|
Относит. удлинение ε=Δl/lо
|
Мгновенное удлинение Δl/l
|
Истинное удлинение ε`=lnl/lo
|
|
0,01 (1%)
|
0,01 (1%)
|
0,01 (1%)
|
|
0,2 (20%)
|
0,167 (16,7%)
|
0,182 (18,2%)
|
|
1,0 (100%)
|
0,5 (50%)
|
0,693 (69,3%)
|
При разных видах деформации выполняются
следующие соотношения: растяжение σ=Еε
(закон Гука), где Е - коэффициент пропорциональности между σ
и
ε,
называемый модулем Юнга; сдвиг τ=Gγ,
где G - модуль сдвига;
объемное сжатие р=В.ΔV/Vo,
где В - модуль объемной упругости. Упругие коэффициенты связаны между собой
соотношениями: G=Е/2(1+μ);
В=Е/3(1-2μ),
где
μ
- коэффициент
Пуассона. При μ=0,5 модуль Е=3G.
Ниже приведены значения модуля Юнга для некоторых материалов в Па (ГПа): медь -
1,2.1011 (120); полистирол - 3.109
(3); мягкая резина - 2.106 (0,002).
При линейной упругости модуль Юнга (Е)
определяют как тангенс угла наклона зависимости σ-ε
(tgα). При нелинейной
упругости он в процессе растяжения чаще уменьшается, и в этом случае пользуются
величиной мгновенного (начального) модуля, который определяют как tg
угла наклона касательной к этой кривой в начале координат. Иногда его
определяют как тангенс угла наклона касательной к этой кривой в точке,
соответствующей данному удлинению. Модуль Юнга является мерой жесткости
материала. Величина, обратная ему (J=1/E),
является мерой деформируемости материала и называется податливостью.
Коэффициент Пуассона при растяжении является отношением уменьшения поперечных
размеров образца к увеличению его продольных размеров. Если цилиндр длиной lo
и радиусом r уменьшается на Δr,
тогда относительное удлинение равно Δl/lо,
а относительное уменьшение радиуса - Δr/rо.
Если объем образца не меняется, то Δl/lо=2(Δr/rо),
т.е. относительный рост длины равен относительному сокращению поперечных
размеров. Тогда коэффициент Пуассона μ=(Δr/rо)/(Δl/lо)=0,5.
В случае идеальной упругости (ненаполненная резина) μ=0,5;
для слабонаполненной резины μ=0,49. В
пластмассах растяжение ведет к изменению структуры материала, включая
расстояния между макромолекулами, и μ=0,2-0,4. В
низкомолекулярных жидкостях упругость отсутствует, и μ=0.
. Термодинамика высокоэластической деформации
Способность обратимо деформироваться на десятки
и сотни процентов является уникальным свойством эластомеров и верным признаком
высокомолекулярной природы исследуемого образца. Механизм проявления
эластичности можно понять, исследуя его термодинамическим методом.
Пусть образец эластомера длиной l0
под действием напряжения f
удлинился на величину dl.
Для упрощения исключим явления вязкоупругости, обеспечивая действующему
напряжению достаточное время для достижения равновесной деформации. Чтобы
предотвратить необратимое перемещение макромолекул, возьмем слабо сшитый
эластомер с редкой сеткой химических связей. Тогда деформация окажется
полностью обратимой и равновесной, не зависящей от времени действия напряжения.
Поскольку эластомеры при деформации практически не меняют объема, работа
деформации обусловлена только действием приложенной силы: dA=fdl,
а для равновесного процесса подведенная к системе теплота dQ=TdS.
Согласно первому закону термодинамики, внутренняя энергия складывается из
работы, совершенной над системой, и теплоты, подведенной к ней:
dU=dA+dQ=fdl+TdS
(1).
Согласно второму закону термодинамики,
внутренняя энергия системы складывается из свободной dF
и связанной
TdS: dU=dF+TdS
(2)
Подставляя (1) в (2), получим dF=fdl.
При постоянных значениях температуры и объема выражение f=dF/dl
раскрывает физический смысл деформирующей силы: она равна изменению свободной
энергии системы в расчете на единицу удлинения. Определив значение dF
из (2) и подставив его в последнее выражение, получим общее выражение для
изменения термодинамических параметров эластомера в процессе деформации:
f=(dU/dl)-T(dS/dl), или
f=fU+fS. (3).
Пространственно-сшитые эластомеры с малой
частотой сетки при температуре существенно выше температуры стеклования Тс
ведут себя как идеальные эластомеры, в которых fU=0.
В идеальном эластомере возникающее при деформации напряжение обусловлено только
изменением энтропии, происходящим за счет развертывания молекулярных клубков,
что повышает ориентированность его структуры. Возникновение ориентации означает
уменьшение беспорядка в системе, т.е. уменьшение энтропии: f=-T(dS/dl).
При изотермическом сжатии газа, свойства которого близки к свойствам идеального
газа, давление также меняется только за счет изменения энтропии:
p=T(dS/dV)
Поэтому и говорят, что упругость эластомера
имеет газовую природу, так как напряжение при деформации обусловлено только
изменением порядка в расположении элементов структуры: в идеальном эластомере -
сегментов (частей) макромолекул, а в идеальном газе - молекул. Очевидно, что
при малой деформации идеального кристалла, структура которого не нарушается,
напряжение возникает только за счет изменения межатомных расстояний
кристаллической решетки.
По экспериментальной зависимости напряжения от
температуры при разных удлинениях, полученной в равновесных условиях
деформации, из уравнения (3) рассчитан вклад изменения свободной энергии и
энтропии в величину напряжения реального эластомера. Оказалось, что в
ненаполненных сетчатых эластомерах при комнатной температуре в широком
диапазоне удлинений величина напряжения обусловлена главным образом изменением
энтропии и лишь на 5-15% - изменением внутренней энергии (рис.3а). Только при
малых удлинениях, когда энтропия меняется еще незначительно, вклад внутренней
энергии в увеличение показателя напряжения достаточно велик. Чем ближе
температура к температуре стеклования Тс, т.е. чем больше эластомер
по свойствам напоминает пластмассу, тем больше роль энергетической составляющей
fU и ее доля в общей
величине силы (fU/f).
Это видно на примере пространственно сшитого полигексилметакрилата (Тс=-3оС),
который при 100ОС ведет себя как эластомер (fU/f=0,26),
а при 30ОС приближается по свойствам к пластмассе, и fU/f
увеличивается до 0,97.
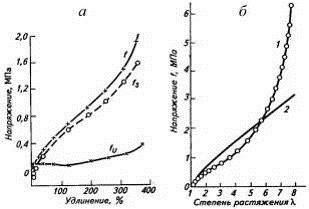
Рис. 3. Экспериментальная кривая (а)
напряжение-удлинение вулканизата НК для равновесных условий (f)
с изменением энтропийной (fS)
и энергетической (fU)
составляющих напряжения и сравнение (б) этой кривой (1) с теоретической кривой
(2), построенной по уравнению f=G(λ-1/λ2).
Представления о существовании длинных гибких
цепных макромолекул позволили применить к анализу деформационных свойств
эластомеров законы статистической термодинамики и установить количественную
связь между структурой макромолекулярного клубка и механическими свойствами.
Для расчетов приняли наиболее простую модель свободносочлененной цепи. Чтобы
учесть заторможенность вращения, ввели понятие сегмента как жесткого элемента
структуры, а соединения между сегментами приняли свободно сочлененными. Тогда
можно подобрать такую длину сегмента l
или такое их число n, чтобы при
общей длине эквивалентной цепи расстояние между ее концами r
было бы таким же, как для реальной макромолекулы. Для идеального эластомера с
учетом зависимости напряжения от изменения энтропии (f=dF/dl)
получили уравнение зависимости напряжения от относительной деформации r/ro
для
отдельной макромолекулы: f=-T(dS/dr)T=(2kT/ro)r/ro,
по которму легко определяется коэффициент пропорциональности между ними G,
называемый модулем эластичности макромолекулы: G=2kT/ro=3kT/nl2.
Из этой формулы следует, что модуль эластичности макромолекулярного клубка
растет с повышением температуры (G-T).
Объясняют это тем, что растянутый клубок стремится вернуться в прежнее (до
деформации) наиболее вероятное состояние, и возвращающая сила тем больше, чем
выше температура и больше интенсивность теплового движения сегментов. Здесь
видна полная аналогия с газом: повышение температуры увеличивает его давление
на поршень благодаря интенсификации теплового движения и росту частоты ударов
молекул по поршню. Поэтому равновесную упругость макромолекул называют «газовой
упругостью». Различие лишь в том, что тепловое движение сегментов направлено на
сокращение клубка, а газа - на расширение.
Статистическая теория высокоэластической
деформации математически описывает экспериментальную кривую только до
деформации 50% (рис. 3б). Несовпадение с экспериментальной кривой при более
высоких деформациях объясняется несовершенством как пространственной сетки
(дефектами структуры), так и самой теории, не позволяющей количественно описать
зависимость свободной энергии dF
от деформации. Поэтому эта теория не может быть использована для
прогнозирования качества изделий. Принципиальное значение статистической теории
состоит в том, что она дает понимание молекулярного механизма проявления
высокоэластичности, объясняет основную причину упругости и появления напряжения
стремлением макромолекул перейти в наиболее вероятное состояние статистического
клубка, когда достигается максимум энтропии и минимум свободной энергии.
В последние годы активизировались работы по
математическому и компъютерному моделированию поведения изделий из эластомеров
для решения прикладных задач повышения ресурса работы по отдельным показателям
их качества. Разрабатываются молекулярные механизмы и микромеханические модели
уменьшения сопротивления качению, повышения сопротивления образованию и
разрастанию трещин и улучшения других показателей качества автомобильных шин,
которые дают возможность прогнозирования срока их службы методом конечных
элементов.
. Релаксационные свойства полимеров
Переход любой системы из неравновесного в
равновесное состояние в результате теплового движения атомов и молекул называют
релаксацией, при этом скорость приближения к равновесию пропорциональна
отклонению системы от равновесия. Следует иметь в виду, что понятие
«равновесный» является относительным, если его характеризовать с точки зрения
времени достижения равновесия. В системе с подвижными сегментами равновесие
устанавливается достаточно быстро, а с малоподвижными элементами структуры
может вообще не достигнуть его. Поэтому принципиальное значение приобретает
оценка деформируемости полимера за заданный отрезок времени.
Если обозначить напряжение в образце σ,
а скорость его релаксации dσ/dt,
то скорость перехода к равновесному состоянию будет пропорциональна напряжению:
dσ/dt=-σ/τ,
где коэффициент пропорциональности 1/τ
зависит от структуры и свойств полимера. После разделения переменных и интегриро-вания
(от 0 до t и от σо
до σ)
получим: σ=σое-t/τ.
Если t=τ,
то σ=σо/е.
Последнее уравнение раскрывает физический смысл константы τ,
которую называют временем релаксации, - это время, за которое начальное
напряжение σо
в деформированном образце уменьшится в е раз. Время релаксации тем меньше, чем
больше скорость релаксации и чем больше скорость теплового движения сегментов,
т.е. оно увеличивается с ростом полярности и потенциального барьера вращения в
макромолекулах и уменьшается с ростом температуры.
