Тепловые и массообменные процессы
Министерство
образования и науки РФ
Федеральное
агентство по образованию
Иркутский
государственный технический университет
Тепловые и
массообменные процессы
Методические
указания к лабораторным работам по курсу
«Процессы
и аппараты химической технологии»
Иркутск 2007
Тепловые и массообменные
процессы. Методические указания к лабораторным работам по курсу «Процессы и
аппараты химической технологии» для студентов очной, заочной и вечерней форм
обучения специальностей:
240401.65 - Химическая
технология органических веществ
240403.65 - Химическая
технология природных энергоносителей и углеродных материалов
240406.65 -Технология
химической переработки древесины.
Составители: Губанов Н.Д.,
Баяндин В.В. – Иркутск, 2007. – 33 с.
Иркутский государственный
технический университет
664074, Иркутск, ул.
Лермонтова, 83
ИСПЫТАНИЕ ДВУХКОРПУСНОЙ ВЫПАРНОЙ
УСТАНОВКИ
Цель работы: изучение
двухкорпусной выпарной установки непрерывного действия, определение количества
выпариваемой воды, коэффициентов теплопередачи по корпусам и удельного расхода
греющего пара.
Приборы и принадлежности:
двухкорпусная выпарная установка, раствор глицерина, денсиметры.
Введение
Выпаривание – это процесс
концентрирования растворов нелетучих веществ, заключающийся в удалении
растворителя путем испарения его при кипении. Движущей силой выпаривания является
разность температур между горячим теплоносителем и кипящим раствором,
называемая полезной разностью температур.
Выпаривание является
одним из наиболее энергоемких процессов химической и пищевой технологии, как по
абсолютному количеству расходуемой энергии, так и по ее удельному расходу на
1кг перерабатываемого раствора. В промышленности в большинстве случаев
выпаривают водные растворы нелетучих веществ.
Выпаривание ведут как под
атмосферным давлением, так и под повышенным или пониженным давлениях. При
выпаривании под атмосферным давлением образующийся вторичный пар удаляется в
атмосферу. При выпаривании под пониженным давлением вакуум в аппарате создают
за счет конденсации вторичного пара в барометрическом конденсаторе. Выпаривание
под вакуумом позволяет снизить температуру кипения раствора, а значит увеличить
разность температур между греющим паром и кипящим раствором, то есть повысить
движущую силу процесса. Это дает возможность уменьшить поверхность теплообмена.
Выпаривание под повышенным давлением позволяет образующийся вторичный пар
использовать в качестве греющего или для других технологических нужд.
В промышленности
применяют как однокорпусные, как и многокорпусные выпарные установки.
Многокорпусные выпарные установки состоят из нескольких соединенных друг с
другом выпарных аппаратов (корпусов). Различают прямоточные и противоточные
многокорпусные выпарные установки.
В прямоточных выпарных
установках греющий пар и выпариваемый раствор движутся прямотоком от корпуса к
корпусу, а в противоточных – навстречу друг другу. Подробнее о многокорпусных выпарных
установках в [1, 2]. В многокорпусных выпарных установках первичным паром
обогревают только первый корпус, а вторичный пар, образующийся в каждом предыдущем
корпусе, используют для обогрева последующего корпуса. Таким образом, в
многокорпусных выпарных установках осуществляется многократное использование
тепла, отдаваемого первичным греющим паром. Это позволяет значительно снизить
расход первичного греющего пара. В качестве горячего теплоносителя в первом
корпусе в основном используют насыщенный водяной пар.
1
Требования безопасности
Перед пуском установки
по наружному осмотру проверить исправность аппаратов, трубопроводов, арматуры,
приборов КИПиА, заземления, защитного отключения, тепловой изоляции. Пуск
установки проводить в присутствии учебного мастера и при его непосредственном
руководстве. Работая на площадках с высокой отметкой и вблизи вращающихся
частей проявлять осторожность и аккуратность.
К выполнению лабораторной
работы студенты допускаются только после прохождения инструктажа по
безопасности труда и пожарной безопасности в лаборатории и на рабочем месте.
2 Описание
установки и порядок выполнения работы
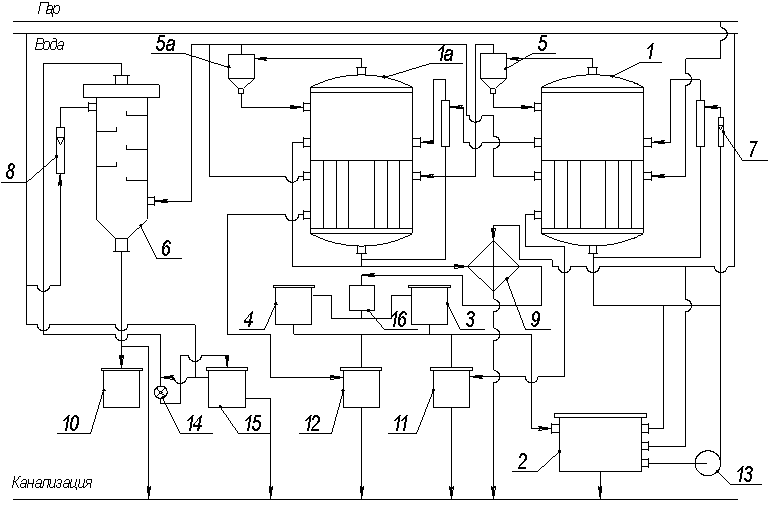
Выпарная установка (рисунок
1) состоит из двух аппаратов 1 и 1а с внутренней греющей камерой и центральной
циркуляционной трубой. Выпарной аппарат состоит из двух основных частей:
греющей камеры, в которой происходит кипение раствора, и сепаратора, в котором
вторичный пар отделяется от раствора. Высота выпарного 1500мм, диаметр 408мм,
поверхность теплообмена 1,5м2 .
Водный раствор глицерина
из бака 2, снабженного указателем уровня, центробежным насосом 13 подается в
выпарной аппарат 1. Расход раствора измеряется ротаметром 7. Обогрев корпуса 1
осуществляется насыщенным водяным паром, поступающим от электропарового котла.
Давление пара в трубопроводе измеряется манометром и регулируется клапанами.
Вторичный пар из корпуса 1 проходит брызгоуловитель 5 и поступает в греющую
камеру корпуса 1а.
Раствор, упаренный до
некоторой концентрации в корпусе 1, под действием разности давлений поступает
в корпус 1а, где упаривается до заданной концентрации. Концентрированный
раствор из корпуса 1а, пройдя через холодильник 9 и фонарь 16, поступает в
сборники упаренного раствора 3 или 4, работающие попеременно. По мере их
заполнения упаренный раствор переливается в бак 2.
|
|
|
I, Iа – выпарные
аппараты; 2 – бак-хранилище; 3, 4 – сборники упаренного раствора; 5, 5а –
брызгоуловители; 6 – барометрический конденсатор; 7, 8 – ротаметры; 9 – холодильник;
10 – барометрический ящик; 11, 12 – сборнки конденсата; 13 – центробежный
насос; 14 – водокольцевой вакуум-насос; 15 – водоотделитель; 16 – смотровой
фонарь,
Рисунок 1 – Схема
двухкорпусной выпарной установки
|
Вторичный пар из корпуса
1а проходит через брызгоуловитель 5а и поступает в барометрический конденсатор
6, орошаемый водой. Расход воды измеряется ротаметром 8. Смесь воды и
конденсата удаляется из конденсатора самотеком через барометрическую трубу в барометрический
ящик 10, а затем в канализацию. Воздух из барометрического конденсатора
отсасывается вакуум-насосом 14. Концентрацию разбавленного и упаренных растворов
в корпусах 1 и 1а определяют ареометром. Давление в аппаратах измеряются
манометрами, вынесенными на щит КИП. Измерение температур конденсата и
поверхности аппаратов производится термометрами сопротивления, работающими в
комплекте с лагометрами. Отбор проб исходного и упаренного растворов производится
через пробоотборники.
