Совершенствование технологии получения адсорбентов из местных полиминеральных глин
МИНИСТЕРСТВО
ВЫСШЕГО И СРЕДНОГО СПЕЦИАЛЬНОГО ОБРАЗОВАНИЯ РЕСПУБЛИКИ УЗБЕКИСТАН
БУХАРСКИЙ
ТЕХНОЛОГИЧЕСКИЙ ИНСТИТУТ ПИЩЕВОЙ И ЛЕГКОЙ ПРОМЫШЛЕННОСТИ
На
правах рукописи
АБДУЛЛАЕВ
НОДИР МАНСУРОВИЧ
совершенствование
технологии получения адсорбентов из местных полиминеральных глин
А 522504 -
«Переработка нефти и газа и ее химическая технология»
Диссертация
на соискание
академической степени магистра
Научный
руководитель: к.т.н. Муродов М.Н.
Заведующий
кафедрой: к.т.н. Адизов Б.З.
Руководитель
отдела магистратуры: доц. Шомуродов Т.Р.
БУХАРА- 2011
г.
ОГЛАВЛЕНИЕ
ВВЕДЕНИЕ
Глава 1. ЛИТЕРАТУРНЫЙ ОБЗОР
1.1
Адсорбционные и каталитические свойства природных алюмосиликатов Узбекистана
1.2
Теория кислотности, отбеливающие и каталитические свойства
природных алюмосиликатов
.3 Физико-химические свойства
Навбахорского бентонита
.4 Адсорбционная активность
бентонита Навбахорского месторождения по нефтяным компонентам
Глава 2. ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
МЕТОДЫ И ОБЪЕКТЫ ИССЛЕДОВАНИЯ
2.1
Методы исследования
2.2
Объекты исследования
ГЛАВА 3.
ИССЛЕДОВАНИЕ ФИЗИКО-ХИМИЧЕСКИХ, КИСЛОТНЫХ И КАТАЛИТИЧЕСКИХ СВОЙСТВ ПРИРОДНЫХ И
АКТИВИРОВАННЫХ БЕНТОНИТОВ
3.1
Изучение влияния способов активации на химический состав природных
алюмосиликатов
3.2
Влияние активации на структуру алюмосиликатов
3.3
Рентгенографические исследования природных и активированных алюмосиликатов
3.4
Структурно-сорбционные свойства естественных и активированных алюмосиликатов
Выводы
Список использованной литературы
ВВЕДЕНИЕ
Всемирный финансово-экономический кризис,
разразившийся в 2008 году и приобретающий сегодня большие масштабы и глубину в
оценках многих международных экспертов и специалистов, получает больше
вопросов, чем ответов о причинах и прогнозах его полнейшего развития.
Каждый из нас должен отдавать себе отчет в том,
что Узбекистан сегодня- это составная часть мирового пространства и глобального
финансового экономического рынка.
Наглядным свидетельством этому являются все
возрастающие наши связи с внешним миром реализация с помощью ведущих развитых стран,
программ по развитию модернизации, техническому и технологическому
переоснащению отраслей экономики, интеграция Узбекистана в международную сферу
торговли, рост импорта и экспорта продукции и товаров.
И вполне закономерно, что глобальный финансовый
кризис и, в первую очередь, его последствия уже сказываются на развитии и
эффективности нашей экономики. Из-за сокращающегося спроса на мировом рынке
снижаются цены на экспортируемую Узбекистаном продукцию, такую как драгоценные
и цветные металлы, хлопок, уран, нефтепродукты, минеральные удобрения и другие.
Актуальность проблемы. На территории Узбекистана
и в определенных ему районов имеются огромные, причем промышленные запасы
природных алюмосиликатов - бентонитовых, каолинитовых, опоковидных и других
видов глин и глинистых образований, обладающих определенными адсорбционными и
каталитическими свойствами. Из-за полиминеральности и высокой
минерализованности они качественно уступают активированным и синтетическим
алюмосиликатам. Поэтому с целью расширения области применения природных
алюмосиликатов в народном хозяйстве необходимы опыты по их активации. При этом,
по-видимому, главное внимание должно быть уделено разработке рационального
способа активации природных алюмосиликатов.
Целъю данной диссертационной работы является
совершенствование технологии получения адсорбентов из местных полиминеральных
глин на основе изучения изменения структурных особенностей, кислотных и
связанных с ними адсорбционных и каталитических свойств, а также установление
взаимосвязи между различными свойствами природных алюмосиликатов различного
химического и минералогического состава и способами их активации, установление
пригодности приготовленных на основе местного сырья адсорбентов и катализаторов
взамен дорогостоящих и дальнепривозных (даже импортных) поглотителей и
катализаторов для удовлетворения потребности интенсивно развивающихся
масложировой (в которой потребность по отбельным глинам за счет импортных
удовлетворяется всего лишь на 12%), нефтеперерабатывающей, энергетической, газовой
и ряда других важных отраслей народного хозяйства республики.
Научная новизна диссертационной
работы заключается в том, что :
- Разработаны различные способы воздействия на
структуру и свойства природных алюмосиликатов. В зависимости от способа обработки
получаются образцы с улучшенными адсорбционными и каталитическими свойствами.
Причем, природные алюмосиликаты в зависимости от вида,
месторождения, минералогического состава по-разному активируются одним и тем же
методом активации. Часто алюмосиликаты
одного
минералогического типа (например, бентонитовые глины), близкие по химическому
составу и некоторым другим показателям, при активации активатором (например,
серной кислотой) реагируют по-разному. Поэтому для каждого месторождения
выявляется свой оптимальный режим теми или иными способами активации.
Поверхность алюмосиликатов покрыта сеткой ионов
гидроксила, которые ответственны за проявление кислых (гидролитическая, от
части обменная кислотность), адсорбционных (если образуются
поверхностно-химические соединения) и каталитических (если реакция протекает по
карбонионному механизму) свойств алюмосиликатов. Причем, содержание гидроксилов
можно изменить путем физических и химических способов активации образцов. С
изменением числа гидроксилов должен меняться комплекс свойств алюмосиликатов. С
этой точки зрения, эксперименты по определению изменения структур, кислых,
адсорбционных, отбеливающих, каталитических и других свойств одного и того же
образца в зависимости от способа обработки представляют интерес для
установления взаимосвязи между изменениями названных свойств алюмосиликатов.
Имеющиеся в литературе материалы, в частности по установлению роли
гидролитической кислотности в катализе и обменной кислотности в процессе
отбеливания, по установлению наличия определенной зависимости между
гидролитической кислотностью алюмосиликата и его емкостью монослоя по парам
воды не позволяют установить каталитические свойства алюмосиликатов на
основании их других свойств.
Практической ценностью работы можно считать, что
в настоящее время положительно решается вопрос относительно изменения кислотных
(адсорбционных и каталитических) центров у природных алюмосиликатов при их
активации. Однако, происходящие при этом количественные изменения, в результате
чего неизбежно происходят качественные изменения в их свойствах, являются
дискуссионными. Поэтому эксперименты, поставленные с целью выявления активации
природных алюмосиликатов, заслуживают определенного внимания, ибо с этим
вопросом связано решение таких теоретических задач, как направленное изменение
свойств алюмосиликатов, и практических задач - получение материалов
(адсорбентов и катализаторов) с заданными свойствами, и их моделирования для
технологических процессов.
1. ЛИТЕРАТУРНЫЙ ОБЗОР
1.1 Адсорбционные и
каталитические свойства природных алюмосиликатов Узбекистана
алюмосиликат адсорбционный
каталитический бентонит
В пределах Узбекистана и Каракалпакской
Республик известно большое количество месторождений с промышленными запасами
алюмосиликатов - глин, глинистых образований и др. [1]. Первая работа по
изучению адсорбционно-отбеливающих свойств бентонитовых глин принадлежит Б.Г.
Запрометову [2]. Систематическое изучение адсорбцион-ных и др.
физико-химических свойств бентонитовых, опоковидных глин, опок, трепелов, цеолит
содержащих пород и др. показало, что они отвечают всем требованиям,
предъявляемым к твердым сорбентам (основное из них - высокие адсорбционные
качества, хорошая избирательность, достаточная прочность гранул, фильтрующая
способность, легкая регенерация, каталитическая активность [3]), и поэтому их
называют природными минеральными сорбентами [4]. Отличительной особенностью
выше названных пород от подобных других регионов бывшего Советского Союза, как
отмечено в [4], являются полиминеральность, высокая минерализация (особенно
карбонатами, сульфатами и хлоридами) с коагуляцией первичных структур,
повышенное содержание окислов железа и преобладание катиона магния над ионами
кальция в обменном комплексе.
Достигнуты значительные успехи в области
изучения поверхностных, адсорбционных и др. свойств природных алюмосиликатов
республики [4]. В противоположность этому их каталитические свойства
практически не изучены. Так, по сведениям М. З.Закирова [5], В. М. Лигуше в
1956 году исследовал каталитическую активность глин много месторождений -
каолиновых и огнеупорных (Лянгар, Ангрен, Сулюкта, Кызыл-Кия и др),
бентонитовых и бентонитоподобных (Келес, Кермине, Кызылтепе и др.) установлено,
что для нефтяной промышленности Узбекистана практический интерес представляет
Кызылтепинское (Азкамарское) и другие месторождение бентонитовых глин.
В 1958 году В. И. Ездаков опубликовал работу, в
которой были изучены каталитические действия глин Азкамарского,
Катта-Курганского, Ингичкинского, Куксайского, Карнапского, Чупанатинского
месторождений на примере реакции бензилирования. Установлено, что реакция
протекает бурно и полно, если взять катализатора 0,023% от массы реагирующего
хлорбензила [6]. Продолжая работы в этом направлении, он установил, что
величина навески глины существенно влияет на глубину полимеризации нефтяных
смол - чем больше навеска при постоянной исходной концентрации смол, тем глубже
процесс полимеризации и меньше растворимость смол. Положение верно для
каолинов, бентонитов, опок, но не применимо к гидрослюдистым и палыгорскитомонтмориллонитовым
глинам, что объясняется, видимо, особенностью их структур. Гомоионные формы
глины, в частности, медной, хромовой, марганцевой форм
полыгорскито-монтмориллонитовой глины Чупанатинского месторождения, обладают
повышенной каталитической активностью [7].
Изучение реакции конденсации нафталина с
моноголоидуксусными кислотами для получения нафтилуксусных кислот в присутствии
глин показало, что внесение катализатора - глины мелкими порциями дает лучший
эффект; при извлечении целевого продукта смолы остаются на глине и кислота
получается совершенно чистой. Наилучшим каталитическим действием в данной
реакции так же, как и в реакции бензилирования, обладает каолин, бентонит и
наименьшим - опока. Термическая активация глин способствовала улучшению выхода
и чистоты целевого продукта [7].
А. С. Султанов и С. А. Бабаходжаева [8],
изучавшие реакции кетонизации уксусной кислоты с помощью естественного и
известкованного нефтеабадского малинового бентонита, обнаружили достаточную
каталитическую активность у глин, обработанных известковым раствором.
Э. А. Ариповым и др. отмечается, что природный
минеральный сорбент является не только поглотителем, но и ускорителем ряда
процессов, если сорбтив не устойчив в данном адсорбционном процессе. Отмечено,
окисление госсипола при адсорбции его из хлороформного ратвора на природных и
обработанных серной и уксусной кислотами азкамарком и келесском бентонитах.
Установлено, что окисляющая способность келесского бентонита почти в два раза
больше, чем окисляющая способность азкамарской глины; кислотная активация
привела к повышению каталитической активности глин, причем глина,
активированная серной кислотой, почти в три раза больше окисляет госсипола, чем
глины, обработанные уксусной кислотой [9].
Таким образом, имеющийся в литературе скудный
материал не дает возможности оценивать алюмосиликаты Узбекистана в качестве
освежителей и катализаторов для промышленности республики. Можно полагать, что
глины и глинистые образования Узбекистана будут уступать по каталитическим (так
же, как и по адсорбционным) свойствам активированным глинам и синтетическим
алюмосиликатам. Отсюда следует необходимость улучшения комплекса свойств глин
республики, что может быть осуществлено путем их активирования и
модифицирования.
В настоящее время почти повсеместно и даже в
промышленных масштабах осуществляются термическая и кислотная; активация глин,
модифицирование их органическими веществами, а также обработка их растворами
минеральных солей. Новыми являются известковый, щелочно-кислотный и кислотно-щелочной
способы активации глин и глинистых образований [3, 4, 7, 10-19].
Проводятся систематические работы по активации и
модификации алюмосиликатов Узбекистана физическими (термо- и гидротермальная
обработка) и химическими (обработка образцов растворами серной кислоты,
извести, солей, поверхностно-активных веществ, водорастворимых
полиэлектролитов), а также комбинированными (кислотно-термическая,
щелочно-поверхностноактивными веществами) способами. Разрабатываются условия
обработки, изучается механизм активации и модификации, исследуются свойства
полученных при этом образцов, а также выявляются области их применения
[4,20,21]. Однако работы по активации глин республики с помощью разлияных
кислот и таких комбинированных способов как кислотно-щелочной и
щелочно-кислотный, до нас не ставились.
Многочисленными исследованиями отечественных и
зарубежных ученых установлено, что природные алюмосиликаты в зависимости от
генетической особенности и минералогического состава одним и тем же методом
активации активируются по разному. Часто алюмосиликаты одного минералогического
типа (например, бентонитовые глины), близкие по химическому составу и некоторым
другим показателям, при активации реагируют с активатором (например, с
кислотой) также не однозначно. Поэтому для каждого месторождения выявляется
свой оптимальный режим активации.
Анализ литературных материалов по напряженному
изменению свойств природных алюмосиликатов позволяет сделать заключение о том,
что при активации и модификации глин происходит сложный физико-химический
процесс, в результате чего меняется химический состав исходного образца (в
связи с этим изменяется и его минералогический состав), его поверхностные
свойства и пористые структуры. Предопределение направленного изменения свойств
глин при их обработке теми или иными способами воздействий связано с знанием
свойств исходного образца, обусловленных структурой глинистых минералов, из
которых состоит природный алюмосиликат. Кроме того, успешное применение как
природных, так и активированных алюмосиликатов в технике в качестве адсорбентов
в адсорбционных процессах или как катализатора в каталитических процессах
связано с решением проблемы регулирования физико-химических процессов,
протекающих на границе раздела фаз.
1.2 Теория кислотности, отбеливающие и каталитические свойства
природных алюмосиликатов
Современные рентгенострутурные, электронно-,
кристалло-, рентгено-, термо-, нейтронографические, ИК-, УФ- и
радиоспектроскопические, химические и другие методы анализов сыграли огромную
роль не только в определении минералогического состава природных
алюмосиликатов, но и позволили установить кристаллохимическую и морфологическую
особен-ность минералов,
входящих в состав природных алюмосиликатов. Показано, что основными
составляющими природных алюмосиликатов являются глинистые минералы.
Согласно классификации глинистых минералов, из
которых, в основном, состоят природные алюмосиликаты, Р. Грима [11], который
среды специалистов, занимающихся исследованием физико-химических и других
свойств глин и глинистых образований, пользуется наибольшей популярностью, они
разделяются на аморфные и кристаллические минералы.
Аморфная фаза глинистых минералов,
или аллофон, состоит, в основном, из геля окислов кремния, алюминия и других
веществ. Один из них - глинозем. Его структура состоит из двух слоев атомов
кислородов или гидроксилов, между которыми в октаэдрической координации
расположены атомы алюминия, находящегося на равном расстоянии от 6 атомов 0 или
6 групп ОН. Обычно расстояние 0-0 равно 2,60 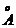 , ОН-ОН 2,94 ,
пространство, доступное для иона в октаэдрической координации, имеет размер
0,61 . Толщина
этого структурного элемента в структуре глинистых минералов равна 5,06. Эта
структурная единица соответствует структуре соединения, называемого
гидраргиллитом
, ОН-ОН 2,94 ,
пространство, доступное для иона в октаэдрической координации, имеет размер
0,61 . Толщина
этого структурного элемента в структуре глинистых минералов равна 5,06. Эта
структурная единица соответствует структуре соединения, называемого
гидраргиллитом 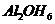 .
.
Второе вещество, входящее в состав
аллофона, - кремнезем, встречающийся иногда в виде геля. Он состоит из
тетраэдрических групп 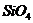 ,
соединенных между собой в гексагональную сеть состава
,
соединенных между собой в гексагональную сеть состава 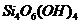 . Расстояние
0-0 в тетраэдрическом слое равно 2,55 ; пространство, доступное для иона,
в тетраэдрической координации имеет размер примерно 0,55; толщина
этого структурного элемента в структурах глинистых минералов равна 4,93. Каждый из
них имеет расстояние от центра до центра примерно 2,1[3, 11, 22].
. Расстояние
0-0 в тетраэдрическом слое равно 2,55 ; пространство, доступное для иона,
в тетраэдрической координации имеет размер примерно 0,55; толщина
этого структурного элемента в структурах глинистых минералов равна 4,93. Каждый из
них имеет расстояние от центра до центра примерно 2,1[3, 11, 22].
Кристаллическая фаза глинистых минералов
состоит из двух выше приведенных структурных элементов. Первый состоит из двух
слоев плотно упакованных атомов кислорода или ОН - групп с атомами алюминия,
железа и магния между ними в октаэдрической координации, второй - из
кремнезекислородных тетраэдров.
Различные совокупности этих
структурных элементов образуют отдельные разновидности глинистых минералов [3,
11, 22]. Например, двухслойный тип кристаллической решетки (1:1), состоящий из
пакетов, которые содержат один слой кремнекислородных тетраэдров, характерен
для глинистых минералов каолинитовой группы (каолинит, накрит, галлуазит и
т.д.). к трехслойному типу решетки (2:1), который образован из двух слоев
кремнекислородных тетраэдров и расположенного между ними одного слоя
алюмокислородных октаэдров, относятся монтмориллонитовые (монтмориллонит,
сиконит, вермекулит, нонтронит и др.) и иллитовые (иллит, слюда и т.п.) группы.
Наличие аморфных веществ обычно
незначительно и не Вов всех глинах. Глинистые минералы в природе встречаются
всегда с примесями; мономинеральные глины редки и все равно в той или иной
степени содержат различные примеси [3, 4, 11, 23].
Химичекими, спектроскопическими,
изотопными и другими методами выявлено, что на поверхности природных
алюмосиликатов (следовательно, и на поверхности глинистых минералов, а также
глин) имеются гидроксилы, связанные с атомами кремния и алюминия в
соответствующих структурных единицах - кремнекислородных тетраэдрах и
алюмокислородных октаэдрах (реже тетраэдрах). Поверхностные гидроксилы
природных алюмосиликатов при их взаимодействии с молекулами сорбтива в ряде
случаев проявляют протонодонорную функцию. Происходящую при этом частичную
протонизацию водорода можно рассматривать как проявление кислотной природы
гидроксильных групп [4, 23]. Поверхностные гидроксилы природных алюмосиликатов
обладают слабокислотными свойствами и играют решающую роль при адсорбции
веществ, способных и донороакцепторному взаимодействию [23], а также при
каталитических процессах, протекающих по карбоноионному механизму [24].
На основании результатов работ,
посвященных изучению химии поверхности [25-27] адсорбционно-каталитических
свойств [28,29] силикагелей, силикатов, алюмосиликатов можно заключить, что
водород группы ОН, принадлежащие атому кремния в тетраэдрической координации,
обладает относительно слабым кислотным свойством, чем водород гидроксильной
группы принадлежавшей атому алюминия, а октаэдрических (даже в тетраэдрических)
координациях.
В настоящее время различают три вида
поверхности у природных алюмосиликатов [30].