Время релаксации τ
зависит от температуры и полярности полимера, однако оценить величину их
влияния можно только путем сопоставления времени релаксации с временем действия
внешней силы t. Соотношение
τ/t, которое называют
критерием Деборы и обозначают D,
определяет все механические свойства полимера. Чем меньше D,
тем быстрее релаксирует и более податлива система. Очень малое значение D
характерно для низкомолекулярных жидкостей. Однако при очень длительном времени
действия на полимер деформирующей силы значение D
окажется небольшим даже при большом времени релаксации, и полимер проявит
текучесть, как если бы это была жидкость. Поэтому ряд эластомеров, например
бутилкаучук, при хранении текут, т.е. обладают свойством хладотекучести. Таким
образом, при малом значении критерия Деборы D
полимеры обнаруживают свойства жидкостей, а при большом значении D
- свойства твердых тел. При этом понятия «твердый» и «жидкий» зависят не только
от химической структуры полимера, но и от времени действия деформирующей силы.
К способам изучения релаксационных свойств полимеров относят релаксацию
напряжения, ползучесть, кривые напряжение-деформация и многократные деформации.
При релаксации напряжения образец быстро
деформируют на заданную величину и сохраняют в деформированном состоянии,
измеряя зависимость напряжения от времени (рис. 4а). В момент фиксации
начальное напряжение соответствует состоянию, когда макромолекулярные клубки
развернулись в процессе деформации, а узлы флуктуационной сетки не успели
распасться и перегруппироваться. Постепенно в напряженном образце происходит
распад узлов флуктуационной сетки, а макромолекулярные клубки все более
свертываются. Когда напряжение в образце упадет до нуля, структура его
становится такой же, как и до растяжения (кривая 1).
Поскольку деформация растяжения не изменилась,
сворачивание клубков и перегруппировка узлов флуктуационной сетки
сопровождались процессом течения макромолекул. После освобождения из зажимов
динамометра такой образец не сократится, так как вся эластическая деформация в
нем перешла в деформацию течения. Чем более полярен полимер, тем более пологой
оказывается кривая релаксации и медленнее падает напряжение. Сетка химических
связей сетчатых эластомеров практически не релаксирует. Напряжение релаксирует
до тех пор, пока не сосредоточится на узлах химической сетки, достигнув
равновесного значения, и не разгрузятся все узлы флуктуационной сетки (кривая
2). Химические связи препятствуют необратимому перемещению клубков молекул, но
не мешают перемещению сегментов. Если образец освободить из зажимов
динамометра, то он с течением времени полностью восстановит свои первоначальные
размеры, и напряжение в нем упадет до нуля.
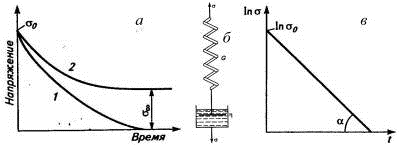
Рис. 4. Релаксация напряжения в линейном (1) и
сетчатом (2) эластомере (а), модель Максвелла (б) и релаксация напряжения в
этой модели (в).
кристаллический полимер стеклование
деформация
Полностью обратимая деформация развивается в
идеально упругой стальной пружине, а полностью необратимая - при нагружении
поршня, помещенного в идеальную жидкость. Последовательное соединение пружины и
поршня является простейшей моделью вязкоупругого тела, которую называют моделью
Максвелла (рис. 4б). Пружина характеризуется модулем G,
жидкость в поршне - вязкостью η,
а напряжение экспоненциально снижается со временем: σ=σое-t/τ
или после логарифмирования lnσ=lnσо-t/τ.
В координатах lnσ-t
это прямая линия, тангенс угла наклона которой к оси абсцисс tgα=1/τ,
что используют как один из методов экспериментального определения времени
релаксации (рис.4в). Напряжение в образце упадет до нуля при времени
наблюдения, намного превышающем время релаксации (τ=1/tgα).
Количественные зависимости между структурой и свойствами эластомеров сложны,
поэтому находят феноменологические закономерности с помощью моделирования.
При ползучести образец быстро нагружают и следят
за деформацией, которая усложняется все возрастающим напряжением в расчете на
постоянно уменьшающуюся площадь поперечного сечения (рис. 5а). Под действием
приложенной нагрузки макромолекулярные клубки развертываются, а часть сегментов
перемещается, ориентируясь в направлении действия силы, что приводит к смещению
и клубков относительно друг друга (кривая 1). Таким образом, развиваются
одновременно и обратимая (высокоэластическая), и необратимая (вязкотекучая)
деформации. После разгрузки образец частично (на величину высокоэластической
деформации) сократится за счет свертывания клубков макромолекул, но сохранит
остаточную, вязкотекучую деформацию. В сетчатом эластомере ползучесть
развивается до нагружения сетки химических связей, которые исключают взаимное
перемещение макромолекул, Поэтому при ползучести образца резины не возникает
необратимой деформации, а после разгрузки образец сокращается до первоначальных
размеров (кривая 2).
Модель Максвелла (рис.5б) не отражает основной
особенности кривой ползучести - наличия участка замедленного развития упругой
деформации. В реальном полимере упругая деформация развивается не как в
пружине, а замедленно, так как перемещение сегментов тормозится вязким сопротивлением
среды.
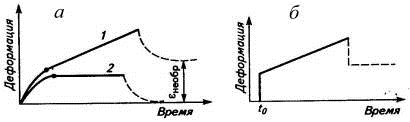
Рис. 5. Ползучесть в линейном (1) и сетчатом (2)
полимерах (а) и кривая ползучести для модели Максвелла (б). Пунктиром
обозначена часть кривых сокращения образца после прекращения действия силы;
точкой обозначены переходы к линейному участку кривых.
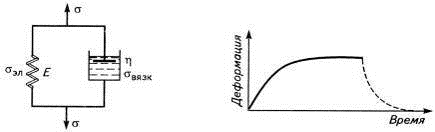
Это отображается моделью Кельвина-Фойгта (рис.
6), где пружина и поршень соединены параллельно, и напряжения на них
складываются: σ=σэл+σвязк.
По закону Гука σэл=Gε,
а по закону вязкого течения Ньютона σвязк=η.dε/dt.
Отсюда σ=Gε+η.dε/dt,
а после интегрирования получим: ε=σ(1-е-t/τ).
Кривая по этому уравнению не учитывает необратимую деформацию и поэтому хорошо
моделирует ползучесть сетчатого эластомера.
Ползучесть линейного полимера хорошо описывается
также объединенной механической моделью, сочетающей модели Максвелла и
Кельвина-Фойгта (рис. 7). К моменту времени t
общая деформация складывается из мгновенно упругой составляющей (1-й элемент,
пружина), замедленно упругой и эластической (2-й элемент) и вязкой (3-й
элемент, поршень):
εобщ=σ/G1+σ(1-е-t/τ)/G2+σt/η3.
При рассмотрении модели выявляется различие
между вязкостью (η3)
и микровязкостью (η2)
полимера. Первая характеризует вязкое сопротивление перемещению макромолекул, а
вторая - перемещению их сегментов.
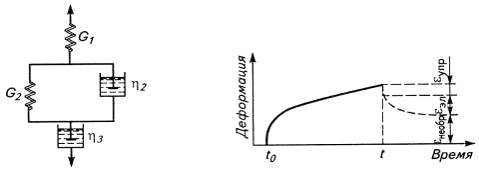
Рис.7. Объединенная механическая модель
вязкоупругого поведения полимера и кривая ползучести для этой модели.
Для снятия кривой напряжение-деформация образец
помещают в динамометр, один из зажимов которого передает нагрузку на
силоизмеритель и неподвижен, а другой перемещается с постоянной скоростью.
Такой режим изучения релаксационных явлений более сложен по сравнению с
релаксацией напряжения и ползучестью, но его широко применяют при определении
механических свойств полимеров. При малых деформациях кривая линейна, отличаясь
от закона Гука зависимостью ее наклона от скорости деформации, что является
проявлением вязкоупругости, а не гуковской упругости (рис. 8).
На начальном участке кривой растяжения
пространственно сшитого эластомера (область I)
напряжение возрастает из-за сопротивления узлов флуктуационной сетки, не
успевающих распадаться (рис. 9а).
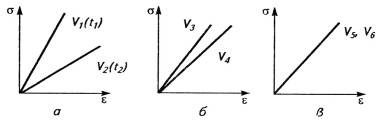
Рис.8. Влияние скорости действия силы V
на деформационные кривые полимеров, отличающихся сильным (а) и умеренным (б)
проявлением релаксационных свойств и идеальной упругостью (в); V1>V2,
V3>V4,
V5>V6.
При дальнейшем росте деформации напряжение
растет медленнее (область II)
из-за распада узлов флуктуационной сетки, облегчающего перемещение и ориентацию
сегментов. При дальнейшей деформации (область III)
напряжение растет за счет ориентации макромолекул, сопровождающейся у стереорегулярных
каучуков кристаллизацией, до точки разрыва образца (1). При замене растяжения
сокращением с той же скоростью перегруппировавшиеся узлы флуктуационной сетки
не успевают восстановиться, и напряжение в образце будет меньше, чем при
растяжении. Площадь под кривой напряжение-деформация является мерой работы
деформации, которая в цикле растяжение-сокращение превращается в теплоту из-за
внутреннего трения сегментов.
Потери механической энергии (гистерезисные
потери) измеряются площадью петли гистерезиса, образованной кривыми растяжения
(1`) и сокращения (3), а само явление несовпадения кривых называется
гистерезисом. Кривые нагрузки и разгрузки совпадают при очень большой скорости
деформации (2), когда не успевают распадаться узлы флуктуационной сетки, деформация
мала (хрупкое разрушение), и критерий D
велик, или при медленной равновесной деформации (4). В этом случае при большой
продолжительности процесса, когда критерий D
мал, все изменения надмолекулярной структуры успевали восстановиться. При
промежуточных значениях скорости деформации или времени цикла гистере-зисные
потери достигают максимума при критерий D=1.
Площадь петли гистерезиса уменьшается от цикла к циклу до предельной величины
(рис. 9б).
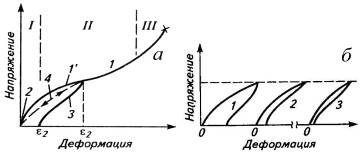
Рис. 9. Зависимость напряжение-деформация для
сетчатого эластомера (а) и уменьшение площади петли гистерезиса при повторении
циклов деформирования (б): 1-стандартная скорость растяжения (I,
II, III-области
структурных изменений); 2- большая скорость растяжения; 3 и 1` -петля
гистерезиса; 4-нагружение и разгрузка в равновесных условиях.
Многократные циклические деформации проводятся в
области линейной вязкоупругости, что позволяет менять в широких пределах
скорость действия силы (частоту) при малой величине деформации. Для перемещения
зажимов с образцами при циклическом режиме деформации используют
электромагнитные приводы или кривошипные механизмы, обеспечивающие
синусоидальное изменение напряжения и деформации. Можно задать синусоиду
деформации и измерять синусоиду напряжения или наоборот.
