислородсодержащих
окислителей и ионов металлов с низким перенапряжением водорода / А.И. Маршаков,
А.П. Высоцкий, Ю.Н. Михайловский // Защита металлов. 1998. Т. 24. №2. С.
189-198.
38. Михайловский Ю.Н. Активация анодного
растворения железа и его пассивация в растворах, содержащих перекись водорода /
Ю.Н. Михайловский, Н.Б. Лукина // Защита металлов. 1983. Т. 19. №6. С. 864-871.
39. Маршаков А.И. Соотношение скоростей
электродных реакций при коррозии цинка в растворах оксокислот и роль
анодноактивных интермедиатов / А.И. Маршаков, Т.И. Соколова, Ю.Н. Михайловский
// Защита металлов. 1947. Т. 33. №1. С. 35-42.
40. Варенко Е.С. Кинетика анодной
ионизации меди в сульфатном и хлоридном электролитах / Е.С. Варенко, С.В.
Нефедова // Вопросы химии и хим. технол. - 1981. - Т. 65. - С. 6-12.
41. Колотыркин Я.М. Аномальное
растворение металлов. Экспериментальные факты и их толкование / Я.М. Колотыркин
// Защита металлов. - М.: 1984. - Т. 20, № 1. - С.14-24.
42. Маршаков И.К. Кинетика анодного
растворения меди в универсальной буферной смеси / И.К. Маршаков, Н.А. Матюхина,
И.В. Протасова // Конденсированные среды и межфазные границы. - 2004. - Т. 6, №
4. - С. 381-386.
43. Маршаков И.К. Аномальное растворение
меди и серебра при катодной поляризации в кислых средах / И.К. Маршаков, Л.Е.
Волкова, Н.М. Тутукина, И.В. Крейзер // Вест. Воронеж. гос. ун-та. Сер.: Химия.
Биология. Фармация - 2005. - №2. - С. 43 -53.
44. Кондрашин В.Ю. Парциальные
электродные процессы при знакопеременной поляризации медного электрода / В.Ю.
Кондрашин // Вест. Воронеж. гос. ун-та. Сер.: Химия. Биология. Фармация - 2011.
- №1. - С. 44-48.
Приложение
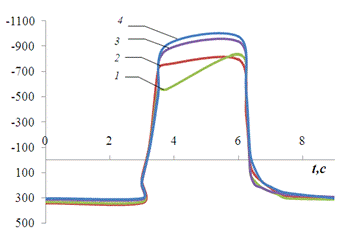
Рис.П.1.
Хронопотенциограмма реверсивной поляризации медного электрода в размешиваемом
растворе 0,19 М Na2SO4 + 0,01 M H2SO4
+ 0,001 M Fe3+:
iА = 0,02 мА/см2, iК = 1(1), 2(2), 3(3), 4(4)
мА/см2.
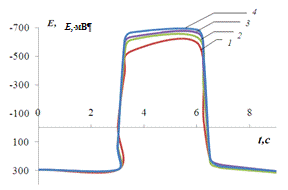
Рис.П.2.
Хронопотенциограмма реверсивной поляризации медного электрода вразмешиваемом
растворе 0,15 М Na2SO4 + 0,05 M H2SO4
+ 0,001 M Fe3+:
iА = 0,02 мА/см2, iК = 1(1), 2(2), 3(3), 4(4)
мА/см2.
Рис.П.3.
Хронопотенциограмма реверсивной поляризации медного электрода в размешиваемом
растворе 0,19 М Na2SO4 + 0,01 M H2SO4
+ 0,005 M Fe3+:
iА = 0,02 мА/см2, iК = 1(1), 2(2), 3(3), 4(4)
мА/см2.
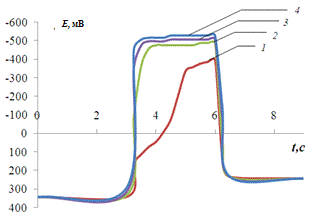
Рис.П.4.
Хронопотенциограмма реверсивной поляризации медного электрода в размешиваемом
растворе 0,15 М Na2SO4 + 0,05 M H2SO4
+ 0,005 M Fe3+:
iА = 0,02 мА/см2, iК = 1(1), 2(2), 3(3), 4(4)
мА/см2.
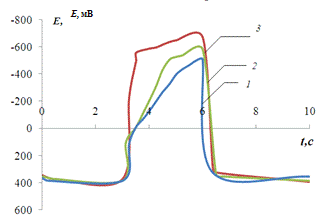
Рис.П.5.
Хронопотенциограмма реверсивной поляризации медного электрода в размешиваемом
растворе 0,2 М Na2SO4 + 0,001 M H2SO4
+ 0,005 M Fe3+:
iА = 0,02 мА/см2, iК = 1(1), 1,5(2), 2(3)
мА/см2.
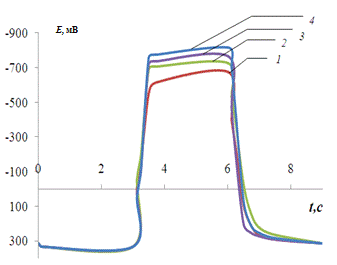
Рис.П.6.
Хронопотенциограмма реверсивной поляризации медного электрода в размешиваемом
растворе 0,18 М Na2SO4 + 0,02 M H2SO4
+ 0,001 M Fe3+:
iА = 0,02 мА/см2, iК = 1(1), 1,5(2), 2(3)
мА/см2.
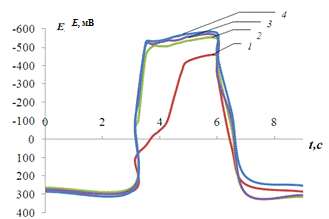
Рис.П.7. Хронопотенциограмма реверсивной
поляризации медного электрода в размешиваемом растворе 0,18 М Na2SO4
+ 0,02 M H2SO4 + 0,005 M Fe3+:
iА = 0,02 мА/см2, iК = 1(1), 1,5(2), 2(3)
мА/см2.