Перед пуском установки необходимо:
Закрыть все воздушные краны и вентили на линии вакуума, паровой линии и линии
раствора. Проверить наличие исходного раствора в баке 2. Проверить наличие воды
в водопроводе.
Пуск установки
производить следующим образом: Открыть вентили на отборных устройствах
манометров и вакуумметров. Подать воду в холодильник упаренного раствора. Затем
заполнить аппарат 1 исходным раствором из бака 2 с помощью наноса 13 через
ротаметр 7 до верхней красной черты и подать пар на установку. Продуть
межтрубное пространство греющей камеры первого корпуса по обводной линии. После
продувки направить конденсат через конденсационные горшки и нагреть раствор
до кипения. После того, как раствор в корпусе 1 начнет кипеть, следует создать
вакуум во втором аппарате. Под действием разности давлений раствор из корпуса 1
начнет самотеком переливаться в корпус 1а. Перелив раствора производить до тех
пор, пока уровень в корпусе 1 не достигнет нижней красной черты. После этого
первый аппарат заполнить до верхней красной черты и процесс повторить. Когда во
втором аппарате уровень раствора достигнет красной черты, заполнение системы
прекращают и начинают процесс выпаривания. Далее необходимо продуть греющую
камеру второго аппарата, выпустив часть пара по обводной линии. Затем конденсат
направить через конденсационный горшок. Установить по ротаметру указанный
преподавателем расход исходного раствора в первый корпус. Начать подачу
раствора из корпуса 1 в корпус 1а. для чего открыть регулирующий вентиль на
линии раствора между корпусами с таким расчетом, чтобы уровень в первом
аппарате не опускался ниже красной черты. Одновременно с началом подачи
раствора подать воду в барометрический конденсатор. Расход воды установить по
ротаметру 8. Подключить к корпусу 1а сборники упаренного раствора. Пустив,
таким образом, всю установку, обязательно при непрерывной подаче раствора в
аппараты, дать ей поработать 40-45 минут. После этого приступить к замерам. Измерения
производятся через каждые 10-15 минут 3-4 раза. Результаты наблюдений сводятся
в таблицу 1.
Таблица 1
Измеренные и рассчитанные
параметры
|
Наименование величины
|
Значение и момент измерения
|
Среднее значение
|
|
Расход исходной смеси, кг/с
|
|
|
|
Расход воды на барометрический
конденсатор, кг/с
|
|
|
|
Давление греющего пара, Па
|
|
|
|
Температура греющего пара, оС
|
|
|
|
Температура исходного раствора, оС
|
|
|
|
Давление в корпусе 1, Па
|
|
|
|
Температура вторичного пара в
первом корпусе, оС
|
|
|
|
Температура кипения раствора в
корпусе 1, оС
|
|
|
|
Давление во втором корпусе, Па
|
|
|
|
Температура вторичного пара в корпусе
1а, оС
|
|
|
|
Температура кипения раствора во
втором корпусе, оС
|
|
|
|
Концентрация исходного раствора, %
масс.
|
|
|
|
Концентрация раствора после
первого корпуса, % масс.
|
|
|
|
Концентрация раствора после второго
корпуса, % масс.
|
|
|
|
Температура поверхности первого
аппарата, оС
|
|
|
|
Температура поверхности второго
аппарата, оС
|
|
|
|
Температура холодной воды, 0С
|
|
|
|
Температура смеси в барометрической
трубе, 0С
|
|
|
|
Температура воздуха, 0С
|
|
|
3
Обработка опытных данных
3.1
Материальный баланс установки
Количество воды,
выпариваемой в первом корпусе
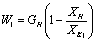 ,
,
где 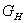 - количество исходного раствора, кг/с;
- количество исходного раствора, кг/с;
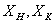 1 - концентрация исходного и
упаренного раствора после корпуса 1, % масс.
1 - концентрация исходного и
упаренного раствора после корпуса 1, % масс.
Количество воды,
выпариваемое во втором корпусе
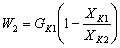 ,
,
где 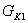 - количество раствора, поступающего из
первого во второй корпус, кг/с;
- количество раствора, поступающего из
первого во второй корпус, кг/с;
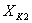 - конечная концентрация раствора во втором
корпусе, % масс.
- конечная концентрация раствора во втором
корпусе, % масс.
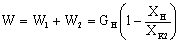
3.2 Тепловой
баланс установки по корпусам
Расход тепла на
выпаривание в первом корпусе
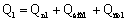
Количество тепла,
затраченное на нагрев исходного раствора от начальной температуры tH до температуры кипения tK1 определяется по уравнению
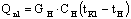 ,
,
где CH – удельная теплоемкость исходного
раствора, Дж/кг К.
Количество тепла, которое
необходимо затратить на выпаривание воды в первом корпусе
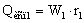 ,
,
где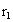 – удельная теплота парообразования при
давлении в корпусе 1, Дж/кг.
– удельная теплота парообразования при
давлении в корпусе 1, Дж/кг.
Расход тепла на
выпаривание во втором корпусе
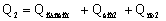
Количество теплоты,
выделяющееся при охлаждении выпариваемого раствора от температуры кипения в
первом корпусе tK1 до температуры кипения tK2 во втором корпусе
определяется по формуле
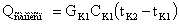 ,
,
где СК1 -
удельная теплоемкость раствора, поступающего из первого во второй корпус, Дж/кг
К.
Количество тепла,
затраченное на испарение воды во втором корпусе
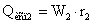 ,
,
где 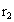 - удельная теплота парообразования при
давлении во втором корпусе, Дж/кг.
- удельная теплота парообразования при
давлении во втором корпусе, Дж/кг.
Потери тепла в окружающую
среду первым и вторым корпусом установки можно определить по уравнениям
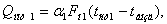
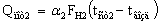 ,
,
где a1, a2 –
суммарные коэффициенты теплоотдачи от стенки аппарата к воздуху, Вт/м2 К;
FH1, FH2
– наружная поверхность выпарных аппаратов (FH1 = FH2=1.92 м2);
tвозд., tст1, tст2 – температура
воздуха и стенок аппаратов, оС.
Суммарные коэффициенты теплоотдачи
конвекцией и излучением от стенки аппарата к воздуху вычисляются по формуле
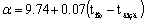
3.2
Коэффициенты
теплопередачи по корпусам
Коэффициенты теплопередачи
определяют из основного уравнения теплопередачи для выпаривания
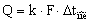 ,
,
где Q – расход тепла на выпаривание в
корпусе, Вт;
k – коэффициент теплопередачи, Вт/м2
К;
F – поверхность теплообмена, м2;
Δ tпол – полезная разность температур (движущая
сила процесса).
Коэффициенты теплопередачи
вычисляют по формулам
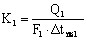 ,
, 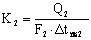 .
.
Полезные разности
температур по корпусам равны
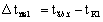 ,
, 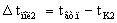 ,
,
где tгр.п.– температура греющего пара, оС;
tвт.п.– температура вторичного пара первого
корпуса, оС.
3.3
Расход
греющего пара
Расход первичного пара находят по уравнению
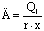 ,
,
где r – удельная теплота конденсации
греющего пара, Дж/кг;
x – паросодержание (степень сухости)
греющего пара (x=0,95).
Расход греющего пара на 1 кг выпаренной воды определяют по формуле:
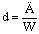 .
.
ИЗУЧЕНИЕ ПРОЦЕССА РЕКТИФИКАЦИИ
Цель работы: изучение
процесса ректификации в тарельчатой колонне непрерывного действия, определение
числа теоретических тарелок и среднего КПД тарелок.
Приборы и
принадлежности: установка ректификации, хроматограф ЛХМ-8 МД, бутылочки с
пробками, смесь этиловый спирт – вода концентрацией 5 – 40 % массовых.
Введение
Ректификация –
процесс разделения жидких гомогенных смесей путем взаимного обмена
компонентами между жидкостью и паром, полученным испарением разделяемой смеси.
Этот процесс основан на различной летучести компонентов, составляющих смесь,
т.е. на различии в температурах кипения компонентов при одинаковом давлении.