Поверхность первого рода -
поверхность скола, представляющая собой край кристаллической решетки или
частиц. Энергия этой поверхности определяется числом атомов с
некомпенсированными электровалентными связями, и поэтому эта поверхность
является наиболее активной. По мнению Э. А. Арипова [4, 31], этот вид
поверхности является не только активным, но и ответственным за число свободных
радикалов (или число парамагнитных центров) у глинистых минералов и глин.
Поверхность второго рода - базальные поверхности внутри пакета; она находится в
поле влияния плоскостей. Поверхность третьего рода - тоже базальная; она
представляет внешнюю поверхность частиц (или кристаллов) и поэтому является
свободной от влияния соседней поверхности, следовательно, по энергетическому
состоянию отличается от поверхности второго рода.
Если рассматривать поверхности
второго и третьего родов без учета изоморфных замещений атомов решетки, то эти
поверхности должны в силу своего кристаллохимического строения являться
электронейтральными. Однако, как отмечено в [3, 11, 22], в реальных глинистых
минералах всегда имеет место изоморфное замещение кремния на алюминий в
тетраэдрической координации и алюминия на магний, железо и др. в октаэдрической
координации, при которых освобождаются отрицательные заряды по одному от
каждого освобождаются.
Таким образом, активные
адсорбционные центры у алюмосиликатов самые разнообразные. Однако, приписывать
предпочтению одного из этих факторов процесс адсорбции или адсорбционного
отбеливания нельзя. Но увеличить долю одного из этих факторов можно путем
активирования и модифицирования глин. Например, увеличение степени дисперсности
образца при активации способствует повышению величин его поверхности первого
рода. Благодаря этому обеспечивается максимальный эффект при контактной очистке
минеральных и растительных масел.
Активная кислотность выражается
величиной рН водной вытяжки образца или его водной суспензии. При внесении
образца в воду в нее переходят присутствующие в образце водорастворимые
минеральные и органические соли, газы (особенно 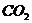 ). Кроме того, имеет место
диссоциация соли алюмо-кремниевых кислот (определение Вернадского В. И. [34]).
). Кроме того, имеет место
диссоциация соли алюмо-кремниевых кислот (определение Вернадского В. И. [34]).
Обменная кислотность определяется
титрованием нейтральной солевой вытажки (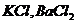 и др.), а гидролитическая
кислотность - титрованием вытяжки, полученной обработкой образца растворами
гидролитически щелочной соли (
и др.), а гидролитическая
кислотность - титрованием вытяжки, полученной обработкой образца растворами
гидролитически щелочной соли ( ),
), 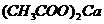 и др.).
и др.).
При вытеснении поглощенных катионов
алюмосиликатами (почва, глина), катионом нейтральных солей в раствор переходит
и некоторое количество алюминия, соли которого, гидролизуясь, дополнительно
обусловливают кислотность. Этот вид кислотности принято называть
обменно-алюминиевой формой кислотностей находят обменно-водородную форму
кислотности образца.
Протонную (бренстедовскую)
кислотность алюмосиликатов, глубоко гидролитическую, определяют отравлением
гидролитически щелочной солью, но только с той разницей, что в этом случае
находят не все подвижные и неподвижные водородные ионы, а по К. В. Топчиевой
[33] - лишь подвижные.
Апротонную (льюисовскую) кислотность
по методу В. Трамбуза [35] определяют титрованием неводной (обычно бензиновой)
суспензии образца этилацетатом или диоксаном в присутствии индикатора
парадиметиламино-азабензола (НДА). Беря в качестве титрующего агента
Н-бутиламин в этих же условиях согласно Тамеле [36], определяется сумма
протонной и апротонной кислотности алюмосиликатов.
Г.А.Бенези [37], измеряя силу
поверхностной кислотности синтетических и природных алюмосиликатных
катализаторов крекинга, показал, что на поверхности катализаторов имеются
кислотные центры, общее количество которых составляет не больше семи процентов
от общей поверхности образца.
С целью приближения к оценке числа
активных центров в условиях катализа К.В.Топчиевой с сотрудниками [38]
разработан импульсный хроматографический метод определения кислотных центров,
основанный на дозировании органического основания-пиридина во
время опыта, с образованием нона пиридина.
О причинах появления кислых свойств
алюмосиликатов (почв, глин и др.) высказаны различные гипотезы. Первая из них
объясняет кислотность наличием поглощенных ионов водорода на поверхности
коллоидных частиц (например, кислые глины) алюмосиликатов. По этой гипотезе,
кроме более прочной связи с частицей алюмосиликатов, ионы водорода ничем не
отличаются от других поглощенных катионов образцами [32, 39].
По второй гипотезе, кислотность
алюмосиликатов обусловлена адсорбированными солями алюминия, которые
обмениваются на катионы нейтрального солевого раствора; образующаяся при этом в
растворе соль гидролизуется и создает кислую реакцию равновесного солевого
раствора [32, 39].
Следующая гипотеза основывается на
наличии гидроксилов на поверхности частиц алюмосиликатов. Допускается, что
водород этих гидроксилов обладает способностью обмениваться на катион других
металлов и, таким образом, водороды ОН группы обусловливают обменную кислотность
[28].
Число поверхностных гидроксилов
зависит от двух факторов - от наличия изоморфного замещения и степени
дисперсности образца. При этом: 1) Чило Он в алюмосиликатах увеличивается с
ростом степени изоморфного замещения 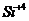 на
на 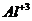 в тетраэдре и на
в тетраэдре и на 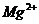 в октаэдре,
т.е. число гидроксилов пропорционально росту отрицательного заряда частиц
алюмосиликата, который повышается с увеличением степени замещения катионов с
большей валентностью на катионы с меньшей валентностью [3, 11, 22, 34, 40, 2].
Число гидроксилов на поверхности частиц тем больше, чем больше степень
дисперсности частиц образца; при диспергировании увеличивается число разрывов
связи
в октаэдре,
т.е. число гидроксилов пропорционально росту отрицательного заряда частиц
алюмосиликата, который повышается с увеличением степени замещения катионов с
большей валентностью на катионы с меньшей валентностью [3, 11, 22, 34, 40, 2].
Число гидроксилов на поверхности частиц тем больше, чем больше степень
дисперсности частиц образца; при диспергировании увеличивается число разрывов
связи 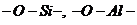 и
и 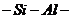 , куда могут
присоединяться наряду с другими молекулами и молекулы воды, водород которых
обладает в этом случае сильно протонизирующей способностью. Причем, этот протон
не входит в состав минерала и обладает высокой подвижностью [41].
, куда могут
присоединяться наряду с другими молекулами и молекулы воды, водород которых
обладает в этом случае сильно протонизирующей способностью. Причем, этот протон
не входит в состав минерала и обладает высокой подвижностью [41].
Многочисленными трудами
отечественных и зарубежных ученых установлено, что природные алюмосиликаты
обладают каталитическими свойствами по отношению к различным органическим
веществам и что у глин разного состава эти свойства проявляются различно.
Рассмотрению явления катализа и роли глинистых катализаторов в крекинге нефти
посвящено много работ.
Главным в этих работах, по-видимому,
является решение вопросов, связанных с каталитическими свойствами
алюмосиликатов, с их структурной особенностью. Предполагают, что на поверхности
частиц алюмосиликатов имеются активные каталитические (адсорбционные) центры.
Так, активные центры согласно И. Легмюра [42], - это энергетически неоднородные
участки на поверхности; Г. С. Тейлора [43] - это взаимнонекомпенсированные
атомы, выступающие в виде пиков; А. Ф. Иоффе [44] - это молекулярные трещины и
ямы, вызывающие понижение прочности и где работа диссоциации будет понижена в
несколько раз (Я. Х. де-Бур [45] считает, что ямы являются активными центрами
при ван-дер-ваальсовой адсорбции); Баландина А. [46] - это участки
кристаллической решетки, способные с молекулами реагентов образовывать
мультиплетный комплекс; Н. И. Кобозова [47] - это ансамбль атомов, которые на
поверхности катализатора удерживаются благодаря адсорбционной силе.
Образование активных цкентров (мест)
катализаторов А. В. Писаржевский [48] связывает с явлением электронной
изоляции. Это послужило основанием для разработки электронно-химической теории
явления катализа, а также процесса адсорбции на неоднородной поверхности
сорбента [49, 50].
В. И. кузнецов [51] поверхность
кристаллических и аморфных алюмосиликатных катализаторов рассматривает как
непрерывно изменяющийся бертоллоид. В основе этого понятия лежит неоднородность
поверхности, которую можно разделять на структурную неоднородность, вызванную
различной активностью разных граней кристаллов, атомов или группы атомов на
ребрах, вершинах граней, на ступенях дислокаций, и на неоднородность, обусловленную
включениями посторонних веществ и образованием новой фазы.
В реакциях превращения углеводородов
самыми наилучшими катализаторами являются кислые катализаторы [52]. В связи с
этим заслуживают внимания данные А. В. Фроста, в которых указано на глубокие
сходства каталитических действий алюмосиликатов с каталитическими действиями
таких кислых катализаторов, как 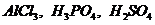 и
и 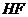 [53].
[53].
В настоящее время в литературе
имеется ряд работ, в которых предпологается модель активного центра
алюмосиликатного катализатора. Одна из таких моделей, как апротонный кислотный
центр тип Льюиса для алюмосиликата, согласно Л. Литтлу [54], состоит из
кремнекислородного тетраэдра и алюмокислородного октаэдра. Рост кислотности
алюмосиликатов проявляется тем сильнее, чем больше степень замещения атома
кремнияв кремнекислородной структурной единице атомом алюминия. Допускается,
что ион алюминии в этой структуре стремится присоединением пары электронов
заполнить р-подуровень. При этом в отсуствии воды образуются кислоты типа
Льюиса, а в ее присутствии - кислоты типа бренстеда по схеме:
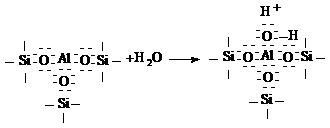
Или
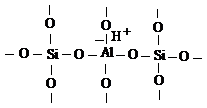
Этот протон, как указывает О. В.
Крылов [41], не входит в состав ОН -группы и обладает высокой подвижностью.
Наличие протона М. М. Мортленд и др. [55] объясняют высокой степенью
диссоциации воды, адсорбированной на центре Льюиса. Причем, воды диссоциируясь,
образует кислоту с подвижным водородом.
Согласно М. Р. Базила и др. [57],
все первичные кислые места на алюмосиликатах представляют собой центры Льюиса,
локализованные на поверхностных атомах алюминия. Бренстедовская кислотность
образуется взаимодействием молекул углеводорода, хемосорбированных на
льюисовских центрах и на соседних поверхностных гидроксильных группах.
Подвижный протон может возникать не
только в четверто координации алюминия, но и в результате электронного смещения
от поверхности силанальной группы к алюминию [58].
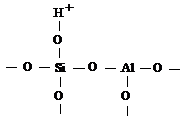
Согласно модели активного центра
алюмосиликата, предложенной А. П. Боллод и К. В. Топчиевой [59], кислотность
связана так с замещением кремния в кремнекислородных тетраэдрах на алюминий с
образованием комплекса 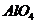 , где
трехвалентный алюминий неспособен полностью насытить все четыре атома
кислорода, в результате чего алюмосиликат проявляет сильные кислотные свойства
по схеме:
, где
трехвалентный алюминий неспособен полностью насытить все четыре атома
кислорода, в результате чего алюмосиликат проявляет сильные кислотные свойства
по схеме:
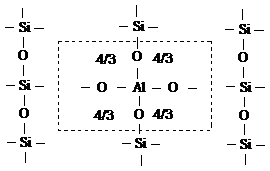
В работе [10] З.Г. Зульфугаровым
предложена модель для активированного килотой монтмориллонита с учетом активных
центров как для гидролитический, так и для обменной формы кислотности по
следующей схеме:
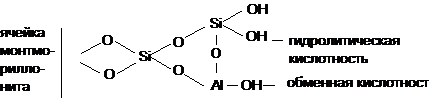
Эта модель рассматривает обменную и
гидролитическую кислотности, отравление ионами щелочных металлов, а также
механизм дегидратации кислого монтмориллонита. Ответственность за отбеливание и
катализ приписывается атому водорода, находящемуся в обменном положении.
Согласно модели А. В. Киселева и В.
И. Лыгина [60], возбужденный протон появляется за счет возникновения парных
кислотных центров апротонного и протонного типа по схеме:
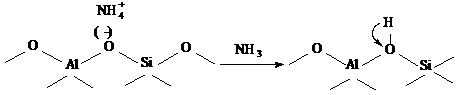
Природа активных центров
алюмосиликатов, согласно М. А. Калико [61], следующая: генерация протона может
осуществиться за счет адсорбированной воды при температурах крекинга:
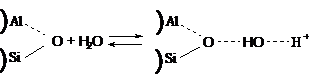
а за счет перестройки гидроксилов -
в хемосорбированную воду:
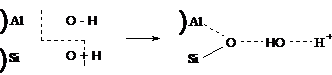
допускается, что из-за химически
ненасыщенной связи между  и
и 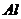 образуется
молкулярный комплекс (например, при разложении кумола), т.е. изопропилобензол
состоящий из адсорбированных молекул воды и молекулы углеводорода, по следующей
схеме:
образуется
молкулярный комплекс (например, при разложении кумола), т.е. изопропилобензол
состоящий из адсорбированных молекул воды и молекулы углеводорода, по следующей
схеме:
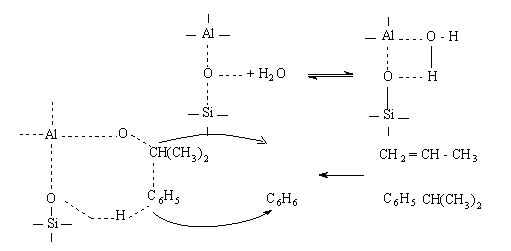
В случае
цеолитов кислотными центрами являются отрицательно в заряженные
тетраэдры  ,
возникающего в результате изоморфного замещения в тетраэдре на , и катионы,
компенсирующие этот заряд, а также кислородные атомы алюмосиликатного каркаса.
Поэтому катионы цеолита, хотя и не являются каталитическим центром, своим
поляризующим действием могут влиять на активность кислотных центров цеолита
[62-69]. Эти взгляды целиком могут быть применены и в случае катализаторов
глинистого происхождения. В аспекте этого представляет интерес механизм
отравления каталитических центров цеолитов. Так, одно- и двухвалентные катионы
полностью компенсируют заряд тетраэдра
,
возникающего в результате изоморфного замещения в тетраэдре на , и катионы,
компенсирующие этот заряд, а также кислородные атомы алюмосиликатного каркаса.
Поэтому катионы цеолита, хотя и не являются каталитическим центром, своим
поляризующим действием могут влиять на активность кислотных центров цеолита
[62-69]. Эти взгляды целиком могут быть применены и в случае катализаторов
глинистого происхождения. В аспекте этого представляет интерес механизм
отравления каталитических центров цеолитов. Так, одно- и двухвалентные катионы
полностью компенсируют заряд тетраэдра  не зависимо от молекулярного
соотношения
не зависимо от молекулярного
соотношения  в цеолите
типа А и Х. в случае же высококремнистых цеолитов типа У, у которых тетраэдра расположены
не через один, а через два тетраэдра , компенсирующие ионы, например,
катион
в цеолите
типа А и Х. в случае же высококремнистых цеолитов типа У, у которых тетраэдра расположены
не через один, а через два тетраэдра , компенсирующие ионы, например,
катион  ,
располагаются асимметрично относительно соседних тетраэдров [62]. Благодаря
этому имеет место катиона по , в результате чего заряд одного будет компенсирован,
а другого - не компенсированным по схеме:
,
располагаются асимметрично относительно соседних тетраэдров [62]. Благодаря
этому имеет место катиона по , в результате чего заряд одного будет компенсирован,
а другого - не компенсированным по схеме:
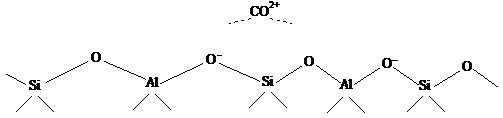
Отрицательный заряд и служит
активным центром алюмосиликата, а из газообразных  и веществ
содержащих аминогруппы
и веществ
содержащих аминогруппы 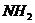 пиридин и
т.п.
пиридин и
т.п.
Содержание воды в катализаторах
крекинга имеет некоторое значение при установлении связи между кислотностью и
химической природой кислотных центров катализатора. Ясно, что после прокалывания,
например, при 800º,
на
поверхности катализатора не остается адсорбционно связанной воды. Однако,
водород присутствует в виде гидроксильных групп, связанных с атомами кремния
или алюминия. Вероятно, водород находится у атома кремния, так как энергия
связи 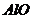 (1541,25
кДж/моль) больше против энергии связи
(1541,25
кДж/моль) больше против энергии связи 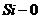 (329,0 кДж/моль). Поэтому при
прокаливании водород удаляется из атома алюминия. Возникающая при этом
вакантность (электронный дефицит) компенсируется за счет миграции водорода от
силанальных групп. И этот водород является кислотным центром бренстедовского
(протонного) типа алюмосиликатного катализатора и у целотов [65, 66].
Установлено, что катализатор с большой величиной протонной кислотности является
одновременно хорошим катализатором в реакциях крекинга углеводородов [67-69].
(329,0 кДж/моль). Поэтому при
прокаливании водород удаляется из атома алюминия. Возникающая при этом
вакантность (электронный дефицит) компенсируется за счет миграции водорода от
силанальных групп. И этот водород является кислотным центром бренстедовского
(протонного) типа алюмосиликатного катализатора и у целотов [65, 66].
Установлено, что катализатор с большой величиной протонной кислотности является
одновременно хорошим катализатором в реакциях крекинга углеводородов [67-69].
Результаты работа Т. Г. Мелликкена и
др. [70, 71] позволяют предположить, что большинство албмосиликатных
катализаторов представляют собой смесь частиц двуокиси кремния и окиси алюминия
с ионами кремния и алюминия в решетке, совместно владеющим ионами кислорода.
Поэтому в смешанной окисной структуре будут проявляться химические свойства
окиси алюминия в ее различных кристаллических формах, тогда как габитусы кристаллов
двуокиси кремния будут играть в определении характера катализатора лишь
второстепенную роль. Бемит, байерит и гидраргиллит представляют собой
окисно-алюминиевые структуры с основными свойствами, и в их кристаллах алюминий
имеет координационное число шесть, будучи связанным с шестью атомами кислорода.
Так как радиус иона алюминия сравнительно невелик (около 0,5), то он
может перейти из состояния, характеризующегося координационным числом 4, в
состояние с координационным числом 5, вступая в коорданиционную связь с
четырьмя, либо с шестью плотно упакованными ионами кислорода. Кислотно-основные
свойства окиси алюминия определяются тем, какое координационное число имеют
ионы алюминия, образующие кристаллическую структуру. Если алюминий имеет
координационное число четыре, он ведет себя как кислота. Соли такой кислоты
имеют кольцевую структуру, состоящую из шести алюмокислородных тетраэдров с
катионом внутри этого кольца. В то же время двуокись кремния в кварце, тридимите
и кристобалите всегда имеет координационное число, равное четырем, причем
существование этих различных кристаллических форм обусловлено различным
расположением кремнекислородных тетраэдров в смесях окислов или в
алюмосиликатах. Стабильная тетраэдрическая структура кремния усилена
соответствующей координацией ионов кислорода вокруг иона алюминия, причем
габитус кристалла стабилизируется такими катионами, как ионы натрия или
алюминия. Так как кристобаллит имеет структуру, аналогичну структуре алюмината
калия, явно выраженные кольца кристобаллита образуются в тех точках
соприкосновения, где ионы кислорода делятся между двуокисью кремния и окисью
алюминия.
