Показатели перекисного окисления липидов и антиоксидантной защиты при интоксикации четырёххлористым углеродом
Введение
окисление отравление тетрахлорметан
Свободно-радикальное окисление (СРО) является необходимым
процессом для нормального функционирования клеток, но вместе с тем это и один
из универсальных механизмов их повреждения [1]. Неконтролируемая генерация активных
форм кислорода, кислородных метаболитов и сбой в функционировании
антиоксидантной системы вызывают окислительное повреждение биомолекул [2], что
приводит к дисфункции клеток и тканей организма. Окислительный стресс играет
ключевую роль в патогенезе многих патологических состояний: гипоксических и
ишемических повреждениях органов; сахарном диабете и его осложнениях; при
интенсивных физических и психоэмоциональных нагрузках, переутомлении, старении,
а также при заболеваниях печени и интоксикациях химическими агентами [3].
Действие многочисленных химических веществ на организм человека сложно и
многообразно. К числу ксенобиотиков с наиболее высокой степенью избирательной
гепатотоксичности и нефротоксичности относятся хлорированные углеводороды,
типичным представителем которых является тетрахлорметан (CCl4) [4]. Имеющиеся
данные свидетельствуют, что развитие CCl4-гепатита сопряжено с интенсификацией
СРО [5].
Важную роль в клинической практике играют антиоксидантные средства,
способные стабилизировать структуру и функции клеточных мембран и поддерживать
структурный гомеостаз организма в условиях патологии [6]. Антиоксидантные
препараты применяются как для профилактики, так и для лечения
свободно-радикальных патологий [2]. Успешность лечения определяется комплексом
мероприятий, включающих в себя, в том числе, восстановление функциональной
полноценности физиологической антиоксидантной системы - активности ферментов и
биоантиоксидантного витаминного статуса [7]. Наиболее токсичные радикальные
продукты ПОЛ удаляются главным образом отдельными биоантиоксидантами (БАО), к
которым, в частности, относится б-токоферол. Поэтому применению БАО, к которым
относятся водо- и жирорастворимые витамины, все больше внимания уделяют
клиницисты и ученые-экспериментаторы [8].
Целью настоящей работы явилось исследование процесса перекисного
окисления липидов и активности некоторых антиоксидантных ферментов при
окислительном стрессе, вызванном токсическим действием тетрахлорметана, а также
изучение состояния про- и антиоксидантного статуса под влиянием б-токоферола.
Исходя из цели, были поставлены следующие задачи:
1. оценить процессы перекисного окисления в плазме крови и гомогенате
почек контрольной группы животных и у животных подвергшихся воздействию
тетрахлорметана;
2. сравнить концентрацию продуктов ПОЛ - МДА и ДК в плазме и почках
у животных с введенным б-токоферолом до воздействия токсиканта и после;
. исследовать активность СОД и каталазы в плазме и гомогенате
почек крыс в норме и в условиях развития патологического процесса, вызванного
введением CCl4;
. определить активность антиоксидантных ферментов в плазме и
почках под действием витамина Е, введенного до и после воздействия
тетрахлорметана.
Работа выполнена на базе кафедры медицинской биологии ИФБиБТ Сибирского
федерального университета.
1. Обзор литературы
.1 Пути генерации активных форм кислорода в организме
Организм существует в аэробных условиях, и кислород является одним из
активных метаболических соединений в тканях. Известно, что О2 обладает высокой
реакционной способностью, но в силу спинового запрета происходит поэтапное
использование потенциальной окислительной способности О2 . Это обеспечивает
ступенчатое восстановление кислорода, которое является энергетически более
выгодным [9].
В тканях организма благодаря наличию сложных ферментативных комплексов со
специфическими электрон-транспортными простетическими и коферментными
группировками восстановление кислорода протекает по 4-х электронному пути с
образованием воды. Это основной путь метаболизма кислорода в организме,
сопряжённый с генерацией энергии. Существуют и другие пути превращения
кислорода, приводящие к образованию реакционно-способных соединений - активных
форм кислорода (АФК). В физиологических концентрациях эти соединения участвуют
в работе регуляторных систем клеток, с которыми связана функциональная
активность клеток, их жизнедеятельность (Рисунок 1) [3].
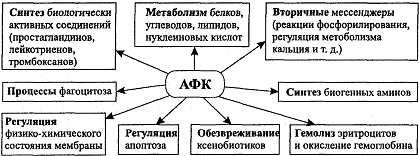
Рисунок 1 - Основные пути участия АФК в метаболических процессах в
организме
АФК - это группа различных соединений радикальной и нерадикальной
природы, которые различаются по продолжительности их существования и
активности. Понятие АФК носит чисто условный характер, эта группа включает
разные по своей химической структуре соединения: молекулы (Н2О2), свободные
радикалы (ОН•, НО•2), ион-радикалы (О2•‾) [10]. Основными компонентами
АФК являются супероксидный анион-радикал (О2•‾), гидроксильный радикал
(ОН•), перекись водорода (Н2О2), синглетный кислород (¹О2), гипогалоиды (окисленные
галогены) - НОСl, НОВr, НОI, пероксильный (алкилдиоксил) (RО2•) и алкоксильный (RО•) радикалы, оксид азота (NО•), пероксинитрит (ОNОО‾). Все эти соединения обладают высокой реакционной
способностью. Хотя понятие АФК с физико-химической точки зрения не совсем
правомочно, тем не менее широкое использование его не вызывает особых
возражений [11].
Образование АФК с химической точки зрения происходит в результате
процесса постепенного восстановления кислорода:
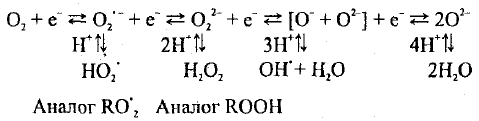 (1.1)
(1.1)
Источником доноров электронов в большинстве биологических систем являются
металлы переменной валентности - Fe++,
Сu+, Со++, Мn++, V++, Сr+ + + +, основными из которых
являются железо и медь [12].
АФК являются продуктами метаболических процессов, ферментативных и
неферментативных, которые в норме протекают в организме [13]. Генерация АФК
происходит по ходу окислительно-восстановительных реакций с участием кислорода
в различных субклеточных структурах клетки и при различных её функциональных
состояниях [14].
Основные источники активных форм кислорода в субклеточных структурах
клетки: митохондриальная электрон-транспортная цепь, плазматическая мембрана
(монооксигеназы, простагландинсинтетаза, НАДФН-оксидаза (фагоцитоз), ПОЛ),
пероксисомы (оксидазы, флавопротеины), эндоплазматический ретикулум и ядерная
мембрана (транспортная система цитохрома Р450, цитохром b558) [13].
Известно, что молекулярный кислород активно участвует в реакциях,
катализируемых оксидазами, оксигеназами (моно- и диоксигеназами). Оксидазы
катализируют реакции окисления различных субстратов, в результате которых
наблюдается прямое восстановление кислорода за счёт присоединения одного, двух
или четырёх электронов с последующим образованием О2•− (ксантиноксидаза),
Н2О2 (оксидазы аминокислот) и Н2О (цитохромоксидаза, аскорбатоксидаза) [9].
Оксигеназы катализируют реакцию активации молекулярного кислорода с
последующим непосредственным внедрением одного или двух атомов кислорода в
субстрат. Вклад этих реакций в общее использование кислорода невелик по
сравнению с дыхательными ферментами митохондрий. Эти оксигеназы участвуют в
метаболизме стеринов, некоторых витаминов, простагландинов, в обезвреживании
ксенобиотиков и т.д. Оксигеназы подразделяются на диоксигеназы и монооксигеназы
и гидроксилазы. Диоксигеназы катализируют реакцию включения всей молекулы
кислорода в обследуемый субстрат, монооксигеназы - одного атома, а второй
переходит в состав образующейся воды. По ходу отдельных ферментативных реакций
могут образовываться О2•− и Н2О2. С диоксигеназными реакциями чаще
связано образование О2•−. Генерация АФК может осуществляться и
неферментативным путём с участием гемсодержащих белков, биогенных аминов,
флавоноидов и т.д. [15].
1.2 Характеристика основных активных форм кислорода
Согласно классификации Ю.А. Владимирова [1], все образующиеся в организме
радикалы в зависимости от происхождения могут быть разделены на природные и
чужеродные. В свою очередь, природные можно разделить на 3 категории:
первичные, вторичные и третичные. Среди причин появления чужеродных форм
радикалов можно выделить 3 основных фактора: радиация, УФ-излучения и
ксенобиотики [3].
Первичные - образуются из молекул за счет реакций одноэлектронного
окисления с участием металлов переменной валентности. Это компоненты
дыхательной цепи, такие, как радикалы убихинона (коэнзима Q), супероксидный
анион-радикал и окись азота. К вторичным, относятся радикалы, образующиеся из
перекиси водорода, липоперекисей и гипохлорита в присутствии ионов Fe2+. Это
гидроксильный радикал, липидные радикалы, участвующие в реакциях цепного
окисления ненасыщенных ЖК цепей липидов биомембран и липопротеинов плазмы
крови. Третичные - радикалы, которые образуются при действии вторичных
радикалов на молекулы антиоксидантов и других легкоокисляющихся соединений.
Первичные радикалы специально вырабатываются организмом и выполняют жизненно
важные функции: переноса электронов в дыхательной цепи (убихинон), защиты от
микроорганизмов (супероксидный анион-радикал), регуляции кровяного давления
(окись азота), тогда как вторичные радикалы оказывают цитотоксическое действие
и, как правило, наносят организму большой вред. АФК могут выполнять функции
вторичных мессенджеров в процессах жизнедеятельности клеток. АФК участвуют в
сигнальной трансдукции, влияя на метаболизм кальция, гидролиз фосфолипидов,
фосфорилирование, модуляцию некоторых факторов транскрипции [1].
Деление активированных кислородных метаболитов на природные и чужеродные
оправдано с позиций их биологической значимости. Действительно, основной
функцией природных АФК является регуляторная, затем защитная, в определённых
ситуациях, как правило, патологических, они могут становиться токсичными и
индуцировать развитие деструктивных процессов, однако такие ситуации являются
исключением, а не правилом [15]. Образующиеся при действии радиации или в
процессе фотодинамической терапии чужеродные АФК, также как радикалы,
возникающие в процессе метаболизма ксенобиотиков, обладают выраженным
цитотоксическим и деструктивным действием [3].
.2.1 Супероксидный анион-радикал
Среди АФК супероксидному анион-радикалу отводят особую роль, так как
считается, что именно он является родоначальником многих других активных форм
кислорода. По оценке Halliwell B. 1 - 3%
поступающего с дыханием кислорода переходит в О2•−, что составляет в год
около 2 кг [16].
Супероксидный анион-радикал или диоксид образуется в результате
одноэлектронного восстановления кислорода. Время жизни его составляет 10-6 с,
что определяет радиус его действия ≈ 0,3 мкм [12].
Основным источником генерации О2•−, как и других АФК, являются
ферментативные системы, к которым относят НАДФН-оксидазу, функционирующую при
состояниях «дыхательного взрыва» в фагоцитирующих клетках, и НАДФН-оксидазу
нефагоцитирующих клеток, ферментативные комплексы системы тканевого дыхания и
микросомального окисления, ксантиноксидазу, альдегидоксидазу,
дигидрооротатдегидрогеназу, флавиновые гидроксилазы,
НАДФН-цитохром-с-редуктазу, ферменты метаболизма арахидоновой кислоты [10].
Уровень спонтанно образующегося О2•− в живых системах колеблется в
пределах 10-12 - 10-11 моль. Наибольшее количество О2•− продуцируется в
фагоцитирующих клетках в процессе «дыхательного взрыва» [17].
Образование супероксидного анион-радикала осуществляется в результате
переноса электрона с цитозольного НАДФН на О2 с образованием
О2•−.НАДФН + 2О2 → НАДФ+ + 2О2▪− + Н+(1.2)
Образующийся в результате «дыхательного взрыва» супероксидный
анион-радикал проявляет бактерицидное, цитотоксическое и иммунорегуляторное
действие [2].
Супероксидный анион-радикал обладает амфотерными
окислительно-восстановительными свойствами. Во многих биологических системах
О2•− может выступать в качестве донора электронов, участвуя в реакциях
восстановления ряда соединений. В физиологических условиях О2•− является
относительно слабым окислителем [9]. Так как супероксидный анион-радикал
обладает зарядом, проникновение его через мембрану затруднено и требует наличия
анионных каналов, которые обнаружены, в частности, в эритроцитарной мембране
[13].
Одноэлектронное восстановление О2•− приводит к образованию
гидродиоксид-иона (НО2▪) и его протонированной формы - перекиси водорода.
.2.2 Перекись водорода
Реакция образования перекиси водорода в физиологических условиях при
нейтральном значении рН протекает в две стадии:
О2•− + Н+ ↔ НО2▪,(1.3)
НО2▪ + О2•− + Н+ ↔ Н2О2 + О2(1.4)
Также образование Н2О2 может осуществляться за счёт непосредственного
двуэлектронного восстановления кислорода [9].
Перекись водорода относится к окислителям средней силы, способным
взаимодействовать с веществами радикальной и нерадикальной природы. Однако, в
физиологических условиях при отсутствии соответствующих катализаторов
(ферментов, ионов металлов переменной валентности) Н2О2 относительно стабильна
и неспособна выступать в качестве окислителя. Перекись водорода, являясь
электростатически нейтральной молекулой, легко проникает через мембраны в
другие клетки и ткани [3].
Физиологический уровень Н2О2 в тканях колеблется в пределах от 10-9 -
10-7 моль. На долю Н2О2 приходится до 13 - 20% кислорода, поступающего в
организм. Наибольшее содержание Н2О2 выявлено в печени (75%), где она активно
используется в процессах дезактивации ксенобиотиков экзогенного и эндогенного
характера [13]. Генерация перекиси водорода в тканях, в основном, связана с
функционированием ферментативных систем. Основным местом генерации Н2О2
являются пероксисомы. В меньших количествах Н2О2 может образовываться и в
других органеллах. В митохондриях за счёт действия ряда оксидаз
(сульфитоксидаза - КФ 1.8.3.1) наблюдается образование Н2О2.
Супероксиддисмутаза является источником перекиси водорода [18].
.2.3 Гидроксильный радикал
Гидроксильный радикал является наиболее токсичным из АФК, проявляя
цитотоксическое и мутагенное действие при состояниях окислительного стресса. ОН▪
обладает наибольшей реакционной способностью среди АФК и фактически может
атаковать любое химическое соединение. Время жизни его колеблется по разным
оценкам от 2 × 10-9 до 8 × 10-9 сек. [12]. Диффузный пробег ОН▪
до вступления в реакцию оценивается расстоянием, равным почти двум его
молекулярным диаметрам (менее 0,001 мкм) [19]. Образование ОН▪, как
одного из наиболее токсических соединений среди АФК, в основном, связывают с
металл-зависимым распадом Н2О2. Генерация гидроксильного радикала происходит в
результате реакции одноэлектронного восстановления перекиси водорода:
Меn + Н2О2 → Меn+1 + ОН▪ + ОН−,(1.5)
Н2О2 + е− ↔ ОН− + ОН▪(1.6)
Экспериментально показано, что в качестве металлов переменной валентности
могут выступать железо, титаний, кобальт, марганец, комплексы ванадия и никеля
[12]. В организме образование ОН▪ связано, в основном, с ионами железа и
меди. Взаимодействие двухвалентного железа с перекисью водорода составляет
основу реакции Фентона:
Fе2+ +
Н2О2 → Fе3+ + ОН▪ + ОН−(1.7)
При наличии в биологической среде Н2О2 и О2•− реакция может
протекать в две стадии. Она представляет модифицированный вариант реакции
Фентона, впервые описанный Haber F. и Weiss J. J.:
Fе3+ +
О2•− → Fе2+ + О2,(1.8)
Fе2+ +
Н2О2 → Fе3+ + ОН▪ + ОН−(1.9)
Суммарная реакция:
О2•− + Н2О2 Їсоль железа→ О2 + ОН▪ + ОН−(1.10)
В настоящее время проведены обширные экспериментальные исследования, в
которых чётко показана генерация ОН▪ в присутствии ионов железа, меди,
О2•− и Н2О2. Роль О2•− в этих процессах сводится к восстановлению
окисленной формы металла [10].
.2.4 Синглетный кислород
Переход кислорода из триплетного состояния в возбуждённое синглетное
состояние связан с обращением спина одного из электронов кислорода. Для
синглетного кислорода в отличие от триплетной формы не существует спинового
запрета и, соответственно, он обладает высокой реакционной способностью, атакуя
биологические молекулы в области двойных связей. Исходя из этого, синглетный
кислород активно участвует в окислительно-восстановительных реакциях, являясь
причиной окислительного повреждения белков, липидов, нуклеиновых кислот [9].