Процесс ректификации
осуществляют в колоннах, представляющих собой вертикальные цилиндрические
аппараты, с контактными устройствами. Наибольшее распространение в
промышленности получили ректификационные колонны, в которых в качестве контактных
устройств используются колпачковые, ситчатые и провальные тарелки [1, 2]. В
ректификационной колонне навстречу друг другу проходят неравновесные по составу
потоки пара и жидкости. Пар в колонне идет снизу вверх, а жидкость стекает
сверху вниз. На каждой тарелке колонны пар и жидкость вступают в контакт. Поскольку
между жидкостью и паром отсутствует равновесие, то на тарелке между ними
происходит обмен компонентами ( массообмен), в результате которого пар
обогащается более летучим (низкокипящим) компонентом, а жидкость – менее
летучим (высококипящим). В ректификационных колоннах массообмен между жидкостью
и паром осуществляется через поверхность контакта паровой и жидкой фаз.
Поверхность контакта фаз на тарелке образуется пузырьками и струями пара при
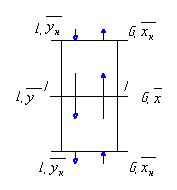
прохождении (барботаже) его через слой жидкости. На рисунке 1 показана схема
ректификационной колонны с провальными ситчатыми тарелками.
Пар, поступающий под
нижнюю тарелку ректификационной колонны из кипятильника 1, имеет низкую
концентрацию низкокипящего компонента. С верхней тарелки колонны пар с высокой
концентрацией низкокипящего компонента уходит в дефлегматор 4, в котором он
полностью конденсируется. Часть образовавшегося конденсата поступает на верхнюю
тарелку для орошения колонны. Эта жидкость называется флегмой Ф. Остальную жидкость
из дефлегматора отбирают в качестве верхнего продукта разделения – дистиллята
Р. Деление конденсата на флегму и дистиллят осуществляется с помощью флегмоделителя
5. Часть жидкости, поступающей в кипятильник с нижней тарелки, отбирают в
качестве кубового остатка W.
Исходную смесь F непрерывно подают на одну из
тарелок в средней части, которая делит колонну на верхнюю укрепляющую 3 и
нижнюю исчерпывающую 2 части. Таким образом, в ректификационной колонне осуществляется
непрерывный процесс разделения исходной смеси F на дистиллят Р с высоким содержанием низкокипящего компонента
и кубовой остаток W с низким его
содержанием. Концентрации получаемых продуктов разделения зависят от числа
тарелок в колонне и от режима ее работы.
1 Требования
безопасности
Перед пуском установки
убедиться в исправности ректификационной колонны, куба-испарителя,
дефлегматора, трубопроводов, арматуры, приборов КИП, заземления, тепловой
изоляции. Проверить наличие воды в магистральном трубопроводе.

1-кипятильник
(куб-испаритель); 2-исчерпывающая часть колонны; 3-укрепляющая часть колонны;
4-дефлегматор; 5-флегмоделитель; 6-контактные тарелки.
Рисунок
1 - Схема работы ректификационной колонны
Пуск установки проводить
в присутствии учебного мастера и под его непосредственным руководством.
Работая на площадках с
высокой отметкой и вблизи вращающихся частей проявлять осторожность и
аккуратность. Включение трубчатых электронагревателей производить стоя на
диэлектрическом коврике. При отборе проб пара необходимо помнить, что на входе
в пробоотборник он имеет температуру около 100оС. К выполнению лабораторной
работы студенты допускаются только после прохождения инструктажа по
безопасности труда и пожарной безопасности в лаборатории и на рабочем месте.
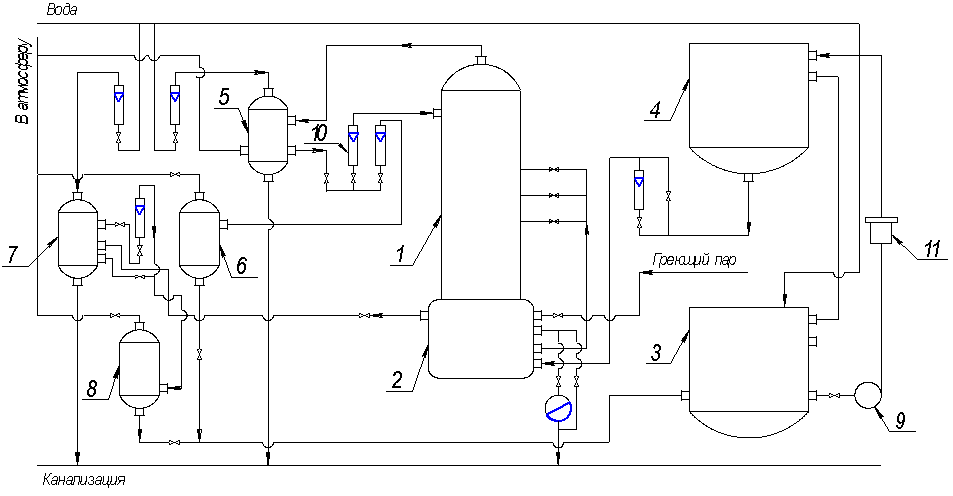
2 Описание
установки и порядок выполнения работы
Исходная смесь хранится в
баке-хранилище 3, из которого насосом 9 через фильтр 11 подается в напорный бак
4. Из напорного бака 4 исходная смесь самотеком через ротаметр поступает в подогреватель,
расположенный в кубе 2, где она нагревается кубовым остатком. Температуру
исходной смеси после подогрева определяют по показанию термометра. В ректификационную
колонну 1 подогретая исходная смесь поступает на 7, 9 или 11 тарелку, считая
сверху. Колонна имеет 12 ситчатых тарелок с сегментными сливными устройствами.
Внутренний диаметр колонны 200мм. Для визуального наблюдения за процессом
барботажа имеется смотровое окно.
С нижней тарелки
жидкость стекает в куб-испаритель 2, имеющий внутри змеевик, обогреваемый
паром. Конденсат греющего пара из змеевика отводится в канализацию через
конденсационный горшок. Расход греющего пара регулируется вентилем, а давление
определяется по манометру. В кубе-испарителя часть жидкости превращается в пар,
а другая отводится в качестве кубового остатка. Кубовый остаток проходит
холодильник 7, где охлаждается водой, и поступает в сборник 8. Из сборника 8
кубовый остаток возвращается в бак-хранилище 3. Сборники 6, 8 и межтрубное
пространство дефлегматора 5 сообщены с атмосферой, что обеспечивает работу
колонны под атмосферным давлением. С верхней тарелки колонны пар, обогащенный
низкокипящим компонентом, поступает в дефлегматор 5, который охлаждается водой.
Расход воды измеряется ротаметром, а температура ее на входе и выходе –
термометрами. Жидкость, образовавшаяся в дефлегматоре после полной конденсации
пара, делится на две части. Одна в виде флегмы подается на орошение колонны, а
другая отбирается в виде дистиллята, который поступает в сборник 6 и далее направляется
в бак-хранилище 3. Количество флегмы и дистиллята измеряется ротаметрами.
Колонна оборудована
пробоотборниками жидкости с тарелок, флегмы, дистиллята, жидкости в кубе, а
также пробоотборниками пара, поступающего на тарелку, и покидающего пенной
слой. Пробоотборники пара снабжены теплообменниками типа «труба в трубе», в
которых проба пара конденсируется, и конденсат собирается в бутылочки с пробками.
На каждой тарелке колонны установлены датчики температуры, работающие в комплекте
со вторичным прибором. Знание температуры жидкости на тарелках позволяет определить
профиль температуры по высоте колонны. Перед пуском установки необходимо подать
поду в дефлегматор, холодильник кубового остатка и в теплообменники на пробоотборниках
пара. Открыть воздушники на дефлегматоре и сборниках.
|
|
|
1 – Ректификационная
колонна; 2 – куб; 3 – бак-хранилище; 4 – напорный бак;
5 – дефлегматор; 6 – сборник дистиллята; 7 – холодильник кубового остатка; 8
– сборник кубового остатка; 9 – насос; 10 – ротаметр; 11 – фильтр
Рисунок 2 – Принципиальная схема установки
ректификации
|
Проверяют, наполнен ли
напорный бак 4 исходной смесью, и, если необходимо, насосом 9 подкачивают ее в
этот бак из сборника – хранилища 3. Из напорного бака заполнить до указанной
отметки куб-испаритель. Открывают вентиль на паропроводе, продувают
конденсационный горшок и начинают прогрев куба-испарителя, следя за давлением
греющего пара по манометру. Начало кипения смеси в кубе определяется визуально
в смотровое окно (над тарелкой появляется пар и капли жидкости).
