Желтые вещества из кобаламинов в присутствии перекиси водорода
Министерство образования и науки РФ
Федеральное государственное бюджетное
образовательное учреждение высшего профессионального образования
Ивановский государственный
химико-технологический университет
Факультет ОХиТ
Кафедра технологии пищевых продуктов
и биотехнологии
Реферат
по курсу "Химия вкуса, цвета и
аромата"
Желтые вещества из кобаламинов в
присутствии перекиси водорода
Выполнил: студент группы 1-128
Шпагилев Н. И.
Проверил: проф. каф. ТПП и БТ
Макаров С. В.
Иваново 2013
Содержание
Введение
Формирование стабильных желтых корриноидов, в том числе по реакции
с H2O2
Структура и электронные свойства гептаметилового эфира дициано - и
аквацианокобириновой кислоты и стабильных желтых форм гептаметилового эфира
дициано - и аквацианокобириновой кислоты
Структура DCSYCbs
Спектрофотометрический анализ
Электронные переходы
Гидросульфид производные кобаламинов
Список литературы
Введение
Первой стадией дегардации корриноидов является образование
"стабилных желтых веществ". Корриноиды стабильны при естественных
условиях, за исключением действия света и др., при которых может быть отделен
5-деоксиаденозил лиганд.
Желтые или коричневые продукты имеют первую главную полосу в
спектре поглощения между 400 и 500 нм, т.е. при малой длине волны, не же ли αβ - полосы в октаэдральных Со (III) - корриноидах. В
отличие от других корриноидов, таких как кобаламин (II) и пятикоординированный
этилкобинамид, эти продукты не могут быть преобразованы в корриноиды обычного
типа (т.е. с типичным спектром) при действии кислорода, гидрооксида, цианида
или света. По этой причине их называют стабильными желтыми формами корриноидов
("стабильные желтые вещества"), добавление цианида к которым смещает
первую главную полосу в длинноволновую область, обычно примерно к 500 нм.
Исследование данных соединений продолжаются уже на протяжении
более шестидесяти лет, но до сих пор остается масса вопросов, связанных как с
их образованием, так и со структурой.
Формирование
стабильных желтых корриноидов, в том числе по реакции СH2O2
Микробная деградация В12 впервые изучена Helgeland, Jonsen и Laland (1959, 1961) и позднее Pfiffner и коллегами. Helgeland и др. обнаружили, что
культуры Aerobacter aerogenes преобразуют В12
в желтые или коричневые продукты, которые были сепарированны на две фракции при
помощи электрофореза. Спектр двух фракций показывал полосы поглощения в воде и
в 0,1 н КСN,
как следует ниже: Пигмент 1, Н2О 474 нм и 342 нм, КСN 500 и 345 нм; Пигмент 2,
Н2О 448 нм и около 310 нм, КСN 482 нм и около 320 нм
(см. Рис.1.).
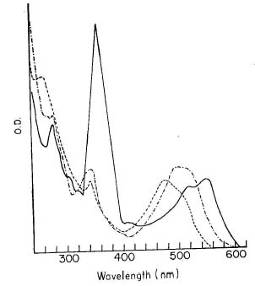
Рис. 1. Спектр поглощения Пигмента 2 в воде (сплошная линия)
и в 0,1 н КСN
(пунктирная линия).
Молярный коэффициент экстинкции для Пигмента 1 при 474 нм
равен 8,4×103 (т.е. сопоставимый с таковым для αβ-полос обычных корриноидов);
соотношение интенсивности высокой и малой энергии полосы было примерно 1 в
первой паре и 1,5 во второй паре. Предполагалось, что в желтых соединениях
происходит разрушение сопряженной системы. Pfiffner и др. обнаружили желтые
соединения при деятельности культур нескольких различных бактерий, из которых Psuedomonas rubescens воздействовала на В12
намного быстрее. Сепарирование продуктов данной реакции электрофорезом и
хроматографией дало четыре основных желтых продуктов (которые были обозначены
А1, А2, В1 и В2). При этом было показано, что А1, А2 и В2 содержат цианид, как
один из лигандов, и то, что нуклеотид в боковой цепи не был затронут, но
бензимидазол, как предполагалось, был свободно координирован. Все три
соединения показывали очень схожие спектры поглощения с полосами при 490 нм и
325-350 нм равной интенсивности. При добавлении цианида в щелочной раствор
происходило смещение полосы низкой энергии в длинноволновую область. Но
пигменты А и В различаются в их реакции с хлорамином-Т. Когда нуклеотид боковой
цепи был отделен гидролизом, спектр поглощения опять показывал две полосы
схожей интенсивности при 440-450 нм и 310 нм. Pfiffner и др. предположили, что
в их желтых соединениях корриновое кольцо каким-либо путем изменено, например
восстановлением, добавлением параллельно одной или более двойных связей,
внедрением нового заместителя в кольцо или образованием изомеров, в которых С10
переносит два атома водорода и С8 (или С13), который утратил атом водорода,
участвует в образовании сопряженной системы двойных связей. Однако прямых
доказательств природы данных структурных изменений не было.
Некоторые стабильные желтые формы корриноидов образуются как
побочный продукт в окислении В12r. Lester Smith был выделен желтый
побочный продукт с полосой поглощения при 458 нм путем окисления кислородом
воздуха В12r. Спектрофотометрическое исследование окисления В12r так же показали рост
полосы при 460 нм, указывающий на образование некоторого продукта,
отличающегося от аквакобаламина. Дальнейшие исследования отражают тот факт, что
В12r окисляется намного быстрее Н2О2, чем
кислородом воздуха, при этом образуется намного больше желтого побочного
продукта (оптическая плотность при 460 нм была приблизительно равна той,
которая была у αβ-полос аквакобаламина).
Эти эксперименты позволили сжать круг вероятных реакций, в которых присутствует
образование стабильных желтых форм корриноидов. Возможными окисляющими агентами
являлись Н2О2, НО2 - и гидроксил
радикал. Взаимодействие может происходить на кобальт ионе, корриновом кольце или
на боковой цепи. Небольшие количества желтых побочных продуктов, образующиеся
при окислении кислородом воздуха, можно вероятно отнести к продуктам
взаимодействия с Н2О2 и др., образующимися на ранних
стадиях. Одним разумным предположением являлось то, что желтое соединение
образуется в результате действия гидроксил радикала на корриновое кольцо.
Последующие работы так же показали, что первая полоса поглощения желтых
побочных продуктов появляется в диапазоне 450-460 нм, но при обработке цианидом
раствор становился красноватым и полоса поглощения смещалась за 486 нм.
Схожие или те же соединения были обнаружены другими учеными,
но при восстановлении корриноидов. Когда растворы В12 были
каталитически восстановлены Pt/Н2 и после повторно окислены,
образовывались побочные продукты реакции, которые имели полосу поглощения при
445-450 нм, которая смещалась при воздействии цианидом в длинноволновую область
480-485 нм. Выход этих соединений был увеличен присутствием НСl или СО во время
восстановления. Beaven и Johnson так же наблюдали коричневые продукты при окислении кислородом
воздуха соединений, образующихся при восстановлении В12 литий
боргидридом или водородом с использованием Pd/BaSO4 катализатора. Эти
коричневые продукты показывают широкий диапазон частот в области 435-450 нм и
дают красноватые производные при взаимодейстии с цианидом. Так было показано,
что формирование желтых соединений может происходить при использовании таких
восстанавливающих агентов как боргидрид натрия или цинка и уксусной кислоты. Другими
учеными были выделены стабильные желтые вещества из реакции В12s c диметилсульфатом.
Возможно, что во всех этих случаях стабильные желтые соединения формируются
вследствие окисления восстановленных корриноидов.
Образование стабильных желтых корриноидов наблюдалось так же
при других условиях, где кобальт (II) корриноиды могут встречаться в виде
интермедиатов, например при фотолизе органокорриноидов и в присутствии
оснований, которые могут активизировать "само-восстановление". Было
сообщено, что фотолиз DBC в воздухе дает желтый побочный продукт, который
стабилен в присутствии кислорода и чей спектр поглощения показывает полосы
приблизительно равной интенсивности при 460 и 330 нм. При этом предполагалось,
что одна из двойных связей в корриновом кольце восстанавливается атомами
водорода, получаемыми из нуклеозидного радикала, образованного в фотолизе. Но
наиболее вероятно, что желтые побочные продукты формируются вследствие
последующего окисления В12r, который образуется при
фотолизе DBC. В данном исследовании так же обнаружено, что фотолиз в воздухе
метилкорриноида, в котором во всех боковых положениях находятся третичные
спирты, дает продукт со спектром, который аналогичен спектру диаквакобинамида в
воде или хлороформе, но стабильное желтое соединение со спектром аналогичным
спектру кобинамид-кобальт (II) наблюдается в этаноле или тетрагидрофуране.
Желтые соединения с полосой поглощения при 455-463 нм,
которая смещается к 485 нм при воздействии цианидом, так же образуются при
взаимодействии В12а с γ-пиколином. Довольно
интересно, что аналоги лактама и 10-хлоролактама не подвергаются этой реакции.
Было первоначально предположено, что сопряжение корринового кольца разрушается
метилированием одного из атомов углерода, но последние исследования показывают,
что похожие реакции могут протекать в присутствии других оснований (НО-,
СН3NH2). Более возможным объяснением, поэтому, является то, что
основание способствует самовосстановлению, в соответствие с чем ион кобальта
восстанавливается, за счет атома водорода на С8 и образуется Со (II) комплекс, который далее
реагирует с воздухом, давая некоторый стабильный желтый корриноид. Это
объясняет, почему аналоги лактама, в которых отсутствует атом водорода при С8,
не вступают в эту реакцию.
Продукты с повышенным поглощением около 450 нм так же
образуются при γ-радиолизе растворов В12
в фосфатном буфере и при недостатке какого-либо другого реагента.
В12 разрушается при действии аскорбиновой кислоты
или витамина С. Эта реакция весьма интересна тем, что существует проблема стабильности
корриноидов в фармацевтических препаратах, содержащих витамин С. Данная реакция
содержит несколько последовательных стадий, при этом может наблюдаться более
чем одна побочная реакция. Было показано, что при взаимодействии В12а
с аскорбатом при рН=4,5-5 в присутствии кислорода при 65ºС, аскорбат полностью разрушает корриноид, высвобождая при этом
простой гидратированный ион кобальта (II). На начальных стадиях реакции происходит
повышение поглощения около 480 нм, в то время как в других исследованиях была
получена серия продуктов реакции аскорбата с В12, которые показывают
более значительное поглощение при 465 нм, чем В12. Во всяком случае
некоторые из представленных продуктов явно являются типичными стабильными
желтыми корриноидами. В12а подвергается быстрому воздействию
аскорбиновой кислоты даже при комнатной температуре (В12 разрушается
более медленно) и скорость увеличивается в зависимости от рН, во всяком случае
в пределе рН=0-7. Распад В12а ингибируется лигандами, такими как CN-, SO32-, NO2-, [FeII (CH) 6] 4 - и
[FeIII (CN) 6] 3-,
но не NCS-, NCO-, NH3, гистидином или различными (неуточненными) аминокислотами,
пуринами или пиримидинами, и так же ингибируется присутствием железа.