В организме пока не обнаружены специализированные системы, участвующие в
генерации синглетного кислорода. Но 1О2 может возникать по ходу ряда
ферментативных реакций, к которым относят каталазу, пероксидазы,
супероксиддисмутазу, липооксигеназу [20]. Образование синглетного кислорода
наблюдается в большинстве пигментных систем при экспозиции их на свету, в
частности, в хрусталике глаза. Молекулы хлорофилла в растениях и
фотосинтетических микроорганизмах являются активными генераторами синглетного
кислорода. Это позволило предложить использовать производный хлорофилла в
качестве сенсибилизаторов при фотодинамической терапии [10].
С помощью хемилюминесценции показано, что стимулированные эозинофилы
могут генерировать 1О2. Существуют сведения в пользу генерации синглетного
кислорода нейтрофилами в процессе «дыхательного взрыва» [18]:
НОСl + Н2О2 → Н2О + Сl− + Н+ + 1О2(1.11)
.3 Перекисное окисление липидов
Высокая агрессивность молекулярного кислорода в отношении органических
радикалов лежит в основе развития цепных процессов свободнорадикального
окисления. Большой вклад в изучение таких процессов перекисного окисления
липидов (ПОЛ) в биологических системах был сделан Б.Н. Тарусовым и его учениками
(Ю.А. Владимиров, А.И. Журавлёв, А.И. Арчаков) [1].
Свободнорадикальное окисление липидов наблюдается во всех тканях аэробных
организмов, главным образом в мембранах и липопротеиновых структурах, и
является вырожденно-разветвлённым цепным процессом. На сегодняшний день теория
и кинетические параметры развития процессов ПОЛ в разных экспериментальных
системах хорошо разработаны и изучены. Весь процесс ПОЛ делят на стадии:
зарождение цепей, развитие цепных реакций и их разветвление, обрыв цепей. Данные
стадии принято описывать последовательностью реакций [9]:
Характерные реакции, протекающие при окислении углеводорода (RH) в присутствии кислорода и
ингибитора (InH)
Зарождение цепей окисления: RH →
R▪
Продолжение цепей окисления: R▪ + О2 → RО2▪
RО2▪
+ RH → RООН + R▪
Вырожденное разветвление цепей: RООН → RО▪ + ▪ОН
RООR′ → RО▪ + ▪ОR′
Обрыв цепей окисления: R▪
+ R▪ → RR
RО2▪
+ R▪ → RООR
RО2▪
+ RО2▪ → RООR + О2
RО2▪
+ InH → RООН + In▪
RО▪
+ InH → RОН + In▪
R▪
+ InH → RH + In▪
RО2▪
+ In▪ → RООIn
Прочие реакции с участием ингибитора RООН + In▪
→ RО2▪ + InH и продуктов его превращений: In▪ + In▪
→ молекулярные продукты
In▪
+ RH → InH + R▪
InH + RООН → In▪ + RО▪
+ Н2О
InH +
О2 → In▪ + НО2▪
RООIn → RО▪
+ InО▪
Примечание. RH - углеводород
(липид); R▪, RО▪ и RО2▪
− алкильный, алкоксильный и перекисный (пероксидный) радикалы,
соответственно; RООН -
органический гидропероксид; InH -
ингибитор; In▪ − радикал ингибитора.
На начальной стадии зарождения цепей из валентно-насыщенных молекул
липидов в биологических системах образуются свободные радикалы. Зарождение
цепей с образованием первичных радикалов R▪ может происходить под действием экзогенных физических
или химических факторов, таких, как ионизирующая радиация, ультрафиолетовое
излучение, ксенобиотики, озон и др. [21]. При ряде патологических процессов
инициирующие окисление радикалы имеют преимущественно эндогенное происхождение
и возникают в результате активации ферментных радикалгенерирующих систем,
включая НАД(Ф)Н-оксидазы, ксантиноксидазу, митохондриальную цитохромоксидазу, NO-синтазы и др. [12]. Образующиеся в
энзиматических реакциях сравнительно малоактивные супероксидный анион- радикал
и оксид азота NO▪, а также пероксид водорода
Н2О2, как правило, не способны непосредственно инициировать процессы ПОЛ, однако
в результате ряда последовательных реакций с участием ферментов и ионов
металлов переменной валентности они могут давать начало высокореакционным
гидроксильным радикалам (ОН▪), гипогалогенным кислотам (НОСl, НОВr), синглетному кислороду (1О2), оксидам азота (NO, NO2▪), обладающим энергией, достаточной для разрыва С -
Н-связей и образования первичных липидных радикалов [22].
В аэробных условиях алкильные радикалы быстро взаимодействуют с
молекулярным кислородом, который выступает в качестве активного акцептора
электронов:
R▪
+ О2 → RО2▪(1.12)
В результате этой реакции образуется пероксидный радикал (RO2▪), который в свою очередь,
атакует липидные молекулы, приводя к возникновению гидропероксида ROOH и нового радикала R▪, индуцирующего продолжение
окислительной цепи:
RО2▪
+ RH → RООН + R▪(1.13)
Важная особенность липидов клеточных структур - большое количество в них
непредельных соединений: моно- и полиненасыщенных жирных кислот и их эфиров,
ненасыщенных алифатических спиртов (сфингозин) и углеводородов (сквален),
полициклических ненасыщенных спиртов гидроароматического ряда (стерины,
витамины группы Д) и др. Присутствие в структурах липидов непредельных
фрагментов способствует их лёгкому окислению, поскольку такие структуры
содержат связи С - Н с относительно низкой энергией, а скорость процесса
пероксидации в значительной мере лимитируется скоростью реакции молекулы RН с перекисным радикалом RO2▪, зависящей от энергии
разрыва С - Н-связи [23]. Принципиальное значение для протекания ПОЛ имеет
накопление в процессе окисления перекисных соединений ROOH. Энергия пероксидной связи О - О в 2-3 раза меньше,
чем энергия связей С - С или С = С, поэтому липидные перекиси - неустойчивые
соединения, легко подвергающиеся дальнейшим превращениям с образованием более стабильных
вторичных продуктов окисления: альдегидов, кетонов, спиртов, низкомолекулярных
кислот (муравьиной, уксусной, масляной), а также эпоксисоединений, многие из
которых токсичны для клеток [9]. Кроме того, перекиси склонны к гомолитическому
распаду:
RООН →
RО▪ + ▪ОН,(1.14)
прямым следствием, которого является разветвление цепей окисления.
Разложение гидроперекисей значительно ускоряется под действием ионов
металлов переменной валентности (прежде всего Fe2+,Cu2+),
облигатно присутствующих в клетках и внеклеточных средах:
RООН +
Fе2+ → RО▪ + ОН− + Fе3+,(1.15)
RООН +
Fе3+ → RО2▪ + Н+ + Fе2+(1.16)
или суммарно:
RООН →
RО▪ + RО2▪ + Н2О(1.17)
Образующиеся в процессе окисления свободные радикалы могут
рекомбинировать с образованием неактивных молекулярных продуктов, однако в
реальных условиях в биологических системах в силу низкой концентрации свободных
радикалов вероятность протекания таких реакций крайне мала, и свободные
радикалы со значительно большей скоростью взаимодействуют с молекулами субстрата
или с ингибиторами, которые всегда присутствуют в клетках и межклеточных средах
[19]. В организмах млекопитающих ингибиторами или антиоксидантами выступает
широкий спектр соединений различной химической природы: витамины Е, К, С,
коэнзим Q, мочевая кислота, каротиноиды и др.
При взаимодействии с данными соединениями образуются неактивные радикалы, не
способные участвовать в продолжении цепи окисления [3].
В организме человека процессы ПОЛ имеют большое значение для обновления
липидов мембран клеток и посредством этого поддержания структурного гомеостаза.
Поэтому разные по своим свойствам антиоксидантные системы необходимы для
поддержания активности ПОЛ на стационарном уровне в условиях значительных
изменений активности образования радикалов. Недостаток поступления облигатных
антиоксидантов приводит к развитию свободнорадикальных патологий - таких, как
авитаминозы Е и К, беломышечная болезнь [24]. Действие внешних прооксидантов
(радиация, ультрафиолет, загрязнители воздуха, гипероксия и др.) и активация
эндогенных механизмов генерации АФК приводят к напряжению механизмов
антиоксидантной защиты и развитию окислительного стресса, который может
проявляться на клеточном, тканевом и организменном уровнях. Окислительный
стресс - важный патогенетический фактор многих заболеваний, особенно
бронхолёгочных и сердечно-сосудистых, связанных с функциональным нарушением
биологических барьеров [11].
Увеличенное образование свободных радикалов в организме и связанное с
этим усиление процессов пероксидации липидов сопровождается рядом нарушений в
свойствах биологических мембран и функционировании клеток. Повреждаются
белковые структуры и липидный бислой в целом [23]. В последнее время ученые
уделяют все большее внимание взаимодействию мембран с нуклеиновыми кислотами в
ядре и митохондриях. По-видимому, одним из результатов пероксидации липидов
может стать повреждение этих макромолекул со всеми вытекающими последствиями.
Наиболее изучены три прямых следствия перекисного окисления липидов [10].
Во-первых, перекисное окисление липидов сопровождается окислением
тиоловых (сульфгидрильных) групп мембранных белков (Pr), в результате
неферментативной реакции SH-групп со свободными радикалами липидов; при этом
образуются сульфгидрильные радикалы, которые затем взаимодействуют с образованием
дисульфидов либо окисляются кислородом с образованием производных сульфоновой
кислоты [1]:
Pr-SH + L· → LH + Pr-S,(1.18)-S· + Pr2-S· →
Pr1-SS- Pr2,(1.19)
Pr-S· + O2 → Pr-SO2· → молекулярные производные (1.20)
Большую роль в патологии клетки играет также инактивация ион-транспортных
ферментов, в активный центр которых входят тиоловые группы, в первую очередь
Ca2+-АТФазы. Инактивация этого фермента приводит к замедлению
"откачивания" ионов кальция из клетки и, наоборот, к входу кальция в
клетку, увеличению внутриклеточной концентрации ионов кальция что, в свою
очередь, ведет к активации фосфолипазы А2, а также высвобождению арахидоновой
кислоты, что может привести к некрозу клеток и тканей [23]. Наконец, окисление
тиоловых групп мембранных белков приводит к появлению дефектов в липидном слое
мембран клеток и митохондрий. Под действием разности электрических потенциалов
на мембранах через такие поры в клетки входят ионы натрия, а в митохондрий -
ионы калия. В результате происходит увеличение осмотического давления внутри
клеток, в митохондриях, что приводит к их набуханию и ведёт к ещё большему
повреждению мембран [18].
Второй результат перекисного окисления липидов связан с тем, что продукты
пероксидации обладают способностью непосредственно увеличивать ионную
проницаемость билипидного матрикса. Так показано, что продукты перекисного
окисления липидов делают липидную фазу мембран проницаемой для ионов водорода и
кальция. Это приводит к тому, что в митохондриях окисление и фосфорилирование
разобщаются, а клетка оказывается в условиях энергетического голода (т.е.
недостатка АТФ). Одновременно в цитоплазму выходят ионы кальция, которые
повреждают клеточные структуры [1].
Третий, самый важный результат пероксидации - это уменьшение стабильности
липидного слоя, что может привести к электрическому пробою мембраны собственным
мембранным потенциалом, т.е. под действием разности электрических потенциалов,
существующей на мембранах живой клетки. Электрический пробой приводит к полной
потере мембраной ее барьерных функций [3].
.4 Метаболизм ксенобиотиков
Многие ксенобиотики, попав в организм, подвергаются биотрансформации и
выделяются в виде метаболитов. В основе биотрансформации по большей части лежат
энзиматические преобразования молекул. Биологический смысл явления -
превращение химического вещества в форму, удобную для выведения из организма, и
тем самым, сокращение времени его действия [7].
Метаболизм ксенобиотиков проходит в две фазы. В ходе первой фазы
окислительно-восстановительного или гидролитического превращения молекула
вещества обогащается полярными функциональными группами, что делает ее
реакционно-способной и более растворимой в воде [25]. Во второй фазе проходят
синтетические процессы конъюгации промежуточных продуктов метаболизма с
эндогенными молекулами, в результате чего образуются полярные соединения,
которые выводятся из организма с помощью специальных механизмов экскреции [26].
Метаболизм одних ксенобиотиков сопровождается образованием продуктов
существенно уступающих по токсичности исходным веществам. В процессе
метаболизма других веществ образуются более токсичные соединения. Примером
такого рода превращений является, в частности, четырёххлористый углерод [27].
Процесс образования токсичных продуктов метаболизма называется
"токсификация", а продукты биотрансформации, обладающие высокой
токсичностью - токсичными метаболитами. Во многих случаях токсичный метаболит
является не стабильным продуктом, подвергающимся дальнейшим превращениям. В
этом случае он также называется промежуточным или реактивным метаболитом [28].
Реактивные метаболиты это как раз те вещества, которые часто и вызывают
повреждение биосистем на молекулярном уровне. Общим свойством практически всех
реактивных метаболитов является их электродефицитное состояние, т.е. высокая
электрофильность. Эти вещества вступают во взаимодействие с богатыми
электронами (нуклеофильными) молекулами, повреждая их [29]. К числу последних
относятся макромолекулы клеток, в структуру которых входят в большом количестве
атомы кислорода, азота, серы. Это, прежде всего, белки и нуклеиновые кислоты.
Реактивные метаболиты либо присоединяются к нуклеофильным молекулам, образуя с
ними ковалентные связи, либо вызывают их окисление. В обоих случаях структура
макромолекул нарушается, следовательно, нарушаются и их функции [30].
Биоактивация далеко не всегда сопровождается повреждением биосубстрата,
поскольку одновременно в организме протекают процессы детоксикации и репарации
[31]. Интенсивность этих процессов может быть достаточной для компенсации
ущерба, связанного с образованием реактивных метаболитов. Тем не менее, при
введении высоких доз токсиканта, повторном воздействии защитные механизмы могут
оказаться несостоятельными, что и приведет к развитию токсического процесса
[22].
І фаза метаболизма в широком смысле может быть определена, как этап
биотрансформации, в ходе которого к молекуле соединения либо присоединяются
полярные функциональные группы, либо осуществляется экспрессия таких групп,
находящихся в субстрате в скрытой форме. Это достигается либо путем окисления
или (значительно реже) восстановления молекул с помощью оксидо-редуктаз, либо
путем их гидролиза эстеразами и амидазами [26].
Фаза II − этап биологической конъюгации
промежуточных продуктов метаболизма с эндогенными молекулами, такими как
глутатион, глюкуроновая кислота, сульфат и т.д. Специфические системы
транспорта конъюгированных дериватов обеспечивают их выведение из организма
[30].
В ходе биопревращений липофильный и, следовательно, трудновыводимый
ксенобиотик становиться гидрофильным продуктом, что обусловливает возможность
его быстрой экскреции.
Основным органом метаболизма ксенобиотиков в организме человека и
млекопитающих является печень, главным образом благодаря разнообразию и высокой
активности здесь ферментов биотрансформации [32]. Кроме того, портальная
система обеспечивает прохождение всех веществ, поступивших в желудочно-кишечный
тракт, именно через печень, до того, как они проникнут в общий кровоток. Это
также определяет функциональное предназначение органа [33]. Тончайшая сеть
печеночных капилляров, огромная площадь контакта между кровью и поверхностью
гепатоцитов, обеспечивающаяся микроворсинками базальной поверхности печеночных
клеток, обусловливают высокую эффективность печеночной элиминации токсиканта на
клеточном уровне [34].
Продукты I фазы метаболизма
поступают в общий кровоток и могут оказывать действие на органы и системы.
Печень выбрасывает в кровь и продукты II фазы метаболизма [26]. Из крови продукты превращения могут захватываться
почками, легкими, другими органами, повторно печенью для экскреции с желчью. С
желчью метаболиты поступают в кишечник, где частично реабсорбируются и повторно
поступать в печень (цикл печеночной рециркуляции) [35].
Несмотря на доминирующую роль печени в метаболизме ксенобиотиков, другие
органы также принимают участие в этом процессе. Почки и легкие содержат энзимы
и I и II фаз метаболизма. Особенно велика роль почек, поскольку в
этом органе имеется специфическая система захвата и катаболизма продуктов
конъюгации, образующихся в печени [7]. Активность других органов, таких как
кишечник, селезенка, мышечная ткань, плацента, мозг, кровь - значительно ниже,
однако наличие энзимов, катализирующих процессы биотрансформации, при
отравлении токсифицирующимися ксенобиотиками, имеет ключевое значение в
развитии патологических процессов в этих органах. В процессе внепеченочного
метаболизма могут образовываться продукты, как аналогичные продуктам
печеночного происхождения, так и отличные от них. Иногда в ходе внепеченочного
метаболизма может осуществляться активация метаболитов, образующихся в печени
[36].