При этом необходимо
полностью открыть вентиль на линии подачи флегмы в колонну и дать колонне
поработать «самой на себя» (за высотой парожидкостного слоя на тарелках
следить в смотровое стекло).
По заданию преподавателя
установить по ротаметрам расходы исходной смеси, флегмы, дистиллята и кубового
остатка. Чтобы расход питания не изменялся, уровень жидкости в напорном баке 4
должен быть постоянным. С этой целью включают насос 9 для непрерывной работы
вплоть до окончания опыта. Все измерения и отбор проб производить после выхода
установки на установившийся режим работы, который определяется по показаниям
вторичного прибора (температура жидкости на тарелках не изменяется). Все полученные
данные записывают в таблицу.
|
Наименование величин
|
Значение
|
Среднее значение
|
2
|
3
|
|
Расход исходной смеси, кмоль/с
|
|
|
|
|
|
Расход дистиллята, кмоль/с
|
|
|
|
|
|
Расход кубового остатка, кмоль/с
|
|
|
|
|
|
Расход флегмы, кмоль/с
|
|
|
|
|
|
Расход воды в дефлегматоре, кг/с
|
|
|
|
|
|
Температура воды, поступающей в
дефлегматор, 0С
|
|
|
|
|
|
Температура воды на выходе из
дефлегматора, 0С
|
|
|
|
|
|
Температура пара, поступающего в
дефлегматор из колонны, 0С
|
|
|
|
|
|
Температура флегмы, 0С
|
|
|
|
|
|
Концентрация, мольные доли:
а) исходной смеси ХF
б) флегмы (дистиллята) Хр
в) кубового остатка Хw
|
|
|
|
|
|
Температура исходной смеси после подогрева,
0С
|
|
|
|
|
|
|
|
|
|
|
|
|
Анализ проб пара и
жидкости проводится методом газовой хроматографии на хроматографе ЛХМ-8 МД или
рефрактометром.
После окончания работы закрывают
вентили на линии греющего пара, прекращают подачу исходной смеси и отбор
дистиллята, закрывают воду на холодильник кубового остатка и теплообменники на
пробоотборниках пара. Воду на дефлегматоре закрывают только после того, как
колонка остынет.
3
Обработка опытных данных
Переводят из массовых
долей в мольные концентрации исходной смеси ХF и дистиллята Хp. Пересчитывают расходы исходной смеси F и дистиллята Р в мольные. Из уравнений
материального баланса колонны находят расход и концентрацию кубового остатка по
уравнениям
W=F – P, Хw=(FXF – PXp)/W,
гдеF, P, W-расходы исходной
смеси, дистиллята, кубового остатка, кмоль/с;
Xf. Xp. Xw-составы исходной смеси, дистиллята,
кубового остатка, мол. доли.
Определяют флегмовое число
R – отношение расхода флегмы к расходу
дистиллята. Рассчитывают количество пара G, поднимающегося по колонне (рисунок 1).
Зная величину G и диаметр колонны, определяют
скорость пара в свободном сечении колонны w. Скорость пара в колонне можно
также рассчитать, определив количество пара из уравнения теплового баланса
дефлегматора [1, 2] (этот расчет может быть использован как проверочный).
По справочным данным
строят на миллиметровой бумаге кривую равновесия  на диаграмме у-х (рисунок 3) и отмечают на оси
абсцисс значения концентраций исходной смеси ХF дистиллята Хр и кубового остатка Хw.
на диаграмме у-х (рисунок 3) и отмечают на оси
абсцисс значения концентраций исходной смеси ХF дистиллята Хр и кубового остатка Хw.
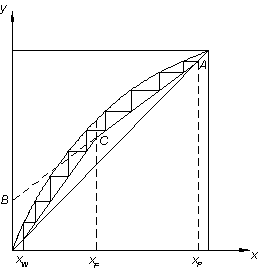
Рисунок 3 –
Определение числа теоретических тарелок
Затем на диаграмму
у-х наносят рабочие линии для укрепляющей и исчерпывающей частей колонны.
Уравнения рабочих линий, получают из уравнений материального баланса. Они имеют
следующий вид: для укрепляющей части колонны
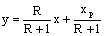 ,
,
для исчерпывающей части
колонны
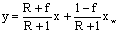 ,
,
гдех, у–концентрация
жидкости и пара в любом сечении колонны, мол. доли;
f – отношение расхода исходной смеси к
расходу дистиллята.
Вычисляют отрезок 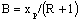 , который отсекает рабочая
линия верхней части колонны на оси ординат (рисунок 3). Через точку А (хр=
ур) и полученный отрезок проводят рабочую линию верхней части
колонны. Через точку Д (хw=
уw) и точку С проводят рабочую линию
нижней части колонны. Между равновесной и рабочими линиями строят ступени
изменения концентраций (рисунок 3). Каждая ступень соответствует одной теоретической
тарелке. Определив число теоретических ступеней nТ, и, зная число реальных тарелок в колонне n, находят среднее значение КПД
тарелки по уравнению
, который отсекает рабочая
линия верхней части колонны на оси ординат (рисунок 3). Через точку А (хр=
ур) и полученный отрезок проводят рабочую линию верхней части
колонны. Через точку Д (хw=
уw) и точку С проводят рабочую линию
нижней части колонны. Между равновесной и рабочими линиями строят ступени
изменения концентраций (рисунок 3). Каждая ступень соответствует одной теоретической
тарелке. Определив число теоретических ступеней nТ, и, зная число реальных тарелок в колонне n, находят среднее значение КПД
тарелки по уравнению
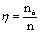 .
.
Величина КПД тарелки зависит от
гидродинамических условий и физико-химических свойств пара и жидкости.
При работе колонны «на себя»
дистиллят не отбирается, т.е. флегмовое число равно бесконечности 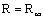 . В этом случае рабочая линия колонны совпадает с диагональю.
. В этом случае рабочая линия колонны совпадает с диагональю.
Лабораторная работа
ЭКСТРАКЦИЯ
Цель работы: практическое
ознакомление с работой противоточной экстракционной установки,
экспериментальное определение движущей силы процесса экстракции и степени
извлечения распределяемого вещества при различных соотношениях потоков.
Приборы и принадлежности:
экстракционная установка, бюретка для титрования проб; 0,1 N раствор щелочи NaOH; индикатор – фенолфталеин; мерные
цилиндры; конические колбы.
Введение
Экстракцией называют процесс
извлечения одного или нескольких компонентов из растворов или твердых тел с
помощью избирательных растворителей (экстрагентов). При взаимодействии раствора
с экстрагентом в нем хорошо растворяются только извлекаемые компоненты и не
растворяются остальные компоненты исходной смеси [1]. Экстракция в системах жидкость-жидкость
представляет собой массообменный процесс, протекающий с участием двух взаимно
нерастворимых или ограниченно растворимых жидких фаз, между которыми
распределяется экстрагируемое вещество.
Процесс жидкостной
экстракции состоит в том, что исходную смесь и экстрагент приводят в тесный
контакт. В результате взаимодействия фаз получают экстракт – раствор
извлеченных веществ в экстрагенте и рафинат – остаточный исходный раствор, из
которого с той или иной степенью полноты удалены экстрагируемые компоненты.
Полученные жидкие фазы (экстрагент и рафинат) разделяются отстаиванием, центрифуированием
или другими гдромеханическими способами. После этого извлекают целевой продукт
из экстракта и производят регенерацию экстрагента из рафината. Основное достоинство
процесса экстакции по сравнению с другими процессами разделения жидких смесей
(ректификация. выпаривание) состоит в том, что он осуществляется при низких рабочих
температурах, чаще всего при комнатной температуре.