Обнаружено, что для реакции между В12 и аскорбиновой кислотой
необходима медь, и она ингибируется цианидом или диэтилдитиокарбамат ионом;
реакция была также катализирована марганцом (II), молибдатом и фторид
ионом, но не ионами кобальта (II), железа (II), магния, цинка или
иодида. Rosenberg исследовал реакцию В12 с аскорбатом при рН 4,7 и 76,6º и показал, что реакция катализируется ионами меди (II) и для нее необходимо
присутствие либо аскорбиновой кислоты (АА), либо дегидроаскорбиновой кислоты (DA) (II) и она может протекать
без присутствия кислорода. Примерно один моль В12 разрушается каждым
добавленным молем CuSO4; но, если медь и АА или DA подогреты до добавления В12, то
количество распавшегося В12 уменьшается. И это весьма интересно в
отношении установления активности интермедиата в этой реакции. Особенности его
активности явно отличаются, если сравнивать с реакцией В12r с О2 или Н2О2,
но потребность в Сu (II) ионах предполагает, что в процессе могут участвовать радикалы. В12
так же подвергается воздействию α-токоферола, но подробное
описание данной реакции отсутствует.
Тиолат анионы (RS-) и HS - могут координировать кобальт (III) ион и восстанавливать
его до состояния кобальт (II). Но и существуют другие реакции, которые до
сегодняшнего момента не полностью изучены. Kaczka и др. обнаружили, что
реакция сероводорода с растворами В12 в этаноле, содержащем каплю
гидроксида аммония, дает красный, кристаллический серосодержащий продукт,
спектр которого очень похож на спектр аквакобаламина (т.е. сера вероятно не
присутствовала в лиганде), при этом сера не находится в форме сульфида,
сульфита или сульфата. Frost и др. сообщили, что В12а был разрушен
различными тиолами, такими как тиогликолевая кислота, цистеин, тиояблочная
кислота и тиосорбит, так же как и аскорбиновой кислотой. Тиосорбит похож на
аскорбиновую кислоту в скорости и полноте, с которой он взаимодействует с В12а
(В12 разрушался намного медленней). Распад В12а
ингибировался добавлением сульфита. Aronovitch и Grossowicz сообщали, что реакция
аквакобаламина или цианоаквакобинамида с гомоцистеином или меркаптоэтанолом
дает желтый продукт с максимумом поглощения при 475 нм. Но, в отличии от В12r, который имеет похожий
спектр, " нет возврата к оригинальному спектру при аэрации, добавлении
перекиси водорода или цианида" и, в отличии от органокобинамидов,
соединение было так же стабильно при взаимодействии с кислородом и светом. Они
предположили, что реакция может касаться как восстановления кобальта вместе с
координацией тиола, так и разрыва сопряжения цепи. Peel получил похожие данные.
При рН=11 тиогликолевая кислота восстанавливает В12, В12а
и цианоаквакобинамид до кобальт (II) комплекса, который может быть реокислен
кислородом воздуха. Но в нейтральном растворе тиогликолат реагирует с В12а,
образуя фиолетовый раствор (предположительно тиолатокобаламин), который после
преобразуется в желто-коричневый. Желтый раствор стабилен на воздухе и первый
максимум поглощения в спектре наблюдался при 490 нм. Дальнейшие изменения
наблюдались так же после часа.
Н2S ведет себя так же как тиолы; в щелочном растворе он
восстанавливает В12а до В12r, но в более нейтральном
растворе он взаимодействует с образованием вначале тиолокобаламина, который
после подвергается неизвестной реакции. Косвенным доказательством является то,
что Н2S будет безвозвратно разрушать нагретый раствор В12.
Другие реакции, которые не включают простую координацию или восстановление,
наблюдались между В12а и тиосульфатом (повышение поглощения около
415 нм) и селеномочевиной (повышение поглощения около 430 нм). В12
так же разрушается смесью тиамина и никотинамида, хотя каждый по отдельности не
действует; в этом случае было показано, что реакция не требует кислорода, но
спектральных изменений для данных продуктов предложено не было.
Факт того, что формирование стабильных желтых корриноидов
часто встречается при условиях, в которых возможно присутствуют свободные
радикалы (Н2О2, О2, фотолиз, γ-радиолиз, тиолы, медь (II) и аскорбиновая кислота) естественно
предполагает их воздействие на корриновое кольцо. Все эти предположения весьма
важны при анализе структур желтых веществ рентгеноструктурным анализом [1].
Структура и
электронные свойства гептаметилового эфира дициано - и аквацианокобириновой
кислоты и стабильных желтых форм гептаметилового эфира дициано - и
аквацианокобириновой кислоты
Сокращения: стабильное желтое производное гептаметилового
эфира кобириновой кислоты, (5R,6R) - Coα,Coβ-дициано-5,6-дигидро-5-гидрокси-гептаметилкоб
(III) иринат-с,6-лактон (DCSYCbs); дицианокобириновая кислоты гептаметилового
эфира (DCCbs); стабильная желтая форма гептаметилового эфира аквацианокобириновой
кислоты (ACSYCbs); аквацианокобаламиновый эфир (ACCbs); дицианокобинамид
(DCCbi); Coα,Coβ-дициано-5,6-дигидро-пентаметилкоб
(III) иринат-a-амид-c,6-лактам (DCLCbs).
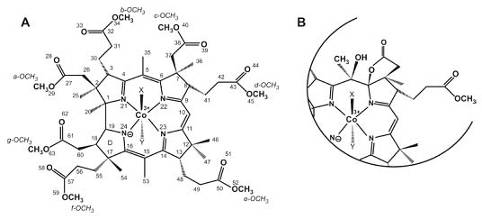
Рис.2. Коррины с Co (III) и используемая нумерация, упомянутая в работе.
"Верхний" лиганд X занимает β-положение в коррине,
"нижний" лиганд Y занимает α-положение в коррине.
В данном стандартном виде коррина "западная"
половина молекулы содержит связь С1-С19 и в "восточной" половине
молекулы связь С10. (А) Дицианокобаламиновый эфир (DCCbs, X = Y = CN-);
При X = H2O, Y = CN - и X = CN-, Y = H2O
возникают два диастереомера аквацианокобаламинового эфира (ACCbs). (В)
Стабильная желтая форма кобаламинового эфира с разрушенным сопряжением на С5.
Дициано-стабильная желтая форма кобаламинового эфира (DCSYCbs) имеет X = Y = CN-,
в то время как аквациано-стабильная форма желтого кобаламинового эфира
(ACSYCbs) содержит два диастереомера - X = H2O, Y = CN - и
X = CN-, Y = H2O.(III) и Co (III) являются классическими
примерами инертных ионов переходных металлов из первой строки d - элементов таблицы
периодической системы Д.И. Менделеева. Время пребывания воды во внутренней
координационной сфере гидратированных ионов Co (III) - 105 с [2],
а для Cr (III) - от 1,8×105 с до 2,2×105 с [3]. Для сравнения, время пребывания в [Co (H2O) 6]
2+ 8,8×10-7 с [4]; в [Fe (H2O) 6]
3+ от 6×10-3 с до 5×10-5 с [5-7] и в [Ni (H2O) 6] 2+ 3,7×10-5 с [4]. Таким образом, одной из примечательных особенностей
химии Co (III) в корринах (витамине B12 и его производных) является
лабильность к замещению лигандов. Вводя лиганд L в комплекс Со (III) с четырьмя N-донорными
(N4) экваториальными
лигандами, он может занимать как X-положение (1).
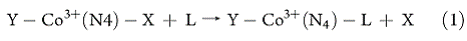
При L=SCN-, X=Y=H2O константа скорости
второго порядка, k2, равна 8,6×10-7 М-1с-1 для
N4= (NH3) 4 [8], но на
девять порядков больше (8,2×102 М-1с-1)
при N4=коррин [9]. Для L=CN-, X=H2O и Y=Me, k2=2×102 М-1с-1
N4= бис
(этилендиамин), но при N4=коррин реакция настолько быстро протекает, что
не может быть даже исследована при использовании метода stopped flow [10].
Степень ненасыщенности экваториальных лигандов, кажется,
является важным фактором в определении скорости замещения аксиального лиганда.
Для L=H2O, X=Y=Cl-, константы скорости изменяются в
соотношении 1: 36: 270 при соответственном варьировании N4 в ряду cyclam
(аббревиатура для 1,4,8,11-тетраазaциклоoтетрадекан), транс [14] диен, cobaloxime
(аббревиатура для бис- (диметилглиоксимата) [11]. Приблизительное
соотношение склонности ионов металла к замещению в коррине, порфирине,
cobaloxime и тетраамминных системах равны соответственно 109: 106: 104: 1
[12,13].
Кинетические данные свидетельствуют о том, что склонность к
замещению аксиального лиганда увеличивается со степенью делокализации цис-π-электронной системы (аммин ≈ cyclam < транс [14]
диена < cobaloxime < порфирин) и обратно пропорциональна размеру
макроциклической полости (порфирин < коррин). Предпологают, что этот
кинетический цис-эффект является следствием делокализации электронной
плотности между экваториальными лигандами и металлом, что делает металл более
слабым, и придает ему более лабильное схожее с Со (II) свойство [12,14,15].
Если это действительно так, то можно ожидать, что склонность к замещению
аксиального лиганда возрастет при увеличении поляризуемости цис-лиганда
и/или при уменьшении размера макроциклической полости и при увеличении
орбитального перекрытия между цис-донорами и металлом.
Цис-эффекты играют важную роль в химии корринов.
Например, в электронных спектрах кобаламинов преобладают π→ π* переходы корринового
кольца, которые сильно зависят от осевого лиганда [15-19]. Длина связи С10-Cl в X-10-ClCbls (Cbl - кобаламин), в котором
водород при С10 заменен Cl, сильно зависит от поляризуемости аксиального
лиганда, X [12]. Замена водорода при C10 на электрон-акцепторные группы, такие
как NO, деактивирует осевое координационное положение в сторону замещения
лиганда [21]. Все это позволяет предположить, что осевая химия лиганда
корринового кобальта может быть изменена при помощи воздействия на электронную
структуру корринового кольца.
Для дальнейшего изучения взаимосвязи между электронной
структурой коррина и взаимодействием кобальта в коррине с экваториальным
лигандом, было подготовлено производное гептаметилового эфира кобириновой
кислоты, (5R,6R) - Coα,Coβ-дициано-5,6-дигидро-5-гидрокси-гептаметилкоб
(III) иринат-с,6-лактон (которое называется стабильной желтой формой
кобаламинового эфира, DCSYCbs, Рис.2), в котором 14-π-е - делокализованная система нормального коррина
превращается в 10 π-е - систему
между N21
и N24, а так же возникает
изолированная двойная связь между N21 и С4. Соединение, первоначально показанное
Gossauer, Grгuning и соавторами [22,23], было охарактеризованно с помощью
масс-спектроскопии, ИК-спектроскопии, 1Н, 13С, 59Co
ЯМР спектроскопии, УФ-спектроскопии, и монокристаллической дифракцией
рентгеновских лучей. Из полученных спектроскопических данных можно сказать, что
уменьшение сопряжения коррина в DCSYCbs повышает твердость Co (III) в коррине.