Некоторые авторы предполагают, что в
прокаленном алюмосиликатном катализаторе присутствует кислотный комплекс типа 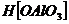 , обладающий
способностью диссоцировать с образованием протонов [72]. Однако, согласно
данным работы А. Г. Облада и др. [71]. Эта структура отвечает ангидриду
метаалюминиевой кислоты
, обладающий
способностью диссоцировать с образованием протонов [72]. Однако, согласно
данным работы А. Г. Облада и др. [71]. Эта структура отвечает ангидриду
метаалюминиевой кислоты 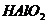 , она не
способна диссоциировать, давая протоны, а потому ее следует считать кислотой
Льюиса, а не кислотой бренстеда.
, она не
способна диссоциировать, давая протоны, а потому ее следует считать кислотой
Льюиса, а не кислотой бренстеда.
Согласно создавшемуся представлению
об активном центре катализатора, углеводород, находящийся в газовой фазе,
отдает электрон льюисовскому кислотному центру, при этом на поверхности
катализатора образуется карбониевый ион, что стабилизирует кислотный центр
катализатора. Стремление алюминия вернуться в состояние, характеризующееся
координационным числом 6, создает движущую силу десорбции. Однако, для
перемещения иона водорода, который также образуется в процессе крекинга,
необходимо одновременно присутствие и сосуществование на двойном центре кислоты
Льюиса и кислоты бренстеда [63]. Транспорт протонов может происходить и без
участия кислоты бренстеда.
Ряд авторов, учитывая возможную
координационную связь у атома алюминия в алюмосиликатах, активным центром в них
считают атом алюминия, имеющий координационное число 3. при взаимодействии
молекул, образующих карбонион с алюмосиликатом, в том числе у цеолитового
крекинга, координационное число у атома алюминия становится равным 4 [28, 36,
70, 71] по схеме:
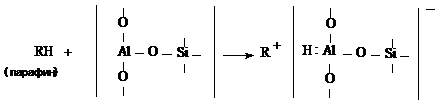
При этом допускают, что обменные
свойства алюмосиликатов обусловлены именно таким атомом алюминия, ибо он,
гидротируясь в водной среде, способствует образованию алюмосиликатного
комплекса, при котором выделяется ион водорода [28, 52].
Таким образом, для алюмосиликатов
характерно наличие протонных и апротонных форм кислотности. Причем, их
содержание, а также соотношение могут регулироваться активацией алюмосиликатов.
Так, при обработке алюмосиликатов (например, глин) в начале, когда концентрация
активатора низка, происходит эквивалентный обмен поглощенных катионов твердой
фазой на катион дисперсионной среды. При этом получается Н-форма глины [4, 21,
31] . Однако, эта форма глины не устойчива во времени и переходит в -форму (или -форма
переходит в Н-форму) по параболическим законам [73]. Поэтому при обработке глин
разбавленными растворами солей алюминия или минеральных кислот образуются
одновременно Н и -формы
глины. При обработке глины раствором кислоты повышенной концентрации происходит
разрушение кристаллической структуры глины и при этом происходит образование
алюмосиликата с повышенным содержанием 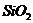 , обладающего кислотными свойствами,
о причине появления которых сказано выше.
, обладающего кислотными свойствами,
о причине появления которых сказано выше.
В работе [70] авторы допускают, что
при кислотной обработке глины, чтобы занаять место иона алюминия в глине, ион
водорода должен ассоциироваться с молекулами воды, тогда как ион анимает
место большего катиона. Это предположение является стратегическим критерием
расположения активного центра с ионами алюминия, находящимися в
непосредственной близости от тетраэдрического кремнезема, и алюминий легко
меняет координационное число от 6 до 4. Если в процессе катализа при
взаимодействии кислого иона алюминия с молекулой углеводорода на поверхности
катализатора образуется ион углеводорода, то возникает вопрос, каким образом
ион углеводорода разлагается и как происходит десорбция продукта. Можно
считать, что тенденция иона алюминия вновь принимать шестерную координацию
является движущей силой десорбции. Следовательно, каталитическая активность
алюмосиликата зависит от легкости, с которой ион алюминия меняет свое
координационное число от 4 до 6. эти предположения подтверждаются работами ряда
авторов [72,].
анимает
место большего катиона. Это предположение является стратегическим критерием
расположения активного центра с ионами алюминия, находящимися в
непосредственной близости от тетраэдрического кремнезема, и алюминий легко
меняет координационное число от 6 до 4. Если в процессе катализа при
взаимодействии кислого иона алюминия с молекулой углеводорода на поверхности
катализатора образуется ион углеводорода, то возникает вопрос, каким образом
ион углеводорода разлагается и как происходит десорбция продукта. Можно
считать, что тенденция иона алюминия вновь принимать шестерную координацию
является движущей силой десорбции. Следовательно, каталитическая активность
алюмосиликата зависит от легкости, с которой ион алюминия меняет свое
координационное число от 4 до 6. эти предположения подтверждаются работами ряда
авторов [72,].
Известна проматирующая роль воды в
каталитическом процессе, если в качестве катализатора используется алюмосиликат
[61,71].
Установлено, что полностью обезвоженный алюмосиликат не активен в реакции
крекинга кумола [71]. Однако, если этот образец снова
гидратировать, его первоначальная активность восстанавливается. Проматирующее
действие небольших количеств воды в реакциях углеводородов объясняется тем, что
вода вызывает десорбцию карбоний-иона, сдвигая равновесие в сторону образования
новых ионов. Большое количество воды приводит к конкуренции за активным центром
между углеводородом и водой, и адсорбируются из них более полярные, т.е. воды,
что приводит к понижению активности катализатора. Однако,
одновременное существование донора протонов (кислоты)и акцептора протонов (основания)
в алюмосиликатах делает их способными для расщепления углеводородов [71].
Вода в алюмосиликатах, повидимому, одновременно служит акцептором и донором
протона, согласно схеме:
повышение
каталитической
активности прокаленных, затем охлажденных до температуры крекинга природных
промышленных алюмосиликатов объясняется неравномерным распределением ОН-групп и
высокой подвижностью протона вновь адсорбированной влаги и их миграцией на
поверхности [70]. Сказанное подтверждается еще и
тем, что при постоянном суммарном значении общей кислотности алюмосиликатного
катализатора с содержанием определенного количества воды протонная кислотность
катализатора имеет наибольшее значение при низких, а апротонная - при высоких температурах
[35, 80].
Есть указание о том, что активность
катализатора уменьшается с ростом радиуса катиона, адсорбированного
алюмосиликатом [81]. Предполагают, что катализ в некоторых алюмосиликатах, в
частности на цеолитах, протекает не на протонных, а не катионных центрах,
имеющих электротстатическое поле сильнее, чем на протонных центрах. Молекула
углеводорода на них поляризуется ( ) или даже отщепляется с
образованием иона карбония.
) или даже отщепляется с
образованием иона карбония.
При оценке активности природных
алюмосиликатов (глин и др.) при крекинге кумола или газойливой фракции исходят
из суммарной величины обменных ионов 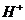 и и отрицательного заряда решетки.
Замечена симбатность между изменением общеобменной кислотности алюмосилиткатов
при их активации и их каталитической активностью в реакциях крекинга [10, 18,
36, и др.].
и и отрицательного заряда решетки.
Замечена симбатность между изменением общеобменной кислотности алюмосилиткатов
при их активации и их каталитической активностью в реакциях крекинга [10, 18,
36, и др.].
Таким образом, у алюмосиликатных
катализаторов имеются каталитические центры, проявление которых, в основном,
зависит от структуры алюмосиликата, наличия воды в нем и от процесса катализа.
Структура природных алюмосиликатов, количество воды в них и связанные с этим
свойства природных алюмосиликатов регулируются воздействием на них физическим и
химическим способами активации. При этом, в зависимости от условий активации,
образуются катализаторы, свойства которых устанавливаются пока опытным путем.
Можно с уверенностью сказать, что при воздействии на природный алюмосиликат
каким-либо методом активации в определенном участке его структуры происходит
изменение, благодаря чему возникает напряженность в структуре в целом, а в
системе появляется сила, направленная на уменьшение напряжения, возникшего в
результате внешнего воздействия на алюмосиликат. Однако, отсутствие прямого
измерения силы возникшего напряжения делает это положение гипотетическим.
Несмотря на это, увеличение адсорбционной и каталитической активности природных
алюмосиликатов при их термо-, термокислотнощелочно, и наоборот, активации
указывает на избыточность внутренней энергии, для компенсации возникающей
внешнего напряжения. Такое предположение согласуется с мнением Д.И.Менделеева о
внутренней подвижности составляющих системы при ее встрече с внешними
молекулами. Другими словами, адсорбенты и катализаторы являются более эффективными,
если в нем созданы благоприятные условия для внутреннего движения. Таким
образом, «катализатор - это приведенное в неустойчивое состояние вещество,
химически возбуждающее субстрат и направляющее химическую реакцию».
1.3
Физико-химические свойства Навбахорского бентонита
Как уже упоминалось, месторождение бентонитовых
глин Навбахор было открыто в 1998 году [45]. Разведочные работы по нему
завершены, запасы утверждены ГКЗ РУз и оно подготовлено к промышленному
освоению.
Вещественный состав бентонитовых глин изучен
авторами [46] комплексными лабораторными методами (химический, термический,
электронно-микроскопический и рентгено-структурный анализы), определена общая
обменная емкость поглощенных оснований, соотношение обменных катионов и изучены
физико-химические свойства. По результатам лабораторных исследований в разрезе
продуктивной толщи месторождения выделены щелочные и щелочно-земельные
разновидности глин, которые сильно отличаются по физико-химическим свойствам
друг от друга. Бентонитовое число (набухаемость) щелочных бентонитов в пробах
колебалась от 42 до 86, в среднем 79мл. Коллоидальность изменялась от 45 до
90,6, в среднем 80,5%. Эти показатели в щелочноземельных бентонитах значительно
ниже, чем в щелочных и составляют в среднем 41 мл и 51%, соответственно.
Объектами нашего исследования были щелочные
бентониты. Химический состав образца приведены ниже.
Химический состав (масс, %)
|
SiO2
|
AI2O3
|
Fe2O3
|
Na2O
|
K2O
|
MgO
|
CaO
|
P2O5
|
TiO2
|
FeO
|
Ппп
|
|
54,20
|
11,78
|
9,29
|
3,36
|
2,53
|
1,77
|
1,34
|
0,15
|
1,37
|
-
|
14,21
|
Как видно из приведенных данных основным оксидом
в минерале является оксид кремния ( 54,20 % масс.), содержание красящих оксидов
тоже значительно: Fe2O3
- 9,29 и TiO2 - 1,37;
оксида алюминия - 11,78% масс. На основании проведенных исследований можно
сказать, что исследуемый образец сложен монтмориллонитом.
Адсорбционные характеристики, полученные из
изотерм адсорбции паров воды и бензола методом БЭТ, (заимствовано из [59]
показывают, что удельная поверхность по воде (237м2/г) больше, чем по бензолу
(67м2/г)). Это обусловлено различием взаимодействий глин с полярными и
неполярными молекулами адсорбата.
Изотермы адсорбции паров воды нового адсорбента
измеренные, при 293К имеют S-образную
форму. В работе [60] сорбционные характеристики, определенные с помощью метода
БЭТ имеют следующие характеристики: ёмкость монослоя (аn)
- 4,81моль/кг; удельная поверхность (S)
- 319м2/г; пределы адсорбции (as)
- 20,2 моль/кг; предельный адсорбционный объем (Vc)
- 0,36 · 103 м3/кг.
Для определения термостойкости адсорбента и выяснения
его поведения в процессе термообработки использован термический анализ (ДТА).
В работе [60] проведены термический анализ,
рентгенографические и электронно-микроскопические (РЭМ) исследования [61,62]
образцов Навбахорского бентонита.
В исследованном образце бентонита (Рис.1.1.),
как показывают рентгенографические данные наблюдается большое количество
максимумов, что отражает сложную кристаллическую слоистую структуру глины.
Преобладает SiO2 с
характерными рефлексами 2q = 210, 270, 36,60 и 39,50. (наиболее
интенсивные рефлексы). Для AI2O3
наиболее характерные рефлексы при 2q = 29,60, в
интервале 42-440 и 47-500.
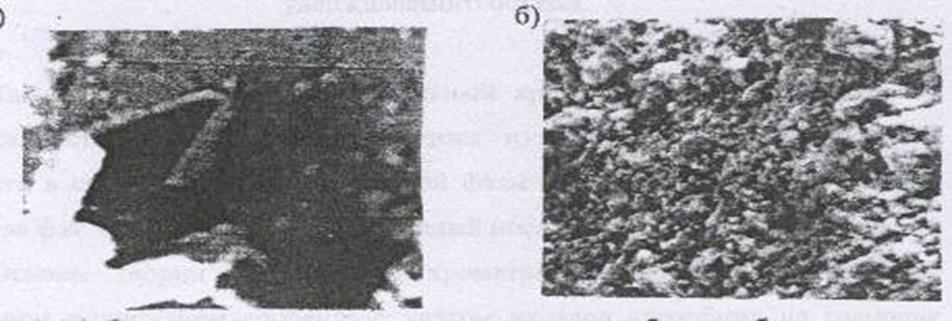
Рис. 1.1. Электронно-микроскопические данные
образцов исходного бентонита
При РЭМ - исследованиях видно, что сами частицы
исходного бентонита имеют зернистую структуру и при больших увеличениях имеют
геометрическую форму, что соответствует кристаллической природе бентонита.
Производство масел из восточных нефтей сильно
отличается от производства масел из старых промышленно освоенных нефтей.
Открытие новых перспективных месторождений высокопарафинистых сернистых,
смолистых нефтей вызывает необходимость разработки новых способов и подходов их
переработки для получения качественных масел.
Для улучшения качеств нефтяных масел в работе
рассматривается вопрос подбора селективных адсорбентов на основе местного
Навбахорского бентонита для удаления нежелательных компонентов масляных
фракций, ухудшающих эксплуатационные свойства.
Поскольку исходный бентонит и продукт его
кислотно-термической активации обладают определенными значениями удельной
поверхности, следует полагать, что они будут обладать поглотительными
способностями.
1.4 Адсорбционная
активность бентонита Навбахорского месторождения по нефтяным компонентам
Для определения емкости бентонита в статических
и динамических условиях из жидкой фазы по различным нефтяным компонентам и
приготовления их модельных растворов выбран циклогексан, как наиболее инертный
растворитель, адсорбционный индекс которого довольно низок 3,30 и который
должен быть высокой степени чистоты.
Циклогексан, полученный любым из существующих
методов, требует тщательной очистки от сопутствующих примесей. Разделить смесь
циклогексана от примесей обычной перегонкой затруднительно. О чистоте
циклогексана судить по коэффициенту преломления также трудно: различное
сочетание циклогексана, бензола, метилциклопентана, а также наличие влаги может
дать значение показателя преломления, близкое к значениям показателя
преломления для чистого циклогексана, в то время как в нем может быть
значительное количество этих примесей. Наиболее правильным критерием чистоты
продукта является температура его кристаллизации, которая для циклогексана
(100-ной степени чистоты) равна 6,520С.
Данные Маскарелли [103] о температурах
кристаллизации системы бензол-циклогексан показывают, что кривая температуры
выпадения циклогексана из раствора при вымораживании падает круто и
эвтектическая точка приходится на температуру - 43,70С, что соответствует
содержанию 24,2% бензола в смеси.
Было показано, что в области высоких
концентраций циклогексана (97 мол. % и выше) коэффициент преломления не является
достоверным критерием чистоты циклогексана, и здесь необходимо определить
температуру кристаллизации. Для этого предложен адсорбционный способ очистки
циклогексана до температуры кристаллизации 6,400С и выше с использованием
силикагелей, активированных углей и цеолитов: наиболее оптимальным адсорбентом
являются активированные угли, в частности угли СКТ и БАУ. Наиболее полную
информацию об углях можно подшепнуть из [93].
В данной работе для очистки циклогексана - до
99,00% степени чистоты использовался уголь БАУ - березовый активированный
уголь.
Очистка циклогексана до требуемых норм
осуществлялась следующим способом: уголь БАУ фр 0,25-0,5мм предварительно
просушенный в сушильном шкафу при температуре 160-1800С в течение 6 часов,
загружался в стеклянную колонку (обыкновенную бюретку с краником), можно в
делительную воронку, и через адсорбент непрерывно пропускался циклогексан с
заранее определенной температурой кристаллизации и собирался в отдельные
приемники. Для каждого фильтрата определялась температура кристаллизации.
Скорость фильтрации циклогексана через адсорбент - 1 капля в 1 сек. По
окончании очистки пробы с одинаковыми температурами кристаллизации сливались в
единую колбу. Составлялся баланс выхода чистого циклогексана.
В работе все растворы готовились на циклогексане
с температурой кристаллизации 6,400С.
Адсорбция в статических условиях. Так как в
состав моторного масла входят углеводороды, сняты изотермы адсорбции н-гексана
и самого моторного масла селективной очистки (продукция Ферганского НПЗ), из
неполярного растворителя циклогексана из жидкой фазы на исходном и
кислотноактивированном образцах бентонита (фр. 0,25-0,5мм). Образцы сорбентов
высушены при температуре 160-1800С в течении 6 часов. При этом условно
допускали, что вся адсорбированная вода удаляется с поверхности сорбента, а
гидроксильный покров бентонита оставался нетронутым.
Растворы готовились 1, 2, 3, 4, 5 %-ной
концентрации. Измерения производились на рефрактометре ИРФ - 22. Предварительно
построен калибровочный график и изучена кинетика адсорбции исследуемых сорбатов
на бентоните. Установлено, что адсорбционное равновесие наступает через 20
часов. Поэтому равновесная концентрация растворов определялась по истечении
этого времени.
Изотермы адсорбции снимали следующим образом: 1г
измельченного адсорбента, предварительно дегидратированного в вышеуказанных
условиях, заливали 10мл растворов сорбатов, н-гексана и трансформаторного масла
в циклогексане различной концентрации. Пробы в закрытых склянках выдерживали
при комнатной температуре (20-250С) при периодическом встряхивании, после чего
определяли равновесную концентрацию.
Величину Гиббсовой адсорбции вычисляли по
общеизвестной формуле:
Cu - Cp
r = ------------ ∙
V где
m
Cu - исходная
концентрация раствора, моль/л
m - навеска
адсорбента, г
V - объем
исследуемого раствора, взятый для исследования адсорбции.
Cp-
равновесная концентрация, моль/л
Из построенных изотерм адсорбции было видно
(рис. 1.2.) , что адсорбция изученных сорбатов в области низкой концентрации
резко возрастает, а затем идет почти параллельно оси абсцисс и имеется у всех
кривых один изгиб.
Причем все они адсорбируются положительно во
всем интервале концентрации, и изотермы адсорбции выражаются простыми
изотермами адсорбции типа изотерм Ленгмюра или Фрейндлиха.
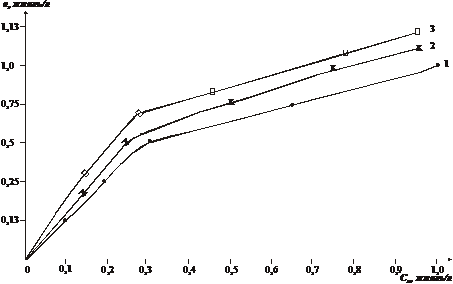
Рис.1.2. Изотермы адсорбции на Навбахорском
бентоните.
- н-гексана на термоактивированном бентоните
- н-гексана на кислотнотермоактивированном
бентоните
- трансформатоного масла на термоактивированном
бентоните
Причем, наибольшую активность имеет бентонит по
трансформаторному маслу, чем по н-гексану. Это можно объяснить
многокомпонентностью трансформаторного масла.