Энзимы, участвующие в метаболизме ксенобиотиков, локализованы в основном
внутриклеточно. Небольшое их количество определяется в растворимой фракции
циотозоля, митохондриях, большинство же связаны с гладким эндоплазматическим
ретикулумом [26]. Методом ультрацентрфугирования гладкий эндоплазматический
ретикулум выделяется из исследуемых клеток в виде фрагментов мембранных
структур, называемых микросомами. Поэтому основная группа ферментов,
участвующих в метаболизме ксенобиотиков, получила название «микросомальные
энзимы» [37].
Часть ферментных систем метаболизма ксенобиотиков локализуются в
жидкостях организма. Прежде всего, это эстеразы плазмы крови, участвующие в
гидролизе целого ряда чужеродных веществ [38].
Разнообразие чужеродных химических веществ, способных подвергаться в
организме метаболическим превращениям, является следствием многообразия
энзимов, участвующих в I фазе
биотрансформации и их низкой субстратной специфичности. Многие из энзимов
существуют в нескольких формах (изоферменты), различающихся по своим
физико-химическим свойствам (молекулярная масса, электрофоретическая
подвижность, абсорбцией света с разными длинами волн), отношению к индукторам и
ингибиторам и активностью в отношении субстратов различного строения [39].
Энзимы I фазы, участвующие в процессе
биотрансформации ксенобиотиков, можно классифицировать в соответствии с типом
активируемой ими реакции [26]:
. Оксидазы смешанной функции: цитохром Р-450 и флавинсодержащие
монооксигеназы;
. Простогландинсинтетазы: гидропероксидазы и другие пероксидазы;
. Алкогольдегидрогеназы и альдегиддегидрогеназы;
. Флавопротеинредуктазы;
. Эпоксидгидролазы;
. Эстеразы и амидазы.
Особое значение для биотрансформации ксенобиотиков имеют микросомальные
энзимы. Как уже указывалось, морфологическим эквивалентом микросом в интактных
клетках является гладкий эндоплазматический ретикулум [6]. В ходе
микросомального окисления часто образуются реакционноспособные промежуточные
продукты. Некоторые из них нестабильны и подвергаются дальнейшему превращению,
другие достаточно устойчивы. К числу таких соединений, подвергающихся
биотрансформации с образованием активных промежуточных продуктов в ходе I фазы метаболизма, относится
тетрахлорметан [40].
.4.1 ЦитохромР-450-зависимая монооксигеназная система
Энзимы рассматриваемой группы, цитохромР-450 зависимые оксидазы (Р-450),
как правило, обладают низкой субстратной специфичностью, вызывая превращения
веществ самого разного строения, и потому часто называются оксидазами смешенной
функции. Р-450 относятся к группе гемопротеинов типа цитохромов b −
пигментов, активно связывающих монооксид углерода [5].
Р-450 представляют собой семейство энзимов, локализующихся в
эндоплазматическом ретикулуме, которые могут быть разделены с помощью иммунологических
и иных методов на несколько подсемейств. Отдельные ткани содержат несколько
различных изоформ Р-450. Встречаются тканеспецифичные формы энзимов [41].
Изоферменты Р-450 часто проявляют перекрестную субстратную специфичность, таким
образом, как правило, более чем один изофермент принимает участие в метаболизме
ксенобиотика [37]. Наличие специфических форм энзимов обусловлено генетическими
механизмами, а повышение содержания в тканях различных изоферментов
индуцируется действием на организм различных ксенобиотиков: лекарств, ядов,
экотоксикантов. Р-450 подвержены не только активации, но и инактивации, как
исходными ксенобиотиками, так и их реактивными метаболитами [26].
Реакции микросомального окисления, протекающие при участии Р-450, как
правило, зависят от содержания O2 и НАДФН в среде. Молекулярный кислород
активируется цитохромомР-450 (или другими цитохормами, например, Р-448) [26] .
Активация осуществляется с помощью НАДФН при участии флавин-содержащего энзима
НАДФН-цитохромР-450 редуктазы. Поскольку донором электронов в превращениях
субстратов, катализируемых этими энзимами, является НАДФН, суммарное уравнение
реакции может быть записано следующим образом [39]:
RH + O2 + HAДФН + Н+ → ROH +
НАДФ+ + Н2О(1.21)
ЦитохромР-450, НАДФН-цитохромР-450 редуктаза и фосфолипиды биологических
мембран, в которые встроены оба энзима, образуют микросомальный
монооксигеназный комплекс. Несмотря на то, что энзимы комплекса связаны с
биологическими мембранами, их свойства могут быть изучены in vitro [42].
Установлены основные закономерности протекания ферментативных процессов с
участием микросомального монооксигеназного комплекса (Рисунок 2) [43].
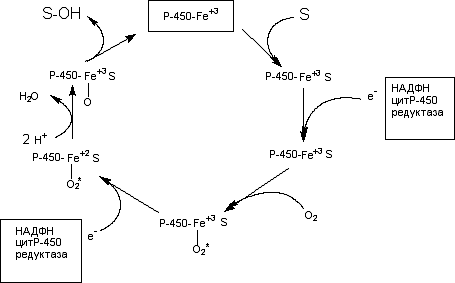
Рисунок 2 − Упрощенная схема превращения субстрата при участии
цитохромаР-450
Как видно из рисунка на начальном этапе ксенобиотик (S) вступает во
взаимодействие с окисленной формой цитохромаР-450. Затем к этому комплексу с
помощью НАДФН-зависимой цитохромР-450 редуктазы присоединяется электрон,
донором которого является восстановленный НАДФН [44]. После этого комплекс
взаимодействует с кислородом. После взаимодействия со вторым электроном (донор −
НАДФН) происходит активация связанного с цитохромом кислорода, который
приобретает способность связывать протоны и образовывать воду. Образовавшаяся
при этом форма цитохромаР-450 гидроксилирует субстрат [7].
Метаболизируемое вещество не связывается непосредственно с геминовой
группой цитохромаР-450. Оно присоединяется к белковой части цитохрома [37].
Процесс превращения ксенобиотиков чувствителен к СО, поскольку это вещество
вытесняет кислород из связи с железом геминовой группы цитохромаР-450.
Некоторые оксидазы резистентны к СО (образование N-оксидов) [22].
Поскольку Р-450 − гемопротеины, их активность отчасти регулируется
процессами синтеза гема, т.е. связана с метаболизмом железа [42]. Нарушение
метаболизма, голодание, понижение соотношения НАДФН/НАДФ+ могут приводить к
снижению активности Р-450 [45].
Окисление ксенобиотиков при участии Р-450 - основной механизм их
биотрансформации в I фазе
метаболизма. Р-450 катализирует окисление практически всех классов органических
молекул. Субстратом для энзимов являются и простые молекулы типа хлороформа и
стероиды и сложные гетероциклические соединения, например антибиотик
циклоспорин [46]. Р-450 могут катализировать не только окисление, но и
восстановление некоторых биосубстратов, например четыреххлористого углерода,
галотана, некоторых других галогенированных углеводородов с образованием
свободных радикалов. Такое необычное превращение реализуется в условия
пониженного парциального давления кислорода в тканях [39].
.4.2 Конъюгация
Превращение молекул в первой фазе биотрансформации усиливает их
полярность, уменьшает способность растворяться в липидах. Уже только благодаря
этому целый ряд чужеродных соединений лучше выделяется с мочой [47]. Эффект еще
более усиливается, когда к образовавшимся в ходе I фазы метаболизма продуктам присоединяются такие эндогенные
вещества, как ацетат, сульфат, глюкуроновая кислота, глутатион и т.д. Как и
энзимы I фазы метаболизма ксенобиотиков,
энзимы II фазы обладают слабой субстратной
специфичностью и участвуют в превращениях большой группы химических веществ
[26].
Рассматриваемую группу энзимов можно классифицировать следующим образом:
. Энзимы, формирующие эфирные или амидные связи с промежуточными
метаболитами:
− ацетил КоА: амин N-ацетилтрансфераза;
− сульфотрансфераза;
− УДФ-глюкуронозилтрансфераза;
. Энзимы, активирующие конъюгацию веществ с глутатионом: глутатион
S-трансферазы;
. Энзимы, активирующие конъюгацию веществ с цистеином:
цистеин-конъюгирующие в-лиазы [7].
В ряде случаев, в ходе метаболизма ксенобиотиков во II фазе также образуются токсичные
продукты (токсификация).
Наиболее важные ферменты второй фазы относятся к классу трансфераз.
Наиболее широка и многообразна активность семейства глутатионтрансфераз,
метаболизирующих тысячи ксенобиотиков. Большинство этих ферментов находится в
гиалоплазме, но один из них локализован в мембранах ЭПС и митохондрий, другой -
в хроматине [26]. Основная реакция - конъюгация с восстановленным глутатионом
(GSH) - протекает в двух вариантах: 1) присоединение к субстрату (алкены и
эпоксиды) полной молекулы GSH, 2) нуклеофильное замещение по электрофильным
атомам С (галоген- и нитроалканы), N (тринитроглицерин), S (тиоцианаты и дисульфиды)
или P (метилпаратион) [48].
R + GSH → HRSG,(1.22)
RX + GSH → RSG + HX(1.23)
При дальнейшем метаболизме глутатионовые конъюгаты переходят в
меркаптуровые кислоты или меркаптаны. Кроме того, глутатионтрансферазы
восстанавливают органические гидроперекиси в спирты и изомеризуют некоторые
стероиды и простагландины [49].
Локализованные в основном в ЭПС уридиндифосфат(УДФ)-глюкуронилтрансферазы
присоединяют остаток глюкуроновой кислоты, а гиалоплазматические
сульфотрансферазы - сульфат к фенолам, спиртам и аминам. Эти ферменты
метаболизируют, например, анилин, фенол, морфин, левомицетин, салицилат,
парацетамол, зидовудин (лекарство против СПИДа), пероральные контрацептивы
[36].
Ацетилтрансферазы метаболизируют путем присоединения ацетила к N-
(сульфаниламиды, противотуберкулезные средства изониазид и n-аминосалициловая
кислота) или к О- (некоторые канцерогены). Мембранные и гиалоплазматические
метилтрансферазы метилируют ОН-, NH2- и SН-группы и метаболизируют, например,
пиридин, тиоурацил, унитиол, кокаин [35].
Функционирование всех ферментов второй фазы ограничивается тем, что они
метаболизируют только те вещества, которые имеют функциональные группы. Именно
поэтому эти ферменты чаще включаются после образования или освобождения
функциональных групп ферментами первой фазы, то есть во второй фазе метаболизма
ксенобиотиков [50]. Однако трансферазы имеют важные достоинства: 1) они есть во
всех клетках, поэтому: 2) функционируют при любых путях поступления
ксенобиотиков в организм, 3) осуществляют или завершают детоксикацию, а иногда
исправляют ошибки первой фазы [39].Совместное функционирование обеих фаз
метаболизма особенно эффективно. В подавляющем большинстве случаев оно
обеспечивает обезвреживание десятков тысяч ксенобиотиков всех химических
классов и самых разных групп: токсических веществ, мутагенов, канцерогенов,
пестицидов (средств для борьбы с вредными растениями и животными), красителей,
растворителей, лекарств и др. [7]. Метаболизм ксенобиотиков происходит в разных
частях клетки, но наиболее активные системы находятся в ЭПС и гиалоплазме. Это
обеспечивает метаболизм или связывание ксенобиотиков на дальних подступах к
наиболее жизненно важным частям клетки - ядру и митохондриям. В результате
увеличивается устойчивость клеток и организма, возникает возможность сохранить
здоровье и жизнь в условиях загрязнения среды [26].
1.5 Механизмы воздействия тетрахлорметана
Как уже отмечалось, развитие цепных радикальных окислительных процессов
сопровождается образованием перекисных (RО2▪) и алкоксильных (RО▪) радикалов. Многие факторы физической и химической
природы могут инициировать зарождение органических радикалов в живых
организмах. В данной работе более подробно следует остановиться на таком
экзогенном химическом факторе как ксенобиотики [51].
Эффективными инициаторами радикалообразования, особенно в клетках печени,
могут выступать монооксигеназы, с этим связано токсичное действие
галогенированных углеводородов, таких как тетрахлорметан и бромбензин [52]. В
клетках тетрахлорметан (ССl4)
метаболизируется монооксигеназной системой с образованием трихлорметильного
радикала (ССl3▪) [3]. Это происходит
благодаря тому, что связь углерод-хлор в молекуле ССl4 сильно поляризована, и углерод имеющий положительный заряд,
легко принимает электрон с активного центра цитохрома Р-450. В результате
последующего гомолитического распада восстановленного интермедиата и образуется
ССl3▪ [53]:
CCl4 +
е− → ССl4−▪ →
ССl3▪ + Cl−(1.24)
Образовавшийся трихлорметильный радикал быстро взаимодействует с
молекулярным кислородом с образованием трихлорметилпероксильного радикала:
ССl3▪ + О2 → ССl3О2▪(1.25)
Трихлорметилпероксильный радикал (ССl3О2▪) способен разрывать С-Н-связи в липидах с
образованием липидных радикалов:
R-Н +
ССl3О2▪ → R▪ + ССl3О2Н(1.26)
Показано, что CCl3O2
▪ очень быстро реагирует с полиненасыщенными жирными кислотами и
еще более активно - с различными антиоксидантами: аскорбиновой кислотой,
в-каротином, б-токоферолом, прометазаном [54]. Радикал CCl3
▪ реагирует с различными субстратами значительно медленнее, чем CCl3O2
▪. Кроме того, CCl3▪ взаимодействует с полиненасыщенными
жирными кислотами и антиоксидантами практически с одинаковой скоростью [55].
Поэтому б-токоферол и прометазан, эффективно ингибируя реакции CCl3O2
• с полиненасыщенными жирными кислотами, не оказывают существенного
влияния на реакции этих субстратов с CCl3▪ [56].
Образующиеся в процессе метаболизма тетрахлорметана радикалы CCl3▪
и CCl3O2▪ вовлекаются в последующие метаболические процессы через реакции
отрыва водорода и галоалкилирование. В результате первой реакции образуется
хлороформ и радикал молекулы донора водорода [57]. В случае, если донором
водорода являются полиненасыщенные ацилы мембранных фосфолипидов, образующиеся
радикалы инициируют процесс перекисного окисления липидов. В результате реакций
галоалкилирования трихлорметильная группа ковалентно связывается с различными
молекулами: липидами, белками, нуклеиновыми кислотами, пиримидиновыми и
пуриновыми основаниями [58]. Так как б-токоферол и прометазан ингибирует
перекисное окисление липидов, инициируемое тетрахлорметаном в микросомах печени
и изолированных гепатоцитах, не оказывая существенного влияния на процессы
ковалентного связывания, был сделан вывод о том, что в реакции отрыва водорода
участвует, главным образом, активный радикал CCl3O2, а галоалкилирование
обусловлено ковалентным связыванием с различными субстратами радикала CCl3▪
[59]. В экспериментах in vitro установлено, что повреждение компонентов
электронтранспортных цепей эндоплазматического ретикулума и инактивация системы
микросомального окисления, нарушение системы кальциевого гомеостаза, секреция
липопротеидов и жировое перерождение печени обусловлены, главным образом,
галоалкилированием [56]. Однако показано, что основной причиной повреждения и
гибели изолированных клеток печени при их инкубации в присутствии
галогензамещенных углеводородов является развитие перекисного окисления липидов
[59].
Повреждающее действие перекисного окисления липидов обусловлено,
по-видимому, двумя механизмами. Во-первых, изменяются структурно-функциональные
свойства мембран в результате потери полиненасыщенных жирных кислот и
накопления окисленных липидов. Во-вторых, образуются низкомолекулярные,
токсичные продукты окисления мембранных липидов [26]. Среди этих соединений
особое значение придается насыщенным и ненасыщенным альдегидам. Так, малоновый
диальдегид, один из конечных продуктов ПОЛ, образует внутри- и межмолекулярные
перекрестные сшивки, в результате происходит инактивация ферментов, в том числе
и не имеющих сульфгидрильных групп, сшивание белков и нуклеиновых кислот.
Получающиеся в этих случаях флуоресцентные хромофоры, так называемые шиффовы
основания, имеют следующую структуру: R-N=CH-CH=CH-NH-R1 [27].
При рассмотрении роли галоалкилирования и перекисного окисления липидов в
патогенетических эффектах галогензамещенных углеводородов следует учитывать,
что в нормальном организме только незначительная часть образующихся пероксидов
липидов вовлекается в процессы радикального разрушения до альдегидов и других
токсичных низкомолекулярных продуктов [30]. Основное количество гидропероксидов
быстро восстанавливается в соответствующие оксисоединения, неспособные
участвовать в дальнейших свободнорадикальных процессах [26]. Установлено, что
образующиеся при ССl4-инициируемом ПОЛ гидропероксиды липидов (LOOH) быстро
восстанавливаются в соответствующие спиртовые группы, не вовлекаясь в
последующие процессы радикальной деградации, а сопряженные двойные связи
(диеновые коньюгаты) сохраняются в молекулах образующихся продуктов (LOH) [40].