Структура
DCSYCbs
Как однозначно установлено с помощью рентгеновской
кристаллографии (рис.3), при взаимодействии DCCbs с аскорбиновой кислотой в
присутствии кислорода атом С5 окисляется с образованием группы ОН, а в боковой
цепи на С6 образуется лактон, предположительно, через образование промежуточное
интермедиата [Co2+O2 ↔ Co3+O2-]
[23].
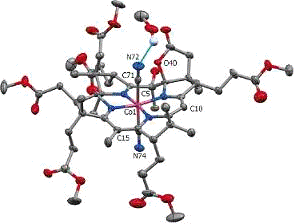
Рис.3. Ассиметрическая единица кристаллической структуры
DCSYCbs. Атомы водорода не изображены и большинство обозначений атомов для
ясности не указаны.
Исследование длин связей С-С и С-N в коррине указывает на
то, что сопряженная система нормального коррина между C5 и С6 была нарушена;
имеется изолированная двойная связь между N21-C4 и триазаметиновая
система с четырьмя сопряженными двойными связями между N22 и N24.
Существуют три сообщения о кристаллической структуре DCCbs
[24-26]. Все три аналогичного качества (R1 = 10-13%). Была выбрана самая
последняя структура для сравнения со структурой DCSYCbs. На рисунке 4 показаны
наложенные друг на друга кристаллические структуры DCSYCbs и DCCbs.
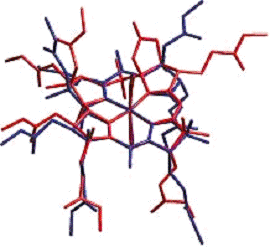
Рис.4. Наложенные друг на друга кристаллические структуры
DCSYCbs (красный) и DCCbs (синий).
Структура DCCbs значительно проще, чем существующая структура
DCSYCbs (R1 = 11,46% и 6,08%, соответственно), поэтому необходимо проявлять
осторожность при сравнении двух структур. Длины связей отличаются примерно на
0,02 Å в среднем из-за того, что некоторые виды связей в DCCbs не были
определены. Заметным исключением является связь C5-C6, которая увеличивается от
1,33 (2) в DCCbs до 1,520 (8) в DCSYCbs из-за расщепления характерной двойной
связи. Валентные углы, в среднем, отличаются на 1º друг от друга, за
исключением угла связи C4-C5-C6 (109,2 (5) º в DCSYCbs и 122 (1) º в DCCbs) и C5-C6-N22 (114,2 (4) º и 126 (1) º, соответственно) за счет добавления группы OH к С5 и формирования
лактона на C6. Угол связи C20-C1-N21 значительно меньше в DCSYCbs (106,3 (4) º), чем в DCCbs (114 (1) º), но причина этого не
выяснена.
Длина связи Co - CN (β) (1,931 (6) Å) в DCSYCbs несколько больше, чем длина связи Co - CN (α) (1,918 (6) Å). Рассмотрение шести
кристаллических структур дицианокобаламиновых эфиров (трех из DCCbs [24-26], в
одной из которых сохранены а и с амиды [27], в другой b положение боковой цепи
занимает свободная кислота [28], и производное норкобириновой кислоты, где
отсутствует метильная группа при С15 [29]) показывает, что связь Co - CN (α) присутствует всегда, но она не значительно больше связи Co-CN (β) (примерно на 0,013 ±0,017 Å). Поэтому длина связи
Co-CN (β), которая ненамного больше Co-CN (α) связи, в DCSYCbs может
отражать стерическое влияние с-лактона.
Существуют две структуры, связанные с DCSYCbs, где с-боковое
положение образует лактам при С6. В 5,6-дигидро-5-гидрокси-гептаметилкоб (III)
иринат-с,6-лактаме [23], длина связи Cо-CN (β) 1,955 (9) и,
безусловно, она больше, чем длина связи Co-CN (α) (1,899 (9) Å). В двух молекулах в элементарной ячейки
5,6-дигидро-гексаметилкоб (III) ирината-a-амида-c,6-лактама длина связи Cо-CN (β) (1.9509 (8) Å и 1.9260 (8) Å) так же больше, чем длина связи Co-CN (α) (1.947 (1) и 1.891 (1) Å). Суммарный угол коррина
(угол между средними плоскостями, проходящими через N21, C4, C5, C6, N22, C9 и
С10-плоскость1 и через N24, C16, C15, C14, N23, C11 и C10 - плоскость 2,
соответственно) в DCSYCbs равен 15,7º и только 5,2º в DCCbs. Это может являться фактором, влияющим на удлинение связи
Cо-CN (β); однако, так как в двух молекулах в элементарной ячейки
5,6-дигидро-гексаметилкоб (III) ирината-a-амида-c,6-лактама суммарные углы
всего лишь 5,9º и 6,0º, то увеличение суммарного
угла коррина не оказывает значительное влияние на удлинение связи Cо-CN (β).
кобаламин перекись желтый корриноид
Угол связи NC-Co-CN в DCSYCbs равен 173,0 (2) º и два лиганда CN - согнуты от "западной" к
"восточной" половине молекулы и не значительно от О40 с,6-лактона
(Рис.2.). Такое искажение является общим для всех дициановых комплексов,
упомянутых выше, и нет никакой разницы в искажении транс-осевых связей между
нормальными корринами и тех, которые содержат лактам или лактон на C6.
Наблюдаемое искажение исходит от тесного контакта C19H и C20Me с β лигандом (рис.5).
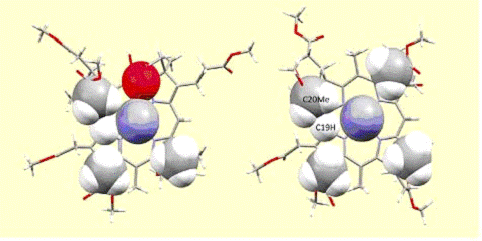
Рис.5. Вид на β положение DCSYCbs
(слева) и DCCbs (справа). Сигнальные метильные группы С46 и С54, С19 водород,
О40 с,6-лактона SYCbs и С37 метильная группа Cbs изображены в соответствии с
моделью заполнения пространства, при использовании стандартных
ван-дер-ваальсовых радиусов этих атомов. Формирование с,6-лактона доставляет
O40 в связывающий карман лиганда β положения коррина
(слева). Хотя расположенный в значительной степени над С5, О40 действительно
имеет Ван дер Ваальсовое взаимодействие с С-донорным атомом координируемого
цианида.
ЯМР спектроскопия.
Несколько групп ученых описали 13С и 1Н
ЯМР распределения Co (I) - DCCbs [30], Co (III) - DCCbs [31-34] и Co (III) -
DCCbs-монокислот [28], - моноамидов [32]. Ernst отмечает единственное значение
для 13С резонанса двух координируемых цианидных лигандов в DCCbs, приблизительно
130 ррm
(CDCl3) или приблизительно 133 ppm (CD3OD) [31], и
впоследствии 130 ррm (CDCl3) [32]. В отношении b-, d-, e-, и
f-моноамид-гексаметиловых эфиров, два 13С резонанса сообщались для
аксиальных лигандов цианида, отличающихся друг от друга от самого меньшего 0,6
ppm в b-моноамиде, до самого большого 4,7 ppm в e-моноамиде (133,9 и 129,2
ppm). Не были даны резонансы для специфичных лигандов каждого диастереомера. В
отношении монокислот [28] наблюдались два резонанса с различными измерениями от
0,63 ppm (130,59 и 131,22 ppm) для b-монокислоты до 4,05 ppm (129,93 и 133,38
ppm) для e-монокислоты. В этом случае авторы предположительно дали слабый
резонанс α-CN лиганда.
В данном ЯМР спектре DCCbs (Рис.6. а. CDCl3)
установлены два цианидовых резонанса при 130,69 и 130,29 ppm, совместимые с их
подобными магнитными и химическими средами, как доказательство
рентгеноструктуры соединения, которое имеет ту же самую длину аксиальной связи
Со-С.
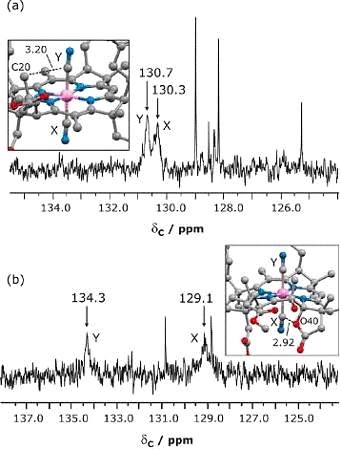
Аксиальные цианид ионы DCSYCbs магнитно уникальны, их
достоинством является расположение в различных карманах связывающего лиганда Со
(III) макроцикла,
доказательством служат вполне разделенные сигналы при 129,1 и 134,3 ppm (Рис.6.
b). Эти сигналы есть в том
же диапазоне химических сдвигов ионов цианида DCCbs и являются такими же
широкими из-за координации атомов углерода к Со (III) иону с его большим
ядерным квадрупольным моментом. Как показано на рис.5, аксиальный ион цианида,
координированный на β-положение (Х лиганд)
макроцикла, испытывает большее стерическое ограничение, чем тот, который
координирован на α-положение (Y лиганд), по
причине внеосевого отклонения. Из рассчитанных с помощью ДПФ магнитных
экранированных тензоров для Со (III) - связанных аксиальных атомов углерода, углерод
у β-CN - более экранирован, чем С у α-CN-. На 13С ЯМР
спектре DCSYCbs наблюдались резонансы при 129,1 и 134,3 ррm, исходя из этого
установлены β - и α-CN - ионы, соответственно.
Никаких дальнодействующих протонных связей аксиального CN - в
DCSYCbs обнаружено не было.13С резонансы аксиального лиганда
дицианокобинамида (DCCbi) были установлены при 140,60 и 141,60 ррm [35]. Интервал двух
резонансов, ΔδCax, в DCSYCbs (5,08 ppm)
гораздо больше, чем в DCCbi (1,00 ppm) или в DCCbs (0,40 ppm). В вакууме
рассчитанные с помощью ДПФ различия химических сдвигов между 13С
резонансами α - и β-CN - DCCbs и DCSYCbs были 3,3
и 5,5 ppm, соответственно, которые хорошо согласуются с экспериментальными
значениями ΔδCax. Большей разницы
химического сдвига аксиального лиганда в DCSYCbs не возникает из-за различий в
длине связи Со-С; экспериментальные (рентгеновские) значения на самом деле
являются эквивалентными в пределах 1σ и в вакууме,
рассичтанные с помощью ДПФ, расстояния между Со-С одинаковы (1,927 Å). Возникновение большего значения ΔδCax связано, весьма
вероятно, с разницей в наклоне двух аксиальных лигандов (см. выше) и с
неэквивалентностью их окружения.
Некоторое из различия между кобаламиновыми эфирами и
кобинамидом связано с разницей в величине магнитной восприимчивости
растворителей, используемых в измерениях (CDCl3 для DCSYCbs и DCCbs,
и D2O для DCCbi, см. ниже), но в кобинамиде оба резонанса
по-прежнему значительно смещены в слабое поле. Это отражает значительную
разницу в электронном взаимодействии между циановыми лигандами и металлом в
двух кобаламиновых эфиров по сравнению с DCCbi (хотя гораздо меньше разницы
между DCCbs и DCSYCbs).