В зависимости от вида контакта между
жидкими фазами экстракторы, как и другие массообменные аппараты, бывают: со
ступенчатым контактом фаз (тарельчатые); с непрерывным контактом фаз
(насадочные).
Методы осуществления жидкостной
экстракции и аппаратура для нее весьма разнообразны. При противоточной
экстракции исходная смесь и экстрагент поступают в экстракционную колонну
навстречу друг другу и движутся в ней противотоком из-за разности в плотностях.
Снизу в колонну подают более легкий продукт, сверху – более тяжелый. Рассмотрим
случай, когда плотность экстрагента больше плотности исходной смеси. Экстрагент
подается сверху и движется вниз, извлекая растворенный в исходной смеси целевой
компонент. Образующийся при этом экстракт выводится из колонны снизу. Первичный
растворитель исходной смеси по мере движения вверх освобождается от целевого
компонента. Очищенный первичный растворитель – рафинат выводится из колонны
сверху. При таком движении жидкостей в верхней части колонны разбавленный
раствор экстрагируемого вещества в первичном растворителе обрабатывается свежим
экстрагентом. Благодаря этому обеспечивается наиболее полное извлечение
экстрагируемого компонента из рафината.
Любой процесс, связанный с переходом
вещества из одной фазы в другую обратим, т.е. молекулы вещества могут
переходить, как в одном, так и в другом направлении. Предел этого перехода –
состояние равновесия между фазами. При полной взаимной нерастворимости
первичного и вторичного растворителей каждый из них присутствует только в одной
фазе. Степень отклонения рассматриваемой двухфазной системы от состояния равновесия
является движущей силой процесса массообмена. Графически движущая сила выражается
отрезком, заключенным между равновесной и рабочей линиями. Равновесия при
экстрагировании можно достигнуть лишь через длительный промежуток времени взаимодействия
сред. Практически в экстракционных аппаратах равновесие не достигается и концентрация
У (действительная, рабочая) отличается от У* (равновесной).
На рисунке 1 представлена схема
процесса противоточной экстракции. Обозначим массовый расход исходной смеси (в
данном случае керосин с бензойной кислотой) через G, содержание извлекаемого компонента в ней через 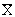 ; массовый расход экстракта
(вода с бензойной кислотой) через L; содержание извлеченного компонента через
; массовый расход экстракта
(вода с бензойной кислотой) через L; содержание извлеченного компонента через 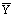 . Концентрации
. Концентрации 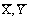 выражены
в относительных массовых концентрациях.
выражены
в относительных массовых концентрациях.
Для противоточного экстрактора
уравнение материального баланса имеет вид
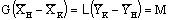 ,
,
где М – масса вещества, перешедшего в
единицу времени из первичного растворителя в экстракт, кг/с.
Рассмотрим сечение I – I экстрактора (рисунок 1) и составим материальный баланс для
верхней части аппарата
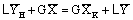
Из этого уравнения определим
концентрацию в любом сечении верхней части аппарата
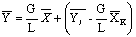 .
.
Мы получили уравнение рабочей
линии процесса экстракции, которое графически выражается прямой линией. В
случае действующего экстрактора значения 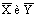 определяются экспериментально и по двум точкам
определяются экспериментально и по двум точкам 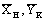 и
и 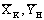 , строится рабочая линия, наклон которой зависит
от соотношения потоков
, строится рабочая линия, наклон которой зависит
от соотношения потоков 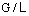 (рисунок 1).
(рисунок 1).
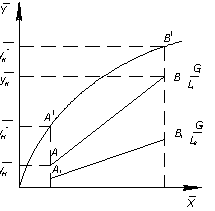
ОС – линия равновесия; АВ – рабочая
линия, прямая, проходящая через точки А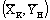 и В
и В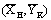 , при соотношениях потоков
, при соотношениях потоков 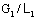 ; А1В1 – рабочая линия, полученная при соотношения
потоков
; А1В1 – рабочая линия, полученная при соотношения
потоков 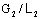 .
.
Рисунок 1- Схема процесса
противоточной экстракции
Положение равновесной линии
определяется физическими свойствами системы и условиями (температура,
давление), в которых находится данная система. Данные по равновесию приведены в
таблице 1.
Таблица 1
Равновесные концентрации бензойной
кислоты
в воде и в керосине
|
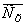 ,
кг/м3 ,
кг/м3
|
0,185
|
0,661
|
1,073
|
1,705
|
|
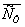 ,
кг/м3 ,
кг/м3
|
0,440
|
0,851
|
1,076
|
Чем больше система отклонена от
равновесия, тем интенсивнее она стремится к нему и наоборот, при малых
отклонениях от равновесия движущая сила мала.
Движущая сила процесса переменна по
высоте аппарата; внизу аппарата она равна отрезку 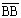 :
: 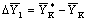 ; вверху аппарата она равна
; вверху аппарата она равна 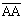 :
: 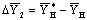 .
.
Средняя движущая сила процесса
экстракции равна среднему арифметическому (если 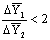 ) или среднему логарифмическому значению.
) или среднему логарифмическому значению.
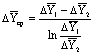
Чем больше движущая сила процесса,
тем больше вещества передается за одно и то же время на одном и том же участке
аппарата из одной фазы в другую 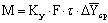 .
.
Изменяя соотношения потоков, мы
изменяем и движущую силу процесса.
1
Требования безопасности
К работе на установке допускаются
студенты, прошедшие в начале семестра инструктаж по технике безопасности в
лаборатории Б-011 и расписавшиеся в книге инструктажей. Студенты обязаны перед
началом работы надеть спецодежду и убрать волосы под косынку. Следует убедиться
в наличии средств пожаротушения и медицинской аптечки. Начинать работу можно
только в присутствии учебного мастера и преподавателя, и только с их разрешения.
Перед началом работы студенты должны отчитаться перед преподавателем о своей
подготовке к работе. Каждый студент должен знать место нахождения средств пожаротушения
и уметь ими пользоваться. При появлении течи через вентили или фланцевые
соединения, прекратить подачу растворителя и экстрагируемой смеси, закрыв все
вентили на линии подачи. В случае поломки ротаметров закрыть вентили на линии подачи
и выключить насосы, если они включены. В случае разлива рабочей смеси на пол,
необходимо немедленно ее убрать в канализацию с помощью тряпки или щетки. По
окончании работы каждый студент обязан проверить и привести в порядок рабочее
место, вымыть пробирки. Запрещается: находиться в верхней одежде или хранить ее
в помещении лаборатории; принимать пищу в лаборатории; уходить с рабочего места
и оставлять включенную установку без присмотра.
2
Описание установки и порядок выполнения работы
Экстракционная установка состоит из
колонного экстрактора 1 (рисунок 2), баков для жидкостей 2, 3, 4, 5, емкостью
0,1668м3, 2-х напорных баков 6, 7, центробежных насосов с
электродвигателями 8, 9, приборов КИП и системы трубопроводов. Колонный экстрактор
состоит из 4-х секций, разделенных между собой ситчатыми тарелками. Размеры
больших секций: высота – 0,282м, диаметр – 0,170м; промежуточных секций: высота
– 0,290м, диаметр – 0,065м. В верхней и нижней секциях имеются по два патрубка
для входа и выхода рабочих жидкостей. В баках 2, 3 находится экстрагент (вода),
в баках 4, 5 исходная смесь (керосин с бензойной кислотой).
Экстрагент из бака 2 или 3,
снабженных указателями уровня, посредством насоса 9 подается в напорный бак 6 и
из него самотеком поступает в верхнюю часть экстрактора 1. Напорный бак 6
служит для поддержания постоянного напора экстрагента, расход которого
замеряется с помощью ротаметров, расположенных на линии подачи экстрагента в колонну.
Исходная смесь из бака 4 или 5, снабженных указателями уровня посредством насоса
8 подается в напорный бак 7, а оттуда в нижнюю часть экстрактора 1. Расход исходной
смеси определяется с помощью ротаметров, расположенных на линии подачи исходной
смеси в экстрактор.