Интересно, что если в экспериментах использовались животные, предварительно
содержащиеся на диете, исключающей содержание витамина Е и селена, доля
гидропероксидов липидов среди продуктов ПОЛ существенно увеличивается.
Напротив, дополнительное введение в пищу селена, витамина Е или других
антиоксидантов приводило почти к полному ингибированию перекисного окисления
липидов [60].
Таким образом, активация процессов ПОЛ под действием галогенированных
углеводородов является причиной развития токсических гепатитов [3]. Перекисное
окисление мембранных липидов приводит к деструкции мембран, в результате цепной
реакции из липидов мембраны образуются новые свободные радикалы, уже вторично
повреждающие другие органеллы клетки, что в конечном итоге и приводит к некрозу
гепатоцитов [61].
.6 Антиоксидантная защита тканей
Активные формы кислорода являются нормальными метаболитами обменных
процессов в организме и выполняют определённую физиологическую роль в
функционировании клетки. Но при определённых состояниях, сопряжённых с
интенсивной генерацией АФК, последние начинают проявлять свою реакционную
способность, связанную с разрушением клеточных структур и токсической
окислительной деструкцией биомолекул тканей - белков, липидов, углеводов и
нуклеиновых кислот [13]. Исходя из высокой реакционной способности АФК, их
содержание в клетках должно поддерживаться на определённом уровне, необходимом
для обеспечения жизненно важных метаболических процессов в клетке [12]. Это
осуществляется за счёт функционирования многокомпонентной антиоксидантной
системы (АОС), разнообразной по механизму действия и химической структуре.
Особенностям функционирования АОС, её классификации, изучению механизма
действия посвящены многочисленные обзоры и монографии [62].
Существует различный подход к классификации АОС. Компоненты
антиоксидантной защиты (АОЗ) в зависимости от их химического строения разделяют
на высокомолекулярные, в основном, белкового происхождения, и низкомолекулярные
соединения. Последние, исходя из растворимости, подразделяют на гидрофобные и
гидрофильные. С позиций механизма действия АОС можно рассматривать как систему,
проявляющую специфическую и неспецифическую антиоксидантную активность.
Действие специфической АОС направлено непосредственно на снижение уровня
оксидантов в тканях путём прямого разрушения и связывания активных форм
кислорода или образующихся радикалов, что приводит к обрыву цепей
свободно-радикальных реакций [13]. В свою очередь, в зависимости от характера
действия выделяют ферментативные и неферментативные компоненты специфической АОС
(Рисунок 3).
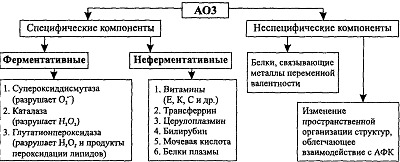
Рисунок 3 - Компоненты антиоксидантной защиты (АОЗ)
Действие неспецифической АОЗ связано со снижением возможности
дополнительной генерации свободных радикалов. Одним из проявлений
неспецифической АОЗ является устранение пула металлов переменной валентности
(железо, медь) за счёт их связывания, в частности, с высокомолекулярными
соединениями (трансферрин, ферритин, лактоферрин, церулоплазмин) и
предотвращение возможности их участия в свободно-радикальных реакциях [63].
Так, при физиологической концентрации ионов меди в плазме крови основная их
масса связывается с белками и только 6% с низкомолекулярными соединениями [13].
Разделение АОЗ на специфическую и неспецифическую достаточно условно.
Так, ряд образующихся комплексных соединений металлов с белками (к примеру,
церулоплазмин) могут проявлять и специфическую антиоксидантную активность (АОА)
[62]. Специфическую АОА проявляют ферментативные и неферментативные её
компоненты, а неспецифическая АОА связана, в основном, с неферментативными
компонентами [9].
К ферментативным элементам относят супероксиддисмутазу, каталазу,
глутатионпероксидазу, фосфолипид-гидропероксид глутатионпероксидазу (КФ
1.11.1.12), селен-независимую глутатион-S-трансферазу (КФ 2.5.1.18), тиол-специфическую пероксидазу
(КФ 1.11.1.7), сульфитоксидазу (КФ1.8.3.1), тиоредоксинредуктазу (КФ 1.6.4.5),
глутатионредуктазу (КФ 1.6.4.2), гемоксигеназу 1 (КФ 1.14.99.3), хинонредуктазу
(КФ 1.6.99.2) [15].
Неферментативная АОС представлена большой, очень разнообразной по своему
химическому строению группой соединений, которые непосредственно нейтрализуют
АФК или косвенно препятствуют их генерации [13].
.6.1 Супероксиддисмутаза
Супероксиддисмутаза (СОД) (супероксид-оксидоредуктаза, КФ 1.15.1.1) -это
ключевой фермент АОЗ, так как при её участии прерывается цепь
свободно-радикальных процессов в начале своего зарождения на стадии
одноэлектронного восстановления кислорода с образованием супероксидного
анион-радикала. Фермент супероксиддисмутаза впервые был описан McCord J. M. и Fridovich I. в 1969 году [64].
СОД осуществляет дисмутацию супероксидного анион-радикала, скорость
которой (2×109 л · моль-1 · с-1 и выше) значительно превышает скорость
спонтанной реакции при нейтральных значениях рН (7×105 л · моль-1 · с-1) [63]. Конечным
продуктом реакции является перекись водорода:
О2▪− + О2▪− + 2Н+ → Н2О2 + О2(1.27)
В настоящее время выделено несколько изоферментных форм СОД. Сu,Zn-СОД (31 кДа) является наиболее распространённой и хорошо
изученной. Она содержится в клетках эукариот (ядра, цитоплазматический матрикс,
пероксисомы, межмембранный просвет митохондрий, лизосомы) и обладает
чувствительностью к воздействию цианидов. Молекула фермента состоит из двух
идентичных субъединиц, каждая из которых содержит в области активного центра
один атом меди и один атом цинка. Cu++ принимает участие в дисмутации супероксидного анион-радикала, а цинк,
по всей вероятности, способствует стабилизации белковой молекулы [22].
Mn-СОД
(40 кДа) является цианрезистентной формой и содержится, в основном, в матриксе
митохондрий и хлоропластов эукариот [52]. Однако предшественник
митохондриальной Mn-СОД синтезируется в цитозоле, в митохондриях происходит
лишь отщепление от него полипептидного фрагмента [65]. В настоящее время этот изофермент
СОД выявлен у бактерий. Фермент состоит из 4 субъединиц, содержащих ион
марганца в области активного центра. Действие Мn-СОД направлено на дисмутацию О2▪ −,
образующегося в результате функционирования системы тканевого дыхания. Обе
формы СОД действуют как внутриклеточные ферменты [3].
В межклеточном пространстве обнаружена высокомолекулярная внеклеточная
(экстрацеллюлярная) СОД (135 кДа), состоящая из 4-х субъединиц. Каждая
субъединица имеет молекулярную массу около 30 кДа и содержит ионы меди и цинка
[62]. Кроме более высокой молекулярной массы, субъединица экстрацеллюлярной СОД
отличается от субъединицы цитоплазматической Cu,Zn-СОД аминокислотным составом,
антигенными свойствами и генным локусом, кодирующим аминокислотную
последовательность апофермента. Каталитическая активность экстрацеллюлярной СОД
значительно ниже, чем у цитоплазматического фермента [63]. Этот Сu,Zn-содержащий гликопротеин является главной изоформой
межклеточных жидкостей - плазмы, лимфы, синовиальной жидкости и в небольших количествах
обнаруживается почти во всех тканях. Экспрессия внеклеточной СОД связана
преимущественно с клетками глии и фибробластами в отличие от Cu,Zn-СОД, которая секретируется фактически всеми клетками
организма [42].
Выделяют ещё одну изоформу СОД - железосодержащий фермент, который
вначале был обнаружен у прокариот. В настоящее время показано, что этот фермент
широко распространён - от бактерий до растений-эукариот семейства Ginkgo biloba, Gingkoaceae, Cruciferae и Nymphaceae и др. Fe-СОД - это димер с молекулярной массой 41 кДа, в области
активного центра которого основное место занимает железо [62].
Содержание изоферментов супероксиддисмутазы в отдельных органах и тканях
экспериментальных животных существенно различается. Наиболее высокий уровень
Cu-,Zn-СОД выявлен в печени (до 350 мкг/г массы ткани), почках (185 мкг/г массы
ткани) и поджелудочной железе (165 мкг/г массы ткани). В сердце, мозге и
эритроцитах содержание этого изофермента составляет около 60-70 мкг/г массы
ткани (клеток). Наименьший уровень супероксиддисмутазной активности выявлен в
мышечной и жировой ткани [22]. Активность Mn-СОД снижается в ряду: сердце,
почки, печень, мозг, поджелудочная железа, мышечная и жировая ткань. В
эритроцитах Mn-СОД отсутствует. В отдельных органах также существуют различия в
распределении супероксиддисмутазной активности. Например, в мозге глиальные
клетки содержат супероксиддисмутазы в шесть раз больше, чем нейроны. С
возрастом, в процессе индивидуального развития организма, удельная активность
супероксиддисмутазы в печени, сердце и мозге мышей снижается, хотя снижение
активности в сердце и мозге компенсируется увеличением количества фермента
[60].
Образующийся в процессе диспропорционирования анион-радикала кислорода
пероксид водорода может утилизироваться с помощью двух ферментов: каталазы и
глутатионпероксидазы (КФ 1.11.1.9).
.6.2 Каталаза
Каталаза (КФ 1.11.1.6) - один из давно известных
ферментов-антиоксидантов, впервые описанный Loew в 1901 году. Каталаза является гемопротеином,
катализирует реакцию расщепления Н2О2. Разложение перекиси водорода происходит
в две стадии:
Каталаза-Fe3+ + Н2О2 →
окисленная каталаза,(1.28)
окисленная каталаза + Н2О2 → Каталаза-Fe3+ + 2 Н2О + О2(1.29)
В окисленном состоянии каталаза может проявлять пероксидазную активность,
участвуя в окислении спиртов и альдегидов. Однако, в физиологических условиях
пероксидазная активность фермента почти в 10 000 раз ниже каталазной [10].
Каталаза содержится в большинстве аэробных клеток. У животных она
присутствует почти во всех тканях организма. Наибольшее ей количество
сконцентрировано в печени, почках и эритроцитах. Содержание каталазы в
щитовидной железе, половых железах, соединительной ткани и ткани мозга
относительно низкое. На субклеточном уровне каталаза, в основном, локализована
в пероксисомах (80%) и цитозоле (20%). Небольшое её количество может
присутствовать в лизосомах и митохондриях. Каталаза относится к внутриклеточным
ферментам и из-за высокой молекулярной массы плохо проникает во внеклеточную
среду, где может быстро подвергаться протеолитическому расщеплению [9]. Поэтому
считается, что внеклеточная защитная роль каталазы незначительна, однако её
повышенное содержание в сыворотке крови при некоторых заболеваниях, таких как
ревматоидный артрит и острый респираторный дистресс-синдром, может служить для
защиты от окисления определённых молекул и структур, в частности, б1-ингибитора
протеиназ. Происхождение сывороточной каталазы при острых воспалительных
процессах не ясно, возможно её выделение из разрушенных клеток [3]. На культуре
лимфоцитов человека было показано, что наличие в культуральной среде даже
небольшого количества каталазы отменяет апоптотическую гибель и возвращает
клеткам Т-линии способность к росту в среде, не содержащей сыворотки [10].
Авторы этой работы предположили, что Т-лимфоциты могут выделять во внешнюю
среду каталазу, что необходимо для их защиты в области воспалительного очага.
Каталитическая активность этого фермента чрезвычайно высокая. Одна
молекула каталазы может разложить за секунду 44000 молекул пероксида водорода.
В присутствии фермента реакция почти не требует энергии активации и ее скорость
определяется скоростью диффузии субстрата к активному центру фермента. Однако
каталаза характеризуется большой величиной Км, и эффективное разложение
пероксида водорода происходит только при его высокой концентрации. Поэтому в
клетке максимальная каталазная активность характерна для мест с высокой
продукцией H2O2, например пероксисомах [22]. Защита каталазы от высоких
концентраций собственного субстрата, по-видимому, обеспечивается НАДФН. Каждая
субъединица тетрамерной молекулы каталазы способна связывать молекулу
восстановленного НАДФН, что предохраняет её от инактивации и повышает
ферментативную активность [3].
.6.3 Глутатионпероксидаза
Глутатионпероксидаза (ГПО) (КФ 1.11.1.9) впервые была описана в 1957 году
Mills G. C. По своей
структуре селенсодержащая ГПО является тетрамером, с молекулярной массой каждой
субъединицы около 19 кДа. Каждая субъединица содержит по одному атому селена,
связанного с цистеиновыми остатками. Фермент локализован, в основном в цитозоле
(около 70%) [9]. В митохондриях млекопитающих находится около 20 - 30% ГПО. В
настоящее время известно пять изоферментных форм селенсодержащей ГПО [23]. В
плазме и молоке обнаружен внеклеточный изофермент, в цитоплазме клеток печени и
кишечника - особый изофермент ГПО-G1, в ядрах спермы содержится специфическая изоформа фермента. Выявлен
изофермент, несодержащий селена и идентичный глутатион-S-трансферазе [3].
ГПО, используя восстановленную форму глутатиона в качестве субстрата,
эффективно расщепляет не только перекись водорода, но и органические
гидроперекисные соединения, включая гидроперекиси полиненасыщенных жирных
кислот. При определённых условиях её роль в обезвреживании Н2О2 в тканях
значительно более весома, чем каталазы, так как действие последней ограничено
пероксисомами. ГПО имеет высокое сродство к Н2О2, что определяет её ведущую
роль в метаболизме перекиси водорода при более низких физиологических
концентрациях каталазы. В некоторых тканях (мозг, сердце, лёгкие), где
активность каталазы низкая, обезвреживание Н2О2 осуществляется, в основном,
ГПО. В условиях ОС, когда резко возрастает концентрация перекиси водорода,
ключевая роль в её расщеплении принадлежит каталазе [22].
В настоящее время выделен изофермент селенсодержащей ГПО, названный «ГПО
гидроперекисей фосфолипидов» или липидгидропероксид ГПО (КФ 1.11.1.12), который
по своей структуре является мономером с молекулярной массой 22 кДа и содержит
один атом селена, связанного с цистеиновым остатком белка. Этот изофермент в
отличие от классической ГПО может восстанавливать гидроперекиси фосфолипидов
без предварительного их гидролиза [9]. Являясь липофильным соединением, он
активно взаимодействует с гидроперекисями фосфатидилхолина, холестерина и
эфиров холестерина в мембранах и липопротеинах низкой плотности. Благодаря
совместному действию с токоферолом ГПО гидроперекисей фосфолипидов эффективное
разрушение продуктов ПОЛ в клеточных мембранах [10].
Активность ГПО зависит от концентрации восстановленной формы глутатиона.
Восстановление образующегося окисленного глутатиона осуществляется за счёт
работы фермента глутатионредуктазы (КФ 1.6.4.2) - цитоплазматического белка,
распределённого в тканях подобно глутатионпероксидазе. Этот фермент
функционально связан с ГПО. Действие глутатионредуктазы зависит от уровня НАДФН
и, следовательно, от состояния пентозофосфатного цикла и активности её
ключевого фермента глюкозо-6-фосфатдегидрогеназы (КФ 1.1.1.49). Она
восстанавливает окисленный глутатион, используя НАДФН, который образуется за
счёт реакций пентозного цикла [12].
Активность и стабильность ключевых ферментов АОЗ взаимосвязаны. Три
основных фермента - СОД, каталаза, ГПО - инактивируются одним из продуктов их
ферментативной реакции. Согласованное действие этих ферментов является
необходимым для защиты их от непосредственного инактивирующего воздействия
образующихся АФК [18]. Так, СОД, разрушая О2▪−, защищает каталазу
от его инактивирующего влияния, при этом снижается вероятность восстановления Fе3+ и возможность образования ОН▪,
который служит прооксидантом ПОЛ. А продукты ПОЛ являются потенциальными
ингибиторами глутатионпероксидазы. Каталаза и ГПО предохраняют СОД от
инактивации, устраняя Н2О2. Поэтому, только при условии сохранения активности и
стабильности всех своих компонентов ферментативная АОЗ способна обеспечить
эффективную защиту организма от токсических форм активированного кислорода
[62].