Среднее изменение в 13C химическом сдвиге
резонансов углеродов корринового кольца, которые отдалены от структурных
изменений в DCSYCbs и не зависят от изменений в резонансе сопряжения в
корриновом кольце (т.е., C1-С3, C11-C19 включительно, n = 12), является сдвигом
на 2,8 ±1,7 ppm в сильном поле в DCSYCbs по сравнению с DCCbi, очевидно
связанным с разницей в величине магнитной восприимчивости двух растворителей.
Аналогичные изменения между DCSYCbs и DCCbs очень малы (|Δδ| = 0,9±0,7 ppm для двух спектров DCCbs [32,34], полученных в
нашей и другой работе), как и для DCLCbs (|Δδ| = 0,8±0,8 ppm).
Аналогичная тенденция наблюдается для метиловых резонансов корринов (т.е. C20,
C25, C47,
C48, C53, C54), с Δδ = - 2,3±0,7 ppm для DCCbi, |Δδ| = 0,4±0,5 для DCCbs, и |Δδ| = 0,2±0,2 ppm для DCLCbs (n = 6).
Переход амидов боковой цепи в метиловые эфиры происходит при
сдвиге в сильном поле 6±1 ppm химического сдвига карбонильного углерода (n = 5,
включая C43), и сдвиге в сильном поле 4,4±1,1 ppm на химическом сдвиге углерода
боковой метильной группы, прилигающей к карбонильной (n = 5, включая C42).
Существует небольшая разница в этих резонансах между DCSYCbs и DCCbs, за
исключением резонансов b-бокового положения, где C32 резонирует при 2,55, 2,48 и 2,56
ppm в сильном поле, в то время как в слабом поле C31 резонирует при 2,11 и 2,02
ppm, причины этого невыяснены.
Значительно большие эффекты обусловлены гидроксилированием на
C5 и циклизацией с-боковой цепи на B кольцо. По сравнению с DCCbi, эти
структурные изменения, сдвиг при С5 и С6 резонансов в сильном поле при 28,8 и
53,7 ppm, соответственно, связаны с изменением в гибридизации этих атомов
углеродов. С4 резонанс перемещается в слабом поле при 11,7 ppm,
предположительно из-за того, что вмешательство сопряжения обеспечивает большую
поляризацию N21-C4 электронной плотности в сторону N. Эффекты незначительны,
однако, на C7 и C8 (1,1 и 2,0 ppm в сильном поле, соответственно). Сравнение 13C
резонансов DCSYCbs и DCCbs показывает очень похожие эффекты. Используя среднее
значение трех данных (этой работы и других) 13C резонанса для DCCbs
установлены Δδ = - 24,6 и - 51,6 ppm
для C5 и C6, соответственно, +15,8 ppm для C4, и незначительное значение +2,0 и
1,7 ppm для C7 и C8, соответственно.
C5 резонанс для DCCbs (41,3 ppm) составляет 37,7 ppm в сильном поле по
сравнению с DCSYCbs, и С6 резонанс (90,6 ppm) составляет 21,4 ppm в сильном поле
(слабый индуктивный отодвигающий эффект лактама по сравнению с лактоном).
Циклизация с-боковой цепи приводит к неожиданному сдвигу (7,0 ppm) в сильном
поле С41 метиленового резонанса (по сравнению с DCCbi; 4,2 ppm по сравнению с
DCCbs) и сдвигу (4,1 ppm) в сильном поле С36 метиленового резонанса (2,0 ppm по
сравнению с DCCbs).
59Co химические сдвиги помогут пролить свет на
природу металла в двух соединениях.59Co резонансы CNCbl, AdoCbl, и
MeCbl в водном растворе, как сообщалось, появляются при 4650, 4480 и 4125 ppm
[36]. Таким образом, в равной степени донорная сила аксиального лиганда и его
взаимодействие с металлом увеличиваются, 59Co резонанс смещается в
сильное поле. Аналогичный эффект был отмечен в cobaloximes, где замена Н2О
на сильные доноры, такие как имидазол и пиридин, приводит к увеличению
экранирования ядра 59Co [37]. В настоящем исследовании, 59Co
резонанс DCCbs наблюдался при 4074 ppm, а для DCSYCbs при 4298 ppm.
Впоследствии определенные с помощью метода ДПФ и кристаллографии аксиальные
связи Со-С, с учетом ошибок каждого метода, оказались равнозначными для двух
структур, экспериментальное различие в 59Co химическом сдвиге для
двух комплексов не может быть отнесено к различиям в Co-CN связывающих
взаимодействиях и указывает на принципиальные различия между взаимодействием Co
(III) иона с цис-макроциклами в представленной паре соединений. Очевидно, что
более делокализованная π-электронная система
коррина в DCCbs по отношению к DCSYCbs способствует формированию более
ковалентного комплекса с ионом металла, на который приходится более сильное
экранирование электронов Co (III) ядра.
Спектрофотометрический
анализ
Электронный спектр DCCbs (в водном растворе) идентичен
таковому, который сообщался ранее [24]. Гауссовская деконволюция (Рис.7. А)
показывает α-полосу при 17,244 см-1 (580 нм)
и последующие три полосы с пониженной интенсивностью и равным интервалом
(1344±32 см-1), D и E полосы при 22 875 и 24 310 см-1 (437 и 411 нм) и γ-полоса при 27 191 см-1 (368 нм) с двумя полосами
меньшей интенсивности по обе стороны при 25 928 и 28 621 см-1 (386 и
349 нм), соответственно.
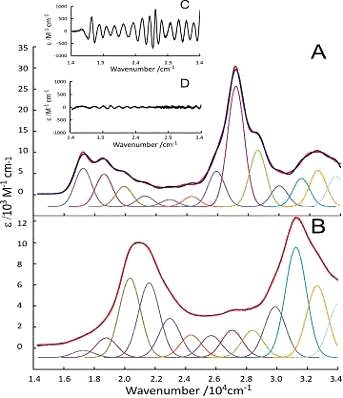
Рис.7. Гауссовская деконволюция электронного спектра 1,88*10-5
M DCCbs (А) в фосфатном буфере (pH=8, 0,1M) и избытке KCN, и (В) 1,62*10-5
M DCSYCbs в деионизированной воде и избытке KCN. Синия линия -
экспериментальный спектр; красная линия - сумма компонентных Гауссовских
функций. Остатки представлены в частях С и D, соответственно.
α-Полоса и три последующие
полосы, включая β-полосу, могут
представлять собой колебательное движение π→ π* электронного перехода с высшей занятой молекулярной орбитали (HOMO) на низшую свободную
молекулярную орбиталь (LUMO), но две полосы по обе стороны от γ-полосы, по всей видимости, соответствуют отдельным электронным
переходам.
Электронный спектр для DCSYCbs в водном растворе и метаноле
идентичен ранее представленному [22]. Спектр имеет сходные черты со спектром
DCCbs, за исключением того, что все пики смещены в сторону более коротких длин
волн и γ-полоса менее интенсивна. Гауссовская деконволюция (Рис.4. В.)
показывает, что α-полоса находится при 20
337 см-1 (492 нм) и имеет последующие четыре полосы с пониженной
интенсивностью и примерно равными расстояниями (1335± 53см-1). Хотя
можно предположить, что эти четыре полосы появляются исключительно за счет
колебательного движения (как в DCCbs), данные ДПФ-моделирования (см. ниже)
показывают, что спектральная область к синей α-полосе особенно сложна и
так же включает в себя несколько независимых синглетных электронных
возбужденных состояний, уникальных для DCSYCbs. D и E полосы наблюдаются при
27 049 и 28 392 см-1 (370 и 352 нм), γ-полоса при 31 241 см-1
(320 нм), с двумя полосами меньшей интенсивности по обе стороны при 29
874 и 32 635 см-1 (335 и 306 нм), соответственно. Кроме того,
наблюдаются два пика низкой интенсивности при большей длине волны, чем α-полоса; они отсутствуют в газовой фазе электронного спектра
DCSYCbs, рассчитанного с помощью ДПФ, и не могут быть отнесены к обычным
синглетным возбужденным состояниям хромофора. Соотношение коэффициента
экстинции γ-полосы, что и у α-полосы, составляет 1,4, что значительно меньше, чем
соответствующий коэффициент (3,1) для DCCbs. π→ π* электронный переход с HOMO на LUMO, соответствующий α-полосе, смещается в
сторону большей энергии (на 3100 см-1), в связи с короткой
делокализованной системой. γ-полоса также сдвигается в
сторону большей энергии в несколько большей степени (на 4050 см-1),
чем α-полоса. Сдвиги в энергии и различиях в интенсивности между DCSYCbs
и DCCbs аналогичны тем, которые наблюдались Eschenmoser [38] для двух
предшественников корриноидов, с и без нарушения, соответсвенно, в сопряженной
системе при С5.
Электронные
переходы
Экспериментальные электронные спектры DCCbs и DCSYCbs (Рис.8)
заметно отличаются, и оба по своему существу сложно связаны с природой и
минимальной симметрией макроцикла, координированного к иону Со (III). Поэтому объяснить
прямое назначение главных переходов трудно, и в течение сорока лет было
использовано множество теоретических методов, предназначенных для описания спектра
витамина В12 и его аналогов [19,39-42].
В этой работе использаволась деконволюция спектров DCCbs и
DCSYCbs на составляющую Гауссовских полос для создания удобной отправной точки,
с которой можно построить четкое представление о ключевых электронных переходах
для каждого комплекса, используя зависящее от времени ДПФ-моделирование [43],
которое особенно удобно для понимания орбитального происхождения электронных
возбужденных состояний хромофора. Кроме того спектр, полученный с помощью этого
метода позволяется объяснить основные и возбужденные синглетные состояния.
В общей сложности 22 возбужденных синглетных состояний были
расположены между 250 и 500 нм для DCCbs и DCSYCbs. Даны основные переходы,
отвечающие за каждое электронное состояние, которые представлены в таблицах 1 и
2.