Зададим синусоиду деформации упругого тела ε=εо.sinωt.
Исходя из закона Гука (σ=Gε),
получим выражение для напряжения, меняющегося по синусоиде без отставания по
фазе (рис.10): σ=Gεоsinωt=σosinωt.
Максимальной амплитуде εо
соответствует максимальная амплитуда σo
(а). Для случая приложения синусоидально меняющегося напряжения (σ=σosinωt)
к идеальной жидкости в соответствии с законом Ньютона имеем
σ=ηdε/dt.
Объединение двух уравнений дает dε/dt=σosinωt/η,
а после интегрирования:
ε=-σocosωt/ηω=σosin(ωt-π/2)/ηω.
Из этого следует (б), что в вязкой жидкости
синусоида деформации отстает от синусоиды напряжения на угол π/2.
Максимальному напряжению соответствует нулевая амплитуда деформации при
максимальной скорости деформации.
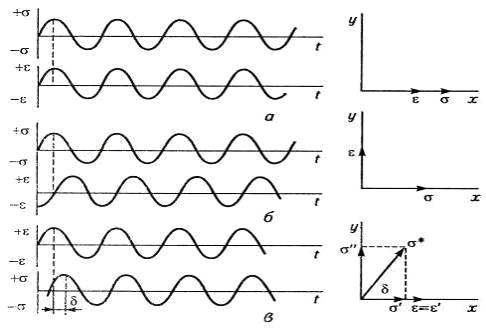
Рис. 10. Синусоиды напряжения и деформации и
сдвиг по фазе между ними при циклической деформации упругого (а), вязкого (б) и
вязкоупругого (в) тел.
У вязкоупругого тела всегда есть угол сдвига фаз
δ
между напряжением и деформацией (π/2 δ 0),
с учетом которого при деформации ε=εо.sinωt
напряжение σ=Gεоsin(ωt-δ)=σosin(ωt-δ).
Если вектор деформации ε
направить по оси х, то вектор напряжения расположится с отставанием на угол δ
(рис.10в), что напоминает диаграмму отставания напряжения от силы тока в
электрической цепи с омическим и индуктивным напряжением. По аналогии и вектор
напряжения можно выразить комплексным числом σ*,
обозначив его проекции на осях х и у соответственно через σI
и σII:
σ*=σI+iσII.
Если задана первоначально синусоида деформации, то вектор деформации
ε
совпадает с его действительной частью εI,
т.е. ε=εI.
С учетом этого получим выражение для модуля вязкоупругого синусоидально
нагруженного тела: σ*/ε=σI/ε+iσII/ε
и далее G*=GI+iGII,
а тангенс угла сдвига фаз tgδ=GII/GI.
Поскольку напряжение выражается комплексным числом, комплексным является и
модуль G*.
Для определения компонентов комплексного модуля G=σ/ε
используют модель Максвелла.
Высота максимума GII
на зависимости их от частоты ω
(рис 11) равна G/2, а точка
перегиба на кривой GI
совпадает с точкой максимума на кривой GII.
Физический смысл комплексного модуля заключается в том, что он состоит из
модуля потерь GII,
являющегося мерой гистерезисных потерь в одном цикле, и модуля накопления GI,
являющегося мерой упругости полимера. Можно сказать, что GI
показывает, сколько энергии накапливает полимер при заданной деформации, а
затем возвращает при разгрузке. Максимальные гистерезисные потери энергии,
соответствующие максимуму GII,
имеют место при частоте ω=G/η,
которая является величиной, обратной времени (ω=1/t),
а η/G=τ.
Отсюда следует, что максимальные гистерезисные потери имеют место при t=τ,
т.е. когда время действия силы, определяемое частотой, совпадает со временем
релаксации полимера, а критерий D=1.
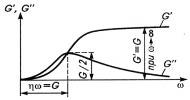
Рис. 11. Зависимость компонентов комплексного
модуля GI
и GII от частоты.
Критерий Деборы D
зависит не только от t
(или ω), но и от τ
при постоянном значении t,
что можно достигнуть путем изменения температуры (рис.12). Практически удобнее
характеризовать гистерезисные потери тангенсом угла потерь (tgδ),
зависимость которого от Т или ω
также выражается кривой с максимумом, расположенным близко к точке, где D=1.
Максимум на кривой tgδ-Т
объясняют следующим образом. При Т<Тс полимер застеклован, и
сегменты макромолекул не могут перемещаться при действии напряжения сдвига.
Таким образом, не затрачивается энергия на
преодоление внутреннего трения, и механические потери (tgδ)
малы. При более высокой температуре, в развитом высокоэластическом состоянии
полимера, когда подвижность сегментов велика, их тепловое движение позволяет им
упруго двигаться вслед за изменением направления сдвига, и потери на внутреннее
трение так же минимальны. В промежуточной области температур (максимум на
кривой) интенсивность теплового движения недостаточна для эффективного
преодоления внутреннего трения, хотя сегменты приобретают некоторую
подвижность. При этом сегменты не могут свободно двигаться, и затрачивается
механическая энергия на выведение их из равновесного состояния и возвращение к
равновесию. Деформация становится вязкоупругой, и вязкая компонента тратится на
преодоление внутреннего трения, что соответствует температурной области II
на термомеханической кривой (рис. 12).
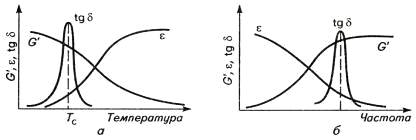
Рис. 12. Зависимость модуля упругости GI,
амплитуды деформации ε и тангенса
угла потерь tgδ
от температуры (а) и частоты деформации (б).
Температурно-частотные зависимости амплитуды
деформации и модуля упругости по сути являются такими же термомеханическими
кривыми полимеров, но полученными при их циклическом нагружении. На этих кривых
температура стеклования Тс совпадает с расположением максимума tgδ.
В точке начала резкого роста GI
или падения ε с увеличением ω,
можно также найти такую частоту действия силы, при которой эластомер начинает
вести себя как твердый стеклообразный полимер. Из этого следует, что любой
полимер с ростом частоты действия силы может оказаться в таких условиях, когда
флуктуационная сетка в нем не успеет перестроиться, и большая деформация не
успеет развиться. Такое явление называют механическим стеклованием.
Приведенные зависимости можно объединить,
построив график зависимости амплитуды деформации от температуры при разных
частотах или от частоты при разных температурах (рис. 13). С повышением
температуры образец при достижении Тс начинает размягчаться, и
амплитуда деформации ε при
заданной частоте действия силы ω1
возрастает. При дальнейшем росте температуры наблюдается переход в область
развитого высокоэластического состояния, и амплитуда деформации практически не
меняется, как и на термомеханической кривой при статическом нагружении. Если
увеличить частоту (ω2),
то при той же Тс флуктуационная сетка полимера не успевает
перестраиваться, и деформация оказывается незначительной. Рост частоты приводит
к росту температуры, при которой в полимере развиваются большие деформации,
т.е. к росту температуры стеклования. Поэтому говорят, что для полимеров
характерна относительность понятия «размягчение».
По рис.13 можно определить зависимость времени
релаксации от температуры. Действительно, точка перегиба на кривой
ε-ω
или точка максимума на кривой tgδ-ω
(в, г) соответствует условию t=τ.
Измерив частоту ω,
при которой происходит перегиб или возникает максимум, можно найти время
релаксации: τ=t=1/ω.
С ростом температуры время релаксации уменьшается, что указывает на рост
подвижности сегментов. Зависимость времени релаксации τ
от температуры Т выражается уравнением Эйринга-Френкеля: τ=АеU/RT,
где U - энергия
активации процесса релаксации. Получив ряд кривых, найдем частоты,
соответствующие температурам в точках максимума, по которым на рис 12 определим
времена релаксации при этих температурах (τ=1/ω).
В координатах lnτ-1/Т
это прямая линия (lnτ=lnA+U/RT),
что позволяет рассчитать энергию активации процесса релаксации как угол наклона
прямой tgα=U/R
(рис.14а).
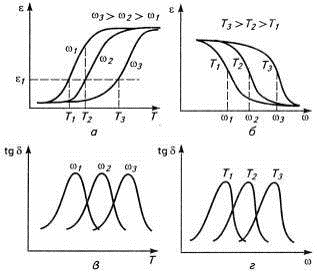
Рис.13. Изменение амплитуды деформации
ε
и тангенса угла потерь tgδ
от частоты ω и температуры Т.
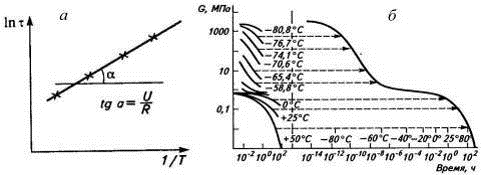
Рис.14. К определению энергии активации процесса
релаксации (а) и построению обобщенной кривой релаксации напряжения при 25оС
(б).
Для полимеров характерна также одинаковая форма
кривых зависимости ε-Т при разных
частотах и ε-ω при разных
температурах (рис.13), т.е. аналогия влияния температуры и частоты на их
механическое поведение. Одна и та же деформация может быть достигнута
изменением либо частоты (времени действия силы), либо температуры, то есть
изменения частоты или температуры оказывают аналогичное действие на
механические свойства. В этом суть принципа температурно-временной
эквивалентности, сформулированного Александровым и Лазуркиным в конце 30-х
годов:
lgto-lgt=β(103/То-103/Т,
где Т - температура, при достижении которой
деформация уменьшается в 2 раза по сравнению с То. Уравнение
отражает эквивалентность действия температуры и логарифма времени, что означает
одинаковое изменение механических свойств полимера при увеличении частоты,
например, в 10 раз, или снижении температуры на 8оС.
На рис. 13 заметна симметрия, позволяющая
«двигать» кривые вдоль оси абсцисс (по шкале времен или частот) вплоть до их
совмещения. Так же совмещаются кривые зависимости модуля упругости от времени
действия силы, полученные при разных температурах и равных временах, Если взять
за основу кривую при 25 оС и ее не двигать, то получим обобщенную
кривую (рис. 14б), подобную термомеханической, которую получили бы при
постоянной температуре, меняя интервал частот (ε-1/ω).
Она является зеркальным отражением кривой ε-Т.
Если измерить величину смещения кривых при разных температурах по шкале времен,
то окажется, что она одинакова для всех полимеров, если исходной температурой
каждого полимера будет температура его стеклования. Это значит, что Тс
является характеристической температурой, определяющей комплекс свойств
полимера, но для сравнения по вязкоупругим свойствам ее выбирать нельзя по
указанной ниже причине.
При Т=Тс свободный объем у всех
полимеров составляет порядка 2,5% от общего объема, что мало для перемещения
сегментов макромолекул при тепловом движении, поэтому прекращается их тепловое
перемещение и не развиваются большие обратимые деформации. Нагревание полимера
приводит к его тепловому расширению, свободный объем увеличивается, и сегменты
могут легко перемещаться (рис. 15).