Поступая в экстрактор,
экстрагируемая смесь и растворитель смешиваются. Благодаря ситчатым тарелкам
достигается более полное смешение рабочих жидкостей. Осуществляется
противоточное экстрагирование, так как экстрагируемая смесь с меньшей плотностью
идет снизу вверх колонн; вода отнимает бензойную кислоту из керосина, выходит
из нижней секции колонны и поступает в бак 2 или 3 через гребенку 10, которая
служит для регулирования положения границы раздела фаз в колонне. Рафинат
(керосин) при выходе из колонного экстрактора по трубопроводу сливается в бак 4
или 5. Для контроля за процессом экстракции в каждой секции колонны имеются
пробники. Для определения состава фаз до и после процесса предусмотрены
пробники на линиях подачи и выхода продуктов. Все бачки и верхние части
трубопроводов имеют воздушники, сообщающиеся с атмосферой.
Открыть вентиль на линии подачи
экстрагента из бака 2 или 3 к насосу 8, открыть вентили на трубопроводе
нагнетания экстрагента в напорный бак 6 и включить насос 8. Когда бак 6 наполнится
и вода по сливной трубе будет сливаться обратно в бак 2 или 3, необходимо
насос выключить Открыть вентиль на линии подачи исходной смеси из бака 3 или 4
к насосу 9, открыть вентили на нагнетательном трубопроводе исходной смеси в напорный
бак 7 и включить насос 9. После наполнения бака 7 по сливной трубе жидкость будет
стекать обратно в бак для исходной смеси. Необходимо насос 9 сразу отключить,
иначе может произойти переполнение бака 7 и разбрызгивание керосина через
воздушный патрубок, расположенный в верхней крышке бака 7.
Регулировочными вентилями
по ротаметрам Р установить заданные преподавателем расходы экстрагента и
исходной смеси в экстрактор и поддерживать их постоянными в течение всего
опыта.
После заполнения полностью колонны и
установления четкой линии раздела фаз (в середине второй или третьей секции)
приступить к отбору проб через пробники П. После отбора проб изменить расход
экстрагента и через 5-10 минут отобрать еще раз пробы через пробники П.
После окончания отбора
проб прекратить подачу исходной смеси, закрыть вентиль на линии слива экстракта
из колонны и продолжать подачу воды в колонну. Поверхность раздела фаз при этом
будет подниматься, и когда она достигнет верха колонны, т.е. весь керосин
сольется из экстрактора, необходимо быстро открыть вентиль на линии слива
экстракта и прекратить подачу экстрагента.
|
|
|
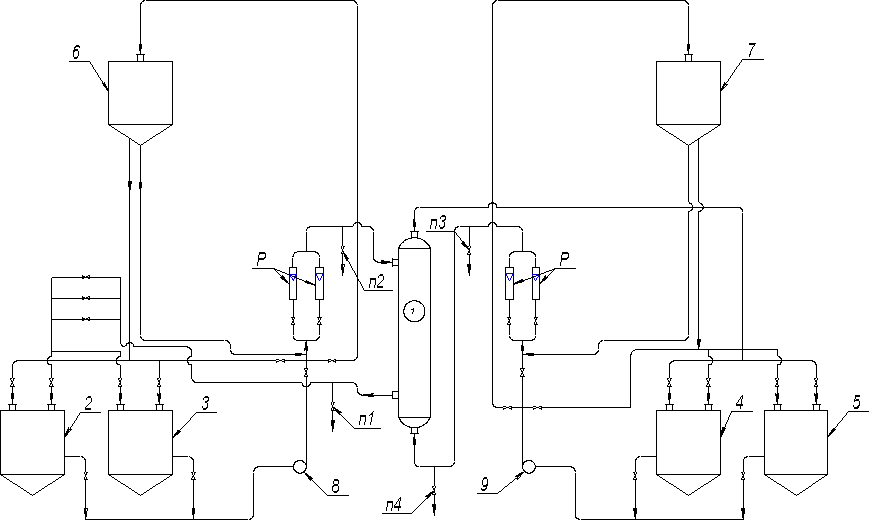
1 – экстрактор; 2, 3 – баки для
воды; 4, 5 – баки для керосина; 6, 7 – напорные баки; 8, 9 – насосы; 10
– гребенка; П – пробники; Р – ротаметры.
Рисунок 2 – Схема
экстракционной установки
|
3
Обработка опытных данных
Содержание бензойной кислоты 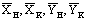 определяем титрованием
отобранных проб 0.1 N раствором NаОН в присутствии индикатора - фенолфталеина.
определяем титрованием
отобранных проб 0.1 N раствором NаОН в присутствии индикатора - фенолфталеина.
Нормальность раствора бензойной
кислоты определяется по зависимости 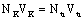
Для титрования берется 20-25 мл
исследуемой жидкости. Содержание бензойной кислоты в исследуемой жидкости
определяется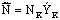 (кг/м3),
где Эк – эквивалент кислоты равный 122.
(кг/м3),
где Эк – эквивалент кислоты равный 122.
Затем необходимо пересчитать объемную
массовую концентрацию в относительные массовые доли по формулам
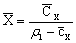
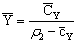 ,
,
Можно принять при рабочих
концентрациях ρ1 - плотность керосина, ρ2 -
плотность воды, кг/м3.
Построить на диаграмме Х – У
равновесную и рабочие линии, соответствующие разным соотношениям . Определить массовый расход первичного растворителя (керосина) 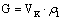 . Определить массовый расход
экстрагента (вода)
. Определить массовый расход
экстрагента (вода) 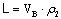 , где VB, VK, ρ2, ρ1 – объемный
расход и плотность воды и керосина. Полученные данные занести в таблицу 2.
, где VB, VK, ρ2, ρ1 – объемный
расход и плотность воды и керосина. Полученные данные занести в таблицу 2.
Убедится,
что при увеличении движущей силы процесса, увеличивается количество переданного
из фазы в фазу вещества М, которое определяется из уравнения материального
баланса. Сравнить конечные концентрации продуктов и сделать вывод о ходе
процесса экстракции, определить степень извлечения бензойной кислоты в обоих
случаях по формуле: 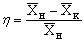
Таблица 2
Опытные и рассчитанные данные
|
Параметр
|
Значения
|
Среднее значение
|
|
Расход исходной смеси, кг/с
|
|
|
|
|
|
Расход экстрагента, кг/с
|
|
|
|
|
|
Относительная массовая концентрация бензойной
кислоты в керосине
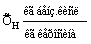
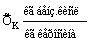
|
|
|
|
|
|
|
|
|
|
Относительная массовая концентрация бензойной
кислоты в воде
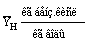
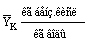
|
|
|
|
|
|
Средняя движущая сила
процесса 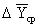
|
|
|
|
|
|
Количество переданного из фазы в фазу вещества М
|
|
|
|
|
|
Степень извлечения бензойной кислоты 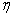
|
|
|
|
|
Лабораторная
работа
АБСОРБЦИЯ
Цель работы: практическое
ознакомление с работой тарельчатого абсорбера, экспериментальное определение
коэффициента массопередачи и сравнение его с рассчитанным теоретически.
Приборы и принадлежности:
абсорбционная установка, бюретка для титрования; 0.1 N раствор соляной кислоты; индикатор-фенолфталеин; конические
колбы.
Введение
Абсорбцией называют процесс
избирательного поглощения газа из смеси газов (или пара из парогазовой смеси)
жидким поглотителем. В абсорбционных процессах участвуют две фазы – газовая и
жидкая. При абсорбции происходит переход вещества из газовой фазы в жидкую,
обратный процесс называется десорбцией, при этом происходит переход вещества из
жидкой фазы в газовую. Все процессы массопередачи обратимы, т.е. в зависимости
от условий, направление перехода распределяемого вещества может быть различным.
Равновесие в процессах абсорбции определяет состояние, которое устанавливается
при продолжительном соприкосновении фаз и зависит от состава одной из фаз,
температуры, давления и термодинамических свойств компонента и поглотителя [1,2].