Таблица 1. ДПФ-рассчитанные возбужденные синглетные состояния (в
вакууме) и главное назначение переходов для DCCbs
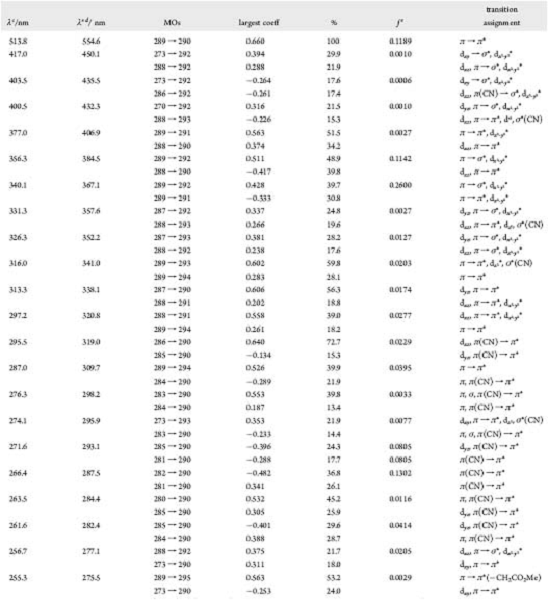
Таблица 2. ДПФ-рассчитанные возбужденные синглетные состояния (в
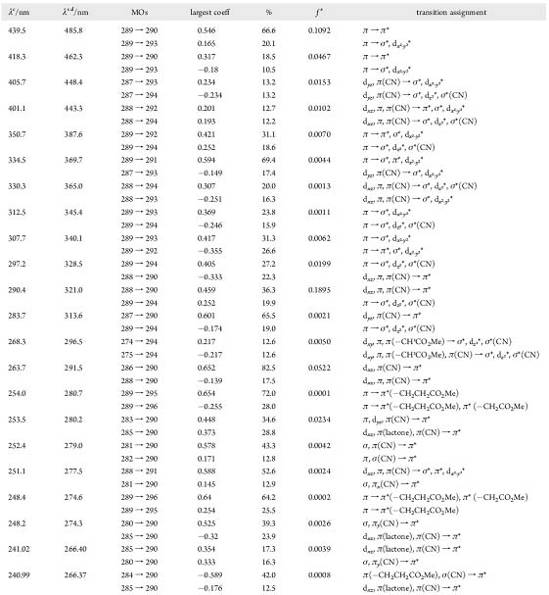
вакууме) и главное назначение переходов для DCSYCbs
Рассчитанные и экспериментальные спектры сравниваются на
рисунке 8.
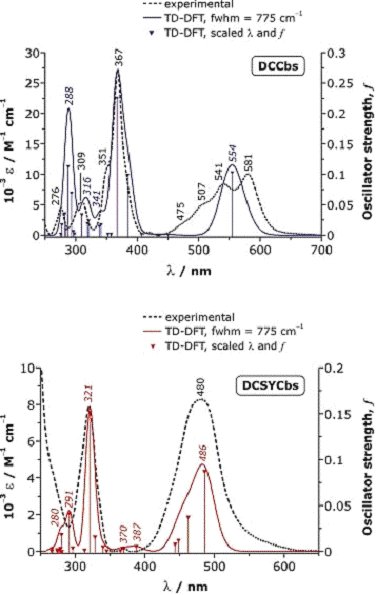
Рис.8. ДПФ электронный спектр (22 синглетных возбужденных
состояний) для DCCbs (вверху) и DCSYCbs (внизу).
Экспериментальные спектры представлены в виде пунктирной
кривой в обоих случаях. Для облегчения сравнения, рассчитанные с помощью ДПФ
спектры были масштабированы в соответствии с экспериментальными γ полосами для обоих соединений (λ и ε коэффициенты масшабирования: 1,0794 и 0,692, соответственно, для
DCCbs; 1,1053 и 0,278, соответственно, для DCSYCbs). Для рассчитанных спектров,
отдельные синглетные состояния показаны как вертикальные линии с
интенсивностями в каждой из единиц оси у; спектральная огибающая представляет
совокупность составных полос, которые имеют ширину 775 см-1 на
половине высоты. Указаны максимумы полос.
Для облегчения спектральных сравнений и распределений полос,
рассчитанный спектр (Рис.8) был масштабирован в соответствии с
экспериментальной энергией γ-полосы и интенсивностью в
каждом конкретном случае.
Для DCCbs форма и основные черты масштабированного
рассчитанного спектра достаточно хорошо соответствуют экспериментальному
спектру при λ> 300 нм. α-полоса в рассчитанном
спектре наблюдается примерно в районе 26 нм от максимума экспериментальной
синей полосы при 555 нм (не смотря на масштабирование всех энергетических
переходов на фактор, который учитывает энергию γ-полосы в соответствии с
экспериментальным значением). Из таблицы 1, α-полоса соответствует
чистому π→ π* переходу, связанному с
переходом электрона с занятой высшей молекулярной орбитали (HOMO 289) на низшую
свободную молекулярную орбиталь (LUMO 290), рисунок 9.
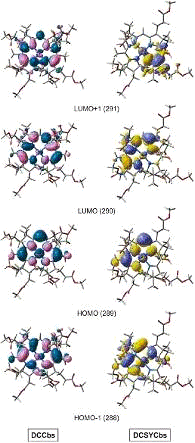
Рис.9. Сравнение граничных молекулярных орбиталей для DCCbs и
DCSYCbs.
Второе рассчитанное возбужденное состояние находится при
вычисленной длине волны 450 нм и возникает в основном из 3dπ→ (σ*, dx2-y2)
переходов. Важный дипольный переход для этого возбужденного состояния
соответствует dxy→ dx2-y2 возбуждению, которое
включает в себя орбитали металлов, которые смешиваются с σ-симметрией МО коррина. Эта полоса загорожена в экспериментальном
спектре DCCbs третьим (475 нм) колебательным обертоном α-полосы, который является результатом слабой силы осциллятора.
Второе возбужденное электронное состояние DCCbs приблизительно приравнивается
параметру поля лиганда ΔO (≈22 222 см-1)
простого октаэдрического Co (III) иона. Эта полоса не была определена в Гауссовской
деконволюции экспериментального спектра (Рис.7) из-за ее малой интенсивности
относительно соседних π → π* полос. Отсутствие некоторых рассчитанных возбужденных синглетных
состояний между 555 и 450 нм наводит на мысль, что β-полоса (541 нм) и полосы низкой интенсивности при 507 и 475 нм в
экспериментальном спектре DCCbs являются колебательной прогрессией α-полосы. Как отмечалось ранее, экспериментальные полосы при 436 и
407 нм соответствуют D и Е полосам коррина, соответственно [19]; из Табл.1, они
соответствуют 3dπ →σ* и 3dπ→π* переходам, которые
являются причиной малой интенсивности. D полоса в
экспериментальном спектре возникает из-за близкого наложения возбужденных
состояний системы при 436 и 432 нм, соответственно (Таблица 1); они не решены с
помощью Гауссовской деконволюции. γ-Полоса для DCCbs (367 нм,
седьмое возбужденное состояние) является смесью π→σ* и π→π* переходов между HOMO и LUMO +1, LUMO +2 коррина (последние не занятые молекулярные
орбитали, которые разрыхляют связи МО лиганда с 3dx2-y2 AO иона
металла). Так как 3d орбитали не сильно смешиваются с HOMO, переходы имеют намного
более значительный π→ π*, чем d-d
характер и сила осциллятора является большой. Две полосы меньшей интенсивности,
обрамленные γ-полосой, отмеченные выше (возбужденные
состояния 6 и 8 при 385 и 358 нм, соответственно, в таблице 1), имеют смешанный
тип, π→ (σ*, dx2-y2), а
так же dπ,π→ π*, и, в отношении полосы
при 358 нм, включают переход к МО со значительным dz2* и σ* (CN) свойством; следовательно, наблюдается некоторое свойство
переноса заряда (с участием осевых и аксиальных лигандов), приводящее к этому
состоянию.
Достаточно интенсивное плечо при 351 нм в экспериментальном
спектре DCCbs не имеет аналогов в рассчитанном с помощью ДПФ спектре. Поскольку
настоящее ДПФ-моделирование не предполагает переходы к колебательным уровням
возбужденного электронного состояния, делают вывод, что плечо при 351 нм,
вероятно, соответствует колебательной прогрессии γ-полосы, частично
совпадающей с малоинтенсивными полосами для возбужденных состояний 8 и 9 (π→ (σ*, dx2-y2)).
Наконец для DCCbs, рассчитанные возбужденные состояния от 319
до 282 нм (с наивысшей интенсивностью при возбужденном состоянии 18, 288 нм)
суммированы в полосу, которая не соответствует экспериментально наблюдаемой полосе
при 276 нм ни по энергии, ни по интенсивности. Считают, что этот близко
расположенных набор возбужденных состояний включает (Табл.1) главным образом
сравнительно несмешанные переходы типа π (CN) → π* и они могут быть названы полосами переноса заряда от лиганд к
лиганду (LLCT), в которых электрон, расположенный первоначально в π-симметричных МО аксиальных лигандов, переносится с π* MO на экваториальный лиганд (корриновый макроцикл, в этом
случае). В ДПФ-моделировании, сообщенном Andruniow и др. [42], этот тип полосы
с переносом заряда дальнего действия (LRCT) наблюдался для возбужденного
состояния, связанного с повышением электрона из аксиального аденозильного
лиганда на π* МО коррина модели производного
аденозилкобаламина. Эти авторы так же обнаружили наоборот неудовлетворительное
совпадение между энергиями (и силами осциллятора) рассчитанных и
экспериментальных полос, связанные с этим СТ состоянием. Похоже, что LRCT
полосы, как правило, трудно определить с помощью ДПФ-методов [42,44,45].
Масштабированный рассчитанный с помощью ДПФ спектр DCSYCbs
показан с наложением на него экспериментального электронного спектра на Рис.8. λ и ε коэффициенты
масштабирования, необходимые для наложения огибающих смоделированной и
экспериментальной γ-полосы, больше, чем те, которые
использовались для спектра DCCbs, и это обычно связано с недостаточным
совпадением между теоретическими и экспериментальными силами осциллятора для
этих соединений. Однако существует четкое совпадение между энергиями
масштабированного рассчитанного возбужденного состояния и экспериментальными
энергиями между 300 и 500 нм. α-полоса масштабированного
теоретического спектра (486 нм) близка по энергии к α-полосе в экспериментальном спектре (≈492 нм), и обычно общая
природа видимого спектра достаточно хорошо воспроизводится в расчетах. В
отличие от различных DCCbs, α - полоса DCSYCbs
является результатом не чистого π → π* перехода (Табл.2) и имеет смешанный характер. Две трети
дипольного переходного момента происходят из π→ π* перехода и другие 20 % в следствии π → (σ*, dx2-y2) перехода. Возбужденные состояния 2-4
расположены между 462 и 443 нм и имеют соотношение интенсивностей (и
энергетический интервал), которое может быть ошибочным для колебательной
прогрессии α - полосы, тем более, что этот вид спектральной
особенности так заметен для DCCbs. Это не тот случай; возбужденные состояния
2-4 являются реальными синглетными возбужденными состояниями хромофора с π→ (σ*, dx2-y2) или d→
d* характером, смешанного с π→ σ* или π→ π* характером. Факт, что 67% α-полосы (возбужденное
состояние 1) содержат строго разрешенный π → π* переход коррина может, однако, допускать достаточно интенсивную
колебательную прогрессию в экспериментальном спектре. Одной из проблем
ДПФ-метода является то, что он не позволяет рассчитывать колебательные
состояния, которые могут быть связанны с электронным возбужденным состоянием
для хромофора. Тем не менее, теперь должно быть ясно, что если вышеупомянутые
синглетные возбужденные состояния (при 462, 448, и 443 нм) DCSYCbs были скрыты
под небольшим расстоянием колебательной прогрессии α-полосы экспериментального спектра, то безликая широкая полоса
будет наблюдаться из-за перекрытия состояний различного происхождения. Кажется,
это является разумным объяснением ключевого различия между спектрами DCSYCbs и
DCCbs в видимой области спектра; в DCSYCbs уменьшение π-делокализации корринового макроцикл приводит к первому
возбужденному синглетному переходу из одного состояния в другое (α - полоса) при более короткой длине волны, чем в DCCbs, и π→ π* и d → d* переходы
показывают сильную степень перекрывания. D полоса DCSYCbs очевидно
состоит из возбужденных состояний 6 и 7 при 370 и 365 нм, соответственно
(Табл.2); они прекрасно совпадают с Гауссовской компонентой (Рис.7) при 370 нм.