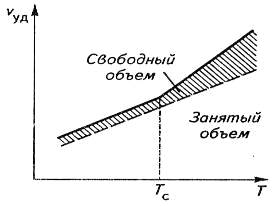
Рис.15. Зависимость удельного объема полимера (Vуд)
от температуры.
Температурное изменение свободного и занятого
объемов.
Поскольку все полимеры в эластическом состоянии
имеют примерно одинаковый коэффициент теплового расширения, приращение
свободного объема при нагревании на одинаковое число градусов выше Тс
оказывается примерно одинаковым. Из этого следует важная закономерность:
релаксационные свойства и способность деформироваться всех полимеров примерно
одинаковы при одинаковом удалении от Тс по шкале температур. Такой
вывод справедлив для интервала температур Тс-(Тс+100оС).
Например, полиметилметакрилат по вязкоупругости при температуре плюс 160оС
приближается к натуральному каучуку при температуре минус 22оС
(табл.3). Возможные различия в их свойствах могут быть обусловлены различиями в
молекулярной массе, которая у каучуков намного выше, чем у пластмасс.
Таблица 3. Характеристические температуры ряда
полимеров
|
Полимер
|
Тс, оС
|
(Тс+50), оС
|
|
Полиизобутилен
|
-75
|
-25
|
|
Полистирол
|
+100
|
+150
|
|
Полиметилметакрилат
|
+110
|
+160
|
|
Натуральный каучук
|
-72
|
-22
|
|
Полидиметилсилоксан
|
-120
|
-70
|
Способность к перемещению под действием тепловой
энергии различна для «свободных» сегментов (время оседлой жизни 10-6-10-4с)
и сегментов, «связанных» узлами флуктуационной сетки (время оседлой жизни 10-10-4с).
Приведенные времена релаксации являются усредненными для группы близких времен.
Поэтому говорят, что в полимерах существует широкий набор времен релаксации,
или непрерывный спектр времен релаксации. Поскольку понятие «сегмент» является
статистическим, спектр времен релаксации непрерывен, что определяется и разной
связанностью сегментов, и разной интенсивностью межмолекулярного
взаимодействия. Непрерывный спектр времен релаксации является наиболее полной
характеристикой физических свойств полимера.
. Стеклование и стеклообразное состояние
Переход полимера в стеклообразное состояние при
охлаждении называется структурным стеклованием и означает фиксацию определенной
структуры и определенного ближнего порядка, которые не меняются при дальнейшем
охлаждении. Фиксация структуры делает стеклообразный полимер неравновесным, что
приводит к зависимости Тс от скорости охлаждения. При медленном
охлаждении сегменты успевают перемещаться даже при приближении к Тс,
и требуется сильно охладить полимер для предотвращения вязких перестроек
структуры. Излом на кривой зависимости удельного объема от температуры (рис.
15) сместится в область более низких температур. Так, выдерживая
поливинилацетат при каждой температуре в одном опыте 0,02 ч, а в другом - 100
ч, получим значения Тс соответственно 32 и 23 оС.
В случае потери способности полимера к
высокоэластической или вязкотекучей деформации при большой скорости действия
силы (механическое стеклование), которая наступает при критерии Деборы D=1,
структура полимера не фиксируется, и тепловое движение сегментов не
прекращается. Однако скорость теплового движения оказывается меньше скорости
действия силы, и заметные деформации не успевают развиваться. Чем больше
скорость действия силы, тем выше Тс при механическом стекловании.
Чем выше скорость охлаждения, тем выше Тс при структурном
стекловании. Это значит, что стеклование является не фазовым (структурным), а
релаксационным переходом. Стеклование определяется не перестройкой
надмолекулярной структуры, а величиной отклика системы на внешнее воздействие.
Этим оно отличается от известных фазовых переходов - кристаллизации или
плавления.
К первой группе способов определения температуры
стеклования Тс относятся методы оценки свойств полимера
(термомеханические кривые и температурные зависимости теплоемкости, удельного
объема и показателя преломления). Вторая группа включает методы определения
температурных изменений подвижности сегментов (радиотермолюминесцентный, ЯМР и
диэлектрический). При малых степенях полимеризации Тс растет с
увеличением ММ до определенного предела, когда начинают перемещаться не
молекулы, а сегменты. Размер сегмента не зависит от ММ, поэтому Тс
остается постоянной. Повышается Тс с ростом полярности и плотности
сшивания полимера, а также гидростатического давления, когда оно уменьшает
свободный объем. Пластификаторы понижают Тс пропорционально их
объемной доле. Несмотря на отсутствие достаточного свободного объема,
стеклообразный полимер деформируется без разрушения на сотни процентов под
действием механических напряжений, увеличивающих свободный объем при неизменной
температуре, но не сокращается самопроизвольно после снятия нагрузки.
На первой стадии деформации (рис.16а) полимер
растягивается упруго за счет увеличения межмолекулярных расстояний или малого
смещения узлов флуктуационной сетки на доли процента или несколько процентов
(область I). На второй стадии
(область II) сегменты
перемещаются и ориентируются в направлении действия силы в вершине какого-либо
микродефекта. На образце возникает шейка, а на кривой - максимум, после чего
напряжение в образце несколько снижается. При дальнейшем растяжении весь
образец постепенно переходит в шейку, и этот процесс сопровождается выделением
тепла. При этом напряжение остается постоянным, на кривой возникает
горизонтальный участок, а величина деформации достигает сотен процентов.
Степень ориентации сегментов в шейке оказывается высокой.
При освобождении из зажимов образец не
сократится самопроизвольно, исчезнет только упругая деформация (доли
процентов), однако при нагревании выше Тс он сократится до длины,
близкой к исходной.
Таким образом, при растяжении стеклообразного
полимера возникает ориентация сегментов, частичное разворачивание
макромолекулярных клубков, а при нагревании макромолекулы свертываются.
Способность стеклообразных полимеров к большим деформациям называют явлением
вынужденной эластичности, а сами деформации - вынужденно-эластическими. После
формирования шейки процесс переходит в третью стадию (область III),
а величина деформации остается как на первой стадии.
Рис.16. Кривая напряжение-деформация для
стеклообразных полимеров (а) и изменение ее формы под влиянием температуры (б):
T1<T2<T3<T4<T5;
1-при T1<Тxp
(хрупкое разрушение); 5-приТ5>Тc
(без образования шейки).
Перемещение сегментов при
вынужденно-эластической деформации полимера происходит под действием
напряжения, однако определенный запас тепловой энергии в нем имеется и при
Т<Тс. С ростом температуры в области ниже Тс запас
тепловой энергии сегментов увеличивается, и требуется меньше внешней
механической энергии для перемещения сегментов, так как предел вынужденной
эластичности (предел текучести σт)
уменьшается (рис.16б). При понижении температуры увеличивается предел
вынужденной эластичности, а сама кривая вырождается, становится неполной.
Разрушение образца может произойти даже до достижения предела вынужденной
эластичности (кривая 1), при очень малых деформациях (доли процента), т.е.
полимер ведет себя как хрупкий, не способный к вынужденно-эластическим
деформациям. Для стеклообразного состояния решающим является вклад механической
энергии в релаксационные процессы и важную роль играет способность полимеров
выдерживать длительное воздействие механической нагрузки.
При нагружении стеклообразного полимера,
например полистирола, мгновенно увеличивается длина образца за счет развития
упругой деформации. Далее реализуется замедленная упругость, характеризующая
развитие вынужденно-эластической деформации, которая качественно аналогична
развитию высокоэластической деформации. При этом возможны два случая: либо
деформация перестает расти (затухающая ползучесть), либо она развивается
непрерывно (незатухающая ползучесть) как за счет истинно необратимой, так и за
счет замедленной вынужденно-эластической деформации без образования шейки.
Полимер может применяться как конструкционный материал в том случае, если под
действием заданной нагрузки в нем развивается только затухающая ползучесть,
позволяющая обеспечить относительное постоянство размеров детали в условиях
эксплуатации.
При исследовании стеклообразных полимеров,
например полиметилмет-акрилата, в условиях циклических деформаций обнаружили
релаксационные α-, β- и
γ-переходы,
на которые затрачивается много механической энергии, переходящей в теплоту за
счет внутреннего трения сегментов. При охлаждении до T<Tc
сегменты теряют подвижность, и потери уменьшаются. На повороты эфирных групп
(комнатная температура) идет меньше энергии, а дальнейшее охлаждение «замораживает»
их. При температуре около минус 267оС наблюдается γ-переход,
связанный с вращением метильных подвесок в сложноэфирных группах полимера. Эти
переходы называются вторичными релаксационными переходами и существенно влияют
на механические свойства полимера, особенно его хрупкость и стойкость к ударным
нагрузкам.
Все стеклообразные полимеры более стойки к удару
и менее хрупки по сравнению с силикатными стеклами. Хрупкость - это свойство
стеклообразных полимеров разрушаться при малых деформациях (до 1%). Полимер
будет хрупким, когда время до разрушения много меньше, чем время релаксации,
что исключает перегруппировку сегментов под действием силы. Хрупкость оценивают
по величине температуры хрупкости Тхр, которую определяют по точке
пересечения зависимостей σт
и σр
от температуры (рис. 17). Интервал вынужденной эластичности (Тс-Тхр)
является областью безопасного применения пластмасс как конструкционного
материала, а охлаждение полимера ниже Тс может привести к переходу в
хрупкое состояние. Температура хрупкости, как и Тс, зависит от
молекулярной массы. Из-за малой молекулярной массы Тс и Тхр
олигомеров совпадают. С ростом молекулярной массы увеличиваются температурные
интервалы вынужденной эластичности (Тс-Тхр) и
высокоэластичности (Тт-Тс), но ухудшается способность
полимеров к необратимым деформациям, что выражается в росте Тт.
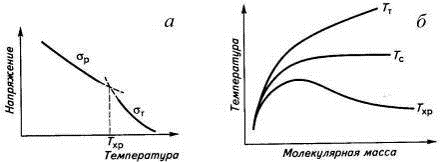
Рис. 17. Температурные зависимости прочности и
вынужденной эластичности полимера (а) и взаимосвязь между молекулярной массой
полимера и температурами его текучести, стеклования и хрупкости (б).
. Механические свойства кристаллических
полимеров
Релаксационные свойства кристаллических
полимеров ярко проявляются в кинетических особенностях процесса их
кристаллизации:
1. Зависимость температуры плавления от
условий кристаллизации: чем меньше скорость кристаллизации и дефектность
кристалла, тем выше температура его плавления. Повышают регулярность кристаллов
«отжигом» (вторичной перекристаллизацией) закристаллизовавшегося полимера.
2. Наличие интервала температур плавления,
который составляет градусы и даже десятки градусов. Сначала расплавится часть
полимера, а при дальнейшем нагревании - другая, более упорядоченная его часть,
что связано с неоднородностью структуры и разным размером кристаллитов.
