Разработка принципиальной технологии извлечения молибдена и кобальта из дезактивированных катализаторов на основе оксида алюминия
СОДЕРЖАНИЕ
ВВЕДЕНИЕ
. АНАЛИТИЧЕСКИЙ ОБЗОР
.1 Молибден, кобальт, никель и их
свойства
.2 Области применения молибдена и
кобальта
. КАТАЛИЗАТОРЫ
.1 Алюмокобальтмолибденовые (АКМ) и
алюмоникельмолибденовые (АНМ) катализаторы
.1.1 Регенерация катализаторов
.1.2 Утилизация отработанных
катализаторов
.2 Способы выделения ценных
компонентов из растворов
.2.1 Сорбционное извлечение
молибдена, кобальта и никеля
.2.2 Экстракционные методы
. ЦЕЛИ И ЗАДАЧИ РАБОТЫ
. ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
.1 Методика проведения экспериментов
.1.1 Материалы и их подготовка
.1.2 Условия проведения
экспериментов
.1.3 Методы анализа
. РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЯ
.1 Выщелачивание молибдена и
кобальта из катализатора на основе Al2O3
.1.1 Отработка процесса разложения
катализаторов на основе оксида алюминия растворами серной кислоты
.1.2 Влияние предварительной
обработки катализатора раствором нитрата натрия перед прокаливанием на
выщелачивание кобальта и молибдена
.1.3 Осветление растворов сульфата
алюминия, полученных при растворении катализаторов в серной кислоте
.2 Сорбция молибдена в статических
условиях из раствора сульфата алюминия при различных рН
.2.1 Сорбция молибдена из раствора
сульфата алюминия
.2.2 Десорбция молибдена раствором
гидроксида натрия
.3 Сорбция кобальта в статических условиях
из раствора сульфата алюминия при различных рН
.3.1 Сорбция кобальта в статических
условиях из раствора сульфата алюминия
.3.2 Десорбция кобальта в
статических условиях одномолярным раствором серной кислоты
.4 Сорбция и десорбция молибдена в
статических условиях
.5 Сорбция и десорбция кобальта в
статических условиях
.6 Сорбция и десорбция молибдена в
динамических условиях
.7 Сорбция и десорбция кобальта в
динамических условиях
.8 Принципиальная технологическая
схема извлечения молибдена и кобальта из катализатора, основанная на
выщелачивании серной кислотой
ЗАКЛЮЧЕНИЯ И ВЫВОДЫ
СПИСОК ИСПОЛЬЗОВАННЫХ ИСТОЧНИКОВ
ПРИЛОЖЕНИЯ
Введение
Развитие химической промышленности всегда
сопровождается образованием значительного количества отходов, что приводит к усилению
техногенного воздействия на окружающую среду и возникновению дополнительных
экологических проблем.
Одной из таких проблем является переработка
металлсодержащих отходов с целью их дальнейшего эффективного использования. В
этой связи определенный интерес представляют отработанные твёрдые
каталитические комплексы [1].
Молибден, кобальт и никель широко используется в
различных областях современной науки и техники (в авиационной, ракетной и
космической технике, нефтехимии, машиностроении, электронике, электротехнике,
приборостроении и др.) и относится к числу стратегических металлов. Спрос на
эти металлы и цены на них в последнее десятилетие постоянно растут.
Катализаторы на основе оксида алюминия или
оксида алюминия с добавкой кремнезема с введенными в них оксидом молибдена в
сочетании с оксидами никеля или кобальта широко используются в процессах
гидрообработки нефтяного сырья.