Если сравнить изотермы адсорбции н-гексана на
исходном и кислотно-активированном образце бентонита, то последний имеет
наибольшую емкость. По значениям емкости монослоя (am)
они располагаются в следующий ряд: исходный бентонит (amмасла
0,6 ммоль/л) > кислотно-активированный бентонит (am
н-гексана 0,5 ммоль/л) > исходный бентонит (am
н-гексана 0,45 ммоль). Установлено что при адсорбции из
неполярной среды (циклогексан) у природных
минеральных сорбентов участвует ½ часть
поверхностных гидроксильных групп. Это обусловлено наличием у природных
минеральных сорбентов активных центров типа центров кислотности Бренстеда у
атома алюминия. Вода, адсорбированная на таких центрах значительно легче
диссонирует таким образом усиливается адсорбционное взаимодействие между
гидроксилом природных минеральных сорбентов и водородом атомов молекул
углеводородов с образованием водородной связи.
Наиболее полное представление о природе
адсорбционных сил, механизме адсорбции и о состоянии адсорбированного вещества
дают термодинамические характеристики процесса адсорбции. Мономолекулярное
взаимодействие в системе сорбат-сорбент может быть оценено и газоадсорбционным
вариантом хроматографии. При этом допускается, что скорость массообмена между
газовой и твердой фазой намного больше, чем скорость инертной фазы
(газоносителя). Газохроматографическим методом измеряется избыточное время удерживания
сорбата в колонке, заполненной сорбентом (в данном случае - бентонитом). Данный
метод позволяет непосредственно определять избыточную величину адсорбции.
Дифференциальная теплота находится из наклона прямолинейных отрезков изотер,
выраженных в координатах LgP
- T-1.
Проведено сравнение энергетических характеристик
адсорбции - теплот адсорбции (Q,
кДж/моль) гомологического ряда н-алканов и углеводородов различной электронной
структуры состава С6 - н-гексана, бензола, циклогексана на исходном и кислотнотермоактивированном
образцах бентонита (фракция 0,25-0,5 мм), определенных вышеуказанным методом.
Как известно, по классификации А.В.Киселева [3],
н- алканы относятся к группе А, т.е. это молекулы со сферически симметричной
электронной оболочкой, как у благородных газов.
Рабочие условия: Хроматограф ЛХМ-7А Дзержинского
ОКБА (РФ), колонка стальная длиной 2м, диаметром 0,4мм, газ-носитель-гелий,
температура колонок - 100,110 и 1200С. В качестве адсорбентов использовались
исходный бентонит и его активированный образец. Как следует из [63], из всех
известных методов улучшения адсорбционных свойств природных минеральных
сорбентов является кислотно-термическая активация. Активирование производили
15% серной кислотой и подвергали температурной обработке при 160-1800С в
течение 6 часов.
В таблице 1.1. приведены значения удельного
удерживаемого объема (Vg
см3/г), который является основной физико-химической константой в храмотографии.
Видно, что Vg
является функцией молекулярной массы сорбата: с ее повышением
значение Vg
увеличивается, а с повышением температуры Vg
для каждого сорбата уменьшается, в связи с тем, что связь сорбат-сорбент
ухудшается и увеличивается количество молекул, переходящих в газовую фазу.
Так, например при температуре колонки 473 К
н-гексана Vg составляет
48,34; н-гептана 95,31 и н-октана - 149,73; инкремент Vg
на одну метиленовую группу в гомологическому ряду 45 см3/г. Такая же картина
наблюдается и для других температур колонки.
Таблица 1.1.
Удельные удерживаемые объёмы н-алканов на
Навбахорском бентоните
|
Сорбат
|
Т,
К пол.
|
Vg, см3/г
|
|
н-Гексан
н-Гептан н-Октан н-Гексан н-Гептан н-Октан н-Гексан н-Гептан н-Октан
|
473
493 513
|
48,34
±0,84 95,31 ±0,42 149,73 ±0,20 40,40 ±1,02 70,64 ±0,58 105,47 ±0,32 33,38
±1,33 57,80 ±0,64 79,80 ±0,54
|
Причем с повышением температуры колонки Vg
для каждого сорбата, как и следовало ожидать, уменьшается ввиду того, что
процесс десорбции превалирует над адсорбцией. Так, например для н-гексана Vg
при 473 К составляет 48,34 см3/г, 493 К - 40,40 см3/г, а при 513 К - 33,38
см3/г.
Таблица 1.2.
Теплоты адсорбции н-алканов С6-С8
|
Сорбат
|
Т,
К
|
1/1000
К
|
TR, мин
|
Lg tR
|
tgα
|
Q, кДж/моль
|
|
н-Гептан
н-Гексан н-Октан н-Гексан н-Гептан н-Октан н-Гексан н-Гептан н-Октан
|
473
493 513
|
2,114 2,028 1,949
|
2,01
2,505 4,05 1,385 2,175 3,34 1,255 2,04 3,175
|
0,27155
-0,661 0,574 -0,6819 0,2655 0,4472 -0,57245 -0,65155 0,4252
|
н-гексан 1,30 н-гептан 1,6 н-октан 1,84
|
н-гексан 33,00 н-гептан 34,42 н-октан 35,68
|
В таблице 1.2. представлены значения температур
колонки, tR мин - время
удерживания компонента, мин; тангенс угла наклона линейной зависимости lg
времени удерживания от обратной величины температуры колонки и рассчитанные на
их основе значения теплота адсорбции исследованных сорбатов (Q,
кДж/моль). Видно, что Q
н-алканов в ряду С6-С8 меняется от 35,65 до 35,68 кДж/моль; инкремент на
метиленовую группу в гомологическом ряду н-алканов равен 1,3 кДж/моль.
Составлена графическая зависимость Vg
и Q от числа
углеродных атомов в молекуле и температуры кипения, которые являются
номограммами. По ним можно вычислить значения Vg
и Q для
гомологического ряда н-алканов.
На предложенном сорбенте снята хроматограмма
смесей н-алканов С6-С8. Пики хроматограмм четкие, симметричные.
Определенные теплоты адсорбции углеводородов,
состава С6- различной электронной структуры: бензола, циклогексана и н-гексана
даны в таблице 1.3.
Сравнивая теплоты адсорбции углеводородов
различной электронной структуры - бензола, циклогексана и н-гексана, можно
сказать, что по значениям теплот адсорбции углеводороды располагаются, как на
исходном, так и обработанном бентоните в следующий ряд: н-гексан <
циклогексан < бензол.
Таблица 1.3.
Теплоты адсорбции углеводородов состава С6
|
Сорбаты
|
Теплоты
адсорбции (Q,
кДж/моль)
|
|
Q1 исх.
|
Q2 обраб.
|
|
Бензол
|
26,00
±0,46
|
13,61
±0,14
|
|
н-гексан
|
20,43
±0,59
|
10,12
±0,14
|
|
Циклогексан
|
22,08
±0,55
|
11,16
±0,12
|
В обоих случаях, как на исходном, так и
термоактивированном бентоните теплота адсорбции бензола выше, что можно
объяснить наличием наряду с дисперсионным взаимодействием в системе сорбат -
сорбент специфического взаимодействия за счет π - электронов
ароматических углеводородов. Причем, сравнивая теплоты адсорбции бензола и
н-гексана можно сказать, что теплота адсорбции π - электронов
ароматического кольца составляет 0,57 кДж/моль.
Из приведенных данных видно, что теплоты
адсорбции изученных сорбатов на обработанном бентоните ниже почти в два раза,
чем на исходном, что можно объяснить концентрированием кристаллической фазы и
уменьшением удельной поверхности бентонита, следовательно, и десорбция
углеводородов будет происходить с меньшими затратами.
Следовательно, бентонит Навбахорского
месторождения можно использовать в качестве сорбента для газоадсорбционной
храмотографии.
Определялась емкость исходного и кислотнотермически
обработанного бентонита по широкому кругу органических веществ, которые могут
встречаться в масляных фракциях: углеводородам и гетероорганическим веществам,
как S-, N-,
и О-содержащие соединения - для первых шести сорбатов (н-гексану, бензолу,
тиофену, пиридину, фенолу, циклогексанкарбоновой кислоте) криоскопическим
методом ( разработка ИОНХ АН РУз [55] ) и двух последних (асфальтенам и смолам)
- по обесцвечиванию их растворов при прохождении через слой сорбента [63].
Сущность криоскопического метода определения динамической емкости сорбентов
заключается в изменении температуры кристаллизации модельных 2% растворов
сорбатов при хроматографировании через 10г. сорбента (фр. 0,25-0,5мм,
стеклянная колонка диаметром 1см, скорость фильтрации 1 капля в 1 сек.).
Так как изменение температуры кристаллизации
раствора пропорционально его концентрации, то, пользуясь экспериментально
найденными значениями температуры кристаллизации, построим «выходные кривые»,
(рис.1.3.) откладывая по оси абсцисс количество жидкости, прошедшей через слой
адсорбента, а по оси ординат - температуру кристаллизации раствора.
При этом площадь фигуры АВЕД, отнесенная к 1кг
(или 100г) адсорбента, соответствует полной динамической емкости до проскока,
на основании которой рассчитывается рабочая активность адсорбента.

Рис. 1.3. Выходные кривые динамической емкости
сорбента, определенные криоскопическим методом.
По площади АВЕД (S1)
и АВСД (S2) выходной кривой
можно графически определить емкость адсорбента (m1)
по формуле:
m1 S1
S1
----- = ------ m1
= ------ M0
M0 S0
S0
где, M0
- количество введенного вещества.
Адсорбция в динамических условиях. Головным
процессом в производстве нефтяных масел является вакуумная перегонка мазута,
все последующие стадии производства сводятся к различным видам очистки
дистиллятного и остаточного сырья от продуктов, ухудшающих эксплуатационные
свойства масел. К таким продуктам относятся смолисто-асфальтеновые вещества, полициклические
ароматические углеводороды, твердые парафины, серо-, кислород -, азотсодержащие
соединения.
Производство масел из восточных нефтей сильно
отличается от производства масел из старых промышленно освоенных нефтей.
Открытие новых перспективных месторождений высокопарафинистых сернистых,
смолистых нефтей вызывает необходимость разработки новых способов и подходов их
переработки для получения качественных масел.
Для улучшения качеств нефтяных масел в работе
рассматривается вопрос подбора селективных адсорбентов на основе местного
Навбахорского бентонита для удаления нежелательных компонентов масляных
фракций, ухудшающих эксплуатационные свойства.
Поскольку исходный бентонит и продукт его
кислотно-термической активации обладают определенными значениями удельной
поверхности, следует полагать, что они будут обладать поглотительными
способностями.
ГЛАВА II.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ. МЕТОДЫ И ОБЪЕКТЫ ИССЛЕДОВАНИЯ
2.1
Методы исследования
В соответствии с поставленной задачей работы
проводили по изучению изменения структуры, структурно-сорбционных
характеристик, отбеливающих, регенерирующих и каталитических свойств природных
алюмосиликатов в зависимости от их способа и режима активации, для чего был
применен комплекс методов анализа - химический, физический и физико-химический
методы исследований.
Химический состав проб был определен
общеизвестным методом силикатного анализа сотрудниками лаборатории
аналитической химии Института Химии АН УзССР (аналитики-
К.С.Маряновская и Л.И. Жданова). На основании валового химического состава были
выведены кристаллохимические формулы глинистых минералов, входящих в состав
образца, по кислородному методу расчета формул по зарядам [90].
Структуры образцов были
охарактеризованы по дифференциально-термическим и рентгенографическим данным,
снятым сотрудниками лаборатории физ.-хим. методов анализа Института Химии АН
УзССР. Кривые ДТА образцов снимались на приборе Курнекова ФПК-55 со скоростью
нагрева 30-40º в мин.,
навеска образцов по 0,5-0,6 г; размер фракции - менее 0,01 мм (оператор Т.
Азизов). Рентгенограммы образцов снимались на дифрактометре УРС-50-ИМ при
режиме трубки 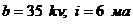 ; скорость
вращения счетчика 4 об/мин.; излучение
; скорость
вращения счетчика 4 об/мин.; излучение 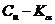 . Для некоторых образцов были сняты
рентгенограммы при режиме:
. Для некоторых образцов были сняты
рентгенограммы при режиме: 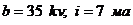 ; вращение образца 0,5 об/мин.;
излучение
; вращение образца 0,5 об/мин.;
излучение 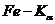 (оператор
М. Казаков).
(оператор
М. Казаков).
Структурно-сорбционные свойства
образцов охарактеризовали через величины их суммарной пористости (Р, %)
суммарного объема пор (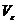 , см3/г) и
удельной поверхности (S, м2/г). Для вычисления Р и были
определены удельные веса образцов по бензолу (истинный удельный вес - d, г/см3) и
по ртути (кажущийся удельный вес - d, г/см3).
Значения удельной поверхности образцов были определены методом тепловой
десорбции азота газохроматическим методом [91]. Кроме того были сняты кривые
кинетики поглощения паров толуола при
, см3/г) и
удельной поверхности (S, м2/г). Для вычисления Р и были
определены удельные веса образцов по бензолу (истинный удельный вес - d, г/см3) и
по ртути (кажущийся удельный вес - d, г/см3).
Значения удельной поверхности образцов были определены методом тепловой
десорбции азота газохроматическим методом [91]. Кроме того были сняты кривые
кинетики поглощения паров толуола при 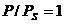 . По данным значениям предельно
сорбированных паров толуола были вычислены статические активности образцов по
толуолу (
. По данным значениям предельно
сорбированных паров толуола были вычислены статические активности образцов по
толуолу (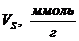 ), удельный
объем пор (
), удельный
объем пор (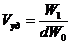 ,
,  , где
, где  - привес за
счет сорбции, г;
- привес за
счет сорбции, г;  - навеска
образца, г;
- навеска
образца, г; 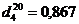 г/см3 -
плотность толуола) и средний радиус пор (
г/см3 -
плотность толуола) и средний радиус пор (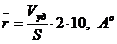 ).
).
Из структурно-механических
параметров образцов были определены объемный вес D, и пористость (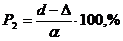 ), а также
механическая прочность (
), а также
механическая прочность (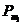 , кг/см2)
гранул и таблеток образцов в размере 3х3 мм по ГОСТу 1140-42 методом
раздавления с применением лабораторного прочностномера.
, кг/см2)
гранул и таблеток образцов в размере 3х3 мм по ГОСТу 1140-42 методом
раздавления с применением лабораторного прочностномера.
Определены различные формы
кислотности. По описанной в [32,] методике находили активную, общеобменную,
обменноводородную, обменноалюминиевую и гидролитическую кислотность образцов и
особенно количество двух н.ш. по методу К. В. Топчиевой [33], по обменной
адсорбции катиона водорода твердой фазы на катион раствора уксуснокислого
натрия, было определено значение протонной кислотности алюмосиликатов.
По методу В. Трамбуза [35],
титрованием неводной бензоловой суспензии образца этилацетатом в присутствии
индикатора Гамета - парадиметиламиноазобензола (ПДА), определяли апротонную
кислотность алюмосиликата. Определением протонной кислотности образцов,
отправленных ионами натрия, вычисляли их кислотный центр. О кислотном центре
судили и на основании спектра поглощения ультрафиолетовых лучей образцами,
отправленными органическими основаниями: из водной среды - n -
диметиламиноазобензосульфокислым натрием (метиловый оранжевый - МО) и неводной
(высушенный бензол) среды - парадиметил-аминоазобензолом
(ПДА). Электронные спектры поглощения таких отравленных образцов снимали на
регистрирующем спектрофотометре СФ-10 в видимой области спектра - 400-700 ммк в
проблемной лаборатории химии природных соединений ТашГУ (оператор В. Б.
Леонтьев). Образцы до отравления органическими основаниями были прокалены до
постоянного веса при 500 и 600º (первая температура для бентонитовых,
а вторая - для каолинитовой и опоковидных глин).
Адсорбционную очистку
асидола-технических нафтеновых кислот и гудрона образцами проводили в
статических и динамических условиях. Были приготовлены 5 и 10%-ные растворы
сорбтивов в циклогексане. Условия контактной очистки: количество адсорбента
(фракции менее 4900 отв/см2) от 10 до 50% от веса нафтеновых кислот вносили в
раствор сорбтива, взбалтывали 15-20 мин., затем фильтровали через складчатый
фильтр; степень обесцвечивания раствора определяли на ФЭК-М по оптической
плотности, применяя каветы с расстоянием между рабочими гранями 10,045 мм, с
использованием светофильтра-1. условие перколяционной очистки:
хроматографировали 10%-ный раствор нафтеновых кислот в циклогексане в
стеклянной колонке диаметром 1 см через слой 2 г адсорбента (фр. 0,16-0,50 мм)
со скоростью одна капля в секунду. После окончания хроматографирования
адсорбент дополнительно промывался циклогексаном. Из элюата отгонялся
растворитель, после чего определялось кислотное число и цвет очищенных
нафтеновых кислот титрованием 0,05 н. спиртовым раствором едкого калия в
присутствии нитрозина синего (до сине-зеленой окраски).
Адсорбционную очистку реактивного
топлива алюмосиликатами проводили также в динамических условиях. Колонку
заполняли 2 г сорбента с размером зерен 0,25 мм. Пропустили через него согласно
описанной в методике.
Адсорбционную очистку реактивного
топлива алюмосиликатами проводили также в динамических условиях. Колонку
заполняли 2 г сорбента с размером зерен 0,25 мм. Пропустили через него топливо.
Отобрав по 50 мл элюата, определяли кислотное число топлива титрованием
спиртовым раствором КИН в присутствии нитрозина синего. Активность адсорбента
выражали по выходу элюата с нулевой кислотностью в граммах поглощенных нафтеновых
кислот на 100 г образца.
Каталитическую активность образцов
изучали в реакциях крекинга кумола. Реакцию разложения углеводорода проводили
на собранной нами по установке с кварцевым реактором проточного типа.
Применялась фракция катализатора 1,0-2,5 мм или гранулы размером 3х3 мм;
отношение катализатора: сырье = 2:1; продолжительность опыта 30 мин.;
температура крекинга 450º;
скорость
подачи сырья 0,5 час-1.
Продукты крекинга анализировались
хроматографически с использованием прибора ЦВЕТ-1 (лаборатория адсорбции
института Химии АН УЗССР, оператор Л. С. Борисова) и ХЛ-4 (лаборатория
гидрокрекинга СредАзНИИНП, оператор Ш. Мурзикова). Режим разделения жидкого
катализата: температура термостата 150º, испарителя - 230º; скорость
подачи газоносителя (гелий) 35 мл/мин; длина колонки 1 м и ток моста 190 ма. В
качестве неподвижной фазы использовали выделенные из битума Избасткентского
месторождения высокомолекулярные ароматические углеводороды, предложенные Л. С.
Борисовой и Н. Д. Рябовой. Они были нанесены на твердую фазу ИНЗ-600 в
количестве 9%. Для разделения газового состава (азот, водород, углеводороды и
др.) длина колонки, наполненной ИНЗ-600, для углеводородов 12 м; температура
детектора - 100º,
а
колонки - 40º,
а
для неуглеводородов длина колонки, наполненной цеолитом NaA (фракция
0,5-0,25 мл) - 2 м; температура колонки - 40º, детектора - 72º.
Индекс активности катализатора
определяли по выходу бензиновой фракции при крекинге керосино-газойливой
фракции согласно ГОСТу МРТУ-38-1-189-65; 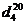 =0,872 г/см3. Температура опыта 450º при
атмосферных условиях, и его продолжительность 30 мин.; объемная скорость 0,7
час-1. до и после опытов катализатор продували азотом в течение 10 мин. со
скоростью 300 мл/мин. Регенерацию катализатора проводили по с перекисью
водорода; расход регенератора 1-2 мл в мин.; температура регенерации 475º. Цикл
крекинга и регенерации катализатора проводили.
=0,872 г/см3. Температура опыта 450º при
атмосферных условиях, и его продолжительность 30 мин.; объемная скорость 0,7
час-1. до и после опытов катализатор продували азотом в течение 10 мин. со
скоростью 300 мл/мин. Регенерацию катализатора проводили по с перекисью
водорода; расход регенератора 1-2 мл в мин.; температура регенерации 475º. Цикл
крекинга и регенерации катализатора проводили.