Переходы, ответственные за D полосу исходят из π-МО коррина, которые
сильно смешиваются с dπ МО металла и π-МО аксиального цианида, так как это было в DCCbs. Назначением МО
этих переходов является смешение σ* и B* орбиталей коррина с
3dx2-y2 и 3dz2 АО металла, а так же с σ* молекулярными орбиталями аксиального цианида, по крайней мере,
для одного из переходов. E полоса для DCSYCbs включает возбужденные состояния 8
и 9 при 345 и 340 нм, которые выгодно отличаются от гауссовской компоненты
экспериментального спектра поглощения при 352 нм. В отличии от полосы D, переходы, ответственные
за эти возбужденные состояния, исходят из высшей занятой молекулярной орбитали,
которая является относительно несмешанной π-орбиталью макроцикла
коррина.
γ-Полоса DCSYCbs при 321 нм
(возбужденное состояние 11) включает, в основном, переход от HOMO-1 до LUMO (36%) и
переход от HOMO до LUMO + 4 уровня (20%). Основной переход полосы имеет
существенный dπ,π, и π (CN) характер для занятой орбитали и чрезвычайно чистый
корриновый π* характер для незанетой орбитали. Таким
образом, γ-полоса DCSYCbs отличается по происхождению
от γ-полосы DCCbs, в которой не наблюдался вклад от π-МО аксиального цианида в занятой или незанятой молекулярных
орбиталях, участвующих в переходе.
Огибающая γ-полосы, рассчитанного с
помощью ДПФ спектра DCSYCbs, является более острой и ассиметричной, чем в
экспериментальной полосе. Ассиметрия полос предполагает наличие плеча умеренной
интенсивности на коротковолновой стороне полосы. Гауссовская деконволюция
экспериментального спектра позволила разъяснить эту неясную полосу, которая
имеет максимум при 306 нм. Судя по интенсивности и отсутствию ДПФ-рассчитанного
аналога, эта полоса, вероятно, возникает из первого колебательного обертона γ-полосы.
В итоге, рассчитанный спектр DCSYCbs имеет множество
малоинтенсивных переходов γ-полосы между 266 и 314
нм; это выражается в двух максимумах при 291 и 280 нм для смоделированной
спектральной огибающей, причем последний виден как плечо на рисунке 8. В этой
области рассчитанная спектральная огибающая заметно отличается по форме от
огибающей DCCbs; например, сильные резкие полосы при 288 нм в расчетном спектре
DCCbs сводятся к плечу в случае DCSYCbs. Рассмотрение назначений переходов
показывает, что орбитальное происхождение переходов совершенно различно. LLCT
переходы для DCSYCbs все еще присутствуют, но в отличие от DCCbs, они, как
правило, связаны с сильно смешанными занятыми орбиталями. Таким образом,
таблица рассчитанных переходов более сложна в анализе для DCSYCbs. Тем не
менее, три особенности заслуживают внимания. Во-первых, чистые π (CN) → π* LLCT переходы, как в
расчетах DCCbs, отсутствуют. Вместо этого, переходы, образующие возбужденные
состояния 14 (292 нм) и 17 (279 нм), исходят из занятых МО, которые включают π-МО аксиального цианида, смешанные с dπ AO или π/σ MO коррина. Силы осциллятора, следовательно, ниже по
сравнению с чистым π (CN) → π* LLCT переходом. Во-вторых, для многих возбужденных состояний при
длине волны <300 нм, переходы связанны с π* MO коррина, смешанные с
одним или более π* компонентами эфирных
боковых цепей. Например, возбужденное состояние 15 (281 нм) связано с π → π* (-CH2CH2CO2Me)
переходом; это связано с переносом электрона на дальние расстояния от π MO (HOMO) к π разрыхляющей МО эфирного
карбонила, присоединенной боковой группы. Силы осцилляторов не высоки. В
рассчитанном спектре DCCbs было только одно такое возбужденное состояние (276
нм, таблица 1), в отличие от четырех рассчитанных для DCSYCbs. Это по-видимому
связано с нарушением π-делокализации коррина, а
также с изменением общей формы и симметрии хромофора в связи с наличием кольца
лактона. В-третьих, кольцо лактона DCSYCbs становится полностью вовлечено в
некоторые возбужденные состояния для желтого производного коррина. Первое из
них при 280 нм (возбужденное состояние 16), где 29% возбужденного состояния
происходит от dxz, π (лактон), π (CN) → π*. Другие возбужденные
состояния с этим переходом, в качестве основного компонента, наблюдаются при
274,3, 266,40 и 266,37 нм.
И, наконец, самая высокая энергия возбужденного состояния для
DCSYCbs (266,37 нм) имеет довольно необычный главный переход (42%), который
включает в себя занятую MO с элементом π-симметрии на боковой цепи
эфира, смешанную с элементом σ-симметрии на осевом
цианидном лиганде.
Выводы.
Цис-эффекты играют важную роль в химии корринов.
Ранее показано следующее: замена Н при C10 в кобаламинах на Cl приводит к тому,
что рКа координированной H2O падает и влияет на кинетику и
термодинамику связывания экзогенных лигандов с ионом металла [14]. Длина связи
С10-Сl
в X-10-ClCbls сильно зависит от поляризуемости аксиального лиганда X [12].
Замена Н при С10 на ЭД-группу дезактивирует аксиальное координационное
положение в сторону замещения лиганда [21]. Для дальнейшего исследования этих цис-эффектов
был приготовлен и описан DCSYCbs, (5R,6R) - Coα,Coβ-дициано-5,6-дигидро-5-гидрокси-гептаметилкоб
(III) иринат-c,6-лактон, из DCCbs. Рентгеновские данные
подтверждают нарушение сопряженной π-системы нормального
макроцикла коррина путем образования гидроксильной группы при C5 и лактона при
C6 на с-боковой цепи коррина. ДПФ-рассчитанные экранирующие тензоры (GIAO
метод) были использованы, чтобы окончательно определить неэквивалентные
химические сдвиги аксиальных цианидных углеродов в обоих производных эфира
кобириновой кислоты.59Co химические сдвиги и сигнальные широты
спектральных линий DCCbs и DCSYCbs существенно отличаются; 59Co ядра
у DCCbs более экранированны (на 224 ррm), чем у DCSYCbs, и ширина линии уже для
родоначальника эфира кобириновой кислоты (DCCbs). Так как сила взаимодействия
между Co (III) и экваториальным макроциклом увеличивается, то νCN аксиально координированного CN - сдвигается в сторону
низких частот; в DCSYCbs и DCCbs νCN наблюдается при 2138 и
2123 см - 1, соответственно, лиганд коррина в DCCbs взаимодействует
сильнее с металлом, чем лиганд в стабильной желтой форме коррина, в связи с
уменьшением сопряжения. ДПФ-методы были особенно полезны при определении
основных и молоинтенсивных полос в электронных спектрах DCCbs и DCSYCbs.
Расчеты позволили разграничить экспериментально наблюдаемые колебательные
прогрессии на α - и γ-полосах для обоих
комплексов, обеспечивая картину спектральных огибающих, обусловленных
переходами чистых безколебательных синглетных возбужденных состояний, дающие
эти полосы [46].
Гидросульфид
производные кобаламинов
Несмотря на то, что окрашенные в пурпурный цвет комплексы
органических тиолов с кобаламинами были тщательно изучены, гидросульфид -
производные кобаламинов упоминаются не так часто. Kaczka и др. получили
кристаллический серосодержащий продукт (λmax = 275, 352, 530 нм)
после барботирования раствора цианокобаламина H2S [47]. George и др.
указывают фиолетово-окрашенный продукт, образующийся при взаимодействии HS-иона
с циано-аквакобинамидом [48]. Dolphin и Johnson использовали HS - ион
в качестве восстановителя в синтезе алкил кобаламинов [49]. Firth и др.
наблюдали максимумы поглощения при 352 нм и 530 нм при смешивании
гидроксикобаламина с разбавленным Na2S [50]. Так как одноатомная
сера имеет важные физиологические функции и так как кобаламин - идеальный
кофактор в метаболизме и транспортировке этой частицы серы, то описание
производного кажется важным. Исследования, описанные здесь, открывают
интересные производные Cbl и Cbi, содержащие стабильный Cо (II).
Коричневые продукты.
Когда гидрокси (или акво) формы Cbl или Cbi смешываются с
неорганическим сульфидом (H2S или HS-) при рН = 5 - 9,
цвет переходит от красного до коричневого в течение 15 мин при комнатной
температуре. В разбавленных растворах, цвет соединений хорошо различим,
Cbl-продукт имеет бордово-коричневую окраску, а Cbi-продукт имеет
оранжево-коричневый цвет. Реакция протекает одинаково хорошо в аэробных или
строго анаэробных условиях. CN-Cbl медленнее реагирует с HS-,
требуется от 45 до 60 мин, чтобы стал виден коричневый цвет соединения при
комнатной температуре. CN-Cbi реагирует по-разному, как описано ниже.
АденозилCbl, метилCb1 или метилCbi не реагируют с HS-, при
отсутствии освещения. ОН-Cbl и OH-Cbi дают те же коричневые продукты, если они
сначала восстанавливаются до стадии зеленого Co (I), с последующим анаэробным
добавлением элементарной серы, растворенной в кипящем этаноле. Все реакции
ускоряются если растворы подогреть.
После завершения реакции (см. промежуточные продукты ниже),
конечный спектры поглощения показывают максимумы или плечи поглощения (sh) при 270, 350, 415 и 528
нм для Cbl-производного и при 263, 330, 434, 490 (sh) и 650 нм для Cbi -
производного (Рис.10).
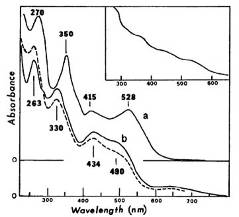
Рис.10. Электронный спектр поглощения в воде. Коричневый
продукт из Cbl (a), коричневый продукт из Cbi (b); пунктирная линия, продукт Cbl в 1,5 н HCI; вставка,
незаряженная фракция Cbi после прохождения через Dowex AG-50-Na+,
pH = 7.
Спектры показывают лишь незначительные изменения, так как
значение рН изменяется от 2 до 10. В 1,5 н. HCl при комнатной температуре Cbl -
производное становится оранжево-коричневым и спектральные изменения такие же,
как и для Cbi-производного (Рис.10). Коричневые продукты являются стабильными
на воздухе и на свету и могут быть подвергнуты очистке без особых мер
предосторожности, хотя длительное хранение их в воде приводит к незначительному
возврату к начальному аквакобаламину. Кристаллизация дает гетерогенный продукт,
содержащий красные полупрозрачные кристаллы и непрозрачные черно-коричневые
гранулы. Продукты содержат серу, которая высвобождается в виде H2S в
присутствии избытка цистеина с выходом 1 моль серы на моль коррина (погрешность
±6%). Продукт из Cbl не заряжен при значениях рН от 3 до 10. Основной продукт
из Cbi является положительно заряженным при рН =5. В дополнении к основному
продукту, приготовление Сbi демонстрирует различные количества (от 0 до 20%)
второго положительно заряженного производного, который, кроме заряда, имеет
свойства, аналогичные свойствам основного продукта.