Для каждой конкретной системы
газ-жидкость при определенной температуре и давлении существует строго
определенная зависимость между равновесными концентрациями, т.е. каждому
значению Х соответствует строго определенное равновесное значение У* и
эту связь можно представить в виде функции 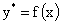
В общем случае эта зависимость
находится опытным путем. Для большего числа систем имеются данные в справочной
литературе. Для разбавленных растворов хорошо растворимых газов равновесная
зависимость хорошо
описывается законом Генри, который имеет вид 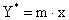 , где
, где 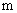 - константа фазового равновесия, величина которой
зависит от природы газа и жидкости и единиц, в которых выражены концентрации.
- константа фазового равновесия, величина которой
зависит от природы газа и жидкости и единиц, в которых выражены концентрации.
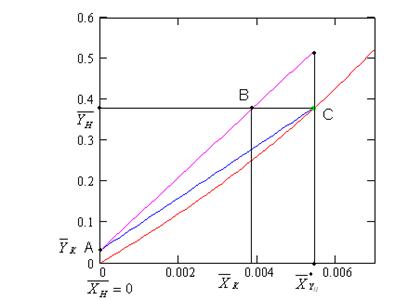
ОС – линия равновесия ; АВ – рабочая линия - прямая,проходящая
через точки (YH, XK) и (YK, XH)
Рисунок 1 - Схема процесса абсорбции
Примем расходы фаз по высоте
аппарата постоянными и выразим содержание поглощаемого газа в относительных
мольных концентрациях. Обозначим: G – расход инертного газа, кмоль/с; YH ,YK -начальная и конечная концентрации компонента
в газовой смеси, кмоль/кмоль инертного газа; L – расход абсорбента, кмоль/сек;
его концентрации XH и XK, кмоль/кмоль абсорбента.
Уравнение материального баланса
абсорбера имеет вид
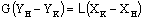 .
.
Выделим любое сечение в аппарате (рисунок
1), например, 1-1, в котором имеются рабочие концентрации X и Y. Напишем материальный баланс для верхней части аппарата по
распределяемому компоненту
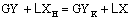
Если исходный абсорбент
не содержит распределяемый компонент (XH = 0), то
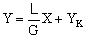 .
.
Полученная зависимость
называется уравнением рабочей линии.
Это уравнение позволяет нам
определить значение рабочей концентрации в любой точке аппарата. Так как G, L – величины постоянные для каждого
конкретного случая, то это уравнение прямой линии и для ее построения
необходимо всего две точки. Можно взять вверху колонны B (XH, YK) и внизу колонны A (XK, YH).
Разность
между рабочей и равновесной концентрациями вещества в данной фазе называется
движущей силой массопередачи ΔY=Y – Y*. Она
непрерывно меняется по высоте аппарата. В частном случае, когда линия
равновесия является прямой, 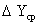 определяется как средняя логарифмическая величина из
движущих сил массопередачи у концов аппарата
определяется как средняя логарифмическая величина из
движущих сил массопередачи у концов аппарата
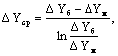 ,
,
где ΔУб , ΔУм – большая и
меньшая разность рабочих и равновесных концентраций на концах аппарата .
Основное уравнение
массопередачи для процесса абсорбции
где М – количество
компонента, передаваемое через поверхность контакта фаз, кмоль/с;
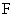 - поверхность контакта фаз, м2.
- поверхность контакта фаз, м2.
Так как поверхность
контакта газа и жидкости зависит от скорости потока, физических свойств фаз и
типа аппарата, то обычно расчеты тарельчатых абсорберов проводят по модифицированному
уравнению массопередачи, в котором коэффициенты массопередачи относят к единице
рабочей площади тарелки
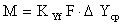 ,
,
где 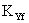 - коэффициент массопередачи, отнесенный к единице площади тарелки, моль/м2(кмоль/кмоль)с;
- коэффициент массопередачи, отнесенный к единице площади тарелки, моль/м2(кмоль/кмоль)с;
F- суммарная рабочая площадь тарелок в
абсорбере, м2;
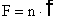 ,
,
где 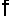 - рабочая площадь тарелке, м2;
- рабочая площадь тарелке, м2;
n - число тарелок в абсорбере.
Рабочая площадь
провальной тарелки может быть принята равной сечению абсорбера. Зная М –
количество поглащенного аммиака (в кмоль/с), и рассчитав F и 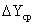 можно определить коэффициент массопередачи
можно определить коэффициент массопередачи
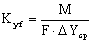 .
.
Коэффициенты
массопередачи определяют по уравнениям аддитивности фазовых диффузионных
сопротивлений
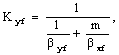
где βyf. βxf – коэффициенты массоотдачи, отнесённые к единице
рабочей площади тарелки, соответственно в газовой и жидкой фазах, кмоль/м2
(кмоль/кмлоь)с.
При абсорбции хорошо
растворимых газов 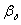 <<
<<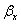 , и в этом случае величиной m/βy
, и в этом случае величиной m/βy можно
пренебречь, т.е.
можно
пренебречь, т.е. 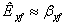 .
Поэтому при абсорбции аммиака водой можно приравнять общий коэффициент
массопередачи частному коэффициенту массоотдачи в газовой фазе.
.
Поэтому при абсорбции аммиака водой можно приравнять общий коэффициент
массопередачи частному коэффициенту массоотдачи в газовой фазе.
1
Требования
безопасности
К работе на установке
допускаются студенты, прошедшие в начале семестра инструктаж по технике
безопасности в лаборатории
Б-011 и расписавшиеся в журнале инструктажей. Студенты обязаны перед началом
работы надеть спецодежду и убрать волосы под косынку. Начинать работу можно
только в присутствии учебного мастера и преподавателя, и только с их
разрешения. Перед началом работы студенты должны отчитаться перед
преподавателем о своей подготовке к работе.
При появлении запаха
аммиака во время работы немедленно прекратить его подачу, перекрыв вентиль
расхода, а расход воды в абсорбер увеличить до полного удаления запаха. В
случае попадания раствора аммиака на кожу или в глаза немедленно обильное промывание
широко раскрытого глаза водой или 0.5-1% раствором квасцов, наложить вазелиновое
или оливковое масло. Необходимо помнить, что высокая концентрация аммиака в
воздухе вызывает обильное слезотечение, боль в глазах, удушье, сильные приступы
кашля, ПДК аммиака в воздухе – 20 мг/м3 . При отравлении аммиаком
через дыхательные пути необходимо вынести человека на воздух, вдыхание теплых
водяных паров, пить теплое молоко с содой. По окончании занятия каждый студент
обязан проверить и привести в порядок рабочее место.
2
Описание
установки и порядок выполнения работы
Абсорбер представляет
собой колонну 1 с внутренним диаметром 0,2м, высотой 1,68м, выполненную из
органического стекла (рисунок 2). В колонне установлены 4 тарелки провального
типа с отверстиями 3,5мм, долей свободного сечения 20%. В верхней части
колонны имеется брызгоотделитель, внутри которого размещены распределитель жидкости
и каплеотборник, уменьшающий унос жидкости уходящими газами. Колонна установлена
на сборнике 2 диаметром 0.44 и высотой 0.7м. Установка снабжена баллоном с
аммиаком 5, системой трубопроводов, арматурой и КИП.
Абсорбент
(вода) подается в верхнюю часть колонны 1. Расход воды регулируется вентилями
по показаниям ротаметра 10. Воздух к установке подается от компрессора, расположенного
в лаборатории Б-03, через коллектор 7. Скорость воздуха замеряется с помощью
диафрагмы 8 и соединенного с ней дифманометра 9. Аммиак из
баллона 5 дросселируется редукционным вентилем, расход NH3 определяется
ротаметром 11. Аммиак поступает в коллектор 7, а затем в смеситель 6; куда
подается и воздух, а затем аммиачно-воздушная смесь подается вниз абсорбера 1.