Мерой каталитической активности
катализаторов служило количество выходящих газообразных продуктов по и бензола
по. Степень превращения кумола выражали в % как отношение количества объема
(массы) газа к теоретическому значению газа, получаемому при 100%-ном
разложении молекул кумола.
2.2 Объекты исследования
Объектами исследования были
сорбтивы, растворители для сорбтивов и сорбенты - природные и активированные
различными способами влюмосиликаты различных месторождений и происхождения.
Сорбтивами были:
1. Технические исходные нафтеновые кислоты
(асидол), взятые из Бакинского нефтеперерабатывающего завода им. Караева;
молекулярный вес 400; кислотное число 221 мг-КОН/г.
2. Кубовой остаток - гудрон (темный по
цвету).
. Реактивное топливо ТС-1 из ФНПЗ с
кислотным числом 8,8 мг-КОН/г.
. Бензол марки ч.д.а. для анализа.
. Толуол марки ч.д.а. для анализа.
. п-диметиаминоазобензосульфокислый
натрий - метиловый оранжевый (МО).
. Парадиметиламиноазобензол (ПДА).
В качестве растворителей для приготовления
растворов нафтеновых кислот, черного хлопкового масла в мисцелле и других
сорбтивов использовлись дистиллированная вода, циклогексан, экстракционный
бензин, бензол, этиловый спирт. Технически циклогексан был очищен адсорбционным
методом с применением активированного угля БАУ. Температура замерзания
очищенного циклогексана была равна 4,46º.
В качестве алюмосиликатов были использованы
глины Азкамарского, Керменинского, Клесского, Дарбазинского, Ангренского,
Огланлынского, Таганского месторождений. Брали также грузинский аскангель,
чешскую, венгерскую отбельные глины, Новокуйбышевские, Уфимские синтетические
алюмосиликаты, алюмосиликат фирмы «Линде», окись люминия, силикагель, цеолиты,
зикеевскую опоку, широкопористое стекло.
Природные глины подвергались термической,
кислотной, кислотно-щелочной, щелочно-кислотной, известковой активации. Перед
активацией пробы доводили до воздушно-сухого состояния. Малые порции измельчали
в ступке или в лабораторной шариковой мельнице до фракции менее 0,16 мм.
Термическая активация. Этот вид активации
проводили как в лабораторных, так и в производственных условиях. Активацию
провели в статических условиях в муфельных печах при 100, 200, 300, 400, 500,
600, 700 и 800ºС с колебанием
температуры ±10º до
постоянного веса при заданной температуре. Термическую активацию больших проб
(одна тонна) осуществляли в Технологической лаборатории института Химии АН УзР
(измельчение до фракции менее 0,16 мм на дизентеграторе) и в катализаторном
цехе опытного завода СредАзНИИНП (сушка при 500-550º).
Кислотная активация. Активацию глин растворами
серной, азотной и соляной кислот провели по методу АзНИИ, заключающемуся в
следующем: образцы глин, после размелчения до фракции 0,16-0,25 мм, помещали в
круглодонную колбу, снабженную обратным холодильником, термометром и
механической мешалкой, оборот которой регулируется электрическим током, и
нагревали на водяной бане при температуре 98-100ºС.
После окончания опыта смесь предварительно промывалась от непрореагировавшей
кислоты. Затем суспензию выливали в воду и промывали через воронки Бюхнера или
подвергали декантации до нейтарльной реакции. Концентрации активаторов были для
серной 5, 10, 15, 25 и 35%. Расход кислот на 100 г сухой глины в соотношении
глина: кислота составлял 75 г; время активации равно 2, 4, 6, 8 и 10 часам с
момента начала кипения раствора активатора.
Кислотно-щелочная активация. С целью проведения
кислотно-щелочной активации ко всей массе активированной кислотой глины при
постоянном перемешивании добавляли раствор едкого натра до нейтральной реакции.
В дальнейшем проводилось промывание и отфильтрация.
Щечлоно-кислотная активация. Сначала в
установке, собранной для проведения кислотной активации, проводили щелочную
активацию, для чего в реактор загружали отмученную от грубых частиц глину и
добавляли 8-15%-ный раствор NаOH
в весовом отношении - глина: NaOH=1:1.
образовавшуюся суспензию нагревали при температуре около 100º
в
течение 5-10 часов. По окончании опыта содержимое реактора декантировали и
разбавляли водой, после чего в реактор добавляли соляную кислоту до полного
растворения осадка, полученного после щелочной обработки глин. Затем, добавляя
водный раствор аммиака, среду доводили до рН 7-8. образующийся при этом
творожистый осадок тщательно отмывали от солей до полного удаления ионов CI-.
Маточный раствор после корректировки содержания в нем щелочи повторно
использовали в процессе активации.
Получение адсорбента и катализатора по В.С.
Комарову [19]. По окончании процесса кислотной активации образцов содержимое
реактора разбавляли водой. Затем кислый раствор отделяли от твердой фазы. Глину
на фильтрате промыли небольшим количеством воды. 1/8 часть от активированной
глины добавляли к кислому фильтрату и тщательно перемешивали. В дальнейшем
фильтрат обрабатывали водным раствором аммиака, а затем промывали. При этом
выпадает осадок, который промывается от ионов и высушивается.
Известковая активация глины. Такой вид активации
разработан Н. Х.Алимухаммедовым и др. [10].
По этому способу тонкоизмельченную глину вносят
в бутыль и доливают насыщенным раствором извести при соотношении Т:Ж=1:15.
перемешивают, оставляют на ночь. Отработанный раствор сливают и заливают свежим
раствором извести. Эту операцию проводят до тех пор, пока глина не насытится
известью. Произвесткованные пробы были любезно представлены Н.
Х.Алимухаммедовым.
Термо-химическая активация. Образцы, полученные
после кислотной, кислотно-щелочной, щелочно-кислотной и других способы
активации, в некоторых случаях дополнительно подвергали термической активации
по описанной выше методике.
Полученные после соответствующей химической
обработки продукты (табл. 2.1) сушили при 105º, измельчали,
просеивали через сито до требуемой фракции или таблетировали на
полуавтоматической таблеточной установке в виде цилиндра в размере 3х4 мм, либо
формовали в гранулы. В последнем случае тесто продавливали через шприц с
отверстием 3 мм и после некоторого высыхания разрезали бритвочкой до размера
4-5 мм и сушили.
Таблица 2.1.
Условие активации природных алюмосиликатов
|
№
|
Вид
и условие активации алюмосиликатов
|
Лабораторный
номер
|
|
Глина
(алюмо-силикат)
|
Условн.
обозначение
|
Вид
активации
|
Условия
активации
|
|
|
|
|
|
Активатор
|
Время
актив., час.
|
Конц.
активатора, %
|
Нейтрализатор
|
|
|
1.
|
Азкамарский
белый бентонит
|
АББАК-6
|
кислотная
|
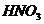 66-161 66-161
|
|
|
|
|
|
2.
|
То
же
|
АББАК-12
|
то
же
|
612-162
|
|
|
|
|
|
3.
|
То
же
|
АББАК-18
|
то
же
|
618-163
|
|
|
|
|
|
4.
|
То
же
|
АББАК-24
|
то
же
|
624-164
|
|
|
|
|
|
5.
|
То
же
|
АББАК-30
|
то
же
|
630-165
|
|
|
|
|
|
6.
|
То
же
|
АББ-К-25
|
то
же
|
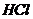 625-200 625-200
|
|
|
|
|
|
7.
|
То
же
|
АББСК-25
|
то
же
|
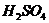 625-201 625-201
|
|
|
|
|
|
8.
|
То
же
|
АББЩК
|
щелочно-кислотная
|
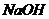 1218247 1218247
|
|
|
|
|
|
9.
|
АСБАК-6
|
кислотная
|
66-166
|
|
|
|
|
|
10.
|
То
же
|
АСБАК-12
|
то
же
|
612-167
|
|
|
|
|
|
11.
|
То
же
|
АСБАК-18
|
то
же
|
618-168
|
|
|
|
|
|
12.
|
То
же
|
АСБАК-24
|
то
же
|
624-169
|
|
|
|
|
|
13.
|
То
же
|
АСБАК-30
|
то
же
|
630-170
|
|
|
|
|
|
14.
|
То
же
|
АСБСК-5
|
то
же
|
65-217
|
|
|
|
|
|
15.
|
То
же
|
АСБСК-10
|
то
же
|
610-218
|
|
|
|
|
|
16.
|
То
же
|
АСБСК-15
|
то
же
|
615-219
|
|
|
|
|
|
17.
|
То
же
|
АСБСК-25
|
то
же
|
625-220
|
|
|
|
|
|
18.
|
То
же
|
АСБСК-35
|
то
же
|
635-221
|
|
|
|
|
|
19.
|
То
же
|
АСБЩК
|
щелочно-кислотная
|
1218-245
|
|
|
|
|
|
20.
|
То
же
|
АСБК-25
|
кислотная
|
625284
|
|
|
|
|
|
21.
|
Келесский
бентонит
|
КБАК-6
|
кислотная
|
66-171
|
|
|
|
|
|
22.
|
То
же
|
КБАК-12
|
то
же
|
612-172
|
|
|
|
|
|
23.
|
То
же
|
КБАК-18
|
то
же
|
618-173
|
|
|
|
|
|
24.
|
То
же
|
КБАК-24
|
то
же
|
624-174
|
|
|
|
|
|
25.
|
То
же
|
КБАК-30
|
то
же
|
630-175
|
|
|
|
|
|
26.
|
То
же
|
КБСК-5
|
то
же
|
65-271
|
|
|
|
|
|
27.
|
То
же
|
КБСК-10
|
то
же
|
610-272
|
|
|
|
|
|
28.
|
То
же
|
КБСК-15
|
то
же
|
615-273
|
|
|
|
|
|
29.
|
То
же
|
КБСК-25
|
то
же
|
625-274
|
|
|
|
|
|
30.
|
То
же
|
КБСК-35
|
то
же
|
635-275
|
|
|
|
|
|
31.
|
То
же
|
КБСК25-2
|
то
же
|
225-240
|
|
|
|
|
|
32.
|
То
же
|
КБСК25-4
|
то
же
|
425-241
|
|
|
|
|
|
33.
|
То
же
|
КБСК25-6
|
то
же
|
625-274
|
|
|
|
|
|
34.
|
То
же
|
КБСК25-10
|
то
же
|
1025-243
|
|
|
|
|
|
35.
|
То
же
|
КБСК25-12
|
щелочно-кислотная
|
1218244
|
|
|
|
|
|
36.
|
Таганский
розо-вый бентонит
|
ТРБАК-6
|
кислотная
|
66-236
|
|
|
|
|
|
37.
|
То
же
|
ТРБАК-12
|
то
же
|
612-264
|
|
|
|
|
|
38.
|
То
же
|
ТРБАК-18
|
то
же
|
618-265
|
|
|
|
|
|
39.
|
То
же
|
ТРБАК-24
|
то
же
|
624-266
|
|
|
|
|
|
40.
|
То
же
|
ТРБАК-30
|
то
же
|
630-267
|
|
|
|
|
|
41.
|
То
же
|
ТРБК-30
|
то
же
|
630-268
|
|
|
|
|
|
42.
|
То
же
|
ТРБСК-25
|
то
же
|
625-269
|
|
|
|
|
|
43.
|
То
же
|
ТРБКЩ-25
|
по
Кома-рову В. С.
|
625276
|
|
|
|
|
|
44.
|
То
же
|
ТРБКЩ
|
щелочно-кислотная
|
1218251
|
|
|
|
|
|
45.
|
Аскангель
|
АСКАК-6
|
кислотная
|
66-176
|
|
|
|
|
|
46.
|
То
же
|
АСКАК-12
|
то
же
|
612-177
|
|
|
|
|
|
47.
|
То
же
|
АСКАК-18
|
то
же
|
618-178
|
|
|
|
|
|
48.
|
То
же
|
АСКАК-24
|
то
же
|
624-179
|
|
|
|
|
|
49.
|
То
же
|
АСКАК-30
|
то
же
|
630-180
|
|
|
|
|
|
50.
|
То
же
|
АСКЩК
|
щелочно-кислотная
|
1218253
|
|
|
|
|
|
51.
|
То
же
|
АСКК-25
|
кислотная
|
625-204
|
|
|
|
|
|
52.
|
Огланлинский
бентонит
|
ОБЩК
|
щелочно-кислотная
|
1218255
|
|
|
|
|
|
53.
|
Дарбазинская
опока
|
ДОПАК-6
|
кислотная
|
66-195
|
|
|
|
|
|
54.
|
То
же
|
ДОПАК-12
|
то
же
|
612-196
|
|
|
|
|
|
55.
|
То
же
|
ДОПАК-18
|
то
же
|
618-197
|
|
|
|
|
|
56.
|
То
же
|
ДОПАК-24
|
то
же
|
624-198
|
|
|
|
|
|
57.
|
То
же
|
ДОПАК-30
|
то
же
|
630-199
|
|
|
|
|
|
58.
|
То
же
|
ДОПСК-5
|
то
же
|
65-211
|
|
|
|
|
|
59.
|
То
же
|
ДОПСК-10
|
то
же
|
610-212
|
|
|
|
|
|
60.
|
То
же
|
ДОПСК-15
|
то
же
|
615-213
|
|
|
|
|
|
61.
|
То
же
|
ДОПСК-25
|
то
же
|
625-214
|
|
|
|
|
|
62.
|
То
же
|
ДОПСК-35
|
то
же
|
635-215
|
|
|
|
|
|
63.
|
То
же
|
ДОПЩК
|
щелочно-кислотная
|
1218235
|
|
|
|
|
|
64.
|
Керменинская
опоковидная
|
КОПАК-6
|
кислотная
|
66-190
|
|
|
|
|
|
65.
|
То
же
|
КОПАК-12
|
то
же
|
612-191
|
|
|
|
|
|
66.
|
То
же
|
КОПАК-18
|
то
же
|
618-192
|
|
|
|
|
|
67.
|
То
же
|
КОПАК-24
|
то
же
|
624-193
|
|
|
|
|
|
68.
|
То
же
|
КОПАК-30
|
то
же
|
630-194
|
|
|
|
|
|
69.
|
То
же
|
КОПСК-5
|
то
же
|
65-223
|
|
|
|
|
|
70.
|
То
же
|
КОПСК-10
|
то
же
|
610-224
|
|
|
|
|
|
71.
|
То
же
|
КОПСК-15
|
то
же
|
615-225
|
|
|
|
|
|
72.
|
То
же
|
КОПСК-25
|
то
же
|
625-226
|
|
|
|
|
|
73.
|
То
же
|
КОПСК-35
|
то
же
|
635-227
|
|
|
|
|
|
74.
|
То
же
|
КОПКЩ-5
|
кислотно-щелочная
|
65228
|
|
|
|
|
|
75.
|
То
же
|
КОПКЩ-10
|
то
же
|
610229
|
|
|
|
|
|
76.
|
То
же
|
КОПКЩ-15
|
то
же
|
615230
|
|
|
|
|
|
77.
|
То
же
|
КОПКЩ-25
|
то
же
|
625231
|
|
|
|
|
|
78.
|
То
же
|
КОПКЩ-35
|
то
же
|
635232
|
|
|
|
|
|
79.
|
То
же
|
щелочно-кислотная
|
1218238
|
|
|
|
|
|
80.
|
То
же
|
ИКОП
|
известковая
|
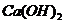 до
насыщения262 до
насыщения262
|
|
|
|
81.
|
Ангренский
каолин
|
АКЩК
|
щелочно-кислотная
|
1218257
|
|
|
|
|
|
82.
|
Дурменская
(Каракуль)
|
ДГАК-18
|
кислотная
|
618-208
|
|
|
|
|
|
83.
|
То
же
|
ДГ-К-18
|
то
же
|
618-209
|
|
|
|
|
|
84.
|
То
же
|
ДГСК-18
|
то
же
|
618-270
|
|
|
|
|
|
85.
|
Зикеевская
опока
|
ЗОПИ
|
известковая
|
до
насыщения285
|
|
|
ГЛАВА 3.
ИССЛЕДОВАНИЕ ФИЗИКО-ХИМИЧЕСКИХ, КИСЛОТНЫХ И КАТАЛИТИЧЕСКИХ СВОЙСТВ ПРИРОДНЫХ И
АКТИВИРОВАННЫХ БЕНТОНИТОВ
3.1 Изучение влияния способов активации на химический
состав природных алюмосиликатов
Химический состав исследованных
природных алюмосиликатов приведен таблицах. Видно, что для изученных
монтмориллонитовых глин свойственно содержание  от 52 до 68%,
от 52 до 68%, 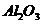 до 23%,
до 23%,  - 10-20% и
п.п.п. - до 17%. Для каолина - 74%, - 22%, - 0,5%; для
опоки - 83%, - 5%, - до 12%.
Содержание окиси магния в глинах республики, как было отмечено в [4], больше,
чем количество окиси кальция в них. Содержание окислов железа колеблется для
монтмориллонитовых глин в пределах 1,01-5,13%, для каолина равно 0,97%, для
опоковидной глины - 3,58% и для опоки - 1,43%.
- 10-20% и
п.п.п. - до 17%. Для каолина - 74%, - 22%, - 0,5%; для
опоки - 83%, - 5%, - до 12%.
Содержание окиси магния в глинах республики, как было отмечено в [4], больше,
чем количество окиси кальция в них. Содержание окислов железа колеблется для
монтмориллонитовых глин в пределах 1,01-5,13%, для каолина равно 0,97%, для
опоковидной глины - 3,58% и для опоки - 1,43%.
Содержание во всех
бентонитах больше, чем в чешской
(обр. 263) глине; то же самое можно сказать относительно содержания глинозема.
Бентонитовые глины, белая - обр. 246
и серая - обр. 216 разновидности Азкамарского месторождения, характеризуются
высоким содержанием окислов щелочно- и щелочноземельных элементов, что
свидетельствует о наличии в них чистого щелочного монтмориллонита; причем,
наблюдается, как уже сказано, преобладание содержания 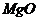 над содержанием
СаО, а также повышенное содержание воды.
над содержанием
СаО, а также повышенное содержание воды.
Опока (обр. 210) от опоковидной
глины (обр. 222), как и следовало ожидать отличается высоким содержанием
кремнезема. Относительно малое содержание глинозема в опоке свидетельствует о
незначительной примеси в ней глинистых минералов.
Мольное соотношение к 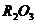 для
изученных нами проб бентонитовых глин колеблется в пределах 4-9,2. это
указывает на преобладание в них глинистого минерала монтмориллонитовой группы.
Завышенное значение : по
сравнению с теоретическим (согласно Р. Гриму [3, 11] оно равно 4) связано с
наличием в них кремнистых пород, главным из которых является халцедон, a - кристобаллит [5].
для
изученных нами проб бентонитовых глин колеблется в пределах 4-9,2. это
указывает на преобладание в них глинистого минерала монтмориллонитовой группы.
Завышенное значение : по
сравнению с теоретическим (согласно Р. Гриму [3, 11] оно равно 4) связано с
наличием в них кремнистых пород, главным из которых является халцедон, a - кристобаллит [5].
На основании данных валового
химического состава, были выведены кристаллохимические формулы (табл. 4) для
бентонитовых глин и каолина по кислородному методу расчета формул по зарядам
[90]. При этом за основу брали теоретические формулы монтмориллонита и
каолинита.
Теоретическая формула
монтмориллонита, согласно [11], следующая:
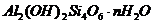 или
или 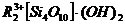
где n -
межслоевая вода. Химический состав монтмориллонита, без учета межслоевой воды,
выражается следующими значениями: = 66,7%, =28,3%.
Мольное соотношение к равно 4.