Промежуточные продукты.
Образование коричневых продуктов включает промежуточные
стадии, характер и длительность которых зависит от природы коррина, рН и
температуры. Результаты, описанные ниже наблюдались при комнатной температуре.
При рН от 3 до 5 OH-Cbl дает временный промежуточный продукт с такой же
абсорбцией, как и для органических тиоловых аддуктов (λmax= 370, 515 и 545 нм). Фиолетовый цвет
превращается в коричневый в течение 15 мин и спектр поглощения распадается, что
показано на рисунке 10, в течение 24 часов. При рН от 5 до 9, фиолетового цвета
не видно, но исходный спектр поглощения показывает максимум при 372 нм, который
исчезает в течение 24 часов. ОН-Cbi - фиолетовый интермедиат, который не
устойчив при низких значениях рН, а в щелочной среде нет никаких доказательств
его существования, поскольку спектр быстро распадается, что показано на рисунке
10. Препараты из ОН-Cbi и H2S содержат различные количества (от 10
до 25%) незаряженного коричневого продукта, который может быть отделен от основного
продукта пропусканием через Dowex AG-50-Na+ (основной продукт
сохраняется). Незаряженных фракции из AG-50-Na+ имеют спектр
поглощения, имеющий конечную плотность и скрытые пики (рис.10). Однако, когда
добавляется CN-, это дает такие же голубые и пурпурные
продукты, как описано ниже, с чистым, хорошо определенным поглощением. Эта
фракция содержит 2 моля серы на моль коррина. Когда ее очищают через Dowex
AG-2-Cl или через фенол, она, по-видимому, преобразуется в основной продукт,
таким образом, после этих процедур, она содержит 1 моль серы на моль коррина.
При этом 98% фракции удерживается AG-50-Na+, и она имеет поглощения,
идентичные основному продукту. Когда Na2S добавляют к раствору
моноциано-Cbi при рН 12, цвет становится сразу же фиолетовый (λmax= 370, 548, 584 нм) и в течение нескольких минут
приобретает насыщенный синий оттенок. Когда смесь нейтрализовали до рН= 7, цвет
соединения становится коричневым и усиливается с получением продукта,
описанного выше.
ЭПР.
Коричневые продукты из Cbl и Cbi показывают характерный ЭПР
спектр Cbl (II) (рис.11А).
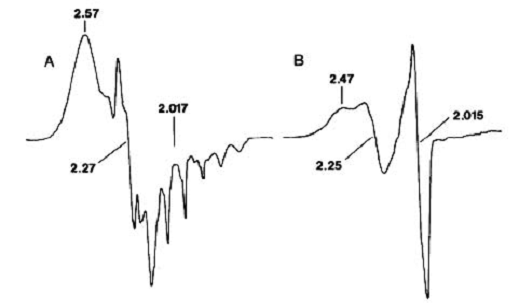
Рис.11. ЭПР спектр. Цифры - g-значения в указанных
точках. (А) продукт из кобаламина; СВЧ-мощность 0,020 мВт; усиление приемника
1x104; интегральный выход Co (II) был больше, чем 90%.
(B) Начальная стадия реакции между ОН-Cbi и NaHS; после 1 часа, смесь
пропускали через колонку Dowex AG-2-Cl для удаления избытка HS - и
замораживали для анализа; СВЧ-мощность 0,2 мВт; усиление приемника 1,6x104;
интегральный выход Co (II) сосатвил 5%.
В таблице 3, наблюдаемые ЭПР параметры сравнивают с
опубликованными значениями.
Таблица 3. Сравнение наблюдаемых и опубликованных ЭПР
параметров.
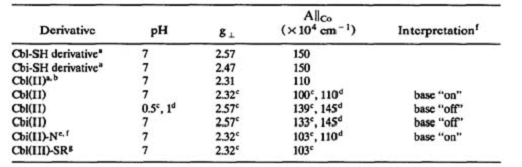
аНайденные значения; b Стандарт Cbl (II) приготовленный
фотолизом аденозил-Cbl; с опубликованные значения Cockle и др. [51]; d значения опубликованные
Bayston и др. [52]; е Cbi (II) в присутствии
азотистого основания, пиридина; f из литературы, структуры интерпретированы, как
имеющие азотистое основание, координированное к кобальту, называемое "base on" и структуры,
основание в которых не координировано к кобальту, "base off "; g тиоловый комплекс Cbl (II).
В начале реакционных смесей, когда интегральный выход Co (II) составил 5%, другой
сигнал равной интенсивности присутствовал при g = 2,015 (рис.11В).
Реакция с CN-.
Коричневые продукты дают уникальную реакцию с щелочным
цианидом. При смешивании с CN - при рН от 11 до 12, цвет сразу же
меняется необычно лазурный. Спектр поглощения синих продуктов из Cbl и Cbi
идентичны и имеют максимумы при 276, 321, 378, 566 и 597 нм. Синие производные
не являются стабильными и в течение 30 мин при комнатной температуре их спектры
преобразуются в спектры дициано-производных (λmax = 367, 540 и 580 нм)
(рис.12). Преобразование показывает изобестические точки при 342, 382, 482 и
592 нм.
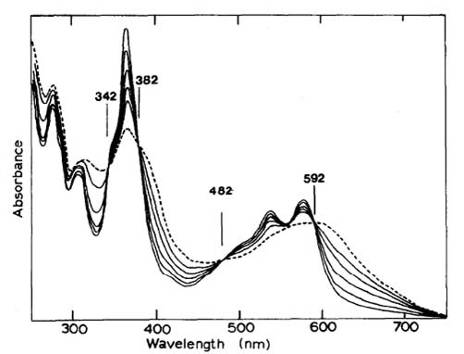
Рис.12. Реакция с CN-. Раствор коричневого продукта из Cbl, 0,01 М которого
реагирует с KCN, спектры записаны при комнатной температуре. Пунктирная линия -
спектр записанный за несколько сетов после смешивания. Другие спектры
записывалились с интервалами в 5 минут, кроме последнего, который был записан
через 10 мин после предпоследнего.
Формирование синих производных и их превращение в дициано -
производные происходит одинаково хорошо в аэробных или анаэробных условиях.
Синие производные более устойчивы в щелочной среде, в которой они образованы;
при рН=8 они преобразуются в фиолетовый примерно вдвое быстрее, чем при рН=12
и, если окрашенный в синий цвет раствор резко подкисляют до рН=4,5, то цвет
становится красным с оптической плотностью моноциано-Cbl (λmax = 360, 515, и 550 нм).
Свойства заряда синих производных дают возможность понять их
особенности. Следующие замечания относятся к производным Cbi, потому что они не
усложнены отрицательно заряженными фосфатными группами. Результаты были
получены с мягким анионитом, DEAE-целлюлоза, потому что полистериновый обменник
(например, AG-2-OH) преобразует синие производные в красно моноциановые формы
(по-видимому при помощи удаления HS - группы):
. Когда синее производное Cbi наносится на DEAE-целлюлозу при
рН 10,5, оно удерживается в дискретной голубой полосе. Когда колонну непрерывно
промывают водой и наблюдают, что синяя полоса превращается в фиолетовую в
течение 30 мин, но не элюируется. Если затем наносится разбавленный KCN,
пурпурная полоса элюируется немедленно.
. Если реакционной смесью (коричневое серосодержащее
производное Cbi + CN-) достигается фиолетовый цвет (λmax= 367, 540, 580 нм) до нанесения на DEAE
целлюлозную колонку, то фиолетовый цвет не сохраняется.
. Как описано выше, продукт синего цвета образуется также при
добавлении Na2S к CN-Cbi при рН 12. Когда эта окрашенная смесь
наносится на DEAE - целлюлозу, сине-фиолетовый цвет сохраняется в дискретной
полосе. Таким образом, показаны два различных фиолетовых соединения с очень
похожими спектрами поглощения - дицианид (незаряженный, стабильный, λmax= 367, 540, 580 нм), образованный в присутствии
избытка CN - и другое соединение (отрицательно заряженное,
нестабильное, λmax = 370, 548, 584 нм),
образованное c
участием CN - и S2-.
Описание механизма образования коричневых производных и их
взаимодействие с CN-.
Алкилтиолы добавленные к кобаламинам в кислой среде дают
аддукты фиолетового цвета. В щелочной среде, один электрон переходит с серы на
кобальт, образуются при этом Cbl (II) и R-Sрадикал, два радикала соединяются, образуя
R-S-S-R [53]. В то время как этот процесс протекает до некоторой степени, когда
HS - связывается с кобаламином в присутствии кислорода, описанные
здесь результаты показывают, что другие реакции приводят к стабильным
производным коричневого цвета, содержащие Co (II).
Спектры поглощения производных показывают несколько
интересных особенностей: первая, хотя производные напоминают Cbl (II) из-за коричневого
цвета соединения, они не имеют характерного поглощения для Cbl (II) (λmax = 311, 472 нм). Вторая, требуемое значение рН
для изменения оптической плотности серосодержащего Cbl-производного, как и для
серосодержащего Cbi-производного, равно примерно нулю, указывающее на то, что
Cbl-производное имеет "base-on" структуру и связь
диметилбензимидазола с кобальтом очень стабильна. Третья, в спектрах серосодержащих
производных наблюдается "наполение" глубоких впадин, обычно
наблюдаемых в спектрах кобаламинов, но при этом впадины в дальнейшем исчезают
так же и в производном Cbi с двумя группами сульфида. Это выравнивание не
связано с влиянием на сопряженные двойные связи системы корринового кольца, так
как при добавлении CN - спектр приобретает исходный вид. Скорее
всего это связано с неорганическими сульфидами координируемые к кобальту, для
сравнения конечное поглощение для восстановленного ферредоксина, который очень
похож на дисульфид Cbi, ниже 600 нм [54]. В то время как ЭСП данные показывают,
что Cbl-производное имеет "base-on" структуру при рН больше нуля,
магнитные параметры (табл.3) и отсутствие суперсверхтонкого расщепления
(Рис.10А) указывают на "base-off” структуру. Нет объяснения столь явному
расхождению. Если предположить, что существует "base-on" структура,
то атом серы предотвращает обычные взаимодействия свободного электрона с атомом
азота.
Описанные наблюдения выявляют несколько новых производных кобаламина,
входящих в комплексное равновесие, полная характеристика которых требует
значительных усилий. Коричневые HS производные, по видимому, не имеют структуру [Co
(II)] - SH, так как их стабильность, ультрафиолетовое и видимое поглощения, а
так же и ЭПР спектры не соответствуют аналогичным свойствам для тиоловых
комплексов, [Co (II)] - SR, сообщенных Cockle и др. [51]. Нижеупомянутые
предварительные объяснения согласуются с выводами (см. схему на рис.13).