В настоящее время в мировой практике широко
распространены в основном алюмокобальтмолибденовые (АКМ) и
алюмоникельмолибденовые (АНМ) каталитические системы. Это катализаторы
гидрогенизационных процессов нефтепереработки, катализаторы гидрообессеривания
нефтепродуктов, которые являются многотоннажной продукцией, поскольку процессы
гидроочистки занимают ведущее место в современных схемах переработки нефти как
в нашей стране, так и за рубежом [2]. Срок службы катализаторов определяется
количеством сырья, пропущенного через 1 кг контакта, и зависит от условий
эксплуатации [3]. Если продукты реакции прочно адсорбируются на поверхности катализатора,
то даже очень хороший сам по себе катализатор оказывается практически
непригодным, так как активность его быстро падает - он отравляется продуктами
реакции [4]. Восстановление каталитической активности производится путём
проведения периодических регенераций вследствие коксоотложений и каталитических
ядов. При регенерации коксовые отложения удаляют путём высокотемпературной
обработки. Однако некоторые изменения в составе катализатора становятся
частично или полностью необратимыми (влияние высоких температур), а со временем
столь значительными, что дальнейшее его использование экономически невыгодно и
он заменяется следующим [3].
Существует два основных принципиальных способа
переработки дезактивированных катализаторов [5] на основе оксида алюминия,
содержащих благородные металлы, первый из которых заключается в растворении
основы - оксида алюминия, второй - на избирательном выщелачивании из
катализаторов ценных компонентов при минимальном растворении носителя.
Представляется, что более перспективным для извлечения молибдена является
второй способ [6]. Однако для извлечения цветных металлов необходимо растворять
основу - оксид алюминия [7].
Целью настоящей работы является разработка
принципиальной технологии извлечения молибдена, а также кобальта из
дезактивированных катализаторов на основе оксида алюминия.
1.
АНАЛИТИЧЕСКИЙ ОБЗОР
.1 Молибден, кобальт, никель и их свойства
Молибден - (Molybdenum), Mo - химический
элемент 6 (VI) группы периодической системы, атомный номер 42, атомная масса
95,94. Металл отличается высокой температурой плавления, низким давлением паров
даже при 2000 °С и малым температурным коэффициентом линейного расширения.
Относительно малое сечение захвата тепловых нейтронов позволяет использовать
молибден в качестве конструкционного материала в ядерных реакторах.
Молибден устойчив на холоду в соляной и серной
кислотах, но медленно корродирует при 80 - 100 °С. В азотной кислоте и царской
водке при нагревании он быстро растворяется. Хорошим растворителем молибдена
служит смесь 5 объемов HNO3,
3 объемов H2SO4
и
2 объёмов воды.
Наиболее характерны соединения молибдена высшей
степени окисления, равной 6. известны также соединения, отвечающие степеням
окисления 5, 4, 3 и 2.
Молибденовые кислоты.
Известно несколько молибденовых кислот, например: молибденовая кислота H2MoO4
(бесцветный мелкокристаллический порошок). Она малорастворима в воде,
растворяется в соляной и серной кислотах. Соли называются нормальными
молибдатами. Все нормальные молибдаты, за исключением солей щелочных металлов, аммония
и магния, малорастворимы в воде. Молибденовая кислота H2Mo2O7
(белое кристаллическое вещество), изополикислота Н2[Мо4О13]
(сильная кислота, существующая в водных растворах).
Изополикислоты и их соли.
Нормальные молибдаты натрия, калия, аммония устойчивы в щелочных растворах. При
подкислении растворов в интервале рН 7,5-2 образуются полимерные анионы
изокислоты Mo7O24-6,Mo8O26-2,
Mo6O20-4
и
др.
Парамолибдат аммония (NH4)6Mo7O24∙nH2O
является распространенным конечным продуктом при переработке рудных концентратов.
В результате термического разложения этой соли получается чистая трехокись
молибдена.
Кобальт и никель - элементы триад, они входят в
семейство железа. Эти элементы по свойствам близки и существенно отличаются от
остальных шести элементов: Ru-Rh-Pd,
Os-Ir-Pt.
Большая часть железа верхних слоёв земли
находится в виде кислородных соединений, из которых наиболее важны промышленные
руды этого металла - лимонит (Fe2O3
H2O),
гематит (Fe2O3)
и магнетит (Fe2O3
FeO). Кроме
того, скопления железа встречаются в виде минерала сидерита (FeСO3),
а также в соединениях с серой и мышьяком. Для кобальта и никеля наиболее
характерно совместное нахождение с последними двумя элементами (и отчасти
сурьмой) в виде минералов типа ЭS,
ЭАs2,
ЭАsS и т.д., примерами
которых могут служить пентландит (FeS
NiS) и кобальтин (CoAsS).