количество отрицательных валентностей равно 22. они составлены из валентности
10 кислородных атомов и двух гидроксильных групп.
Теоретическая формула каолинита,
согласно [11], следующая: 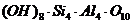 или
или 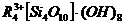 .Количество
отрицательных валент-ностей составлено 10 атомами
кислорода и 8 группами гидроксила и в сумме равна 28. химический состав
каолинита следующий: =46,54, =39,50 и =13,96%. Из
данных, приведенных в табл. 3, видно, что химический состав ангренского каолина
(обр. 256-АК) сильно отличается от состава каолинита и характеризуется низким
содержанием (примерно на 45%) глинозема, а содержание кремнезем в 1,6 раз
больше теоретического. По сведениям Р. Грима [11], такие породы, относящиеся к
каолинитовой группе, называются анокситом. Согласно Хентриксону (сведения Р.
Грима [11]), аноксит состоит из каолинитовых структурных слоев, между которыми
статически расположены двойные кремнекислородные тетраэдрические слои. В
кремнекислородных слоях тетраэдры каждой сетки направлены к середине слоя так,
что тетраэдры противоположных сеток в средней плоскости имеют общие вершины, в
которых расположены общие для них атомы кислорода.
.Количество
отрицательных валент-ностей составлено 10 атомами
кислорода и 8 группами гидроксила и в сумме равна 28. химический состав
каолинита следующий: =46,54, =39,50 и =13,96%. Из
данных, приведенных в табл. 3, видно, что химический состав ангренского каолина
(обр. 256-АК) сильно отличается от состава каолинита и характеризуется низким
содержанием (примерно на 45%) глинозема, а содержание кремнезем в 1,6 раз
больше теоретического. По сведениям Р. Грима [11], такие породы, относящиеся к
каолинитовой группе, называются анокситом. Согласно Хентриксону (сведения Р.
Грима [11]), аноксит состоит из каолинитовых структурных слоев, между которыми
статически расположены двойные кремнекислородные тетраэдрические слои. В
кремнекислородных слоях тетраэдры каждой сетки направлены к середине слоя так,
что тетраэдры противоположных сеток в средней плоскости имеют общие вершины, в
которых расположены общие для них атомы кислорода.
Таким образом, ангренская каолиновая
глина является полиминеральной системой, в которой может находится не только
каолинит, но и другие глинистые минералы этой группы, в частности, аноксит.
Известно, что в глинистых минералах
существуют следующие главные изоморфные замещения: 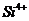 на
на 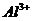 ; на
; на 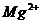 или
или 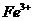 ;
; 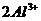 на
на 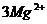 или
или 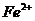 . В
монтмориллонитах и некоторых других гилнах идет также замещение обменоспособных
катионов [3, 11, 22]. Выведенные кристаллохимические формулы для азкамарских
(обр. 246-АББ, 216 АСБ), таганского (обр. 250/ТРБ), келесского (обр. 239-КБ) и
огланлинского (обр. 254-ОБ) бентонитов и аскангеля (обр. 252-АСК) соответствуют
формулам мантмориллонита:
. В
монтмориллонитах и некоторых других гилнах идет также замещение обменоспособных
катионов [3, 11, 22]. Выведенные кристаллохимические формулы для азкамарских
(обр. 246-АББ, 216 АСБ), таганского (обр. 250/ТРБ), келесского (обр. 239-КБ) и
огланлинского (обр. 254-ОБ) бентонитов и аскангеля (обр. 252-АСК) соответствуют
формулам мантмориллонита: 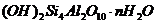 . В
тетраэдрической сетке обр. 246-АББ, 216-АСБ, 239-КБ, 254-ОБ и 256-АК находятся
атомы кремния, а в остальных
. В
тетраэдрической сетке обр. 246-АББ, 216-АСБ, 239-КБ, 254-ОБ и 256-АК находятся
атомы кремния, а в остальных 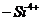 частично замещен на .
частично замещен на .
Из данных таблиц видно, что из-за
отсутствия изоморфного замещения в тетраэдрической координации глин азкамарского
серого, колесского, огланлинского бентонитов наблюдается избыток положительного
заряда этой ячейки, что обусловлено, несомненно, наличием избытка . В глинах,
у которых имеется частичка замещения на наблюдается повышение
отрицательного заряда тетраэдрической структурной единицы, что придает им
дефектность или вакансии (не насыщенность валентных связей).
Вакантные места в октаэдрической
координации глинистых минералов опеределяли как произведение разницы суммы
катионов в октаэдрической координации от двух к валентности алюминия. При этом
выявлено, что величины зараядов октаэдра, в противоположность зарядам
тетраэдра, колеблется в широком диапазоне. Причем, из-за замещения иона высокой
валентности на ион относительно низкой валентности суммарный заряд октаэдра
всегда отрицательный. Другими словами, чем больше замещений, тем больше
значения отрицательных зарядов, т.е. тем больше вакантных мест (или
дефектность). По уменьшению абсолютного значения суммарного заряда
октаэдрической координации монтмориллонита изученные нами глины располагаются в
следующий ряд:
Известно [11], что в
монтмориллонитовых глинах одна шестая часть алюминия в октаэдрической сетке
бывает замещенной на магний. Аналогичная картина наблюдается и в случае
изученных нами бентонитов. Изоморфные замещения на в тетраэдрической и на , и др. в
октаэдрической координации глин создают неуравновешенность заряда в
соответствующих структурных единицах и обусловливают дефект в кристаллической
решетке.
На основании анализа химического
состава глин, их кристаллохимической формулы и зарядов решетки можно допустить,
что большая часть связано с
ионами , входящими
в структуру монтмориллонита. Часть окиси железа входит в октаэдрическую
структурную единицу, а основная масса его находится в виде свободных окислов,
что придает бентонитам желтый, или красноватый, пятнистый оттенки, как это
свойственно азкамарскому белому, келесскому бентонитам и ангренскому каолину. 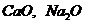 и др. в
бентоните находятся в поглощенном виде катионы, которые компенсируют
отрицательный заряд глинистого минерала.
и др. в
бентоните находятся в поглощенном виде катионы, которые компенсируют
отрицательный заряд глинистого минерала.
Обычно монтмориллонит не содержит 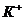 . Однако
наличие
. Однако
наличие 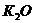 в
бентонитах и в других глинах связано с присутствием слюды, полевого шпата [11]
.
в
бентонитах и в других глинах связано с присутствием слюды, полевого шпата [11]
.
Итак, в октаэдрической координации
всех изученных проб имеется изоморфное замещение на соответствующий катион 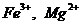 . Изоморфное
замещение на в
тетраэдрической координации наблюдается в случае аскангеля, азкамарского белого
и таганского бентонитов. В октаэдрической сетке каолина , в противоположность
бентонитам, замещен только на ион . Во всех глинах проявляется дефект
структуры минералов - вакантные места. Причем, последний составляет 75-81% от
общего заряда кристаллической решетки минералов, входящих в состав образца.
. Изоморфное
замещение на в
тетраэдрической координации наблюдается в случае аскангеля, азкамарского белого
и таганского бентонитов. В октаэдрической сетке каолина , в противоположность
бентонитам, замещен только на ион . Во всех глинах проявляется дефект
структуры минералов - вакантные места. Причем, последний составляет 75-81% от
общего заряда кристаллической решетки минералов, входящих в состав образца.
Химическая активация глин привела к
существенному изменению их химического состава, следовательно, и
кристаллохимической структуры. Так, кислотная активация привела к возрастанию в образцах
(табл. 3.1.) При этом содержание окисей алюминия и железа уменьшается;
количество окиси магния практически остается постоянным или уменьшается
незначительно, что свидетельствует о сохранении катиона магния в октаэдрической
координации. Обращает на себя внимание тот факт, что содержание СаО
тоже изменяется незначительно, если активацию глин провести с раствором серной
кислоты.
По-видимому, кальций в данном случае в глинах присутствует в трудно растворимой
в воде форме.
Таблица 3.1.
Изменение химического состава глин после
кислотной активации
|
Лабораторный
номер пробы и ее обозначение
|
Содержание
компонентов на сухую массу, %
|
S (сумма)
|
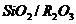
|
|
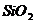 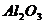 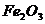 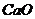 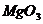 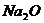 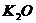 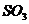 п.п.п. п.п.п.
|
|
|
|
|
|
|
|
|
|
|
|
61
|
АББК-6
|
53,45
|
17,16
|
3,10
|
0,36
|
2,36
|
0,33
|
0,30
|
0,07
|
23,34
|
100,47
|
5,1
|
|
62
|
АББК-12
|
54,34
|
16,52
|
3,02
|
0,17
|
2,28
|
0,32
|
0,20
|
0,07
|
23,05
|
99,74
|
5,2
|
|
63
|
АББК-18
|
54,66
|
16,32
|
2,99
|
0,17
|
2,04
|
0,22
|
0,20
|
0,10
|
23,37
|
100,40
|
5,4
|
|
64
|
АББК-24
|
55,63
|
15,37
|
2,94
|
0,12
|
1,91
|
0,20
|
0,20
|
0,17
|
22,20
|
100,54
|
5,5
|
|
65
|
АББК-30
|
56,41
|
15,28
|
2,06
|
0,06
|
1,87
|
0,20
|
0,20
|
0,21
|
24,70
|
99,88
|
6,3
|
|
00
|
АББК-25
|
54,98
|
18,66
|
2,49
|
0,32
|
2,50
|
0,25
|
0,18
|
сл.
|
20,79
|
100,17
|
5,0
|
|
01
|
АББСК-25
|
62,65
|
17,59
|
1,89
|
0,58
|
2,19
|
0,21
|
0,17
|
0,10
|
14,55
|
99,8
|
5,8
|
Таблица 3.2.
Химический состав глин, подвергнутых щелочной,
кислотной и известковой активации
|
Лабораторный
номер пробы и ее обозначение
|
Содержание
компонентов на сухую массу, %
|
S (сумма)
|
|
|
п.п.п.
|
|
|
|
|
|
|
|
|
|
|
|
235-ДОП-ЩК
|
74,24
|
5,94
|
1,71
|
0,51
|
0,65
|
1,80
|
0,72
|
0,09
|
14,77
|
100,43
|
20,05
|
|
238-КОП-ЩК
|
58,03
|
12,89
|
4,00
|
3,63
|
2,27
|
0,94
|
1,26
|
0,12
|
17,44
|
100,62
|
6,0
|
|
262-ИКОП
|
51,27
|
8,51
|
2,99
|
13,83
|
2,11
|
0,97
|
1,38
|
0,45
|
18,21
|
99,72
|
8,5
|
|
247-АББ-ЩК
|
51,76
|
19,73
|
2,86
|
0,17
|
2,91
|
3,70
|
0,06
|
0,09
|
18,76
|
100,09
|
4,3
|
|
245-АСБ-ЩК
|
59,04
|
15,78
|
5,71
|
0,51
|
2,28
|
2,36
|
1,80
|
0,12
|
12,25
|
99,85
|
4,3
|
|
244-КБ-ЩК
|
59,83
|
15,22
|
4,65
|
0,91
|
2,36
|
1,80
|
1,54
|
0,09
|
13,62
|
100,02
|
5,5
|
|
251-ТРБ-ЩК
|
48,39
|
20,57
|
0,65
|
0,91
|
2,83
|
7,67
|
0,21
|
0,03
|
19,13
|
100,39
|
4,0
|
|
253-АСК-ЩК
|
54,38
|
17,95
|
3,64
|
0,68
|
3,97
|
2,21
|
2,87
|
0,03
|
14,10
|
98,70
|
4,3
|
|
255-ОБ-ЩК
|
70,23
|
8,40
|
0,97
|
1,26
|
1,22
|
1,44
|
0,29
|
0,06
|
11,25
|
100,17
|
13,7
|
|
257-АК-ЩК
|
59,44
|
18,49
|
0,81
|
0,34
|
0,08
|
5,65
|
0,34
|
0,15
|
15,11
|
100,20
|
5,3
|
Мольное отношение кремнезема к сумме полуторных
окислов с ростом концентрации растворов кислот и продолжительности контакта
глин с активаторами увеличивается. Причем, с повышением содержания кислот в
единице объема раствора активатора до определенной концентрации эта величина
увеличивается незначительно; резкое изменение начинается при активировании глин
с применением раствора кислот с концентрацией более 15% в случае серной кислоты
и более 24% - в случае азотной кислоты.
Если сопоставить изменение химического состава
глин (например, азкамарского белого бентонита обр. 165-АББ-АК-30, 200-АББК-25 и
201-АББСК-20) от вида кислоты, то видно, что при прочих равных условиях (время
действия 6 часов, температура активации 100º, концентрация
кислоты 25%) серная кислота действует более активнее, чем другие кислоты, хотя
количество протонов в единице объема раствора серной кислоты меньше, по
сравнению с концентрацией протонов растворов других кислот.
Изменение мольного соотношения у
опоковидных глин (обр. 190-194, 223-227) и опок (обр. 195-199, 211-215) при
кислотной активации более резко выражено, чем у бентонитовых глин. Причем, это
заметнее, если активатором были растворы серной кислоты.
Влияние продолжительности активации
кислотой на химический состав глин изучали на примере келесского бентонита
(обр. 239). Его активировали 25%-ным раствором серной кислоты при температуре
кипения воды в течение 2 (обр. 240-КБ-СК252), 4 (обр. 241-КБ-СК254), 6 (обр.
274-КБ-СК256) и 10 (обр. 243-КБ-СК25-10) часов. Их химический состав совпадает
с химическим составом для образцов, приготовленных в аналогичных условиях И.
Сатаевым и Э.Ариповым [4,]. Поэтому вполне приемлемы в нашем случае выводы,
сделанные этими авторами. Определяя химический состав кислотной вытяжки
келесского бентонита и сопоставляя химический состав активированных глин, они
установили, что оптимальным условием сернокислотной активации глин является
шести часовая обработка глины раствором кислоты. При этом концентрация раствора
активатора должна быть равной 5-10% - это достаточно для вымывания
присутствующих в глине сопутствующих веществ. Применение раствора выше этой
концентрации приведет к резкому изменению , что соответствует началу
разрушения кристаллической структуры монтмориллонита - основного составляющего
бентонита.
Если допустить, что начало резкого
увеличения мольного соотношения от концентрации кислоты можно
принять за начало разрушения кристаллической решетки монтмориллонита, входящего
в состав бентонитовых и опоковидных глин и опок, то не трудно заметить, что
кристаллическая структура глинистого минерала сохраняется в случае азкамарского
серого, келесского бентонитов и керменинской опоки при активации их серной
кислотой с концентрацией до 15%, а при активации их, а также ахкамарского
белого, таганского, огланлинского бентонитов азотной кислотой - до 24%. Эти
результаты дополняют данные, полученные И.Сатаевым, Н. Абдуллаевым и Э.Ариповым
[4,20,21,], установшими условия активации для глин Узбекистана.
В табл. 3.2 приведен химический
состав глин, подвергнутых щелочно-кислотной и известковой активации. В противоположность
кислотной активации, в данном случае не наблюдается существенного изменения
химического состава глин. Оно и понятно, т.к. вещества, которые вымывались из
образцов растворами щелочи, осаждаются кислотой, если, конечно, при этом
образуются трудно растворимые соли. Надо полагать, одним из таких веществ
являются не только силикаты, но и алюмосиликаты. Об этом свидетельствует
сопоставление данных табл. 6 с данными, приведенными в табл. 3. однако, окислы
щелочных и щелочноземельных металлов, как и следовало, ожидать, уменьшаются,
т.к. их соли тоже растворимы в воде кислотой.
Известковая активация (обр.
262-ИКОП) керменинской глины привела к увеличению содержания СаО и ППП, что
согласуется с результатами работ [4,].
Составление по данным химического
состава проактивированных бентонитовых глин кристаллохимические формулы
монтмориллонитовых глин позволили выяснить, что в тетраэдрической координации
всех глин увеличивается атом кремния, благодаря чему повышается и заряд этой
структурной ячейки. Причем увеличение в этой структурной единицы тем
больше, а чем больше контакт глин с кислотой. Так, для келесского бентонита,
активированного 25% в течении
2, 4, 6 и 10 часов (обр. 240-КБСК 25-2, 241-КБСК 25-4, 274-КБСК 25-6 и 243-КБСК
25-10), в
тетраэдрической координации увеличивается от 4,11 до 5,01 и в соответствии с
этим заряд этой структурной ячейки возрастает с 0,11 до 1,01; чем больше
концентрация активатора, т.е. увеличение концентрации с 5 до 35%
привело к повышению в тетраэдре
азкамарского серого бентонита с 4,35 до 5,00, а его заряда от 0,13 до 1,00; в
чем больше разрушающая способность кислоты. Так, значение в тетраэдре
азкамарского серого бентонита, активировнного с 25%-ными растворами , и 30%-ным равно 4,85;
4,12 и 4,15 соответственно.
Аналогичным образом изменяется и
состав и заряд октаэдрической ячейки. С ростом степени разрушения ячейки
повышается степень ненасыщенности этой координации, благодаря ему повышается
число вакантных мест, т.е. увеличивается абсолютная величина заряда этой
структурной единицы монтмориллонита. В соответствии с этим и изменяется
суммарный заряд решеток монтмориллонита (рис. 3.1).
Изучение влияния щелочно-кислотного
способа активации на заряды алюмосиликатов показало, что величина заряда
кристаллической решетки в случае ангренского каолина, огланлинского, келесского
и азкамарского серого бентонитов по сравнению с исходным уменьшается. Наряду с
этим, кроме таганского и азкамарского белого бентонитов, в пробах,
активированных по щелочно-кислотному способу, в тетраэдрических координациях
изоморфного замещения не наблюдается. В значительной степени повышается
содержание обменных катионов, обусловленное, по-видимому, присутствием в
поглощенном состоянии катионов 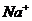 от активатора-едкого натра.
от активатора-едкого натра.
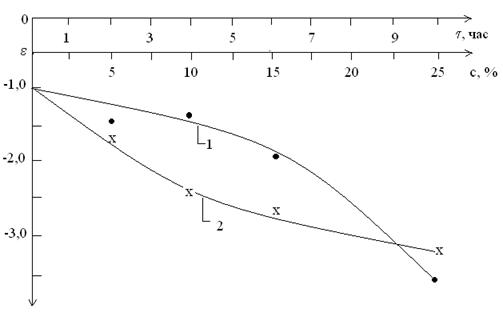
Рис. 3.1. Изменение заряда
кристаллической решетки монтмориллонита (e)
в зависимости от концетнрации (с) , времени (t) активации азкамарского
серого (1) и келесского (2) бентонитов.
Таким образом, заряд элементарной
ячейки обусловлен за счет изоморфных замещений в тетраэдрической и октаэдрической
координациях. Электрононейтральность ячейки алюмосиликата может быть
рассмотрена как компенсация заряда обменными поглощенными катионами. Общий
заряд ячейки алюмосиликата в зависимости от условий химического воздействия на
него в значительной степени изменяется. Изменение заряда ячейки в зависимости
от концентрации и продолжительности времени воздействия активатора идет в
сторону увеличения. Щелочно-кислотный способ активации алюмосиликатов
способствует повышению содержания обменных катионов, свидетельствующему об
увеличении заряда решетки, что подтверждается повышением в незначительной
степени общей суммы (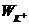 ) катионов в
кристаллической ячейке минерала.
) катионов в
кристаллической ячейке минерала.