Действие неферментативной АОЗ связано с большой группой низко- и
высокомолекулярных химических соединений. Низко- и высокомолекулярные
антиоксиданты обеспечивают надёжную и всестороннюю защиту тканей от
токсического действия прооксидантов за счёт разной их локализации в
субклеточных компонентах клетки и во внеклеточной среде [63].
К высокомолекулярным соединениям, проявляющим неферментативную
антиоксидантную активность, относят церулоплазмин, трансферрин, альбумины
плазмы, липопротеины высокой плотности, гаптоглобин, фибриноген, гемопексин,
г-глобулины, гепарин, цитохром с, тиоредоксин и т.д. [3].
Низкомолекулярными соединениями, проявляющими антиоксидантное действие,
являются аскорбиновая кислота, токоферолы, мочевая кислота, билирубин,
восстановленная форма глутатиона, эстрогены, флавоноиды, каротиноиды, некоторые
аминокислоты и т. д. [11].
Основное назначение АОС в тканях - это защита от токсического воздействия
АФК при нормальном функционировании организма и при состояниях окислительного
стресса. В организме существует определённое равновесие между ферментативными и
неферментативными элементами АОЗ, так как последние могут при ОС выступать в
качестве прооксидантов, являясь источниками АФК. Однако и ферментативные
компоненты АОЗ при ряде патологических состояний могут вносить свой вклад в
генерацию АФК [9].
.7 Альфа-токоферол
Важной особенностью физиологической антиоксидантной системы является
распределение ее компонентов между гидрофильной и гидрофобной фазами клеточных
структур. Жирорастворимые антиоксиданты (токоферол, в-каротин, полифенолы,
убихинон и др.) локализованы в липидных структурах - мембранах, липопротеинах.
Водорастворимые - аскорбиновая кислота, глутатион, эрготионеин, антиперекисные
и протеолитические ферменты - функционируют внутри клетки. Во внеклеточном
пространстве находятся различные металлопротеины (церулоплазмин, трансферрин,
лактоферрин, гемоглобин, альбумин), мочевая и аскорбиновая кислоты [67]. Таким
образом, все клеточные структуры и внеклеточная среда находятся под контролем
АОС.
Ферментативные антиоксиданты - средство внутриклеточной защиты, в
сыворотке, лимфе и других средах их содержание мало. Вместе с тем во всех
водных и липидных фазах организма могут протекать радикальные окислительные
процессы, в защите от которых важную роль играют антиоксиданты - ингибиторы
органических радикалов, среди которых важное место занимают соединения фенольного
типа [3]. Фенольными антиоксидантами принято называть любые соединения вида Аr(ОН)n, в которых одна или несколько гидроксильных групп соединены
с ароматическим ядром (Аr),
при этом молекулы могут содержать несколько фрагментов Аr(ОН)n. Фенольные антиоксиданты (АrОН) эффективно взаимодействуют с гидроперекисными радикалами
жирных кислот и ненасыщенных липидов в реакции:
АrОН + RO2▪ → АrО▪ + RООН(1.30)
По существу в данной реакции не наблюдается исчезновения свободной
валентности, а имеет место только замена гидроперекисного радикала RO2▪ на феноксильный АrО▪, однако при этом достигается
эффект ингибирования свободнорадикального окисления, обусловленный большей
стабильностью АrО▪, который
практически не участвует в реакциях продолжения цепей окисления [15].
В частности, к фенольным антиоксидантам относятся - б-токоферол,
полифенолы, флавоноиды и близкие к ним по строению хиноны - витамины группы К,
убихинон. Их действие усиливают серосодержащие соединения - глутатион,
эрготионеин, цистеин, метионин, а также витамины А и С, в-каротин [24].
Таким образом, эндогенные фенольные антиоксиданты занимают ключевое
положение в антиоксидантной системе организма. Прежде всего, это связано с тем,
что они контролируют целостность и функциональную активность важнейших клеточных
структур - мембран [33].
Витамин Е относится к наиболее сильнодействующим природным антиоксидантам
и является «первой линией обороны» клеточных мембранных фосфолипидов [66].
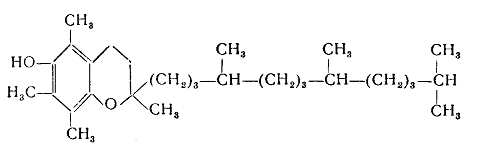
Рисунок 4 - Химическая структура б-токоферола (витамин Е)
При этом в ингибировании ПОЛ участвуют только восстановленные формы
витамина Е, а восстановителем антиоксидантных свойств токоферола являются так
называемые коантиоксиданты. Коантиоксиданты могут быть гидрофильными и
липофильными, природными и синтетическими; к наиболее изученным относятся
убихинол (восстановленная форма коэнзима Q10), аскорбиновая кислота, билирубин, 3-гидроксиантраниловая
кислота [8]. В результате происходит не только восстановление молекулы витамина
Е, но и предотвращается возможность инициации радикалом б-токоферола окисления
липидов [67].
Суточная потребность в витамине Е составляет 4-5 мг [69]. В организме
человека максимальное содержание витамина Е (суммарное количество токоферолов)
обнаружено в надпочечниках, жировой ткани, гипофизе, печени и семенниках [8]. В
клетках б-токоферол преимущественно концентрируется в богатых ненасыщенными
липидами мембранах митохондрий и лизосом, а также эритроцитах, плазматических
мембранах, т. е. там, где измеряется высокое парциальное напряжение кислорода
[70]. В ядрах клеток также выявляются значительные количества б-токоферола,
связанного с негистоновыми белками, предполагается, что ядерный токоферол
защищает хроматин от окислительных повреждений [7].
У млекопитающих витамин Е, поступающий в составе липидов пищи животного
или растительного происхождения, всасывается в кишечнике, после чего включается
в хиломикроны и через лимфатическую систему транспортируется в кровь, откуда
основная его часть переходит в печень [3].
В печени происходит процесс включения витамина Е в состав липопротеинов,
которые затем секретируются в кровь. В крови витамин Е, как и другие
жирорастворимые витамины, переносится липопротеинами, при этом если у мужчин большая
часть витамина связана с липопротеинами низкой плотности, то у женщин - с
липопротеинами высокой плотности [15].
Внутриклеточная транспортировка липофильного б-токоферола осуществляется
в комплексе со специальными б-токоферолсвязывающими (-переносящими,
-ассоциированными) белками. Наиболее хорошо описан и изучен
«б-токоферолпереносящий белок» печени («б-tocopherol transfer protein», б-ТТР) [12]. У животных (крыса, кролик) и человека
б-ТТР содержится в цитозоле гепатоцитов и осуществляет транспорт витамина Е от
липосом к эндоплазматическому ретикулуму, где и происходит синтез липопротеинов
[8].
В числе основных причин высокой эффективности б-токоферола можно назвать
следующие:
. стабилизация липидного бислоя за счет образования устойчивых комплексов
между молекулами б-токоферола и остатками полиненасыщенных жирных кислот,
ограничивающих доступ активных форм кислорода (АФК) к жирнокислотным цепям
[66].
. ярко выраженная антирадикальная активность, проявляющаяся против всех
свободных радикалов кислорода, в том числе в отношении самого реакционноспособного
- супероксиданиона, а также синглетного кислорода [8].
б-Токоферол способен прерывать цепи окисления благодаря переносу водорода
с фенольного кольца на пероксидный радикал с образованием молекулярного
продукта. Образующийся из б-токоферола феноксирадикал является относительно
стабильной и нереакционноспособной структурой за исключением его взаимодействия
с другими пероксидными радикалами RОО▪. При окислении хроманового кольца
и боковой цепи б-токоферола образуется продукт, не являющийся СР. Этот продукт
образует коньюгат с глюкуроновой кислотой и экскретируется из организма с
желчью [68].
Было установлено, что б-токоферол, селенит натрия, ряд фенольных
соединений природного происхождения оказывают выраженное гепатопротекторное
действие при нарушениях функций печени, вызванных гепатотропными ядами,
способствуют снижению нарастающего количества продуктов ПОЛ в крови, желчи,
гомогенатах тканей внутренних органов [69]. В модельных смесях липидов
природного происхождения б-токоферол обеспечивает до 60-85% ингибирующего
эффекта по сравнению с другими природными антиокислителями [7].
2. Материалы и методы
.1 Объект исследования
В качестве объекта исследования использовали взрослых лабораторных белых
крыс, самцов, массой 250 - 350 г, содержащихся на стандартном рационе питания в
виварии. Животные были разделены на 5 экспериментальных групп: 1 - интактные
животные; 2 - крысы, подвергнутые интоксикации ССl4; 3 - животные, которым ежедневно в течение 14 дней per os
вводили б-токоферол в дозе 200 мг/сутки, по истечении двух недель животных
декапитировали; 4 - крысы, которым ежедневно в течение 14 дней перорально
вводили б-токоферол в дозе 200 мг/сутки, затем на 15 день вводили
тетрахлорметан и через 24 часа осуществляли забой; 5 - крысы, подвергнутые интоксикации
ССl4 (в течение 24 часов), затем в
течение 14 дней получавшие витамин Е (200 мг/сутки).
Окислительный стресс индуцировали введением 50% раствора ССl4 в
вазелиновом масле из расчета 250 мкл ССl4 на 100 г веса животного. Забой
животных производили через сутки после введения гепатотоксина (2 и 4
экспериментальные группы).
У декапитированных животных в плазме определяли активность
аланинаминотрансферазы; в плазме крови и гомогенате почек оценивали показатели
ПОЛ по содержанию малонового диальдегида (МДА) и диеновых конъюгатов (ДК),
определяли активность антиоксидантных ферментов - каталазы и
супероксидисмутазы.
.2 Определение активности аланинаминотрансферазы
Аланинаминотрансфераза (АлАТ) - эндогенный фермент из группы трансфераз,
подгруппы аминотрансфераз (трансаминаз), широко используемый в медицинской
практике для лабораторной диагностики повреждений печени.
АлАТ катализирует перенос аминогруппы аланина на альфа-кетоглутаровую
кислоту с образованием пировиноградной кислоты и глутаминовой кислоты. Переаминирование
происходит в присутствии кофермента пиридоксальфосфата - производного витамина
В6.
Определение активности аланинаминотрансферазы проводили с использованием
набора реактивов фирмы «Витал».
Принцип метода:
А) L-аланин + б-кетоглутарат ←АлАТ→
пировиноградная кислота + L-глутамат;
Б) Фотометрическое определение содержания оксалоацетата в пробе на основе
реакции с 2,4-динитрофенилгидразином.
Реактивы (состав набора фирмы ВИТАЛ):
реагент № 1. Субстратная смесь (L-аланин - 0,2 моль/л, Б-кетоглутаровая кислота - 2,0 ммоль/л, Фосфатный
буфер, рН 7,4 - 0,1 моль/л);
реагент № 2. Раствор 2,4-ДНФГ (2,4-динитрофенилгидразин - 1,0 ммоль/л,
соляная кислота - 1,0 моль/л);
калибратор (Пируват натрия - 1,0 ммоль/л);
реагент № 4. Гидроксид натрия 4,0 моль/л.
Флаконы 1, 2 содержат готовые к использованию реагенты. Калибратор готов
к использованию. Содержимое флакона 4 (или аликвоту) развести
бидистиллированной водой в 10 раз.
Ход определения:
Таблица 1 - Ход определения АлАТ
|
Поместить в пробирки
|
Опытная проба, мл
|
Холостая проба, мл
|
|
Субстратно-буферный раствор
|
0,5
|
0,5
|
|
Плазма крови
|
0,1
|
-
|
|
Инкубировать в водяной бане при 37 °С 30 минут
|
|
Раствор 2,4-ДНФГ
|
0,5
|
0,5
|
|
Плазма крови
|
-
|
0,1
|
Через 20 минут (20-25 °С) в пробирки внести по 5 мл 0,4 моль/л раствора NaOH и через 5-10 минут фотометрировать
против холостой пробы в интервале длин волн 500-560 нм (лopt=537 нм) в кювете с толщиной
поглощающего слоя 1 см.
Таблица 2 - Построение калибровочного графика
|
№ пробы
|
калибратор
|
Дист. вода
|
Раствор 2,4-ДНФГ
|
Содержание пирувата в пробе
|
Концентрация каталитической активности АлАТ (С)
|
|
мл
|
мл
|
мл
|
нмоль
|
мкмоль/(с*л)
|
ммоль/(ч*л)
|
|
1
|
0,05
|
0,55
|
0,5
|
50
|
0,278
|
1,0
|
|
2
|
0,10
|
0,50
|
0,5
|
100
|
0,556
|
2,0
|
|
3
|
0,15
|
0,45
|
0,5
|
150
|
0,834
|
3,0
|
|
4
|
0,20
|
0,40
|
0,5
|
200
|
1,112
|
4,0
|
|
5
|
0,25
|
0,35
|
0,5
|
250
|
1,390
|
5,0
|
|
6
|
0,30
|
0,30
|
0,5
|
300
|
1,668
|
6,0
|
|
контр
|
-
|
0,60
|
0,5
|
-
|
-
|
-
|
Через 20 минут (20-25 °С) в пробирки внести по 5 мл 0,4 моль/л раствора NaOH и через 5-10 минут фотометрировать
против дстиллированной воды при длине волны 500-560 нм (лopt=537 нм) в кювете с толщиной поглащающего
слоя 1 см.
Расчет активности фермента в сыворотке крови производят по калибровачному
графику.
. Рассчитать по калибровочному графику коэффициент k:
k = С
/ Е,(2.1)
Е = (Екал - Ек),(2.2)
где Екал - экстинция калибровочной пробы;
Ек - экстинция контроля;
С - каталитическая концентрация АлАТ (активность) в мкмоль/(с*л) или
ммоль/(ч*л) в калибровочной пробе.
. Каталитическая концентрация АлАТ (активность) рассчитывается в
мкмоль/(с*л) или ммоль/(ч*л) по формуле:
АлАТ = Епр × k,(2.3)
где АлАТ - каталитическая концентрация (активность);
k -
коэффициент, расчитанный по калибровочному графику;
Епр - экстинция опытной пробы, измеренной против холостой пробы.
При активности АлАТ, превышающей 3 ммоль/(ч*л) (0,834 мкмоль/(с*л)),
сыворотку (плазму) крови следует развести. При расчете учесть степень
разведения.
.3 Определение содержания гемоглобина
Содержание гемоглобина в эритроцитах определяли стандартным
гемоглобинцианидным методом. Гемоглобин крови при взаимодействии с
железосинеродистым калием (красная кровяная соль) окисляется в метгемоглобин,
образующий с ацетонциангидрином гемиглобинцианид, оптическая плотность которого
при 540 нм пропорциональна концентрации гемоглобина в образце крови.
Реактивы:
. стандарстный раствор гемоглобина с известной концентрацией (120 г/л);
. набор реагентов для определения гемоглобина в крови “ГЕМОГЛОБИН-АГАТ”;
Ход определения:
К 5 мл трансформирующего раствора добавляли 20 мкл крови. Все тщательно
перемешивали и оставляли на 10-15 мин при комнатной температуре для полного
развития окраски. Также подготавливали пробу со стандартным раствором
гемоглобина с концентрацией 120 г/л. Измерение оптической плотности опытной и
калибровочной проб проводили на ФЭКе при длине волны 540 нм в кювете с толщиной
10 мм против трансформирующего раствора. По оптической плотности гемоглобина в
калибровочной пробе проводили расчет содержания гемоглобина в опытных пробах:
Нb = Dо × 120 / Dх
,(2.4)
где Hb - концентрация гемоглобина, г/л;
Dо -
оптическая плотность опытной пробы;
Dх -
оптическая плотность пробы со стандартным раствором Hb;
- концентрация Hb в стандартном растворе, г/л.
.4 Определение содержания белка
Для определения содержания белка использовали микробиуретовый метод
(Кочетков, 1971).
Реактивы:
. стандартный раствор белка (сывороточного альбумина);
. биуретовый реактив (реактив Бенедикта) - 17,3 г цитрата натрия и 10 г
карбоната натрия растворяют при подогревании в небольшом количестве воды, в
раствор добавляют 1,73 г сульфата меди, растворённого в 10 мл воды, и доводят
водой до 100 мл.
. 6%-ный раствор NaOH.
Ход определения:
К 2 мл физиологического раствора, содержащего 10 мкл гомогената,
добавляют 2 мл 6%-ного NaOH и 0,2 мл реактива Бенедикта. Раствор хорошо
перемешивают и через 15 мин фотометрируют при 330 нм на спектрофотометре.
Предварительно строят график по стандартному раствору белка, по которому,
построив линию Тренда, и определяют концентрацию белка в растворе.
.5 Определение содержания диеновых конъюгатов
Определение содержания первичных продуктов ПОЛ - диеновых конъюгатов,
осуществляли спектрофотометрически в экстрагированной гептановой фазе липидов
при 233 нм.