. Температура плавления Тпл
превышает на несколько градусов и даже десятков градусов в зависимости от
скорости нагревания или охлаждения температуру кристаллизации Ткр
полимеров. Кривые охлаждения и нагревания кристаллического полимера образуют
петлю, похожую на петлю гистерезиса при намагничивании и размагничивании
железного сердечника. Несовпадение температур и гистерезис кристаллизации -
следствие релаксационных процессов при создании кристаллической структуры (рис.
18).
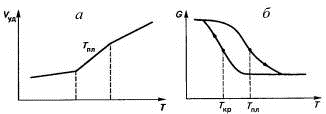
Рис. 18. Температурная зависимость удельного
объема (а) и модуля упругости (б) полимера в области плавления (нагревание и
охлаждение показаны стрелками).
4. Протяженность интервала температур, в
котором происходит плавление, зависит от Ткр. Чем выше Ткр
и ближе она к Тпл, тем медленнее идет кристаллизация, меньше
возникает дефектов в кристаллической структуре и однороднее по размерам
оказываются возникшие кристаллиты. Все это обуславливает сужение интервала
температур плавления с ростом Ткр, то есть с уменьшением
переохлаждения кристаллизующегося расплава. Отсюда - необходимость правильного
выбора метода их определения.
Температура плавления ряда полимеров ниже
комнатной, а НК при Тпл=28 оС имеет максимальную скорость
кристаллизации при -25 оС. Полимеры со стереорегулярным строением,
не способные кристаллизоваться при заданной температуре, легко кристаллизуются
при растяжении вследствие ориентации макромолекул и, следовательно,
упорядочения в расположении сегментов. В растянутом полимере возникает кристаллическая
структура, которая ограничивает подвижность сегментов, повышает его жесткость и
модуль упругости. С увеличением аморфной части повышается способность полимера
к деформации. Такое сочетание жесткости и податливости у кристаллического
полимера делает его менее хрупким и придает ему определенное сходство со
стеклообразными полимерами по механическому поведению.
Кривая напряжение-деформация кристаллического
полимера делится также на участки, отражающие стадии процесса растяжения (рис.
19). Первая стадия (линейный участок I)
характеризует полностью обратимую деформацию неразрушенной структуры
кристаллитов, которая приводит к увеличению свободного объема полимера и
достигает нескольких или нескольких десятков процентов. Вторая стадия (участок II)
начинается после достижения предела текучести σт,
совпадающего с моментом микроразрушения в наиболее дефектном месте, проходит
через все стадии образования шейки при неизменном напряжении с выделением тепла
за счет внутреннего трения и сопровождается перестройкой кристаллической
структуры.
По форме кривой эта стадия напоминает
пластическую деформацию металлов, поэтому напряжение в образце и называют
пределом текучести σт.
Формирование шейки на образце проходит через стадии рекристаллизации сегментов,
частичного разрушения кристаллитов и пластической деформации кристаллитов и
сферолитов. Третья стадия (участок III)
включает деформацию сформировавшейся шейки вплоть до разрыва образца. Предел
текучести увеличивается с ростом скорости деформации или со снижением температуры,
что может привести к разрушению образца на второй стадии деформации до
завершения формирования шейки (кривая 2), и даже к хрупкому разрушению при σт
ниже предела прочности (кривая 1). Кривая 4 имеет S-образную
форму и может быть получена и для частично кристаллического полимера
(полиэтилена низкой плотности) при малой скорости деформации, и для
эластомеров, кристаллизующихся при деформации (вулканизатов НК). В первом
случае кристаллический полимер, подобно аморфному эластомеру (рис. 16), растягивается
без образования шейки вплоть до разрыва, а во втором - при достижении
определенного удлинения (около 400%) вследствие интенсивной кристаллизации
резко возрастает напряжение. Вблизи точки разрыва напряжение в вулканизате
кристаллизующегося эластомера может в несколько раз (иногда на порядок)
превышать напряжение в некристаллизующемся эластомере с сохранением S-образной
формы кривой.
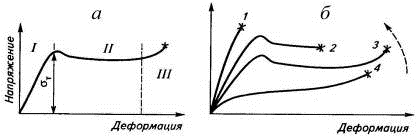
Рис. 19. Кривая напряжение-деформация
кристаллического полимера (а), и изменение ее формы под влиянием температуры и
скорости деформации (б): стрелка - направление уменьшения температуры или роста
скорости деформации; звездочки - точки разрыва образцов.
По релаксации напряжения и ползучести
кристаллические полимеры при температуре ниже Тпл и напряжениях ниже
σт,
когда шейка не может образоваться, аналогичны стеклообразным полимерам.
Повышенная доля аморфной части увеличивает при релаксации в условиях Т>Тс
падение напряжения, а при Т>Тпл, когда полимер расплавлен,
напряжение может быстро упасть до нуля. Предельная деформация ползучести так же
больше, чем в аморфных полимерах, а при превышении некоторого предела
напряжения ползучесть становится незатухающей. Этот процесс аналогичен
растяжению без образования шейки. В нагруженном кристаллическом полимере со
временем может произойти разрастание наиболее опасного микродефекта с
образованием шейки, и он может внезапно быстро удлиниться в десятки раз, но не
разрушиться полностью.
Полимер при этом упрочняется, и ползучесть
прекращается, однако самопроизвольное удлинение детали или конструкции под
нагрузкой равносильно ее разрушению. Ползучесть в кристаллизующемся эластомере
определяется тем, достигает ли общая эластическая деформация величины,
необходимой для начала кристаллизации, или нет. В первом случае ползучесть
развивается как в некристаллизующемся эластомере, а во втором - кристаллиты
прекращают дальнейшее ее развитие. Циклические деформации, как и в
стеклообразных полимерах, выявляют ряд максимумов потерь в области Т<Тс,
которые дают важную информацию о структуре кристаллических полимеров. Максимум
при Т=Тс показывает долю аморфной части, а широкий размытый максимум
в области Тс<Т<Тпл вблизи Тпл связан с
нарушениями кристаллической решетки и наличием проходных цепей между ламелями.
Высокоэластическая деформация эластомеров,
вынужденно-эластическая деформация стеклообразных полимеров и пластическая
деформация кристаллических полимеров приводят к развертыванию макромолекулярных
клубков и ориентации макромолекул в направлении действия силы. В ориентированных
эластомерах можно охлаждением до температуры ниже Тс зафиксировать
это состояние. Все ориентированные полимеры имеют одно общее свойство: их
прочность и модуль упругости при растяжении в направлении ориентации намного
больше, а в перпендикулярном направлении - намного меньше, чем у исходного
неориентированного полимера.
Кристаллический полимер (полипропилен) при
нормальной температуре и скорости растяжения дает типичную кривую 1 (рис.20).
После нагрева пленки до температуры на 10-20 оС ниже Тпл
получим кривую 2 в режиме вынужденно-эластической деформации без образования
шейки. Если охладим ориентированную пленку до комнатной температуры и повторим
ее растяжение, то получим кривую 3 с сильно выросшей прочностью. Аналогично
этому при растяжении хрупкой пленки из полистирола получим исходную кривую 4,
затем - кривую 5 его ориентационной вытяжки и кривую 6 ударостойкого полимера.
Последнее подтверждается возросшей площадью под кривой σ-ε
при
20 оС, которая является мерой работы разрушения образца.Ориентированные
полимеры получают в промышленности в результате ориентационной вытяжки исходных
неориентированных волокон или пленок при минимальных напряжениях и таких
значениях температуры и скорости, чтобы предотвратить образование шейки.
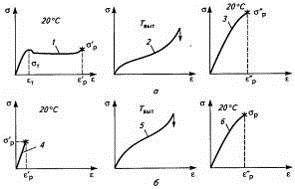
Рис 20. Влияние вытяжки кристаллического (а) и
аморфного (б) полимеров на растяжение их пленок: 1,4-растяжение исходных
пленок; 2,5-ориентационная вытяжка пленок; 3,6-растяжение ориентированных
пленок.
Это достигается максимальным приближением
температуры к Тс или Тпл или даже нагреванием чуть выше
температуры размягчения. Чем больше увеличивается прочность полимера в
направлении ориентации, тем больше она снижается в перпендикулярном
направлении. Для обеспечения равнопрочности пленок их ориентируют в двух
взаимно перпендикулярных направлениях. Такие пленки являются хорошим
упаковочным материалом и при нагревании частично сокращаются (релаксируют),
плотно охватывая упакованный предмет.
. Теории разрушения и долговечность полимеров
Прочность характеризует свойство материала
сопротивляться разрушению под действием механических напряжений. Прочность
полимеров - это их важная техническая характеристика. Расчеты показали, что
теоретическая прочность у неориентированных полимеров составляет 20-50 МПа, а
после ориентации увеличивается примерно в 10 раз. Однако экспериментально
определенная прочность как напряжение, вызывающее разрушение образца, во много
раз меньше ее теоретического значения. Это является следствием существования в
их реальной структуре дефектов, обусловленных методикой получения полимерного
материала и появившихся в результате изготовления образца для испытаний. В
последнем случае чаще всего возникают поверхностные дефекты, которые наиболее
опасны как концентраторы дополнительных напряжений.
Перенапряжения на дефектах инициируют рост
трещин, приводящих к разрушению образца. Чем более опасен дефект, тем больше
перенапряжения и меньше прочность. Гауссова симметрия типичных кривых
распределения всех полученных значений прочности позволяет оценивать ее как
среднее из них, соответствующее максимуму на кривой (рис. 21). Чем тоньше
образец, тем меньше площадь его общей поверхности и меньше вероятность
нахождения на ней дефекта. Поэтому для разрушения тонких образцов требуется
большее напряжение и одновременно у них расширяется кривая распределения
(разброса) значений прочности, так как даже малый дефект может больше снизить
прочность. Зависимость прочности от площади поперечного сечения или формы
образца объясняют проявлением масштабного фактора.
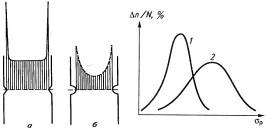
Рис.21. Концентрация напряжений с острой (а) и
сглаженной (б) вершинами на микродефектах образцов и кривые распределения
значений прочности: 1- волокна большого диаметра; 2- очень тонкие волокна.
Накопление механической энергии в деформируемом
образце в виде энергии упругости провоцирует зарождение на наиболее опасном
дефекте магистральной трещины, которая затем разрастается, разделяя образец на
части. В процессе роста трещины энергия, запасенная в образце, тратится на
образование новой поверхности и на всевозможные перемещения структурных
элементов по пути ее движения, которые приводят к рассеянию энергии за счет
внутреннего трения в виде теплоты. Наиболее простым является разрушение при
малой деформации, когда вся запасенная энергия образца идет на образование
новой поверхности. Для этого случая Гриффитом предложена теория хрупкого
разрушения, так как наименьшие деформации, разрушающие полимер, наблюдаются при
переходе его из стеклообразного состояния в хрупкое:
Fp=(2aE/πlo)1/2,
где Fp
-
прочность,
a - удельная энергия поверхности, возникшей при
разрыве;
lo
-
длина микродефекта, E-модуль
упругости (модуль Юнга).