3.2 Влияние активации на структуру алюмосиликатов
Термографическое изучение
естественных и активированных алюмосиликатов. Видно, что
для ДТА азкамарского белого (кривая 1), серого (кривая 2), келесского (кривая
3), таганского (кривая 4), огланлинского (кривая 13) бентонитов, опоковидной
глины (кривая 7), а также цеолита (кривая 12) характерно наличие при
температурах 120-200º,
480-700º и
850-930º
соответственно трех эндоэффектов. На кривых ДТА остальных образцов имеется
только один эндоэффект при температуре 150-200º. Термическая
реакция при температуре 100-150º (1 эндоэффект) обусловлена удалением
адсорбционно-связанной (межслоевой) воды. Второй эндоэффект, который по
сравнению с первым проявляется относительно слабо, обусловлен выделением
структурной (конструкционной) воды. Появление третьего эндоэффекта для
бентонитовых и опоковидных глин, основным составляющим которых из глинистых
минералов является монтмориллонит, дискутируется. Оно связано либо с наличием в
образцах бейделлита, либо с аморфизацией минералов [3, 11, 22, ].
Химическая активация существенным
образом влияет на форму кривых ДТА глин. На кривых ДТА кислотноактивированных
глин, как правило, отчетливо проявляется первый эндоэффект. Второй и третий
эндоэффекты не проявляются или проявляются очень слабо. Причем, если они
проявляются, то при относительно низких температурах.
Кривая ДТА келесского бентонита,
активированного серной кислотой различной концентрации. Видно, что с ростом
концентрации активатора второй и третий эндоэффекты исчезают. Аналогичные
результаты были получены авторами работ [4, 69,] при
сернокислотной активации келесского и азкамарского бентонитов. Авторами было
установлено, что по легкости изменения эндоэффекты бентонитов могут быть
расположены в ряд: III > II > I. Другими
словами, площадь эндоэффектов, связанных с термическими реакциями, в которых
первостепенную роль играет структура минерала, уменьшается быстрее с
увеличением концентрации кислоты. Эти выводы подтверждаются и нашими
исследованиями.
В предыдущем параграфе этой главы
отмечалось, что серная кислота лучше действует на глины. Проводимые работы по
анализу ДТА проб, полученных активацией азотной кислотой, показали, что в
результате активации глин на их кривых ДТА сохраняются все эндоэффекты;
наблюдается увеличение интегральной площади, как это было и при сернокислотной
активации, если концентрация активатора не превышает 10%, первого эндоэффекта.
Такое явление, по-видимому, следует
объяснить исходя из декатионирования глин при кислотной активации, в результате
чего образуется их водородная форма. Для кислых (Al и Н) форм
глин характерно увеличение интегральной площади термической реакции выделения
адсорбционно связанной воды [4, 20, 31,]. На этом основании можно заключить,
что при приготовлении Н-форм глин лучше пользоваться азотной кислотой, т.к. на
кривой ДТА образца бентонита, обработанного 30%-ным раствором имеются все
три эндоэффекта, свойственные для исходной глины (рис. 4, кривая 6). Поэтому
вряд ли целесообразно говорить о том, что увеличение интегральной площади
первого эндоэффекта на кривых ДТА глин кислотной активации обусловлено
повышением содержания свободной кремневой кислоты на поверхности частиц глин.
Если учесть, что их образованию предшествует разложение (растворение глин
кислотой) и осаждение продуктов разложения в кислой среде, то следует полагать,
что свободные кремневые кислоты в глинах должны быть больше при их
сернокислотной активации, т.к. разрушающее действие последней значительно
превосходит таковое у других кислот.
Щелочно-кислотная и
кислотно-щелочная активация привели тоже к исчезновению или, во всяком случае,
к ослаблению интегральной площад второго эндоэффекта на кривых ДТА глин.
Существенное изменение наблюдается в первом эндоэффекте: происходит углубление
глубины эффекта и смещение максимума эффекта в сторону низкой температуры (в
пределе 20º).
Такое
явление обусловлено, по-видимому, увеличением степени дисперсности исходных
глин в результате их активации и повышением количества сорбированных паров воды
продуктами активации. Сказанное подтверждается данными по п.п.п., приведенными
в табл. 3 и 6. например, п.п.п. для исходного таганского бентонита (обр.
250-ТДБ) составляет 10-20%, а для активированного (обр. 251-ТРБ-ЩК) - 19, 13%,
т.е. содержание воды увеличивалось примерно в 2 раза.
3.3 Рентгенографические исследования природных и активи-рованных алюмосиликатов
Рентгенограммы исследованных глин
подтверждает их поли-минеральность. На рентгенограммах бентонитовых глин
имеются линии монтмориллонита (16,0-14; 1,70-1,63; 1-65-163; 1,52-1,48 Аº(, интенсивность
которых примерно в 7 раз больше интенсивности остальных линий. На этом
основании можно заключить, что количество монтмориллонита в бентонитах
превышает 70%. Кроме линии монтмориллонита на рентгенограммах бентонитов
имеются линии кварца (3,53-3; 1,82-1,81; 1,55-1,54; 1,38-1,37 Аº) гидрослюды
(1,79; 2,44; 2,12; 1,37 Аº).
на
рентгенограммах опок и опоковидных глин явно выражены линии кварца; наряду с
ними имеются линии, характерные для монтмориллонита, гидрослюды и гипса (3,02 Аº).
Рентгенограмма ангренского каолина
характеризуется линиями каолинита (7,01; 3,53; 2,54; 2,31; 2,32; 1,99; 1,49 Аº). поскольку
обогащенный каолин был обожжен при 700º в течении 2-х часов, то,
естественно, что на рентгенограммах имеется лини 3,38 Аº, характерная
для муллита. Характерной особенностью для рентгенограмм каолина, как всех
рентгенограмм других глин, является наличие интенсивной линии отражения
рентгеновых лучей с a=4,08 Аº, соответствующей
криталлолиту.
Химическая обработка глин привела к
изменению их рентгенограммы. Например, интенсивность линий, характерных для
монтмориллонита, на рентгенограммах бентонита увеличивается с повышением
концентрации активатора - серной кислоты (до 10%), а затем уменьшается. при
этом интенсивность линий, характерных для кварца, сильно увеличивается. В
некоторых случаях появляются новые линии 14,23; 2,12; 1,83; 1,65; 1,53; 1,37 Аº. Кроме того,
исчезает линия с межплоскостным расстоянием около 3,17 Аº,
характерная для щелочных бентонитов, что свидетельствует о появлении кислых
форм глин [4, 10].
Аналогичная картина наблюдается и
при кислотной активации опоковидной глины и опоки. Исчезновение
линии монтмориллонита на рентгенограммах этих глин наблюдается при активации их
растворами кислот с концентрацией выше 15%. Эти результаты совпадают
с данными Н.Ф.Абдуллаева [69].
Увеличение интенсивности линий,
характерных для глинистых минералов, от концентрации кислоты связано с
обогащением образцов (несомненно, преимущественно в Н-форме) за счет удаления
растворимых в воде и в разбавленных растворах кислот солей или полуторных
окислов металлов, а также тех веществ, с помощью которых склеивались первичные
частицы глины во вторичные [4]. Уменьшение интенсивности линии монтмориллонита
и увеличение интенсивности линии , которые имеют место для образцов,
приготовленных растворами минеральных кислот относительно высокой концентрации,
связано, в основном с разрушением структуры глинистого минерала. Однако,
наличие линии с d=10,13; 2,54 Аº и др.,
характерной для монтмориллонита, а также линии, свойственной гидрослюде, у
образцов, полученных обработкой исходных глин растворами кислот высокой
концентрации, свидетельствует о сохранении алюмосиликатного каркаса,
принадлежащего глинистым минералам монтмориллонитовой группы. На этом основании
можно делать вывод о том, что при обработке бентонитовых и опоковидных глин и
опок, основным составляющим, которых из глинистых минералов является
монтмориллонит, кислотой сохраняется структура, свойственная для
алюмосиликатного каркаса глинистых минералов. Этот вывод вполне согласуется с
данными других авторов [4, 20, 69].
Анализ рентгенограмм продуктов,
полученных щелочно-кислотной активацией глин, показал, что они отличаются от
рентгенограмм исходного сырья. Например на рентгенограммах образца,
приготовленного щелочно-кислотной активацией ангренского каолина, имеются линии
12,38; 8,75; 7,15; 4,10; 3,70; 3,28; 2,98; 2,62, характерные для цеолита типа NaA
. На этом основании можно заключить, что обработка каолина обожженного при
температуре образования метакаолинита - 700º, раствором
щелочи
с последующей
обработкой реакционной массы нейтрализацией соляной кислоты и доведением
среды до нейтрального состояния привела к изменению не только начальной
структуры каолина, но и к формированию нового минерала, структура которого сходна
со структурой цеолита. Исходя из сложности химического состава этого образца и
предполагая различные его вариации в процессе формирования кристалла, можно
утверждать, что в обр. 262-АК-ШК цеолит является цеолитом группы содалита
(цеолит NaA тоже входит
в эту группу), для описания состава которых имеется много вариантов [64].
Резюмируя изложенные в этом
параграфе материалы, можно заключить, что в результате химической активации
существенным образом меняется структура минералов, входящих в состав исходных
глин. При внесении исходных глин в раствор кислоты в первую очередь происходит
смачивание частиц образца раствором (скорее всего растворителем) активатора.
Затем происходит растворение растворимых в воде или разбавленных растворах
кислота солей и других веществ, всегда присутствующих в образцах. Это приводит
к некоторому обогащению исходного образца основными компонентами - глинистыми
минералами. Дальнейшее повышение концентрации активатора приводит, как
отмечается Э.А.Ариповым в [4], к растворению веществ, с помощью которых
склеивались мелкие фракции образцов в агрегат. Разрушение агрегатов, надо
полагать, как справедливо отмечается в [11], происходит с края частиц.
Последующее повышение содержания активатора в единице объема раствора приводит
к разрушению кристаллической структуры глинистого минерала - монтмориллонита.
При этом имеет место существенное изменение на термограммах. Изложенный здесь
механизм влияния кислоты на природные алюмосиликаты (в основном глины,
состоящие из монтмориллонита) находится в согласии с выводами, сделанными в [69] в
результате изучения механизма кислотной активации бентонитов методом
ИК-спектроскопии и в [4,], полученными при электронномикроскопическом изучении
этого процесса.
Механизм щелочно-кислотной активации
является сложным. Основным механизмом, примелемом в данном случае, является
механизм, описанный в [19], сущность которого заключается в растворении глины в
растворах щелочей, сопровождающимся появлением алюмосиликатов, структурные и
химические составы которых отличаются от материнской породы, и в разложении
этих продуктов кислотой, в результате чего образуются соли минеральных кислот и
кремневая кислота, которая через некоторое время желатинируется за счет
конденсации 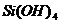 . Чтобы соли
алюминия не уходили из реакционной смеси, реакционную массу нейтрализуют
аммиаком. При этом из раствора осаждаются гидроокись алюминия и гель кремневой
кислоты.
. Чтобы соли
алюминия не уходили из реакционной смеси, реакционную массу нейтрализуют
аммиаком. При этом из раствора осаждаются гидроокись алюминия и гель кремневой
кислоты.
Очевидно, эти новообразования будут
влиять на физико-химические и другие свойства природных алюмосиликатов.
3.4 Структурно-сорбционные
свойства естественных и активированных алюмосиликатов
Приведены
значения общей пористости (Р, %), суммарного объема пор (, см3/г) и
удельной поверхности по азоту (S, м2/г), а также удельного объема
пор (Vуд., см3/г),
среднего радиуса пор (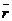 , Аº), статической
активности (Vs, моль/г) по
толуолу. Видно, что структурно-сорбционным характеристикам природные глины
приближаются к чешской отбельной глине. Так, значение общей пористости
колеблется в пределах 36-68: (для чешской глины пробы ЧЕШОГ она равна 59%), от
0,25 до 0,68 см3/г. Однако, величина удельной поверхности природных глин меньше
таковой у чешской глины. Такое различие имеется в адсорбционных свойствах. Так,
сорбционная активность по толуолу бентонитовых глин ниже, чем у чешской.
Сорбционная емкость опоки и опоковидной глины приближается к чешской.
, Аº), статической
активности (Vs, моль/г) по
толуолу. Видно, что структурно-сорбционным характеристикам природные глины
приближаются к чешской отбельной глине. Так, значение общей пористости
колеблется в пределах 36-68: (для чешской глины пробы ЧЕШОГ она равна 59%), от
0,25 до 0,68 см3/г. Однако, величина удельной поверхности природных глин меньше
таковой у чешской глины. Такое различие имеется в адсорбционных свойствах. Так,
сорбционная активность по толуолу бентонитовых глин ниже, чем у чешской.
Сорбционная емкость опоки и опоковидной глины приближается к чешской.
Комбинированные
способы активации не привели к существенному изменению структурно-сорбционных
параметров природных алюмосиликатов. В противоположность этому кислотная
активация природных алюмосиликатов привела к резкому увеличению удельной
поверхности, среднего радиуса пор и т.д. При этом наблюдается следующее:
а) с ростом концентрации кислоты
значение удельной поверхности увеличивается, а в случае бентонитовых глин оно
проходит через максимум; при активации их в оптимальных условиях;
б) относительное изменение значения
больше при сернокислотной активации, а также больше у бентонитовых глин, чем у
других. Это и понятно, т.к. серная кислота, как было показано в предыдущих
разделах, действует более активнее, чем другие минеральные кислоты, а
бентониты, по сравнению с опоковидной глиной, больше подвергаются разрушению.
На этом основании можно допустить наличие корреляции между приростом значений
структурно-сорбционных характеристик и количеством ушедших из образцов окислов
металлов при их кислотной активации.
Действительно было найдено, что чем
больше количество вышедших из глин кислотой 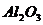 , тем больше значение удельной
поверхности активированных глин (рис. 9).
, тем больше значение удельной
поверхности активированных глин (рис. 9).
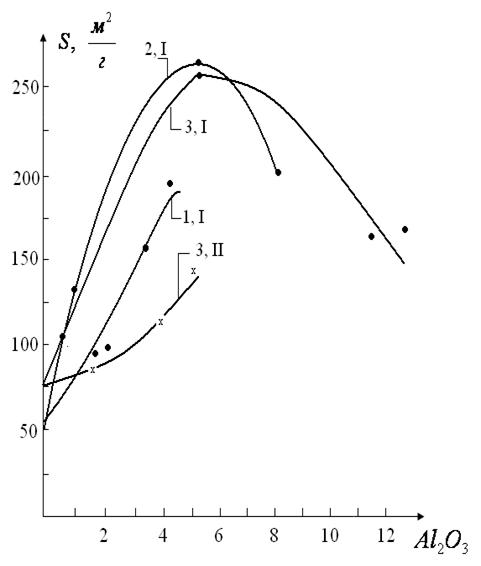
Рис.3.2.
Изминение удельной поверхности дарбазинского опока(1), келесского(2),
азкамарского серого (3), бентонитов от каличества вымытых АL2O3 при их
активации серной(1) и азотной (II)кислотами.
Аналогичная картина наблюдается и
между значениями S и 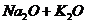 . Оказывается, чрезмерное разрушение
(увеличение вымытых окислов металлов) структур глинистых минералов
монтмориллонитовой группы, их которых состоят исходные глины, отрицательно
сказывается на увеличении удельной поверхности образцов или их других
адсорбционно-структурных характеристиках.
. Оказывается, чрезмерное разрушение
(увеличение вымытых окислов металлов) структур глинистых минералов
монтмориллонитовой группы, их которых состоят исходные глины, отрицательно
сказывается на увеличении удельной поверхности образцов или их других
адсорбционно-структурных характеристиках.
Если улучшение
адсорбционно-структурных свойств природных глин при их кислотной активации
обусловлено высвобождением адсорбционных центров глин в результате удаления из
них ранее адсорбированных веществ (соли, поглощенные катионы), то уменьшение
является, несомненно, результатом разрушения кристаллической структуры глин.
Другими словами, изменение структурно-сорбционных центров глин при их кислотной
активации объясняется изложенным в предыдущих параграфах механизмом кислотной
активации.
Наличие связи между величиной
структурно-сорбционных свойств и количеством ушедших веществ, которые
отравляюще действовали на адсорбционные центры глин, подтверждается еще и
работами по изучению влияния термической активации на свойства глин. Так, с
повышением температуры увеличивается количество ранее сорбированных веществ.
Благодаря этому увеличивается величина удельной поверхности образца; при
высокой температуре, когда выделяется кристаллизационная вода и происходит
спекание образца - S уменьшается.
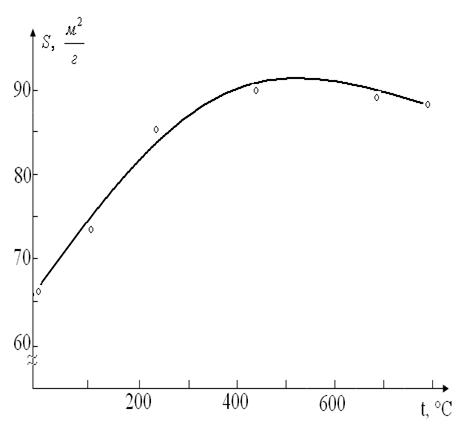
Рис.3.3. Изминение удельной
поверхности (S, м2 /г )
азкамарской от температуры ее сушки
Известно, что скорость установления
адсорбционного равновесия в системе поры сорбтива-сорбент устанавливается тем
быстрее, чем больше пористость образца [4, 11]. Изучение кинетики сорбции паров
толуола естественными и активированными глинами подтвердило это положение. Так,
основная масса сорбтива сорбируется за 18-30 дней, причем насыщение парами
толуола кислотой активированных образцов идет быстрее, чем у естественных и
других, полученных комбинированными способами активации. Однако, количество
предельно сорбированных паров толуола у образцов, приготовленных по
кислотно-щелочному, щелочно-кислотному способу активации, больше, чем у
кислотно-активированных проб и приближается к значениям соотношения
адсорбтив-сорбтив (статической активности) для силикагеля и окиси алюминия, что
позволяет судить о наличии адсорбции пара толуола не только в порах, но и в
катионных кислотных цетнрах алюмосиликата.
Итак, природные алюмосиликаты,
которыми мы располагали, являются природными минеральными сорбентами,
обладающими пористой структурой. При активации изменяются их
структурно-сорбционные свойства. Причем, их изменение больше у бентонитовых
глин, чем у опоковидных. Серная кислота больше изменяет пористость структуры,
чем другие кислоты.
ВЫВОДЫ
. С помощью химических, физических,
физико-химических методов исследования изучено влияние термической, кислотной
(серная, соляная, азотная кислоты), кислотно-щелочной, щелочно-кислотной и
известковой активации на изменения комплекса свойства природных
алюмосиликатов-бентонитовых Азкамар.
. По кислородному методу расчета
формул по зарядам выведены кристаллохимические формулы для исходных и
активированных бентонитовых глин и каолина. Установлено, что: а) изоморфное
замещение на в
тетраэдрической координации имеется в случае азкамарского белого, таганского
розового бентонитов, а в остальных - азкамарского серого, келесского,
огланлинского бентонитов - отсутствует; б) большая часть в глинах
представлена ионами в
октаэдрической координации; основная масса окисла железа находится в виде
свободных окислов, а 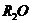 и RO в виде
поглощенных катионов; в) кислотная активация привела к возрастанию в тетраэдре
всех глин и заряда в целом тем больше, чем больше концентрация активатора и чем
больше время контакта образца с активатором, кислоты по влиянию на глины
располагаются в ряд
и RO в виде
поглощенных катионов; в) кислотная активация привела к возрастанию в тетраэдре
всех глин и заряда в целом тем больше, чем больше концентрация активатора и чем
больше время контакта образца с активатором, кислоты по влиянию на глины
располагаются в ряд 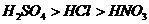 ; г)
щелочно-кислотной способ активации алюмосиликатов способствует повышению
содержания обменных катионов, незначительному увеличению заряда решетки.
; г)
щелочно-кислотной способ активации алюмосиликатов способствует повышению
содержания обменных катионов, незначительному увеличению заряда решетки.