Принцип метода:
Вследствие р-р переходов спектры коньюгированных гидроперекисей
полиненасыщенных жирных кислот характеризуются интенсивным поглощением в
ультрафиолетовой области спектра с максимумом при 232-234 нм (Каган и др.,
1986).
Реактивы:
. гептан-изопропанольная смесь в соотношении 1:1;
. 0,74%-ный водный раствора KCl.
Ход определения:
Определение содержания диеновых коньюгатов (ДК) проводили в плазме крови
и гомогенате почек. Для этого липиды экстрагируют стократным избытком смеси
растворителей. В гомогенизатор вносят 0,1 мл плазмы или гомогената, добавляют 5
мл изопропилового спирта и тщательно растирают до получения гомогенной
суспензии. Содержимое гомогенизатора количественно переносят в мерную
центрифужную пробирку, в которую затем добавляют 5 мл гептана.
Экстракт центрифугируют в течение 10 мин при при 3000 об/мин. (1700g).
Надосадочную фракцию переносят в градуированную пробирку и добавляют 1/5 объема
раствора KCl для отмывки липидного экстракта от нелипидных примесей. После
тщательного встряхивания образовавшаяся эмульсия расслаивалась на две
прозрачные фазы. В гептановом экстракте (верхняя фаза) измеряют спектрофотометрически
содержание сопряженных диенов в кювете с длиной оптического пути 1,0 см против
гептана. Расчёт количества ДК производят с учетом молярного коэффициента
экстинкции:
С (моль / г ткани) = D · F · / K · с,(2.5)(моль / л плазмы) = D · F · / K
· v,(2.6)
где С - концентрация ДК;- оптическая плотность гептанового экстракта;-
фактор разведения, равный 121,2;- коэффициент молярной экстинкции для ДК при
длине волны 233 нм, равный 27000 М-1см-1 (Паранич и др., 1993);
с - концентрация белка, г/л;
v - объём
образца плазмы, л.
.6 Определение содержания малонового диальдегида
Определение вторичного продукта ПОЛ - малонового диальдегида, проводили
по качественной реакции с тиобарбитуровой кислотой.
Принцип метода:
В липидных системах в результате процессов перекисного окисления липидов
образуется малоновый диальдегид (МДА), взаимодействие которого с
2-тиобарбитуровой кислотой приводит к образованию хромогена с максимумом
поглощения в красной области видимого спектра при длине волны 532 нм (Ko,
Godin, 1990).
Реактивы:
. 0,9%-ный раствор NaCl (физиологический раствор);
. 30%-ный раствор трихлоруксусной кислоты (ТХУ);
. 0,1М раствор динатриевой соли этилендиаминтетраацетата (ЭДТА)
(С10Н14N2Na2O8 * 2H2O, Mr = 372,24г/моль);
. 1%-ный раствор 2-тиобарбитуровой кислоты (ТБК);
. 0,05 н раствор NаОН;
Ход определения:
К 0,2 мл плазмы добавляют 0,8 мл физиологического раствора и 0,5 мл
раствора ТХУ, перемешивают и ставят в ледяную баню на 2 часа. Затем
центрифугируют 15 мин при 3000 об/мин. (1700g). Аналогичным образом готовят
контрольную пробу, в которой эритроциты заменяют равным объемом
дистиллированной Н2О. 1 мл супернатанта переносят в другую пробирку, добавляют
0,075 мл ЭДТА и 0,25 мл раствора ТБК, приготовленного на 0,05 н растворе NаОН.
Содержимое перемешивают и ставят пробирки в кипящую водяную баню на 15 мин.
Затем пробирки охлаждают до комнатной температуры.
Для определения концентрации МДА в ткани печек использовали 3 мл
гомогената и 1,5 мл раствора ТХУ, соответственно объём остальных реактивов
увеличивали в 3 раза.
Измеряют поглощение опытной пробы против контрольной на спектрофотометре
при 532 нм 600 нм в кюветах с толщиной слоя 1,0 см. Расчёт содержания МДА
производят с учётом коэффициента молярной экстинкции образовавшегося хромогена,
равного 1,56*105 М-1см-1 (Стальная И.Д., 1977):
С (моль / г ткани) = D532 - D600 · F / К · m,(2.7)(моль / л плазмы) =
D532 - D600 · F / К,(2.8)
где С - содержание МДА;- оптическая плотность опытной пробы при длине
волны 532 нм;- оптическая плотность опытной пробы при длине волны 600 нм;-
фактор разведения, равный 9,94;
К - коэффициент молярной экстинкции образовавшегося хромогена, равный
1,56·105 М-1см-1;- масса образца, г.
.7 Определение активности супероксиддисмутазы
Принцип метода основан на ингибировании реакции аутоокисления адреналина
в щелочной среде в присутствии супероксиддисмутазы (СОД), вследствие дисмутации
супероксидных анион-радикалов, которые являются продуктом одного из этапов
окисления и, по-видимому, одновременным участником его последующих стадий [Практикум
по свободнорадикальному окислению, 2006; Сирота, 1999].
Об интенсивности аутоокисления адреналина, судят по динамическому
нарастанию поглощения при длине волны 347 нм, обусловленному накоплением
продукта окисления, не описанного ранее в литературе, и опережающим по времени
образование адренохрома (с максимумом поглощения при 480 нм).
Реактивы:
. 0.1 % (5.46 мМ) аптечный раствор адреналина;
. бикарбонатный буфер, pH=11 (2.12 г Na2CO3, 0.168 г NaHCO3, 0.074 г ЭДТА
растворяли в 200 мл дистиллированной воды; pH доводили до нужного значения
добавлением NaOH);
. этанол-хлороформная смесь (2:1).
Ход определения:
В качестве источника СОД используют эритроцитарный гемолизат, плазму
крови или гомогенаты тканей. В пробирку вносят 50 мкл плазмы или гомогената и
450 мкл дистиллированной воды, предварительно охлажденной до 0°С. В пробирки
добавляют по 250 мкл этанол-хлороформной смеси. Полученная суспензия интенсивно
перемешивается и центрифугируется при 8000 об/мин в течение 15 минут.
Супернатант используется для определения активности СОД. Измерение проводится в
кювете с длиной оптического пути 10 мм. Последовательность внесения реагентов в
пробу представлена в табл.2.3. Исходная концентрация адреналина в пробе равна
230 мкМ.
После внесения адреналина в холостую пробу, содержащую бикарбонатный
буфер, содержимое кюветы тщательно и быстро перемешивается. Изменение
оптической плотности регистрируется через каждые 30 секунд в течение 3 минут.
Аналогичным образом обрабатывается и опытная проба. Для расчета активности СОД
используют показатели величины поглощения контрольной и опытной проб.
Таблица 3 - Порядок внесения реагентов в пробу
|
Реагенты
|
Контрольная
|
Опытная
|
|
0,2 М бикарбонатный буфер, рН 11
|
3 мл
|
3 мл
|
|
Дистиллированная вода
|
0,05 мл
|
-
|
|
Супернатант (плазма)
|
-
|
0,05 мл
|
|
5,46 мМ раствор адреналина
|
0,15 мл
|
0,15 мл
|
Активность фермента выражают в Единицах активности на грамм Нb или белка (за Единицу активности СОД
принято 50% ингибирование реакции окисления адреналина). Расчет активности СОД
производят по формуле:
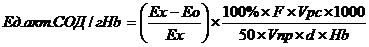 ,(2.9)
,(2.9)
где 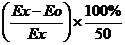 - Ед. активности на мл плазмы, гомогената;
- Ед. активности на мл плазмы, гомогената;
Ео - оптическая плотность опытной пробы;
Ек - оптическая плотность холостой (контрольной) пробы;
- коэффициет пересчета на единицу активности СОД;
F -
коэффициент разведения материала в пробе, равный 15;рс - объем реакционной
среды, равный 3,15 мл;
Vпр -
объем вносимого образца (плазма, гомогенат), равный 0,05 мл;
d -
длина оптической пути кюветы (1 см);
Нb - содержание гемоглобина, г/л (либо
с - концентрация белка, г/л);
- коэффициент для расчёта результата на грамм Нb или грамм белка.
.8 Определение активности каталазы
Каталаза катализирует разложение пероксида водорода по реакции:
Н2О2 → 2 Н2О + О2(2.10)
О ходе реакции судят по изменению концентрации пероксида водорода в пробе
спектрофотометрическим методом при длине волны 230 нм, максимуме поглощения
Н2О2. В качестве источника фермента можно использовать гомогенаты тканей,
клетки крови, биологические жидкости, субклеточные органеллы.
Реактивы:
. 1 М трис-НСl буфер, рН 8,0,
содержащий 5 мМ ЭДТА. Для приготовления буферного раствора 12,1 г триса и 0,186
г динатриевой соли этилендиаминтетрауксусной кислоты помещают в мерную колбу на
100 мл и доводят до метки дистиллированной водой.
. 1 М фосфатный буфер, рН 7,0. Для его приготовления берут 14,13 г
фосфата натрия двузамещённого, помещают в мерную колбу на 100 мл и доводят до
метки дистиллированной водой.
. 10 мМ раствор пероксида водорода (0,03%).
Ход определения:
Для определения активности каталазы готовят две пробы - контрольную и
опытную в соответствии с таблицей.
Таблица 4 - Содержание проб для определения активности каталазы
|
Компоненты инкубационной пробы
|
Контрольная проба, мл
|
Опытная проба, мл
|
|
1 М трис-НСl
буфер с 5 мМ ЭДТА, рН 8,0
|
0,15
|
0,15
|
|
Н2О2
|
-
|
2,70
|
|
Н2О
|
2,85
|
0,15
|
|
Гомогенат, плазма
|
0,06
|
0,06
|
Реакцию запускают внесением в пробу, находящуюся в спектрофотометрической
кювете с расстоянием между рабочими гранями 1,0 см, гомогената, плазмы или
гемолизата. Содержимое кюветы перемешивают и определяют начальную оптическую
плотность. За ходом реакции следят в течение 3 минут. Регистрацию оптической
плотности проводят при комнатной температуре.
Для приготовления гемолизата к 0,1 мл упакованных эритроцитов или 0,1 мл
гомогената ткани приливают 2 мл охлаждённой до 0оС дистиллированной воды. Далее
для определения активности каталазы в эритроцитах полученный 1мл гемолизата
разводят до 100 мл водой (или 5 мкл на 10,6 мл дистиллированной воды). Расчет
активности каталазы производят по формуле:
А = (ДЕ / мин ÷ е ·с) · F · 1000,(2.11)
где А - активность фермента в моль/мин · г Нb или моль/мин · г белка;
ДЕ / мин - изменение оптической плотности в минуту;
е - коэффициент молярной экстинкции пероксида водорода, при л =230 нм
равный 0,071 М-1·см-1;
с - концентрация белка, г/л;
F -
коэффициент учитывающий разведение супернатанта (21) и эритроцитов (2121).
2.9 Статистическая обработка результатов
Для обработки полученных результатов использовалась программа
Statistica 6. Обработку результатов проводили с помощью подсчета медианы и
интерквартального разброса (С25 и С75 процентели). Проверку гипотезы о
статистической достоверности двух выборок проводили с помощью
непараметрического критерия Манна-Уитни для независимых выборок с достоверностью
p<0,05.
3. Результаты исследования
Об оценке функционального состояния АОС судили на основании определения
про- и актиоксидантного статуса. Прооксидантный статус оценивался по содержанию
продуктов ПОЛ: ДК и МДА. Антиоксидантный статус оценивался по активности СОД и
каталазы. Данные параметры были исследованы в плазме и почках подопытных
животных.
.1 Уровень аланинаминотрансферазы
Аланинаминотрансфераза синтезируется внутриклеточно, и в норме лишь
небольшая часть этого фермента попадает в кровь. Наиболее высокая активность
АлАТ выявляется в печени и почках, меньшая - в поджелудочной железе, сердце,
скелетной мускулатуре. При повреждении или разрушении клеток, богатых АлАТ
(печень, почки), происходит выброс этого фермента в кровяное русло, что приводит
к повышению его активности в крови. Поэтому в качестве показателя степени
токсического поражения печени и почек в плазме измеряли активность АлАТ
(Рисунок 5).
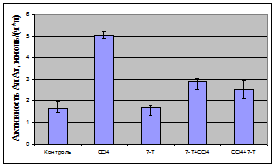
Рисунок 5 - Активность АлАТ в плазме до введения ССl4, после введения и под влиянием
б-токоферола (* р<0,05)
Активность аланинаминотрансферазы, в группе подвергшейся воздействию ССl4, повышается в 3,1 раз (р<0,05),
по сравнению с контрольной группой. Показатели АлАТ у животных получавших в
течение 14 дней б-токоферол находятся на одном уровне со значениями контроля. В
четвёртой группе, где крысам перед введением тетрахлорметана две недели
ежедневно per os давали витамин Е, активность энзима превышает значения
контроля и группы с введённым б-токоферолом на 71% (р<0,05). Тогда как, по
сравнению с группой «CCl4»
фермент проявляет значительно меньшую активность - активность в группе «б-Т+CCl4» на 43% ниже чем в «CCl4» (р<0,05). В случае, когда
витамин Е в течение 14 дней вводили после отравления тетрахлорметаном, активности
трансаминазы превышала значения контроля на 35%, относительно группы «ССl4» активность АлАТ снижалась в 2
раза, однако данные изменения не достоверны.
Таким образом, значительное повышение активности АлАТ после отравления
свидетельствует, о том, что введение 50% раствора тетрахлорметана вызывает
разрушение плазматических мембран. Данные, что приём витамина Е до и после
введения токсиканта снижает активность аланинаминотрансферазы указывают на
нефро- и гепатопротекторное действие б-токоферола, которое очевидно связано со
способностью витамина стабилизировать клеточные мембраны.
.2 Показатели про- и антиоксидантной системы в плазме при интоксикации
тетрахлорметаном
В плазме крови б-токоферол является основным и вероятно единственным
липидрастворимым антиоксидантом. Основным переносчиком витамина Е в плазме
крови являются липопротеины. Плазма крови - первая реагирует на недостаток
токоферола в организме, что является важным признаком при диагностике и лечении
ряда заболеваний.
Концентрация МДА и ДК (Рисунок 6, 7) в плазме у животных с введённым
четырёххлористым углеродом достоверно превышает значения контрольной группы, на
67,3% и 40% соответственно (р<0,05), что подтверждает активацию процессов
перекисного окисления.
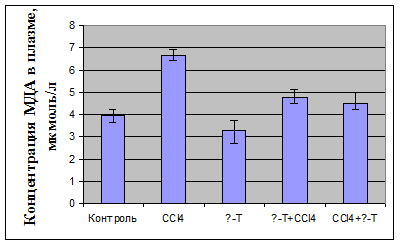
Рисунок 6 - Концентрация МДА в плазме до введения ССl4, после введения и под влиянием
б-токоферола (* р<0,05)
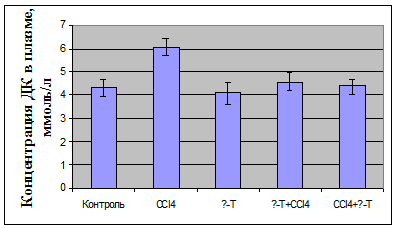
Рисунок 7 - Концентрация ДК в плазме до введения ССl4, после введения и под влиянием
б-токоферола (* р<0,05)
Уровень продуктов ПОЛ у крыс, которым ежедневно в течение 14 дней вводили
200 мг б-токоферола, снижался по сравнению с контролем - МДА на 18%
(р<0,05), ДК на 4,3%. При введение витамина Е до интоксикации наблюдалось
повышение концентрации МДА относительно нормы - на 17% (р<0,05),
концентрации диеновых конъюгатов на 5% (р<0,05). Однако, концентрация продуктов
перекисного окисления не достигла такого высокого уровня, который наблюдается
при воздействии только токсикантом.
Применение витамина после поражения ядом, способствовало небольшому
снижению МДА и ДК по сравнению с группой «б-Т+CCl4»: концентрация МДА ниже на 6%, ДК - на 4%
(р<0,05). Относительно контроля концентрация МДА в этой группе выше на 12%
(р<0,05), ДК - на одном уровне с нормой.
Если сравнивать соотношение показателей групп «б-Т» и «б-Т+CCl4», то видно, что концентрация МДА
плазмы в четвёртой группе повышается по сравнению с «б-Т» на 32% (р<0,05),
тогда как концентрация ДК превосходит значение этой группы только на 9%, но эти
значения не достоверны.
Введение витамина Е оказывает явный положительный эффект при отравлении
тетрахлометаном, т.к. значения концентрации и ДК и МДА достоверно снижаются по
сравнению с группой «CCl4»
- ДК на 24% («б-Т+CCl4» относительно
«CCl4») и 27% («CCl4+б-Т» относительно «CCl4») (р<0,05), МДА на 28% и 32% соответственно
(р<0,05).