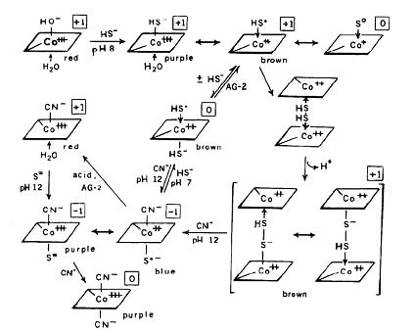
В ходе обсуждения ниже, и на рисунке 13, структуры
представляют собой производные Cbi, но они могут быть также применены (при
необходимости) к производным Cbl с "base-on" структурой. Временный
фиолетовый промежуточный комплекс наблюдали при рН от 3 до 5, и он является HS
- аддуктом, [Co (III)] - HS-. На начальной стадии, когда 5%
кобальта - Co (II), второй сигнал при g=2,015, вероятно, связан с образованием
HSрадикала, что указывает на присутствие [Co (II)] - HS. Реакция Cbi
(I) с элементарной серой
указывает на то, что частицы [Co (I)] - Sº существуют по крайней
мере временно. Таким образом, на начальных стадиях реакции возможно следующее
равновесие: [Co (III)] - HS - ↔ [Co (II)] - HS↔ [Co (I)] - Sº и медленное соединение двух радикалов дает димер [Co (II) - S-S - [Co (II)]. Предложенные
электронные сдвиги аналогичны тем, которые происходят в гидрид [Co (III)] - H - ↔ [Co (II)] - H↔ [Co (I)] - H+ [55], и в
молибденсодержащих гидроксилазах, Mo (VI) - S2 - ↔ Mo (IV) - Sº [56]. Структура предлагаемого димера аналогична структуре
гептаметилового эфира коб (II) ириновой кислоты [Co (II)] - I - [Co (II)] [57]. Структура синего
производного CN-h
[Co (II)] - S, полученного при
добавлении CN, имеет отрицательный заряд и нестабильна. Результаты показывают,
что синее производное может быть образовано двумя различными способами, путем
добавления CN - к коричневому серосодержащему производному и
добавлением S2 - к моноцианокобинамиду при рН 12. Последняя реакция
дает первый промежуточный продукт фиолетового цвета, который, в частности,
превращается в синий цвет в течение нескольких минут; фиолетовым промежуточным
соединением, как полагают, является CN - [Co (III)] - S2-, которое подвергается
электронному сдвигу и образуется синее производное. Структура, CN - [Co (II)] -
S-, является нестабильной
и электронный сдвиг легко дает CN - [Co (III)] - S2. Таким образом, синий
продукт из реакции CN - с коричневым серосодержащим производным Cbi
стал фиолетовым с той же скоростью, что и при взаимодействии в растворе с
избытком CN - или при промывании водой через DEAE-целлюлозу. Не были
найдены условия для стабилизации синих производных или отрицательно заряженных
фиолетовых промежуточных соединений, и они не были извлечены из DEAE-целлюлозы
и дополнительно не описаны. Поскольку сульфитоCb1 обнаружен как
"естественная" форма витамина в тканях животных [58], возможно, что
наоборот серосодержащее производное является природной формой, и что оно
преобразуется в сульфито Cb1 во время процедуры извлечения. Равновесие между [Co (III)] - SH и [Co (I)] - Sº, как предполагается, может наблюдаться в организме бактерий,
многие из которых содержат большое количество кобаламина, что может
способствовать метаболизму серы [59].
Список
литературы
1. J. M. Pratt. Inorganic chemistry of Vitamin B12;
John Wiley & Sons: London, New York, 1972.
2. A. C.rutenberg, H. J. Taube. Chem. Phys. 1952, 20,
825-826.
. J. Hunt, R. A. Plane. J. Am. Chem. Soc. 1954, 76, 5960-5962.
. T. J. Swift, R. E. Connick. J. Chem. Phys. 1962, 37, 307-320.
. D. Fiat, R. E. Connick. J. Am. Chem. Soc. 1968, 90, 608-615.
. H. W. Dodgen, G. Liu, J. P. Hunt. Inorg. Chem. 1981, 20,
1002-1005.
. M. Grant, R. B. Jordan. Inorg. Chem. 1981, 20, 55-60.
. W. G. Jackson, S. S. Jurisson, B. C. McGregor. Inorg. Chem.
1985, 24, 1788-1790.
. E. A. Betterton. Ph. D. Thesis, University of the Witwatersrand,
Johannesburg, 1982.
10. M. S. A. Hamza, C. Ducker-Benfer, R. van Eldik. Inorg. Chem.
2000, 39, 3777-3783.
. C. K. Poon. Coord. Chem. Rev. 1973, 10, 1-35.
. K. L. Brown, S. Cheng, X. Zou, J. D. Zubkowski, E. J. Valente,
L. Knapton, H. M. Marques. Inorg. Chem. 1997, 36, 3666-3675.
13. L. Knapton, H. M. Marques. Dalton Trans. 2005, 889-895.
14. L. Knapton, H. M. Marques. Dalton Trans. 2005, 889-895.
. C. B. Perry, M. A. Fernandes, K. L. Brown, X. Zou, E. J.
Valente, H. M. Marques, Eur. J. Inorg. Chem. 2003, 2095-2107.
. P. Day. Coord. Chem. Rev. 1967, 2, 99-108.
. C. Giannotti. In B12; Dolphin, D., Ed.; Wiley: New York, 1982;
393 - 430.
. J. M. Pratt. In Chemistry and Biochemistry of B12; Banerjee, R.,
Ed.; John Wiley & Sons: New York, 1999; 113 - 164.
19. T. Andruniow, P. M. Kozlowski, M. Z. Zgierski. J. Chem. Phys. 2001,
115, 7522-7533.
20. C. B. Perry, H. M. Marques, S. Afr. J. Chem. 2005, 58, 9-15.
. H. M. Marques, L. Knapton, X. Zou, K. L. Brown. J. Chem. Soc.,
Dalton Trans. 2002, 3195-3200.
22. A. Gossauer, B. Gruning, L. Ernst, W. Becker, W. S. Sheldrick.
Angew. Chem., Int. Ed. Engl. 1977, 16, 481-482.
. B. Gruning, G. Holze, T. Jenny, P. Nesvadba, A. Gossauer, L.
Ernst, W. S. Sheldrick. Helv. Chim. Acta 1985, 68, 1754-1770.
. A. J. Markwell, J. M. Pratt, S. S. Shaikjee, J. G. Toerien. J.
Chem. Soc., Dalton Trans. 1987, 1349-1357.
. K. Kamiya, O. J. Kennard. Chem. Soc., Perkin Trans.1 1982,
2279-2288.
. A. Fischli, J. J. Daly. Helv. Chim. Acta 1980, 63, 1628-1643.
27. B. Dresow, G. Schlingmann, G. M. Sheldrick, V. B. Koppenhagen.
Angew. Chem., Int. Ed. Engl. 1980, 19, 321.
. B. Krautler, C. Caderas, R. Konrat, M. Puchberger, C. Kratky. Helv. Chim.
Acta 1995, 78, 581-599.
. A. R. Battersby, I. Grgurina, P. R. Raithby, E. Egert, K. Harms,
G. M. Sheldrick, Acta Crystallogr., Sect. C 1989, C45, 1589-1593.
30. R. Benn, R.
Mynott. Angew. Chem., Int. Ed. Engl. 1985, 24, 333-335.
1. L. Enrst. Liebigs Ann. Chem. 1981,
376-387.
. L. Ernst, H. Maag. Liebigs. Ann. 1996, 323-326.
. L. Ernst. J. Chem. Soc., Perkin Trans.1 1984, 2267-2270.
. A. Gossauer, K. - P. Heise, H. Gotze, H. H. Inhoffen. Liebigs
Ann. Chem. 1977, 1480-1499.
35. K. L. Brown, J. M. Hakimi. Inorg. Chem. 1984, 23, 1756-1764.
. A. Medek, V. Frydman, L. Frydman. Proc. Natl. Acad. Sci. U. S.
A. 1997, 94, 14237-14242.
. C. Tavagnacco, G. Balducci, G. Costa, K. Taschler, von
Philipsborn, W. Helv. Chim. Acta 1990, 73, 1469-1480.
. A. Eschenmoser, R. Scheffold, E. Bertele, M. Pesaro, H.
Gschwend. Proc. Roy. Soc. A 1965, 288, 306-333.
. A. Veillard, B. J. Pullman. Theor. Biol. 1965, 8, 307-316.
40. L. L. Ingraham, J. P. Fox, N. Y. Ann. Acad. Sci. 1969, 153,
738-743.
. G. N. Schrauzer, L. P. Lee, J. W. Sibert. J. Am. Chem. Soc.
1970, 92, 2997-3005.
. T. Andruniow, M. Jaworska, P. Lodowski, M. Z. Zgierski, R.
Dreos, L. Randaccio, P. M. Kozlowski. J. Chem. Phys. 2009, 131, 105105.
. R. E. Stratmann, G. E. Scuseria, M. J. Frisch. J. Chem. Phys.
1998, 109, 8218-8224.
44. A. Dreuw, M. J. Head-Gordon. Am. Chem. Soc. 2004, 126,
4007-4016.
. D. J. Tozer. J. Chem. Phys. 2003, 119, 12697-12699.
. S. M. Chemaly, К. L. Brown, M. A. Fernandes, O. Q. Munro, C. Grimmer, F. M.
Marques. J. Inorg. Chem. 2011, 50, 8700-8718.
7. E. A. Kaczka,
D. E. Wolf, F. A. Kuehl, and K. Folkers, J. Am. Chem. Sot. 1951, 73, 3569.
48. P. George, D. H. Irvine, and S. C. Glauser, Ann. N. Y. Acad.
Sci. 1960, 88, 393.
. D. H. Dolphin and A. W. Johnson, Chem. Sot. Proc. 1963, 311.
. R. A. Firth, H. A.0. Hill, J. M. Pratt, R.G. Thorp, and R.J.P.
Williams, J. Chem. Sot. A, 1969, 381.
. S. A. Cockle, H.A. Hill, S. Ridsdale, and R. J. P. Williams, J.
Chem. Sot. Dalton, 1972.297.
. J. H. Bayston, F.D. Looney, J. R. Pilbrow, and M. E. Winfield,
Biochemistry 9, 1970, 2164.
53. G. N. Schrauzer and J.W. Sibert, Arch. Biochem.
Biophys. 1969, 130, 257.
. B. B. Buchanan and D.I. Amon, Adu. Enzymof. 1970, 33, 119.
. G. N. Schrauzer and R.J. Holland, J. Am. Chem. Sot.
1971, 93, 406O.
. R. C. Wahl and K.V. Rajagopalan, J. Biol. Chem. 1982, 257, 1354.
. J. P. Glusker, in B12, D. Dolphin, Ed., John Wiley & Sons,
1982, Chap.3.
. J. A. Begley and C.A. Hall, J. Chromafog. 1979, 177, 360.
. J.I. Toohey. J. Inorg. Chem. 1993, 49, 189-199.