Формула правильно описывает ряд закономерностей,
в частности влияние глубины дефекта (специального надреза) на прочность. Расчет
по этой формуле удельной энергии поверхности a приводит к
значениям, которые примерно в 100 раз превышают экспериментально найденные
значения. Различия объясняются тем, что даже при хрупком разрушении полимеров
относительно велика доля энергии, затрачиваемой на перемещение структурных
элементов вблизи поверхности разрушения, т.е. на некоторое деформирование
полимера растущей трещиной.
Часто в вершине трещины развивается
вынужденно-эластическая деформация (рис. 22) - перемещение и ориентация
сегментов, их последующее разрушение и формирование ориентированных тяж
(волокон). Трещина, заполненная ориентированными тяжами полимера, называется
микротрещиной (а), которая, в отличие от обычной трещины, способна потом
«залечиваться». Ориентация полимера в микротрещинах изменяет показатель
преломления, и в местах их скопления из-за сильного рассеивания света возникает
металлический блеск. Поэтому микротрещины еще называют трещинами серебра.
Полимеры в высокоэластическом состоянии к моменту разрыва достигают
значительных деформаций, что сильно влияет на механизм их разрушения.
Первоначальная трещина с острой вершиной постепенно расширяется, но не растет,
так как накопленная механическая энергия из-за низкого модуля еще мала, перенапряжения
частично релаксируют, а полимер в вершине трещины ориентируется и упрочняется,
образуя множество тяжей (б).
Ориентированные тяжи формируются в вершине
трещины до тех пор, пока напряжение в уменьшающемся поперечном сечении
достигает критического значения для быстрого прорастания магистральной трещины,
разрушающей образец. В высокоориентированном полимере очень велико
сопротивление разрастанию трещин поперек образца, в десятки и сотни раз
превосходящее сопротивление разрастанию продольных трещин. Поэтому трещина
постоянно меняет направление роста (в), а в месте разрушения образуется
«бахрома» из мельчайших ориентированных волокон.
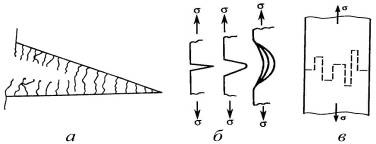
Рис. 22. Строение и распространение трещин в
хрупком полимере (а), эластомере (б) и высокоориентированном полимере (в).
Разрушение полимеров длительно действующей
нагрузкой происходит при напряжениях, значительно меньших критического
значения. Поэтому, кроме прочности, материал характеризуют долговечностью -
временем, в течение которого он не разрушается под действием заданного
напряжения. Тепловая энергия распределяется неравномерно, случайные флуктуации
и сосредоточение энергии в определенный момент на более напряженной цепи
приводят к ее разрыву. Со временем число разрывов накапливается, и образующийся
дефект разрастается настолько, что происходит зарождение магистральной трещины,
приводящей к разрушению образца. Такой механизм разрушения заложен в
кинетической теории прочности: медленное накопление термомеханодеструкции
отдельных макромолекул создает условия для разрушения всего образца. Чем больше
напряжение σ, действующее на
образец, тем больше вероятность разрыва химической связи при данной
температуре. С другой стороны, при одинаковом напряжении вероятность разрыва
химической связи тем больше, чем выше температура, т.е. чем значительнее
флуктуации тепловой энергии, что отражено в формуле Журкова:
τр=τое(Uo-γσ)/RT,
где Uo
- энергия активации разрыва связи при напряжении, стремящемся к нулю;
τр
- время ожидания разрыва связей (долговечность полимера);
τо
- период тепловых колебаний атомов (для твердого тела 10-12-10-13с).
Уравнение предполагает линейную зависимость lgτр
от σ
или от обратной температуры 1/Т, при которой прямые сходятся в одной точке,
называемой полюсом (рис.23).
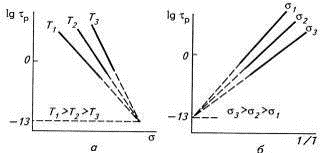
Рис.23. Зависимость долговечности τр
от напряжения σ при разных
температурах (а) и от обратной величины температуры (1/Т) при разных
напряжениях (б).
Увеличение напряжения облегчает преодоление
энергетического барьера механодеструкции. Произведение γσ
есть величина энергии, которая снижает энергетический барьер разрыва связи, а γ
- структурный коэффициент, характерный для данного полимера. Понижением
напряжения можно достигнуть такого его значения (рис. 24а), при котором разрушение
полимера практически не будет ускоряться. Напряжение, при котором долговечности
напряженного и ненапряженного полимеров равны, называется безопасным.
По уравнению Бартенева, для эластичных сшитых
полимеров (резин):
τр=Вσ-b,
где В и b
- константы, зависящие от природы полимера. Константа В отражает еще
температурную зависимость долговечности:
τр=Воσ-bеU/RT.
В соответствии с этим уравнением lgτр
линейно уменьшается с ростом lgσ
(рис. 24б). Достоверные зависимости долговечности от напряжения или температуры
для разных полимеров помогают создать базу для прогнозирования
работоспособности изделий и конструкций из них. Линейная зависимость lgτр
от σ
или от lgσ
позволяет вести интерполяцию, т.е. определение долговечности в пределах
экспериментально исследованного интервала напряжений, а также экстраполяцию за
его пределами.
Динамическая усталость (утомление) полимера -
это снижение его прочности под действием многократных периодических нагрузок
или деформаций, с чем приходится часто сталкиваться в технике и быту. Например,
сумка из поливинилхлоридной пленки растрескивается по месту сгиба, хотя
остальная часть ее не имеет признаков разрушения. Резина амортизатора,
подвергающаяся периодическим деформациям, разрушается намного раньше, чем в
напряженном состоянии, но без динамических нагрузок.
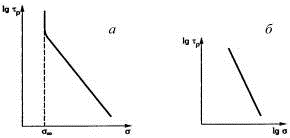
Рис.24. Зависимость долговечности от напряжения
для пластмасс (а) и сетчатых эластомеров (б).
Существуют два основных режима нагружения
полимеров при испытании на динамическую усталость: εо=const
и εср=const
(режим 1, аналогичный условиям опыта по релаксации напряжения) и σо=const
и σср=const
(режим 2, аналогичный испытанию на ползучесть). При утомлении по первому режиму
снижается как σср,
так и амплитудное значение напряжения σо.
Одновременно развивается и процесс утомления, проявляющийся в снижении
прочности. Когда прочность окажется равной суммарному напряжению (σср+σо),
произойдет разрушение. Сопротивление полимера утомлению удобно характеризовать
не временем до разрушения, а числом циклов деформации до разрушения Np.
При испытании по второму режиму со временем увеличивается как εср,
так и амплитудное значение деформации εо.
Как и в первом режиме, при достижении прочностью образца величины заданного
суммарного напряжения происходит разрушение. Число циклов деформации до
разрушения является мерой динамической выносливости.
Рассмотрим поведение пластмассы или жесткой
резины в разных режимах утомления. Чтобы испытания прошли в разумно короткий
срок, зададим в режиме 1 достаточно большое значение амплитуды деформации
εо. Поскольку
пластмасса - жесткий материал, и ее модуль велик, то в образце возникает
значительное напряжение σо,
что обусловит большую работу деформации. Приближенно можно считать работу
деформации А=σоεо/2.
Чем больше подводимая в каждом цикле работа, тем быстрее в полимере идут
структурные изменения, и развивается процесс его утомления. Поэтому значение Np
будет невелико, т.е. образец разрушится быстро. В режиме 2 даже при большом
заданном значении σо
из-за большого модуля упругости пластмассы величина εо
и работа деформации А в каждом цикле окажутся небольшими (образец мало
деформируется), а значение Np
будет большим.
При испытании динамической выносливости мягкой
резины по режиму 1 зададим также большую амплитуду
εо. Однако из-за
меньшего модуля резины, чем у пластмассы, в образце развиваются небольшие
напряжения, в каждом цикле к нему подводится небольшая работа, и он долго не
разрушится. Обратная картина имеет место при испытании мягкой резины по режиму
2. Задаем большое σо
и при малом значении модуля резины получим большое εо
и большую работу А, подводимую в каждом цикле. Таким образом, пластмассы при
циклических нагрузках лучше работают в режиме σо=const
и быстрее разрушаются в режиме εо=const,
а резиновые (низкомодульные) образцы более долговечны при испытании в режиме
постоянной деформации (εо=const).
Снижение прочности при утомлении обусловлено
многими факторами, роль которых зависит от типа полимера и условий испытания:
1. Механохимическая деструкция перенапряженных
макромолекул как следствие микронеоднородности микроструктуры (трещины и
механические включения, захлесты и переплетения макромолекул).
2. В стеклообразных и кристаллических
полимерах - в результате накопления и разрастания микродефектов (мелкие трещины
или пустоты, микротрещины, ослабляющие поперечное сечение образца).
. Процессы перегруппировки
надмолекулярных структур, ориентации и рекристаллизации, которые необратимо
изменяют размеры образца, «разнашивают» его в процессе утомления.
. Разогрев, достигающий в шине 100 оС
при больших тепловыделениях и затрудненности отвода тепла (летом при быстром
движении автомобиля). Разогрев может приблизить пластмассу к области D=1,
где есть максимум механических потерь, что может привести к тепловому разрушению
образца.
. Процессы окисления (старения),
протекающие при утомлении в результате подвода механической энергии и
саморазогрева образца.
По аналогии с долговечностью, число циклов до
разрушения (Nр)
жесткого полимера линейно зависит от амплитуды напряжения. При малой амплитуде
напряжения (σ=σ∞)
Nр
становится очень большим. Поэтому безопасной амплитудой напряжения считают
20-30% прочности полимера:
σ∞=(0,2-0,3)σр,
а кривую на рис.25а называют кривой Веллера.
Аналогичная зависимость для эластомеров выражается эмпирической формулой
Резниковского (рис.25б):
где σp
- прочность полимера;
β - коэффициент
выносливости, показывающий темп снижения прочности при утомлении. Чем больше β,
тем лучше сопротивляется полимер развитию усталостных процессов в нем. Даже
менее прочный полимер, имеющий большее значение β,
может быть динамически более выносливым. Резина, поведение которой
характеризуется кривой 2, при малых амплитудах напряжения может выдерживать
больше циклов до разрушения, чем более прочная резина (кривая 1), но обладающая
меньшим коэффициентом выносливости β.
Прогноз работоспособности полимеров по приведенным зависимостям эксплуатации
весьма приблизителен.
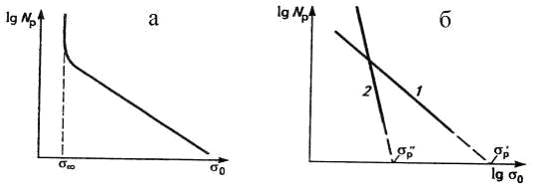
Рис. 25. Зависимость числа циклов до разрушения
(lgNр)
от амплитуды напряжения для пластмассы (а) и резины (б).
Линейная зависимость статической и динамической
долговечности от времени или от числа циклов до разрушения соблюдается в
сравнительно узком интервале напряжений. Поэтому прогнозы работоспособности
полимеров в разных условиях эксплуатации весьма приблизительны.