На первые сутки после введения четырёххлористого углерода наблюдается
снижение активности антиоксидантных ферментов по сравнению со здоровыми
животными: СОД снижается на 42%, каталаза - на 40% (р<0,05) (Рисунок 8, 9).
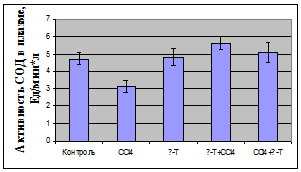
Рисунок 8 - Активность СОД в плазме до введения ССl4, после введения и под влиянием
б-токоферола (* р<0,05)
В плазме животных, которым вводили б-токоферол, наблюдается повышение активности каталазы по сравнению с контрольными
показателями на 13% (р<0,05).
Активность СОД в группе «б-Т» не изменяется по сравнению с нормой. Относительно
группы «CCl4», уровень активности ферментов в
группе «б-Т» выше: СОД на 35%, каталазы на 48% (р<0,05).
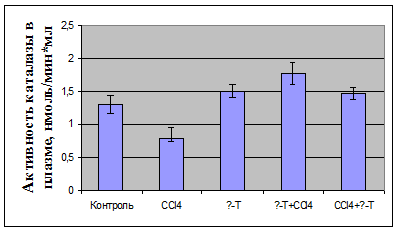
Рисунок 9 - Активность каталазы в плазме до введения ССl4, после введения и под влиянием
б-токоферола (* р<0,05)
Предварительное применение витамина Е способствует активации СОД и
каталазы после воздействия CCl4.
Относительно контроля активность СОД повышается на 17%, а каталазы на 26%
(р<0,05). По сравнению с животными, которым вводили CCl4 активность СОД повышается на 44% (р<0,05),
активность каталазы - на 55% (р<0,05). Исходя из этого, можно предположить,
что б-токоферол оказывает на каталазу более стимулирующее влияние, чем на
активность СОД.
Применение витамина в течение 14 дней после влияния тетрахлорметана,
приводит к тому, что активность антиоксидантных ферментов снижается
относительно группы «б-Т+CCl4»:
СОД на 10%, каталазы на 17%. Однако, данные значения не достигают значений
контрольной группы.
3.3 Показатели про- и антиоксидантной системы в почках при интоксикации
тетрахлорметаном
Почки чрезвычайно сложный орган, как в плане морфологии, так и
физиологии, основные функции которого − экскреция продуктов метаболизма
из организма, регуляция водного и электролитного баланса. Поскольку основные
транспортные и концентрационные процессы происходят в проксимальном отделе
канальцев, именно этот отдел нефрона наиболее часто повреждается токсикантами.
Важной функцией почек, сказывающейся на нефротоксичности ряда веществ,
является их способность метаболизировать ксенобиотики. Хотя интенсивность
метаболизма значительно ниже, чем в печени, здесь определяются те же
ферментативные системы, и напряженность биотрансформации достаточно высока.
Хотя многие ксенобиотики одновременно метаболизируют с образованием активных
радикалов и в печени и в почках, повреждение органа, по всей видимости,
обусловлено действием той части общего количества вещества, которая
метаболизирует именно в почках. Близость метаболических процессов, протекающих
в печени и почках, обусловливает практически одинаковую чувствительность этих
органов ко многим ксенобиотикам (хлорированные углеводороды, токсины бледной
поганки, паракват и др.).
Через сутки после введения животным тетрахлорметана наблюдается повышение
уровня МДА в гомогенате почек в 2 раза (р<0,05) по сравнению с контролем,
концентрация диеновых коньюгатов увеличивается на 40% (р<0,05) (Рисунок 10,
11).
Применение б-токоферола снижает содержание продуктов перекисного
окисления по сравнению с показателями интактных животных: МДА на 14%
(р<0,05), ДК на 4% (р<0,05).
Применение витамина Е до и после воздействия ядом ведёт к незначительным
изменениям концентрации продуктов ПОЛ относительно контроля. В группе «б-Т+CCl4» содержание МДА выше нормы на 5,1%
(р<0,05), показатели ДК равны норме, но эти изменения недостоверны. При
применении токоферола после отравления, уровень МДА снижается на 7%
(р<0,05), ДК - на пол процента (р<0,05).
Концентрация продуктов ПОЛ четвёртой и пятой групп превышает показатели у
животных, которым в течение двух недель вводили только б-токоферол. В группе с
введённым витамином Е до ССl4 МДА
на 20% (р<0,05), ДК на 4% выше, в группе «CCl4+б-Т» МДА выше на 8%, ДК на 4% относительно «б-Т»
(р<0,05).
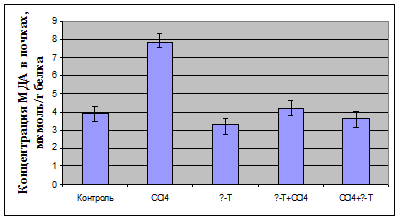
Рисунок 10 - Концентрация МДА в почках до введения ССl4, после введения и под влиянием
б-токоферола (* р<0,05)
Концентрация МДА в группе «б-Т+CCl4» на 52% ниже (р<0,05), чем в группе, где животные подвергались
только воздействию токсиканта, ДК ниже на 40% (р<0,05). В пятой группе
уровень ДК и МДА также значительно ниже показателей группы «ССl4»: ДК на 40% (р<0,05), МДА на 54%
(р<0,05).

Рисунок 11 - Концентрация ДК в почках до введения ССl4, после введения и под влиянием
б-токоферола (* р<0,05)
Применение витамина Е после интоксикации ведёт к уменьшению МДА в почках
относительно показателей группы «б-Т+CCl4» на 5% (р<0,05), а концентрация диеновых конъюгатов остаётся на том
же уровне.
Активность антиоксидантных ферментов в почках под влиянием
четырёххлористого углерода спустя 24 часа после воздействия ССl4 достоверно понижается (Рисунок 12,
13). Так активность СОД относительно контроля снижается на 25% (р<0,05),
активность каталазы - на 26% (р<0,05).
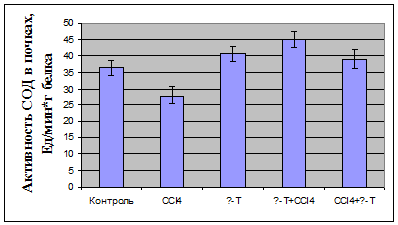
Рисунок 12 - Активность СОД в почках до введения ССl4, после введения и под влиянием
б-токоферола (* р<0,05)
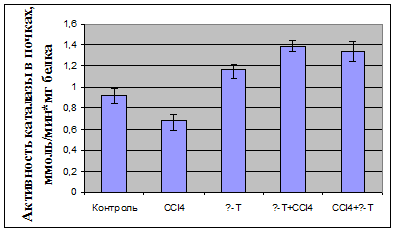
Рисунок 13 - Активность каталазы в почках до введения ССl4, после введения и под влиянием
б-токоферола (* р<0,05)
При воздействии б-токоферола на здоровых особей активность
супероксиддисмутазы и каталазы увеличилась относительно контроля: СОД на 10%
(р<0,05), каталазы на 21% (р<0,05).
Предварительное введение витамина Е до воздействия ССl4 привело к активации ферментов.
Относительно нормы активность супероксиддисмутазы повысилась на 19%
(р<0,05), относительно группы «б-Т» на 10% (р<0,05). Активность каталазы
в этой группе превышает контрольные значения на 34% (p<0,05), а также превышает активность, у животных с
введённым б-токоферолом - на 16% (p<0,05).
При применение б-токоферола после отравления тетрахлорметаном активность
СОД ниже, чем в группе «б-Т+CCl4»
(на 13%), активность каталазы - на 3%. Уровень СОД по сравнению с животными,
которые просто получали витамин Е снижен на 4%, а активность каталазы,
наоборот, выше на 13%, но данные изменения не достоверны. Уровень активности
супероксиддисмутазы данной группы выше значений контроля на 6%. Активность
каталазы превышает значения группы интактных животных на 32%, но полученные
изменение не достоверны.
Таким образом, общая «картина» про- и антиоксидантной системы такова:
. концентрация продуктов перекисного окисления липидов, как в плазме, так
и в гомогенате почек, после отравления животных тетрахлорметаном, в разы
превышает значения интактных животных, что подтверждает активацию процессов
СРО. Данное повышение МДА и ДК происходит на фоне ингибирования активности СОД
и каталазы под влиянием яда. В ходе метаболизма ксенобиотика образуется
огромное количество свободных радикалов, которые повреждают мембраны клеток и
запускают каскад перекисного окисления липидов. Такое «поведение» энзимов можно
объяснить тем, что ферментативная антиоксидантная система не справляется с
таким стремительным увеличением повреждающих радикалов, что приводит к быстрой
инактивации пула ферментов, следовательно, ведёт к истощению АОС. Ещё одной
причиной снижения активности ферментов, вероятно, является то, что
высокореактивные радикалы, образующиеся в ходе метаболизма тетрахлорметана (трихлорметильный
радикала (ССl3▪) и трихлорметилпероксильный
радикал (ССl3О2▪)), вступают в реакцию с
белковой молекулой фермента, инактивируя его. Так, активность каталазы может
быть снижена за счёт окисления Fe3+,
входящего в состав гема. В результате, в окисленном состоянии каталаза
проявляет гораздо меньшую активность.
. Под воздействием альфа-токоферола, концентрация МДА в плазме и почках
(р<0,05) и ДК в почках (р<0,05) ниже контрольных значений, ДК в плазме на
уровне нормы. Возможно, такое влияние можно объяснить стабилизирующим действием
витамина на структуру мембран клеток, и уменьшением в них естественной
активности ПОЛ, либо такой эффект является следствием проявления
антиоксидантных свойств самого токоферола, а так же активацией им ферментов
АОЗ.
Наряду с такими изменениями концентрации продуктов ПОЛ, в рассматриваемой
группе наблюдается увеличение активности каталазы в плазме и почках
относительно нормы, активность СОД плазмы остаётся на уровне контроля. СОД в
гомогенате почек выше активности фермента у интактных животных. Такое
«поведение» ферментов можно объяснить влиянием на них витамина Е, так в
литературе имеются данные, что альфа-токоферол оказывает стимулирующее влияние
на синтез гема, а следовательно на синтез гемсодержащих ферментов, к которым и
относится каталаза. Следствием этого является ликвидация перекисей ненасыщенных
жирных кислот и липидов, повреждающих клеточные и субклеточные мембраны.
Вероятно, токоферол оказывает стимулирующее влияние на супероксиддисмутазу, так
ивестно, что у крыс, содержащихся на витамин Е-дефицитной диете, снижается
активность СОД.
. Применение альфа-токоферола до воздействия ксенобиотика ведёт к
значительному снижению концентрации ДК и МДА в плазме и почках относительно
животных, подвергшихся влиянию только ССl4, но концентрация продуктов ПОЛ не достигает значений нормы.
Активность антиоксидантных ферментов при предварительном приёме витамина Е
увеличивается относительно контроля и группы животных принимавших только
токоферол. Данные результаты могут свидетельствовать о проявлении
мембраностабилизирующих свойств витамина. Альфа-токоферол выступает ловушкой
радикалов, что препятствует стремительному развитию СРО, следовательно, это
ведёт к снижению продуктов ПОЛ, а также способствует защите самих ферментов от
воздействия радикалов, что поддерживает их высокую активность.
. При использовании витамина Е после воздействия токсиканта, концентрация
МДА (р<0,05) и ДК в плазме ниже, по сравнению со значениями группы с
предварительным введением витамина Е, при этом МДА не достигает значений
контроля, тогда как концентрация диеновых коньюгатов равна норме. В почках
наблюдается снижение МДА по отношению к группе «б-Т+CCl4» (р<0,05), также концентрация малонового
диальдегида ниже контрольных значений, но не достигает значений группы «б-Т».
Концентрация ДК находится на одном уровне со значениями контрольной группы.
Причиной столь незначительных колебаний концентраций диеновых конъюгатов в
исследуемых группах, наряду с более выраженными изменениями малонового
диальдегида, является неустойчивость данного продукта ПОЛ. ДК относятся к
первичным продуктам перекисного окисления липидов, тогда как МДА - вторичный интермедиат.
Активность супероксиддисмутазы и каталазы в плазме и почках ниже
активности этих ферментов в группе с предварительным использованием
альфа-токоферола. При этом уровень активности СОД и каталазы не достигает
значений интактных животных.
Заключение
Оценка состояния про- и антиоксидантного статуса в условиях токсического
воздействия на живой организм остаётся важным вопросом в связи с ухудшающейся
экологической обстановкой.
Четырёххлористый углерод - высокореакционное соединение, сегодня всё ещё
широко применяется в промышленности, как растворитель (жиров, смол, каучука и
др.), для получения фреонов, как экстрагент, в медицине. Тетрахлорметан являлся
стандартным наполнителем переносных огнетушителей для советской бронетехники.
Четырёххлористый углерод вступает во взаимодействие с эндогенными
липидами биологических мембран, запуская реакции перекисного окисления, в
результате которых целостность биомембран нарушается. Как токсический агент,
проникая, в частности, внутрь митохондрий, он оказывает воздействие на процессы
энергетического обмена клетки, что может быть связано не только с действием
целой молекулы тетрахлорметана, но и с продуктами его метаболизма. Установлено,
что центральное место в метаболизме CCl4 занимает радикал ССl3▪
образующийся на первой стадии трансформации в системе цитохромаР-450.
Для предупреждения и ограничения патологических процессов в клетке
применяются средства неферментативной защиты, к которым относится
альфа-токоферол. В данной работе оценивается влияние витамина Е на процессы
перекисного окисления липидов и определяется активность антиоксидантных
ферментов при применении его до отравления ядом и после.
Результаты, полученные в работе, подтверждают данные имеющиеся в научной
литературе о повреждающем действии тетрахлометана на клеточные мембраны. Под
воздействием ксенобиотика активируются процессы СРО, что приводит к накоплению
в тканях продуктов перекисного окисления. Радикалы, образующиеся в результате
биотрансформации четырёххлористого углерода, вступают в реакции не только с
липидами, но и с белками, что приводит к повреждению ферментов антиоксидантной
системы и снижению их активности.
Нами установлено, что под влиянием витамина Е наблюдается снижение
продуктов липопероксидации в исследуемых тканях относительно нормы. Возможно
такое влияние можно объяснить стабилизирующим действием витамина на структуру
мембран клеток, либо такой эффект является следствием проявления
антиоксидантных свойств самого токоферола, а так же активацией им ферментов
АОЗ.
Наряду с такими изменениями концентрации продуктов ПОЛ, в группе
получавшей только токоферол наблюдается увеличение активности каталазы в плазме
и почках относительно нормы. Активность СОД плазмы остаётся на уровне контроля,
СОД в гомогенате почек выше активности фермента у интактных животных. Вероятно,
это обусловлено тем, что альфа-токоферол оказывает регулирующее влияние на
синтез гема, и, следовательно, на синтез гемсодержащих ферментов, к которым
относится каталаза.
Исходя из полученных результатов, можно сказать, что альфа-токоферол в
нашем эксперименте повышает активность антиоксидантных ферментов на фоне
воздействия четырёххлористого углерода. Особенно это выражено в группе,
получавшей витамин до интоксикации ядом. При этом в группах с применением
альфа-токоферола до и после введения ксенобиотика, наблюдается снижение
концентрации продуктов перекисного окисления, но данные значения не достигают
контрольных. Положительное влияние альфа-токоферола подтверждается изменениями
в активности аланинаминотрансферазы, согласно которым повреждение клеточных
мембран значительно уменьшается под действием витамина.
Таким образом, применение альфа-токоферола в качестве средства
неферментативной защиты остаётся очень актуальным. Использование витамина можно
рекомендовать в качестве профилактического средства для персонала промышленных
предприятий и вредных производств, в частности, где в технологии используется
четырёххлористый углерод. Однако нельзя забывать, что альфа-токоферол это
фармакологический препарат и его дозировка должна быть подобрана врачом. Прием
больших доз витамина может привести к проявлению им своих прооксидантных
свойств - запуску процессов пероксидации липидов.
Список использованных источников
1 Владимиров, Ю.А. Свободные радикалы в биологических
системах / Ю.А. Владимиров // Соросовский образовательный журнал. − 2001.
− Т. 6, № 12. − С. 13-19.
Ельцова, Л.В. Изучение фармакологической активности
производных пирроло[1,2-а]бензимидазола, проявляющих антиоксидантные и
антирадикальные свойства: автореф. дис. канд. биол. наук: 14.03.06 / Ельцова
Лариса Витальевна. - Волгоград, 2010. - 25 с.