Смесь движется снизу вверх, а сверху вниз по тарелкам стекает вода. На
тарелках образуется газо-жидкостный слой. Вода поглощает абсорбтив (аммиак) и
поступает в сборник 2. Воздух, содержащий газ, выбрасывается в атмосферу. В
нижней части абсорбера находится пробоотборник для отбора пробы жидкости, поглотившей
аммиак. Концентрация аммиакта в воде определяется титрованием пробы 0.1 HCℓ. Для
определения гидравлического сопротивления на 2-х тарелках служит дифманометр
12.
При выполнении работы
строго соблюдать очередность подачи. Сначала необходимо подать воду в абсорбер,
установить заданный расход и поддерживать его постоянным. Затем подать воздух в
колонну, установить и поддерживать постоянную его скорость в колонне и замерить
перепад давления на 2-х тарелках. И только потом открыть редукционный вентиль и
подать аммиак в абсорбер. Установить расход аммиака не более 10-20 делений по
ротаметру, через 3-5 минут отобрать пробу жидкости внизу колонны и прекратить
подачу аммиака, затем воздуха и не ранее 10-15 минут закрыть вентили на линии
подачи воды в абсорбер.
Все измеренные величины
занести в таблицу.
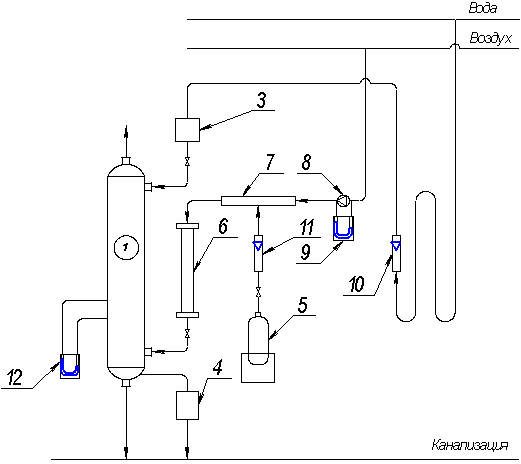
1 – абсорбер; 2 –
сборник; 3 – фильтр; 4 – гидрозатвор; 5 – баллон с аммиаком; 6 – смеситель; 7 –
коллектор; 8 – диафрагма; 9 – дифманометр; 10,11 – ротаметры; 12 – дифманометр;
В – вентили
Рисунок 2 - Схема
абсорбционной установки
3
Обработка
опытных данных
Таблица
1
Опытные
и рассчитанные данные
|
Наименование величин
|
Значение
|
Среднее значение
|
|
1
|
2
|
3
|
|
Расход воды; кмоль/с
|
|
|
|
|
|
Скорость воздуха в колонне; м/с
|
|
|
|
|
|
Расход воздуха в колонне; кмоль/с
|
|
|
|
|
|
Расход аммиака, кмоль/с
|
|
|
|
|
|
Перепад давления на 2-х тарелках;
Па
|
|
|
|
|
|
Концентрация аммиака в исходной смеси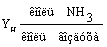
|
|
|
|
|
|
Концентрация аммиака в воздухе на
выходе из колонны 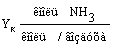
|
|
|
|
|
|
Концентрация аммиака в воде на выходе
из колонны 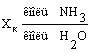
|
|
|
|
|
|
Движущая сила массопередачи
|
|
|
|
|
|
Коэффициент массопередачи
Kyf (опытный)
|
|
|
|
|
|
Коэффициент массоотдачи
βyf (расчетный)
|
|
|
|
|
Расходы воды, воздуха и
аммиака необходимо выразить в мольных единицах. Концентрацию аммиака в воздухе
YH определяем исходя из расходов
аммиака и воздуха в кмоль NH3/кмоль воздуха.
Конечная концентрация
аммиака в воздухе YK рассчитывается
из уравнения материального баланса
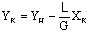 .
.
Начальная концентрация
аммиака в воде XH=0,
так как вода, поступающая в абсорбер, аммиака не содержит аммиак.
Конечная концентрация
аммиака в воде XK определяется
титрованием отобранной пробы 0.1 н раствором HCℓ. Определив в результате титрования нормальность
раствора, выразить концентрацию поглощаемого газа в Cх [кмоль/м3] и пересчитать в относительные мольные
единицы по формуле
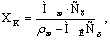
где Мж, МК
– молекулярные массы воды и аммиака, кг/кмоль;
ρж –
плотность воды, кг/м3.
Расход воздуха в колонне
определяется по уравнению расхода.
Рассчитав концентрации
YH, YK, XH, XK, строим рабочую линию по 2 точкам
(XH , YK), (XK, YH ).
Линия равновесия
определяется по уравнению 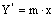 ,
где m=1.825 – найдена экспериментально для
водных растворов NH3. Наносим рабочую и равновесную линии
на график, определяем
,
где m=1.825 – найдена экспериментально для
водных растворов NH3. Наносим рабочую и равновесную линии
на график, определяем 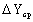 . Зная количество поглощенного
аммиака, определяем коэффициент массопередачи Kyf. Коэффициент массоотдачи βyf определяем из критерия Нуссельта диффузионного
. Зная количество поглощенного
аммиака, определяем коэффициент массопередачи Kyf. Коэффициент массоотдачи βyf определяем из критерия Нуссельта диффузионного
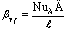 ,
,
где Д – коэффициент
молекулярной диффузии компонента в газе, м2/с;
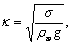 ℓ – характерный линейный размер, в данном случае ℓ =κ -
капиллярная константа, м.
ℓ – характерный линейный размер, в данном случае ℓ =κ -
капиллярная константа, м.
где 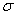 - поверхностное натяжение на границе
газ - жидкость, н/м .
- поверхностное натяжение на границе
газ - жидкость, н/м .
Критерий Нуссельта диффузионный
для провальных тарелок рассчитывается по уравнению 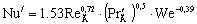 ,
,
где критерий Рейнольдса 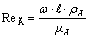 ;
;
критерий Прандтля 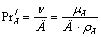 ;
;
критерий Вебера 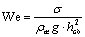 .
.
Высота столба жидкости
на тарелке hст, рассчитывается из гидравлического сопротивления
орошаемой тарелки ΔРт, измеренного U-образным дифференциальным манометром, м.
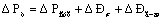 ,
,
где ΔР сух –
сопротивление сухой тарелки, Па;
ΔРσ-сопротивление
тарелки, вызываемое силами поверхностного натяжения, Па;
ΔРг-ж -
сопротивление газожидкостного слоя на тарелке, Па.
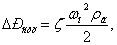
где ζ – коэффициент
сопротивления тарелки (ζ=2.1);
ωо –
скорость газа в отверстиях тарелки (ωо =ω/ƒсв),м/с;
ƒсв -
свободное сечение тарелки (ƒсв=0.2).
Сопротивление тарелки,
вызванное действием сил поверхностного натяжения, определяют по уравнению
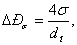
где dо – диаметр отверстий тарелки, м.
Сопротивление
газожидкостного слоя принимают равным статическому давлению слоя
ΔРг-ж=gρжhст
Определив критерий
Нуссельта диффузионный, рассчитывают βyf и сравниваем Kyf и βyf.
Сделать вывод о
сопоставимости полученных значений коэффициентов.
Список литературы
1.
Дытнерский Ю.И.
Процессы и аппараты химической технологии: Учебник для вузов. Изд. 3-е. В 2-х
кн. / Ю.И. Дытнерский. – М.: Химия, 2002. –кн.1. - 400 с.: ил. -кн. 2. -368
с.: ил.
2.
Павлов К.Ф. Примеры
и задачи по курсу процессов и аппаратов химической технологии. 9-е изд., пер. и
доп. / К.Ф. Павлов, Н.Г. Романков, А.А. Носков. – Л.: Химия, 1981. - 560 с.
3.
Руководство к
практическим занятиям в лаборатории процессов и аппаратов химической
технологии: Учеб. пособие для вузов. / Под. ред. чл.-корр. АН СССР П.Г. Романкова
- 6-е изд., перераб. и доп. – Л: Химия, 1990. - 272 с.: ил.