. Термо- и рентгенографическими
методами установлено, что а) с ростом концентрации кислоты и времени активации
на кривых ДТА уменьшается быстрее интегральная площадь III
эндоэффекта, а затем II эндоэффекта. увеличение
площади I эндоэффекта
и смещение ее в сторону более низких температур наблюдается с использованием
относительно разбавленных растворов и при комбинированных способах
(кислотно-щелочной и щелочно-кислотной) обработки исходных глин; б) с
повышением концентрации кислоты (до 10%) интенсивность линий монтмориллонита
увеличивается, а затем уменьшается, но полностью не исчезает и у образцов,
приготовленных применением концентрированных растворов (30% 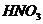 , 35%
, 35% 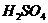 , 20% ); в) на
рентгенограммах образцов щелочно-кислотной активации каолина обнаружены линии,
характерные для цеолита-содалита.
, 20% ); в) на
рентгенограммах образцов щелочно-кислотной активации каолина обнаружены линии,
характерные для цеолита-содалита.
. Комбинированный способ активации в
противоположность кислотной активации не привела к заметному изменению
структурно-сорбционных характеристик (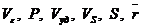 ). Их изменение больше у
бентонитовых глин, чем у опоковидных. Серная кислота больше изменяет пористость
исходных образцов, чем другие кислоты. Установлено, что с ростом концентрации
активатора и контакта обработки кислотой образцов увеличивается, а в случае
бентонитовых глин - проходит через максимум.
). Их изменение больше у
бентонитовых глин, чем у опоковидных. Серная кислота больше изменяет пористость
исходных образцов, чем другие кислоты. Установлено, что с ростом концентрации
активатора и контакта обработки кислотой образцов увеличивается, а в случае
бентонитовых глин - проходит через максимум.
. Показано, что адсорбционное
равновесие адсорбент-пары толуола устанавливается тем быстрее, чем больше
пористость адсорбента.  по толуолу
у образцов, приготовленных комбинированным способом активации больше, чем у
кислотно-активированных и равна таковой у силикагеля и окиси алюминия.
по толуолу
у образцов, приготовленных комбинированным способом активации больше, чем у
кислотно-активированных и равна таковой у силикагеля и окиси алюминия.
. Величины протонной и апротонной
кислотности, как следовало ожидать, зависят от вида исходного сырья и способа
их обработки. Природные глины и их продукты кислотной активации обладают
большей кислотностью, по сравнению с окислами алюминия и кремния, но меньшей по
отношению к синтетическому алюмосиликату - катализатору. Установлено, что
величина кислотных центров у глин тем больше, чем больше отрицательный заряд
октаэдрической координации монтмориллонита. Причем, эти величины больше, чем
жестче условия кислотной активации глины, что является дополнением к правилам
В.Трамбуза, утверждающего повышение апротонной кислотности от температуры
активации алюмосиликата.
. Исследование каталитическая
активность природных и активированных алюмосиликатов в реакции крекинга кумола
и разложения бертолетовой соли. Показано, что природные алюмосиликаты, как и
следовали ожидать, не обладают достаточным каталитическим действием. При их
химической обработке получаются катализаторы, по активности крекингу кумола не
уступающие промышленным катализаторам и окими марганца-катализатора разложения
бертолетовой соли. Был наблюден большой выход (65-70%) индивидуальных
углеводородов в катализате при крекинге кумола с азкамарским и огленлинским
бентонитами 25%-ной сернокислотной и ангренским каолином щелочно-кислотной
активации. Установлено, что: а) чем больше заряд структуры монтмориллонита, тем
больше он обладает каталитической активностью; б) образец, обладающий большей
протонной кислотностью (или числом кислотных центров) является лучшим
катализатором крекинга. Предположено, что крекинг кумола на кислот
алюмосиликате происходит на электронноненасыщенном алюмокислородном полиэдре 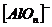 с
образованием иона карбония за счет перехода атома водорода, активированного под
действием двойной связи бензольного кольца с третичного атома углерода в .
с
образованием иона карбония за счет перехода атома водорода, активированного под
действием двойной связи бензольного кольца с третичного атома углерода в .
Список
использованной литературы
1.
И.А.Каримов
«Мировой финансово-экономический кризис, пути и меры по его преодолению в
условиях Узбекистана», Узбекистан, Ташкент. 2009 г.
.
Запрометов Б.Г., Колмаков Б.И. Бюллетень САГУ, вып. 16,23 (1934).
.
Грим Р. Минералогия и практическое использование глин, М., Изд-во «Мир» (1967).
.
Арипов Э.А. Природные минеральные сорбенты, их активирование и модифицирование,
Ташкент, Изд-во «Фан»УзССР (1970).
.
Закиров М.З. Полезные ископаемые эоценовых отложений. Приташкентского района,
Ташкент, Изд-во «Фан»УзССР (1966).
.
Ездаков В.И. К вопросу применения глин различной минералогической прирлды в
качестве катализаторов бензилировании. Тр.Уз.Гос.ун-та.новая серия, 85 (1958).
.
Ездаков В.И. Физико - химические исследования природных сорбентов. Автореферат
докт.дисс.Киев (1968).
.
Султанов А.С., Бобохаджаева С.А., ДАН УзССР 3,28 (1967).
.
Арипов Э.А. и др. . ДАН УзССР 6,26 (1968).
.
ЗульфугаровЗ.Г. Исследование физико - химических свойств и отбеливающей
способности глин месторождений Азербейджанской ССР и гумбрина, Баку, изд-во Ан
Азерб.ССР (1957).
.
Грим Р. Метрология глин.М.. Изд-во «Ил» (1955).
.
Овчаренко Ф.Д. Гидрофильность глин и глинистых минералов, Киев, Изд-во АНССР
(1961).
.
Мерабишвилли М.С., Бентанитовые глины, М., Госгеолтехиздат (1968).
.
Овчаренко Ф.Д., и др. Коллоидная химия палигорскита, Киев, Изд-во Ан УССР
(1963) .
.
Поверхностные явления на алюмосиликатах. Сб. статей, Тбилиси, «Мецниерба»
(1965).
.
Круглицкий Н.Н. Физико - химические основы регулирования свойств дисперсий
глинистых материалов, Киев, Изд-во «Науководумка» (1968).
.
Гафуров Р.Г., Арипов Э.А. « Регенрация трансформаторного масла азкамарскими
бентонитами » В.сб. «Вопросы естественных наук» . Т-1, часть-1. Тр. ТашГУ и
БухГПИ. Ташкент- Бухара ( 1968).
.
Курбанбаева Т., Гафуров Р.Г., Саломов Х.Т.«Адсорбционное осветление хлопкового
масла с помощью глины Кермине». Там же, стр. 88.
.
Арипов Э.А., Гафуров Р. Г. «Кислотности и адсорбционно-отбеливающие способности
местных природных минеральных сорбентов». Материлы 15 научно- теоретической
конференции Бух ГПИ им. С . Орджоникидзе, Бухара (1970) .
.
Курбанбаева Т., Арипов Э.А., Гафуров Р.Г.Адсорбционное осветление хлопкового
масла природными сорбенлтами». Известия ВУЗ СССР «Пищевая технология» 3, 175
(1971).
.
Арипов Э.А., Курбанбаева Т., Гафуров РюГ «Адсорбенты для темных нафтеновых
кислот (асидола и кубовых остатков ). Известия ВУЗ СССР «НЕФТЬ и ГАЗ», 10,51
(1972).
.
Гафуров Р.Г., Арипов Э.А «О возможности удленения срока службы
трансформаторного масла природными минеральными сорбентами». ДАН УзСССР 5,30
(1972).
24. Г. И. Те с л е к к о. Сб. «Бентониты
Узбекистана». Ташкент. Изд-во
АН УзССР, 1963, стр. 26.
. А. Г. С е и д о в. Сб. «Вопросы минералогии
осадочных образова
ний», кн. 3-я и 4-я. Изд-во Львовского ун-та, 1956, стр. 613.
. Е. Я. Штейгельберг, Г. И. Ту р к е в и ч, М.
А. К нр д о. Сб.
«Исследование и использование глин». Изд-во Львовского ун-та, 1958,
стр.430.
. М. П. Г а б и н е т. Сб. «Исследование и
использование глин». Изд-
бо Львовского ун-та, 1958, стр. 491.
. Е. Ф. По л у эк то в а. Бентонитовые глины
Украины, сб. 4. Киев,
Изд-во АН УССР, 1960, стр. 31.
. М. Ф. В и к у л о в а. Сб. «Физические методы
исследования осадоч
ных пород и минералов». М., Изд-во АН СССР, 1962, стр. 14,
. Е. К. Лазаренко. Сб. «Вопросы минералогии
осадочных обра
зований», кн. 3-я и 4-я. Изд-во Львовского ун-та, 1956, стр. 345.
31. И.. Н. А н т и п о в-К аратаев, Б. К-
Бруновский. Коллоидный журнал, 2, 351, 1936.
. Н. И. Н. Антипов-Каратаев, Б. К. Бруновский,
А. А. Ро-д е. Сб. «Почвенный поглощающий комплекс и вопросы земледелия». Изд-во
Всесоюзной академии сельскохозяйственных наук, М.-Л., 1937.
33. Б. Б. Звягин. Электронография и структурная
кристаллография
глинистых минералов. М., Изд-во ЛГУ, 1964.
. С. Лазарев, В. И. Р а д у ш е в, А. А. Нырков.
Сб. «Вопро
сы минералогии осадочных образований», кн. 3-я и 4-я. Изд-во Львовского
ун-та, 1956, стр. 337.
35. Д. М. К-Мак-Эвен. Монтмориллонитовые
минералы. В кн.: «Рентгеновские методы определения н кристаллическое строение
минералов глин».ИЛ, 1955.
36. А. Д р и ц. Сб. «Исследование и
использование глин». Изд-во
Львовского ун-та, 1958, стр. 686.
37. Е. Г. Куковский'и др. Бентонитовые глины
Украины, сб. 4.
Киев, Йзд-во АН УССР, 1960, сто. 15 и 41.
38. Ю. С. Л е б е д е в, Е. Я. М а р ч е и к о,
Р. Г 3 и з о в а. Бентонито
вые глины Украины, сб. 4. Киев, Изд-ао АН УССР, 1960, стр. 24.
. Ю.С. Лебедев. Рентгенография минерального
сырья, сб. I. М.,
Геолтехиздат, 1962, стр. 39.
. Н. И, Горбунов. «Почвоведение», № 10, 1952.
. В. П. Казанцев. Записки Всероссийского
минералогического
общества, сер. 2, 63, 464. 1934.
. Методическое руководство по петрографо-мипера
логическому изучению глин. Под руководством М. Ф. Викуловой. Труды ВСЕГЕИ.М.,
Госгеолтехиздат, 1957.
43. Ф. Д. О в ч а р е п к о. Гидрофильность глин
и глинистых .минералов. Киев, Изд-во АН УССР, 196!, стр. 130.
44. Н. И. Г о р б у н о в, И. Г, Ц ю р у п з, Е.
А, Ш у р ы г и н а. Рентгсногра/ймы, термограммы и кривые обезвоживания
минералов, встречающихся в почвах и глинах. М., Изд-во АН СССР, 1952, стр. 61.
45. Петров. Сб. «Исследование и использование
глин». Изд-во
Львовского ун-та им. Ив. Франко, 1958, стр. 84.
46. Б. В. Дерягин, Н. И. Захраева, М. В. Талаев,
В. В. Ф н-липповский. Определение удельной поверхности порошкообразных тел по сопротивлению
фильтрации разреженного воздуха. Изд-во АН СССР, 1957.
47. М. М. Дубинин, Физико-химические основы
сорбционной техники. Изд. 2-е, ОНТИ, 1935.
. Дж. В. Мак-Бэн. Сорбция газов и паров твердыми
телами.ОНТИ, 1934.
49. С. Брунауэр. Адсорбция газов и паров. ИЛ.
1948.
50. В. Киселев. Структура адсорбентов и энергия
адсорбции.
Автореферат докторской диссертации. М., 1950.
51. Б. А. Л я л к и н д, С. В. К а п а ц и и с к
и н, Г. А. К у с т о в а, А. А.М а с л о в а. Сб. «Получение, структура и
свойства адсорбентов». Л , Изд-во АН СССР, 1959, стр. 15
52.
С. Комаров Н. Ф. Ермоленко. Сб. Научных работ Института физико- органической
химии АН БССР, №7, 57, 1959.
.
М. М. Дубинин. Физико - химчксеские основқ собрционной техники. Изд. 2 -
е. ОНТИ, 1935.
.
С.Брунауер. Адсорбция газов и паров. ИЛ, 1948.
.
В.Т.Быков. Сорбционные свойства и структура природных сорбентов дальневосточных
месторождений. Автореферат докторской диссертации М., 1951.
.
Ф.А.Слисаренко, ОЕ.М.Тимофеева. Сб. «Природные минеральные адсорбенты ». Киев,
Изд - во АН УССР, 1960, срт.252
.
Г.Д. Берншнейн. Фильтрация смазочных масел в автотракторных дизелях. Изд -во «
Советская наука », 1940.
.
И.В.Брай. Регенерация трансформаторных.масел. «Химия », 1966.
.
В.С.Иванов. Руководящие указания по очистке и регенерации изоляционных,
турбинных и смазочных масел. ГЭИ, 1944.
.
С.М.Раховская, Н.Н.Грязев .Сб. «Природные минеральные сорбенты ».Киев, Изд -во
АН УССР, 1960, стр. 262.
.
О.К.Давтян, С.И.Бурштейн. Бентонитовые глины Украины, сб. 3.Киев, Изд - во АН
УССР, 1959, стр 149.
.
В.Е.Ефимов. Природные сорбенты Дальнего Востока. Труды Дальневосточного филиала
АН СССР, сер. Хим.., вып. 4, 116, 1960.
.
С.Г.Телетов.Сб. « Природ ные минеральные сорбенты ». Киев, Изд -во АН УССР,
1960, стр. 216.
.
С.М.Раховская. Исследование сорбционных свойств при родных сорбентов Поволжья в
условиях очистки энергетических масел. Автореферат кандидатской диссертации,
1958.
.
С.М.Фридман. Сб. « Природные сорбенты ». М.., «Наука », 1967, стр. 207.
.
В.П.Астафьев. Осушение доменного дутья и других газов минеральными гелями.
ОНТИ, НКТП, 1936.
.
И.Е.Ходанович. Оператор по очистке и сушке горючих газов. М.-Л.,Гостоптехиздат,
1948.
.
А.А.Коуль, Ф.С.Ризенфельд. Очистка газа. М.., Гостоптехиздат, 1962, стр. 260.
.
К.С.Зарембо, Г.И. Нусинов. Очистка. Осушка и одоризация природных газов.
Гостоптехиздат, 1947.
.
Р.Г.Гаджиева, В. Ф. Негреев. Сб .«Бентонитовые глины (гильаби) Азербайджана ».
Баку, Изд - во АН АзССР , 1951, стр, стр 81.
.
Д.И.Ожерельев, В.С.Масляев, Е.А.Усикова. Сб. « Природные минеральные сорбенты».
Киев, Изд - во АН УССР, 1960. стр .238
.
Фозилов С.Ф., Гафуров Р., Вахидовв
Н. Нефт
махсулотларини тозалашда махалий сорбентлардан фойдаланиш. «Фан ва ишлаб
чикариш интеграциясини жаддаллаштириш муаммолари» Республика илмий-амалий
анжуман материаллари. Бухоро 2007 78 бет.
.Комаров
В.С. Адсорбенты и их свойства. Минск, «Наука и техника», 1977,248 с.
.Грим
Р.Э. Минералогия и практическое использование глин. М., Изд-во «Мир», 1967.
.Ребиндер
И.А. Поверхностные явления в дисперсных системах. Физико-химическая механика.
Избр. Труды. М., Наука, 1979, 382с.
.П.П.
Дмитриев, Н.Х. Алимухаммедов, Т.Т. Адылова, Б.Н. Хамидов, Н.Д. Рябова. Об
адсорбционных свойствах некоторых природных и синтетических алюмосиликатов.// В
сб. Адсорбенты для анализа и разделения нефтепродуктов. Ташкент. Изд. «Фан»
Узб.ССР. 1970. С.50-55.
.
П.П. Дмитриев, Н.Х. Алимухаммедов. В сб. Адсорбционные свойства природных и
синтетических сорбентов. Ташкент. Изд. «Фан» Узб.ССР. 1969.
.Опоковидная
глина Кермине. Ташкент, ФАН Узбекской ССР, 1970, 96с.
.Опоковидные
глины Узбекистана и Казахстана. Труды Ташкентского института инженеров
железнодорожного транспорта. Ташкент, 1970, С.78.
.Физико-химические
свойства минеральных сорбентов. Ташкент ФАН, 1973, С.98.
.Нарметова
Г.Р., Хамидов Б.Н., Каландаров Д.А., Муродов М.Н. Абдурахманова Ф.
Адсорбционная способность бентонита по отношению к нефтяным компонентам./ «IV
конгресс Нефтегазопромышленников России» Материалы конференции.
Нефтепереработка и нефтехимия., Уфа. 2003 г. -С. 132.
.Мирзаев
А.У., Чиникулов Х. Новое месторождение бентонитовых глин Навбахорского
месторождения. // Геология и минеральные ресурсы, 1999. № 5, С. 23-30.
.Муродов
М.Н., Нарметова Г.Р. Адсорбенты на основе местных минеральных сорбентов для
очистки нефтяных масел. // Узб.хим.журн, 2004. № 4, С. 38-40.
.Рябова
Н.Д., Адылова Т.Т. Криоскопический метод определения селективности и
сорбционной емкости адсорбентов типа молекулярных сит// Узб.хим.ж., 1961 г.,
№5, с.27-31.
.Нефтепродукты:
методы испытаний. М., Стандартгиз. 1961, -С.980.
.Нефтепродукты:методы
испытаний.М.,Стандартгиз.1966(1 и 2т) -С.396.
.Нефтепродукты:методы
испытаний. М.,Стандартгиз.1961(1 и 2т) -С.416
.Б.М.Рыбак.
Анализ нефти и нефтепродуктов. М., Гостоптехиздат, 1962, -С.888.
.Исагулянц
В.И., Егорова Г.М. Химия нефти. М., Химия, 1965, -С.517.
.Нарметова
Г.Р., Аликулова М., Каландаров Д., Муродов М. Исследование нефтей и продуктов
их переработки хроматогарфическими методами. «Ўзбекистон
минерал
хом-ашёларини кимёвий қайта
ишлашнинг долзарб муаммолари» мавзусидаги Республика илмий анжумани маъруза
тезисларининг тўплами.
Ташкент, 2003, 8-9 бет.
.Рябова
Н.Д. Адсорбенты для светлых нефтепродуктов. Ташкент, ФАН, 1975, -С.141.
.Абдукаримов
Р.С., Убайдуллаев Б.Х. Адсорбенты для очистки авиакеросина. Матер. конф.
«Современное состояние процессов переработки нефти». Уфа, 2004, -С.140.
.
Муродов М.Н. Технология адсорбционной очистки трансформаторных масел
Навбахорским бентонитом. Автореф. дисс.
канд. техн. наук,
Ташкент, 2005, -С.26.
.Голдштейн
Дж, Нюбери Д. И и др. Растровая электронная микроскопия и рентгеновский
микроанализ. М., Мир, 1984, -С.162-169.
.Нарметова
Г.Р., Каландаров Д.А., Муродов М.Н. Рентгенографическое и микроскопическое
исследование исходного и модифицированного бентонита. Матер. конф.
«Современное состояние процессов перера-ботки нефти., Уфа, 2004, -С.271-272.
.Хамидов
Б.Н.,Нарметова Г.Р., Исмаилов И, Мурадов М., Каландаров Ж. Разработка
технологии получения пластичных смазок на основе местного сырья и идентификация
их состава./ «Ўзбекистонда кимё таълими, фани ва технологияси» Республика
илмий-амалий конференцияси. Тезислар тўплами, Тошкент. 2002 й. 237-238 б.