. Реология расплавов и растворов полимеров
Реология изучает свойства полимеров в
вязко-текучем состоянии и является теоретической основой их переработки. Зная
основы реологии, можно рассчитать скорость движения расплава полимера при
заполнении пресс-форм и обеспечить получение изделий нужного качества.
Некоторые полимеры невозможно перевести в вязко-текучее состояние из-за
склонности к термодеструкции, и их перерабатывают в виде растворов. Полную
реологическую характеристику раствора или расплава полимера дают зависимости
напряжения сдвига τ (Н/м2)
от скорости сдвига γ (с-1),
которые называют кривыми течения (рис. 26). Наиболее простая - пропорциональная
зависимость между ними, представляющая собой закон Ньютона (τ=ηоγ),
когда вязкость ηо
(Па.с) остается постоянной. Обычно с ростом напряжения сдвига
скорость течения растет быстрее, чем это следует из закона Ньютона, и такие
полимеры называют псевдопластичными жидкостями. Системы с наполнителями,
образующими достаточно упругие цепочечные структуры, сначала не текут, т.е.
напряжение сдвига растет, а скорость течения остается нулевой. Возникает
некоторое напряжение сдвига - предел текучести, после которого система течет
либо как ньютоновская, либо как неньютоновская жидкость. Полимеры, течение
которых начинается при любом напряжении сдвига, называются вязкими (кривые 1 и
2), а полимеры, обладающие предельным напряжением сдвига - пластичными (кривые
3 и 4).
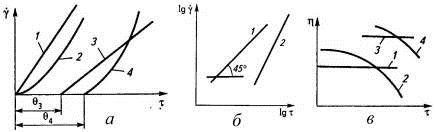
Рис. 26. Кривые течения полимеров (а, в) и
зависимости скорости сдвига от напряжения сдвига в логарифмических координатах
(б): 1-ньютоновская жидкость; 2-псевдопластичная жидкость; 3-идеально
пластичное тело; 4-неидеально пластичное тело.
Деформация идеально упругого тела описывается
законом Гука (деформация пропорциональна приложенному напряжению). Деформация
идеально вязкого тела описывается законом Ньютона (скорость деформации
пропорциональна приложенному напряжению). Большинство тел не являются идеально
упругими или идеально вязкими, и для них типична кривая течения 2. Для ее
описания применяется формула τ=ηоγп,
названная степенным законом течения. Для сравнения его с законом Ньютона
необходимо изобразить экспериментальные данные в логарифмических координатах:
lgτ=lgηо+lgγ
(закон Ньютона)
lgτ=lgηо+nlgγ
(степенной закон).
Оба уравнения в логарифмических координатах
выражаются прямой линией (рис. 26б), но тангенс угла наклона первой кривой
равен единице, а второй - n,
называемый индексом течения, который мало зависит от напряжения сдвига и
считается постоянным при изменении скорости сдвига на 2-3 порядка.
Ускорение течения с ростом напряжения сдвига
обусловлено изменениями структуры полимера, приводящими к снижению начальной
ньютоновской вязкости до эффективной вязкости, которое называют аномалией
вязкости. Для определения вязкости используют вискозиметры, подразделяющиеся по
конструкции на капиллярные и ротационные. В технологической практике для оценки
вязкости применяют также показатель текучести расплава, который определяют в
граммах полимера, прошедшего через стандартный капилляр за определенное время
при определенных значениях температуры и давления. По теории Эйринга-Френкеля,
жидкость течет перескоком отдельных молекул в соседнее положение, если оно
свободно, т.е. для этого необходима «дырка» по соседству с «горячей» молекулой.
Молекулы перемещаются в направлении действия напряжения, а «дырки» - в
обратном. Поскольку вязкое течение полимеров осуществляется путем
последовательного перемещения сегментов, то макромолекула может двигаться
только рептационным способом (движением пресмыкающихся) как бы по коридору,
образованному совокупностью «дырок». Сегменты перестают совершать тепловые
перемещения, когда общий объем всех «дырок» (свободный объем) становится равным
2,5% от общего объема полимера.
С увеличением скорости течения полимера растет
(до 500%) эластическая деформация цепей и удлиняются (в 6 раз)
макромолекулярные клубки. При некотором предельном значении упругой энергии
сегменты перестают участвовать в перескоках, поскольку напряжение сдвига
уравновешивается упругой силой клубка, стремящегося вернуться в исходное
положение. Это сопровождается у полимера с узким ММР потерей контакта струи
расплава со стенками канала и явлением скачкообразного роста его расхода,
получившим название срыва струи. В полимере с широким ММР только часть наиболее
высокомолекулярных клубков начинает перемещаться в потоке как единое
образование, снижая потери энергии на внутреннее трение и увеличивая скорость
течения, что по закону Ньютона указывает на снижение вязкости. Поэтому полимеры
с узким ММР вадут себя как ньютоновские жидкости, а с широким ММР - как
псевдопластичные жидкости с аномалией вязкости.
В растворах полимера общая деформация и
ориентация клубков может и не приводить к потере ими способности к течению. При
некотором напряжении сдвига, когда надмолекулярные структуры предельно
разрушены, структура раствора больше не меняется, и вязкость снова перестает
зависеть от напряжения сдвига. При этом можно получить так называемую «полную
кривую течения» (рис. 27). В областях I
и III раствор течет, как
ньютоновская жидкость, но надмолекулярная структура этих состояний настолько
различна, что по вязкости они могут разгичаться в тысячи раз.
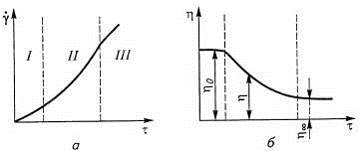
Рис. 27. Зависимость скорости сдвига γ
(а)
и вязкости η (б) от напряжения
сдвига τ:
I и III
- области напряжения сдвига, соответствующие течению с постоянной вязкостью; II
- область структурной вязкости.
По теории Эйринга-Френкеля, вязкость экспоненциально
зависит от температуры: ηо=АеU/RT,
а энергия активации вязкого течения определяется из зависимости lnηо-1/Т.
Для полиэтилена она равна 46-53, полистирола - 92-96, поливинилхлорида - 146 и
ацетата целлюлозы - 292 кДж/моль. Это значит, что с ростом температуры вязкость
быстрее снижается у поливинилхлорида, чем у полиэтилена. Но даже у полиэтилена
вязкость уменьшается почти в 10 раз при повышении температуры на 60-80оС.
Поэтому при переработке расплавов полимеров стремятся повышать температуру,
насколько это позволяет склонность к термодеструкции. У ацетата целлюлозы
энергия вязкого течения сопоставима с энергией разрыва химической С-С-связи
(250-334 кДж/моль). Это значит, что при переработке расплава ацетата целлюлозы
течение и термодеструкция пойдут с примерно равными скоростями, поэтому во
избежание термодеструкции пленку следует получать из раствора полимера. В
отличие от энергии активации, вязкость полимера определяется совокупными
затратами на перемещение всех сегментов макромолекулы и поэтому зависит от их
числа. Вязкость олигомеров пропорциональна ММ (ηо=КМW),
а с ростом ММ и гибкости макромолекул возникают их зацепления и захлесты,
усиливающие зависимость вязкости от ММ: ηо=КМW3,4
(рис. 28).
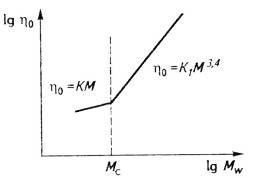
Рис. 28. Зависимость начальной ньютоновской
вязкости от среднемассовой молекулярной массы в двойных логарифмических
координатах.
Если построить экспериментальные зависимости
начальной ньютоновской вязкости ηо
от среднемассовой молекулярной массы МW
полимеров в двойных логарифмических координатах, то точка перегиба на них
показывает значение молекулярной массы (Мс), при которой возникает
флуктуационная сетка, образованная узлами переплетений или ассоциатами
сегментов (табл. 4). Вязкость расплавов промышленных полиамидов и полиэфиров
(ММ десятки тыс.ед.) находится в диапазоне десятков Па.с,
полиэтилена и полипропилена (ММ до сотен тыс.ед.) - 103-105
Па.с, а каучуков (ММ от сотен тыс до миллионов ед.) - 106-107
Па.с, что обуславливает большие затраты энергии на их переработку.
Рост Тт с увеличением ММ уменьшает область вязкотекучего состояния,
что ограничивает возможности переработки полимера, поскольку небольшие
колебания температуры приводят либо к потере текучести, либо к заметной
термодеструкции.
Таблица.4. Молекулярная масса возникновения
флуктуационной сетки ряда промышленных полимеров:
|
Полимер
|
Мс, тыс. ед.
|
Полимер
|
Мс, тыс. ед.
|
|
Полистирол
|
31,2
|
Полиизобутилен
|
15,2
|
|
Полиметилметакрилат
|
27,5
|
Цис-полиизопрен
|
10,0
|
|
Поливинилацетат
|
24,5
|
1,4-Полибутадиен
|
5,9
|
Эластичность расплавов следует учитывать при
переработке полимеров в изделия, т.к. может проявить себя в следующих
нежелательных формах:
· аномалия вязкости, постепенное
нарастание напряжений в полимере и потеря текучести при больших напряжениях
сдвига - прямое следствие эластических деформаций;
· сокращение длины и «разбухание»
струи, искажение формы струи из капилляра вискозиметра или головки экструдера,
затрудняющее расчет отверстий для требуемого профиля экструдата, - прямое
следствие релаксации эластических деформаций при выходе из капилляра.
Как следует из изложенного, при синтезе и
переработке полимеров большое значение имеют процессы их взаимодействия с
низкомолекулярными жидкостями. Взаимодействие между макромолекулами и
молекулами растворителя, называют сольватацией. При наличии сродства между ними
происходит самопроизвольное диспергирование друг в друге (растворение),
начинающееся с быстрого проникновения в фазу полимера молекул растворителя.
Макромолекулы за это время не успевают перейти в фазу растворителя, и полимер
набухает, поглощая растворитель. Увеличение массы (Qм)
или объема (Qо)
полимера в результате набухания его в определенных условиях (форма и размеры
образца, продолжительность, температура и др.) называют степенью набухания:
Qм=(mн-mо).100/mо%;
Qо=(Vн-Vо).100/Vо%.
Растворитель быстро проникает по механизму
капиллярного всасывания в области рыхлой упаковки макромолекул, раздвигая их
(внутриструктурное набухание). Одновременно с заполнением пор, пустот и
каналов, растворитель медленно диффундирует в надмолекулярные образования
полимера и разрушает их (межструктурное набухание).
Скорость набухания υ
зависит от скорости диффузии растворителя в полимер и может оцениваться углом
наклона зависимости степени набухания от времени к оси абсцисс или определяться
по увеличению массы Δm
(υм)
или степени набухания ΔQ
(υQ)
образца полимера за данный отрезок времени Δτ:
υм=(m2-m1)/(τ2-τ1)=Δm/Δτ;
υQ=(Q2-Q1)/(τ2-τ1)=
ΔQ/Δτ.