Меньщикова, Е.Б. Окислительный стресс. Прооксиданты и
антиоксиданты: учеб. / Е.Б. Меньщикова, В.З. Ланкин, Н.К. Зенков, И.А. Бондарь,
Н.Ф. Круговых, В.А. Труфакин. - М.: Фирма «Слово», 2006. - 556 с.
Андреещева, Е.М. Оксидативный статус и активности
аконитатгидратазы и NADP-изоцитратдегидрогеназы в гепатоцитах крыс в норме и
при токсическом гепатите / Е.М. Андреещева, Т.Н. Попова, В.Г. Артюхов, Л.В.
Матасова // Вестник ВГУ. Сер. химия, биология. - 2008. - № 2. - С. 74-77.
Левенкова, М.В. Некоторые регуляторные свойства
глюкозо-6-фосфатдегидрогеназы из печени крыс в норме и экспериментальном
токсическом гепатите / М.В. Левенкова, Т.Н. Попова, А.В. Семенихина, С.В.
Назарова // Вестник ВГУ. Сер. Химия. Биология. Фармация. - 2004. - № 1. - С.
134 - 138.
Агарков, А.А. Регуляция активности глутатионредуктазы из печени
крысы при токсическом гепатите и действии гепатопротекторов: автореф. дис.
канд. биол. наук: 03.00.04 / Агарков Александр Алексеевич. - Воронеж, 2009. -
24 с.
Гонский, Я.И. Роль нтиоксидантной системы в патогенезе
токсического гепатита / Я.И. Гонский, М.М. Корда, И.Н. Клищ // Патол. физиол.
эксп. тер. - 2007. - № 2. - С. 43-45.
Катикова, О.Ю. Особенности витаминного статуса у больных с
заболеваниями печени различной этиологии. Возможности витаминотерапии / О.Ю.
Катикова, Е.В. Ших // Рос. журн. гастроэнтерол. гепатол. колопроктол. - 2009. -
Т. 19, № 3. - С. 21-31.
Дубинина, Е.Е. Продукты метаболизма кислорода в
функциональной активности клеток (жизнь, смерть, созидание и разрушение): учеб.
/ Е.Е. Дубинина. - СПб.: изд-во Медицинская пресса, 2006. - 400 с.
Зенков, Н.К. Окислительный стресс. Биохимический и
патофизиологический аспекты: науч. изд. / Н.К. Зенков, В.З. Ланкин, Е.Б.
Меньщикова. - М.: МАИК. Наука, 2001. 343 с.
Осипов, А.Н. Активные формы кислорода и их роль в организме /
А.Н. Осипов, О.А. Азизова, Ю.А. Владимиров // Успехи биол. химии. − 1990.
− Т. 31. − С. 180-208.
Владимиров, Ю.А. Свободные радикалы в живых системах / Ю.А.
Владимиров, О.А. Азизова, А.И. Деев // Итоги науки и техники. Биофизика. −
1991. − Т. 29. - С. 176.
Владимиров, Ю.А. Свободные радикалы и антиоксиданты / Ю.А.
Владимиров // Вестник РАМН. − 1991. − № 7. - С. 43 - 51 .
Frey P.A. Radical mechanisms of enzymatic catalysis.
Annu Rev. Biochem. 2005.
V.70. P. 121 - 148.
Меньщикова, Е.Б. Окислительный стресс: Патологические
состояния и заболевания: учеб. / Е.Б. Меньщикова, Н.К. Зенков, В.З. Ланкин,
И.А. Бондарь, В.А. Труфакин. - Новосибирск: АРТА, 2008. - 284 с.
16 Halliwell B. Oxygen toxicity, oxygen radicals,
transition metals and disease. / B. Halliwell, J.M.C. Gutteridge // Biochem
J.-1984.-Vol.219.-P.1-14.
17 Янковский, О.Ю. Токсичность кислорода и биологические
системы. Эволюционные, экологические и медико-биологические аспекты: учеб. /
О.Ю. Янковский. - СПб.: изд-во Игра, 2000. - 294 с.
Владимиров, Ю.А. Перекисное окисление липидов в биологических
мембранах / Ю.А. Владимиров, А.И. Арчаков // Вестник РАМН. - 1998. - № 43. - С.
51 - 53.
Болдырев, А.А. Роль активных форм кислорода в
жизнедеятельности нейрона / А.А. Болдырев // Успехи физиол. наук. - 2003. - Т.
34, № 3. - С. 21-34.
20 Combined glutathione-S-transferase Ml and T1
genetic polymorphism and tacrine hepatotoxicity Text. / T. Simon, L.
Becquemont, M. Mary-Krause [et al.] // Clin. Pharmacol. Ther. 2000. - Vоl. 67, N 4. - P. 432-137.
Lee, V.M. Zonal location of compensatory hepatocyte
proliferation following chemically induced hepatotoxicity in rats and humans
Text. / V.M. Lee, R.G. Cameron, M.C. Archer // Toxicol.Pathol. 1998. - Vol. 26,
N 5. - P. 621-627.
22 Костюк, В.А. Биорадикалы и биоантиоксиданты: монография. /
В.А. Костюк, А.И. Потапович. − Минск: БГУ, 2007. - 174 с.
Барабой, В.А. Механизмы стресса и перекисное окисление
липидов / В.А. Барабой // Успехи современной биологии. - 1991. - Т. 111, вып.
6. - С. 923 - 931.
Воскресенский, О.Н. Биоантиоксиданты - облигатные факторы
питания / О.Н. Воскресенский, В.Н. Бобырев // Вопр. мед. химии. - 2004. - № 4.
- С. 21-26.
Лим, В.Г. Влияние четыреххлористого углерода на перекисное
окисление липидов и показатели системы иммунитета / В.Г. Лим, П.Ф. Забродский,
В.В. Васильев // Химическая и биологическая безопасность. Саратов. воен.-мед.
ин-т. 2006. - № 3. - С. 34 - 41.
Куценко, С.А. Основы токсикологии: учеб. пособие / С.А.
Куценко. - СПб.: Фолиант, 2004. - 720 с.
27 Babich, M.M. Metformin-induced acute hepatitis
Text. / M.M. Babich, I. Pike, M. L. Shiffman // Am. J. Med. 1998. - Vol. 104, N
5. - P. 490-492.
Diclofenac-associated acute cholestatis hepatitis
Text. / H. Hackstein, W. Mohl, W. Puschel [et al.] / Z. Gastroenterol. 2001. - Vol. 36, N 5. -
P. 385-389.
Буклис, Э.Р. Коррекция трофологического статуса у больных
циррозом печени / Э.Р. Буклис // Рос. журн. гастроэнтерол. гепатол.
колопроктол. - 2003. - Т. 13, № 5. - С. 53-57.
Пентюк, А.А. Поражение печени ксенобиотиками. / А.А. Пентюк,
Л. В. Морозов, О.В. Паламарчук // Вестн. Винницкого мед. ун-та. - 2004. - № 9. - С. 125-134.
Cholestatic hepatitis ascribed to the use of
thiabendazole Text. / I.A. Eland, S.C. Kerkhof, D. Overbosch [et al.] / Ned.
Tijdschr. Geneeskd. 1998. - V. 142, N23.-P. 1331-1334.
Bjorkman, D. Nonsteroidal anti-inflammatory
drug-associated toxicity of the liver, lower gastrointestinal tract and
esophagus Text. / D. Bjorkman // Am. J. Med. 1999.-Vol. 105, N5.-P. 17-21.
Acute hepatotoxicant exposure induces TNFR-mediated
hepatic injury and cytokine/apoptotic gene expretion Text. / T.L. Horn, T.D.
O'Brien, L.B. Schook [et al.] // Toxicol. Sci. 2008. - Vol. 54, N 1. - P. 262-273.
Безбородкина, Н.Н. Морфометрия митохондриального аппарата
гепатоцитов нормальной и цирротически изменённой печени крыс / Н.Н. Безбородкина,
С.В. Оковитый, М.В. Кудрявцева, О.В. Кирик, И.В. Зарубина, Б.Н. Кудрявцев //
Цитология. - 2008. - Т.50, № 3. - С. 228-236.
Костюк, В.А. Влияние производных о-бензохинона на
свободнорадикальные процессы, инициируемые в микросомах печени крыс
четыреххлористым углеродом / В.А. Костюк // Биохимия. − 1999. − Т. 56, №1. − С. 109 − 114.
A comparison of apoptosis and necrosis induced by
hepatotoxins in HepG2 cells Text. / T.O'Drien, G.Babcock, J. Cornelius [et al.]
/ Toxicol. Appl. Pharmacol. 2007. - Vol. 164, N 3. -
P. 280-290.
Арчаков, А.И. Окислительная модификация цитохромаР-450 и
других макромолекул в процессе их обновления / А.И. Арчаков, В.Г. Згода, И.И.
Карузина // Вопр. мед. химии. - 1997. - Т. 43, № 1. - С. 3-27.
38 A new drug responsible for microvesicular
steatosis: ticlopidine Text. / A.J. Remy, B. Heran, G. Galindo [et al.] /
Gastroenterol. Clin. Biol. 1999. - Vol. 23, N 1.-P. 151-152.
Мясоедова, К.Н. Новое в изучении цитохромовмР-450 / К.Н.
Мясоедова // Биохимия. - 2008. - № 9. - С. 1199-1205.
Костюк, В.А. Роль ковалентного связывания и перекисного
окисления липидов в повреждении печени четыреххлористым углеродом / В.А. Костюк
// Биохимия. −
1991. − Т. 56, № 10. − С. 1878−1885.
Drug-and estrogen-induced cholestasis through
inhibition of the hepatocellular bile salt export pump (Bsep) of rat liver
Text. / B. Stieger, K. Fattinger, J. Madon [et al.] // Gastroenterology. 2006. - Vol. 118, N 2. -
P. 422-430.
Воробьёва, О.В. Влияние экспериментального цирроза печени у
самок крыс на патологию и регенерацию печени у потомства: автореф. дис. канд.
медицинских наук: 14.00.15/ Воробьева Ольга Васильевна. - Ульяновск, 2007. - 23
с.
43 Dourakis, S.P. Sex hormonal preparations ,and the
liver Text. / S.P. Dourakis, G. Tolis // Eur. J. Contracept. Reprod. Health. Care.
2008. -Vol. 3, N l.-P. 7-16.
Петренко, А.Ю. Влияние препаратов эмбриональных тканей
человека на интенсивность перекисного окисления липидов при остром токсическом
гепатите у крыс / А.Ю. Петренко, О.В. Оченашко // Проблемы криобиологии. -
2001. - № 2. - С. 66 -71.
Кравченко, Л.В. Характеристика острого токсического действия
четырёххлористого углерода как модели окислительного стресса / Л.В. Кравченко,
Н.В. Трусов, М.А. Ускова, И.B. Аксенов, Л.И. Авреньева, Г.В. Гусева, М.А.
Васильева, А.В. Селифанов, В.А. Тутельян // Токсикологический вестник. − 2009. − № 1.
- С. 12 − 18.
Cuncic C., Detich N., Ethier D. Vanadate inhibition of
protein tyrosine phosphatases in Jurkat cells: modulation by redox state. J.
Biol. Inorg. Chem. 2006. V.4. N3. P. 354 - 359.
Hallfrisch J., Muller D., Singh V. Vitamin A and E
intakes and plasma concentrations of retinol, betacarotene, and
alpha-tocopherol in men and women of the Baltimore Longitudinal Study of Aging
// Am. J. Clin. Nutr. - 2003. - Vol. 60. - P. 176-182.
Bearman, S.I. Veno-occlusive disease of the liver
Text. / S.I. Bearman // Curr. Opin. Oncol. 2003. - Vol. 12, N2.-P. 103-109.
Bond, G.R. Population-based Incidence and Outcomc of
Acetaminophen Poisoning by Type of Ingestion Text. / G.R. Bond, L.K. Hite //
Academic Emergency Medicine. 1999. - Vol. 6, N 11. - P. 1115-1120. .
Free radicals and reactive oxygen species in
programmed cell death Text. / K.M. Anderson, T. Seed, D. Ou, J. E. Harris //
Med. Hypotheses. 1999. - Vol. 52, N 5. - P. 451-463.
Hepatic histopathology of a vitamin A overdose in
mouse liver Text. / T. Shintaku, T. Murata, K. Yamaguchi [et al.] // J.
Electron. Microsc. 2007. -Vol. 47, N3.-P. 263-267.
Fazio V.M., Barrera G., Martinotti S., Farace M.G.,
Giglioni B., Frati L., Manzari V., Dianzani M.U 4-Hydroxynonenal, a products of
cellular lipid peroxidation, which modulates c-myc and and globin gene
expression in K562 erythroleukemic cells // Can-cer Res. 2002. Vol.52.
P.4866-4871.
Coe, J.E. Estrogen-induced hepatic toxicity and
hepatic cancer: differences between two closely related hamster species Text. /
J.E. Coe, K.G.I Shak, M.J. Ross//Liver. 1998.-Vol. 18, N5.-P. 343-351.
Bloomer, J.R. Liver metabolism of porphyrins and haem
Text. / J.R. Bloomer // J. Gastroenterol. Hepatol. 2001. - Vol. 13, N 3. - P. 324-329.
Забродский, П.Ф. Влияние дихлорэтана на показатели системы
иммунитета и перекисного окисления липидов / П.Ф. Забродский, В.Г. Лим, Н.М.
Трошкин // Вестник новых медицинских технологий. − 2005. − Т. 12, № 2. - С. 15 − 19.
Dianzani M.U. Biochemical aspects of fatty liver // in
Hepatotoxicology (Meeks R.G., Harrison S.D., Bull R.J. - eds). 1998. CRC Press
Inc. London. P. 327-399.
Dianzani M.U., Paradisi L., Parola M., Barrera G.,
Rossi M.A. Action of the aldehydes derived from lipid peroxidation on isolated
liver plasmamembranes // in Free radicals, Lipoproteins, and Membrane Lipids
(Crastes de Paulet ed). 2006. Plenum Press. New York. P.171-181.
Beall, H.D. Mechanisms of action of quinine-containing
alkylating agents inqol-directed drug development Text. / H.D. Bell, S.I.
Winski // Front. Biosci. -2007. Vol. 1, N 5. - P. 639-648.
Dianzani M.U. Free radicals in physiology and
pathology // Boll Soc It Biol Sper. 2007. Vol.68. P.491-511.
Сазонтова, Т.Г. Значение баланса прооксидантов и
антиоксидантов - равнозначных участников метаболизма / Т.Г. Сазонтова, Ю.В.
Архипенко // Патологическая физиология и экспериментальная терапия. - 2007. - №
3. - С. 2-18.
Скакун, Н.П. Поражение печени ССl4: науч. изд. / Н.П. Скакун, Г.Т. Писько, И.П. Мосейчук. -
М.: НИИТЭХИМ, 1989. - 94с.
Дубинина, Е.Е. Антиоксидантная система плазмы крови / Е.Е.
Дубинина // Укр. биохим. журнал. - 1992. - Т. 64, № 2. - С. 3-14.
Дубинина, Е.Е. Выделение и свойства супероксиддисмутазы
плазмы крови человека / Е.Е. Дубинина, В.В. Туркин, Г.А. Бабенко, В.А. Исаков
// Биохимия. - 1992. - Т. 57, № 12. - С. 1892-1901.
Дубинина, Е.Е. Характеристика внеклеточной
супероксиддисмутазы / Е Е. Дубинина // Вопр. мед. химии. - 1995. - Т. 41, № 6.
- С. 8-12.
65 Acute hepatic necrosis in a patient with cyproterone
acetate Text. / A. Lombardi, P. Ferrazza, F. Castaldi [et al.] // G. Chir.
2005. - Vol. 19, N 4. -P. 161-163.
66 Балаболкин, М.И. Применение витаминов с антиоксидантным
действием в комплексной терапии сахарного диабета / М.И Балаболкин, Е.М.
Клебанова, В. М. Креминская // Лечащий врач. - 2007. - № 10. - С. 52-55.
67 Floreani A., Baragiotta A., Martines D. et al. Plasma antioxidant levels
in chronic cholestatic liver diseases // Aliment. Pharmacol. Ther. - 2009. - Vol. 14, N 3 - P.353-358.
Сорокина, И.В. Роль фенольных антиоксидантов в повышении
устойчивости органических систем к свободно-радикальному окислению: науч. изд.
/ И.В. Сорокина, А. Крысин, Т.Б. Хлебникова. - Новосибирск: изд-во РАН, 1997. -
230 с.
Ших, Е.В. Особенности витаминного статуса и пути его
коррекции / Е.В. Ших // Сonsilium medicum. - 2007. - Т. 5, № 4. - С. 12-18.
Тутельян, В.А. Витамины и микроэлементы в клинической
фармакологии: науч. изд. / В.А. Тутельян. - М.: Палея-М, 2007. - 360 с.