Степень набухания, при которой появляется
горизонтальный участок кривой, называется максимальной (Qмакс)
или равновесной. Полимер набухает значительно медленнее в парах, чем в
жидкости, но максимальная степень набухания при этом не изменяется. Скорость
проникновения растворителя от поверхности вглубь зависит от степени
термодинамического сродства растворителя и полимера, температуры, уровня
межмолекулярного взаимодействия в полимере и других условий процесса. В начале
процесса набухания концентрация растворителя уменьшается от поверхности к
центру, образец сильно деформируется, и в нем возникают внутренние напряжения,
вызывающие разрыв наиболее растянутых участков макромолекул с образованием
свободных радикалов, способных инициировать реакции деструкции. Интенсивная
окислительная деструкция со снижением прочности полимера, усиливающаяся с повышением
температуры, наблюдается в том случае, если сам растворитель легко окисляется.
Процесс набухания можно представить как одностороннее смешение, а набухший
полимер - как две находящиеся в равновесии фазы с поверхностью раздела между
ними.
Неограниченное набухание самопроизвольно
переходит в растворение, что характерно для линейных аморфных полимеров с
невысокой степенью полимеризации. Степень набухания, после которой начинается
растворение, должна быть достаточной для полной сольватации макромолекул и их
отделения от остальной массы набухающего полимера. Вокруг набухающего образца
образуется слой раствора полимера. Диффузия макромолекул постепенно выравнивает
их распределение по всему объему растворителя с образованием однофазной
гомогенной системы (рис. 29а). В точке а скорость растворения становится равной
скорости набухания, а в точке в она начинает превышать скорость набухания, и
масса образцов уменьшается. Между этими точками образцы имеют максимальную
степень набухания Qмакс
в течение времени Δτ. Оба
показателя уменьшаются со снижением ММ и разветвленности макромолекул и
межмолекулярного взаимодействия в полимере. Ограниченное набухание не
сопровождается растворением из-за их низкого термодинамического сродства или
химического связывания макромолекул в сетчатую структуру (рис. 29б).
С ростом густоты сетки степень и скорость
набухания снижаются. Своеобразный ход кривой 3 объясняется экстракцией из
набухающего сетчатого полимера растворимых низкомолекулярных компонентов смеси
и их растворением. На участке c-d
растворение заканчивается. Степень набухания Q1
соответствует максимуму набухания сетчатого полимера с растворимым компонентом,
Q2
- чистого сетчатого полимера, а Q1-Q2
- количеству растворившегося компонента.
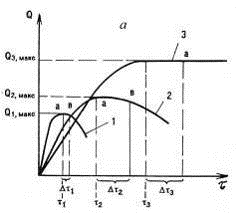
Рис.29. Кинетика набухания линейных (а) и
сетчатых (б) полимеров: 1а-с небольшой ММ; 2а-с большой ММ или разветвленных;
3а-с большой ММ и сильным межмолекулярным взаимодействием; 1б-редкосетчатых;
2б-густосетчатых; 3б-сетчатых с экстрагируемыми компонентами.
Увеличение объема полярных полимеров при
набухании в полярных растворителях сопровождается уменьшением объема
(контракцией) всей системы, которое обусловлено ориентацией и уплотнением при
сольватации молекул растворителя на поверхности макромолекул, а также
заполнением микропор внутри аморфных областей. Контракция V
прямо пропорциональна интегральной теплоте набухания qинт,
т.е. V/qинт=const,
и может быть определена по эмпирическому уравнению:
V=αm/(β+m),
где m
- масса растворителя, поглощенного при набухании 1 г полимера; α
и β-константы.
Истинные растворы полимеров и растворы
низкомолекулярных веществ различаются не только явлением набухания, но также
рядом отклонений от классических законов и уравнений термодинамики. Любое
растворение сопровождается уменьшением свободной энергии системы при смешении
компонентов. Особенностью же растворения полимеров является очень большая роль
энтропии, так как введение растворителя в полимер повышает вероятность
изменения конформации макромолекул. Изменение энтропии ΔSсм
при растворении определяется уравнением Флори-Хаггинса:
ΔSсм=-R(n1lnφ1+n2lnφ2),
где R-универсальная
газовая постоянная;
n1
и n2 -
число молей компонентов; φ1
и φ2 -
их объемные доли. Теплота смешения ΔНсм
при данной концентрации связана с плотностью энергии когезии следующим
уравнением:
ΔНсм/(Vφ1φ2)=[(ΔЕ1/V1)1/2-(ΔЕ2/V2)1/2]2,
где V-общий
объем смеси;
ΔЕ1 и ΔЕ2-изменение
энергии когезии в процессе смешения; V1
и V2-объемы
компонентов;
φ1
и φ2-объемные
доли компонентов. Отношение ΔЕ/V
называется плотностью энергии когезии (плотностью энергии межмолекулярного
притяжения), а величина (ΔЕ/V)1/2
- параметром растворимости δ.
Таким образом, ΔН=V1V2(δ1-δ2)2.
Если δ1-δ2=0
(δ1=δ2),
то
ΔН=0,
и при растворении главную роль играет энтропийный фактор.
Термодинамическое сродство полимера и
растворителя, определяющее способность к растворению и набуханию, зависит от
строения макромолекул полимера и молекул растворителя. Эмпирическое правило
«подобное растворяется в подобном» подтверждается тем, что обычно неполярные
полимеры легко растворяются в неполярных растворителях и не растворяются в
полярных, а полярные полимеры растворяются в полярных растворителях.
Взаимодействие функциональных групп или атомов приводит к возникновению
донорно-акцепторных и иных связей, образующих устойчивые комплексы макромолекул
полимера с молекулами растворителя, способные их отделить друг от друга и
перевести в раствор. Например, ароматические полимеры вследствие подвижности π-электронов
бензольного ядра образуют π-комплексы
с молекулами ароматических или хлорсодержащих растворителей. Оценивают
термодинамическое сродство компонентов по степени снижения их химических
потенциалов, которые определяют путем измерения давления пара растворителя над
раствором, осмотического давления, и другими методами.
Абсолютную вязкость растворов (η)
можно определить в капиллярном вискозиметре и рассчитать по уравнению Пуазейля:
η=πΔРR4τ/8LV,
где R и L-радиус
и длина капилляра; ΔР-разность
давлений на концах капилляра; V-объем
шарика с раствором; τ-время
истечения раствора. На практике чаще пользуются не абсолютной, а удельной
вязкостью, которую рассчитывают по формуле:
ηуд=(ηс-ηо)/ηо,
где ηс
и ηо
- вязкость раствора определенной концентрации и растворителя соответственно.
Отношение удельной вязкости к концентрации
называют приведенной вязкостью. Экстраполяцией прямой зависимости приведенной
вязкости (ηуд/с)
от концентрации (с) к оси ординат можно найти значение характеристической
вязкости [η], под которой
понимают вязкость бесконечно разбавленного раствора. Макромолекулы в таком
растворе рассматриваются как обособленные, изолированные друг от друга.
Зависимость между характеристической вязкостью раствора и молекулярной массой
полимера выражается уравнением Марка-Хаувинка-Куна: [η]=КМα,
где
Различают разбавленные и концентрированные
растворы. В разбавленных растворах концентрация с<1/[η],
при которой макромолекулы находятся на расстояниях, превышающих их собственные
геометрические размеры. В концентрированных растворах с>1/[η],
и макромолекулы растворенного полимера могут взаимодействовать друг с другом,
что приводит к резкому возрастанию их вязкости. Из-за высокой вязкости к
концентрированным растворам относят часто растворы с концентрацией от долей
процента (для жесткоцепных полимеров) до 1% (для гибкоцепных полимеров).
Появление пространственной сетки из лабильных ассоциатов непрерывно меняющегося
состава, взаимосвязанных «проходными» макро-молекулами, приводит не только к
аномалии вязкости растворов, но и другим специфическим эффектам, связанным с
переходом их в твердое состояние (студни и гели). Например, концентрированные
растворы ацетатов целлюлозы представляют собой трехмерную сетку, в ячейках
которой находится растворитель. Тип растворителя определяет структуру и густоту
сетки. Таким образом, ассоциаты можно рассматривать как зародыши новой фазы.
Аномально высокая вязкость и быстрый ее рост с
увеличением концентрации являются отличительной особенностью растворов
полимеров. Чем лучше полимер растворяется в жидкости, тем больше уровень его
сольватации, снижение межмолекулярного взаимодействия и затруднения к свертыванию
макромолекул в плотные клубки. В «плохих» растворителях сольватация выражена
слабо, что широко используется на практике для снижения вязкости растворов
путем добавления «нерастворителя». Растворы полимеров способны рассеивать свет,
что обусловлено непрерывным изменением их концентрации в микрообъемах и
соответствующим изменением показателя преломления вследствие образования и
распада ассоциатов. По величине светорассеяния разбавленных растворов, в
которых макромолекулы свернуты в клубки диаметром менее 0,1 длины волны света,
можно определить ММ полимера. Растворы полимеров способны также избирательно
поглощать световые лучи. По ультрафиолетовым и инфракрасным спектрам поглощения
можно судить о присутствии в полимерах сопряженных двойных связей и определенных
атомных групп, характеризующих строение макромолекул.
Литература
1. Тугов И.И., Кострыкина Г.И. Химия
и физика полимеров. - М.: Химия, 2009. - 432с.
. Бухина М.Ф., Курлянд С.К.
Морозостойкость эластомеров. - М.: Химия, 2009. - 176с.
. Иванов В.С. Радиационная химия
полимеров. - Л.: Химия, 2008. - 320с.
. Попов А.А., Рапопорт И.Я., Заиков
Г.Е. Окисление ориентированных и напряженных полимеров. - М.: Химия, 2007. -
232с.
. Привалко В.П. Молекулярное
строение и свойства полимеров. - Л.: Химия, 2006.-238с.
. Зуев Ю.С., Дегтева Т.Г. Стойкость
полимеров в эксплуатационных условиях. - М.: Химия, 2006. - 264с.
. Бухина М.Ф. Техническая физика
эластомеров. М.: Химия, 2004.-224с.
. Тадмор З. Гогос К. Теоретические
основы переработки полимеров. М.: Химия, 2004. - 628с.
. Бартенев Г.М., Зеленев Ю.В. Физика
и механика полимеров. М.: Высшая школа, 2003. - 390с.
. Аскадский А.А., Матвеев Ю.И.
Химическое строение и физические свойства полимеров. - М.: Химия, 2003. - 248с.
. Казале А., Портер Р. Реакции
полимеров под действием напряжения. Л.: Химия, 2003. - 440с.
. Догадкин Б.А., Донцов А.А.,
Шершнев В.А. Химия эластомеров. - М.: Химия, 2001. - 376с.
. Стереорегулярные каучуки. / Под
ред. У. Солтмена; Пер. англ. Под ред. З.З. Высоцкого. - М.: Мир, 2001. - 1004с.
. Розовский А.Я. Гетероцепные
химические реакции. М.: Наука, 1980. - 324с.