В химически чистом состоянии железо, кобальт и
никель могут быть получены восстановлением их окислов водородом или
электролизом растворов солей. Все три элемента представляют собой блестящие
белые металлы с сероватым (Fe,Co)
или серебристым (Ni)
оттенком. Железо и никель легко куются и прокатываются. Кобальт более твёрд и
хрупок. В отличие от других металлов Fe,
Co и Ni
притягиваются магнитом.
По химическим свойствам железо, кобальт и никель
являются металлами средней активности. В отсутствие влаги они при обычных
условиях заметно не реагируют даже с такими типичными металлоидами, как О, S,
Cl и Br.
Однако при нагревании взаимодействие со всеми ними протекает довольно
энергично, особенно если металлы находятся в измельчённом состоянии.
Располагаясь в ряду напряжений между железом и
оловом, кобальт и никель стоят ближе к олову. Поэтому оба металла растворяются
в разбавленных кислотах медленнее железа. Устойчивость к действию
концентрированной HNO3
по ряду Fe-Co-Ni
быстро уменьшается. Сильные щёлочи на все три элемента не действуют.
Кислородные соединения двухвалентных элементов
семейства железа образуют ряд закисей общей формулы ЭО. Относящиеся сюда окислы
- чёрная FeО,
серо-зелёная СоО и зелёная NiO
- практически нерастворимы в воде и щелочах, но легкорастворимы в кислотах.
Водородом при нагревании они могут быть восстановлены до металла, причём
лёгкость такого восстановления по ряду Fe-Co-Ni
несколько увеличивается.
Отвечающие окислам ЭО гидраты закисей Fe,
Co и Ni
общей формулы Э(ОН)2 могут быть получены только косвенным путём. Все
они практически нерастворимы в воде и обычно употребляемых растворах сильных
щелочей, но легкорастворимы в кислотах. С химической стороны рассматриваемые
гидраты характеризуются, следовательно, основными свойствами.
Общим методом получения гидроокисей Э(ОН)2
является взаимодействие растворов соответствующих солей Fе
и его аналогов с сильными щелочами. Образующиеся при этом объёмистые осадки -
белый Fe(ОН)2,
розово-красный Co(ОН)2
и яблочно- зелёный Ni(ОН)2
- сильно отличаются друг от друга по отношению к кислороду воздуха. Тогда как Ni(ОН)2
с ним не реагирует, а Co(ОН)2
окисляется лишь медленно до коричнево-бурого Со(ОН)3, гидрат закиси
железа быстро переходит в буро- красный гидрат окиси железа Fe(ОН)3.
Гидратированные ионы Э.. окрашены в
цвета: бледно-зелёный (Fе..),
розово-красный (Со) и ярко-зелёный (Ni..).
Те же окраски характерны для образованных ими кристаллогидратов солей.
Напротив, в безводном состоянии отдельные соли окрашены различно, причём цвета
их не всегда совпадают с собственной окраской ионов Fе2+
(бесцветный), Со2+ (красноватый) и Ni
2+
(жёлтый),
а зависят также от природы аниона.
Аммиакаты трёхвалентного
кобальта отличаются большой устойчивостью, и жёлтый комплексный катион [Co(NH3)6]3+
образует хорошо кристаллизующиеся соли с рядом анионов, простые соли которых
для Со3+ неизвестны. Желто-оранжевый ион [Co(NH3)6]3+
характеризуется эффективным радиусом 2,5 А и константой нестойкости 3 - 10 -33.
Длина связи кобальта с азотом аммиака равна 1,99 ± 0,08 А, а данные для
ее силовой константы очень противоречивы (5,4 и 1,1). Ион этот не разрушается
концентрированной серной кислотой, а его термическая диссоциация наступает лишь
около 200 °С. Замещение в нём аммиака на другие нейтральные молекулы или
кислотные остатки ведёт к образованию множества разнообразных комплексных
соединений трёхвалентного кобальта, большинство которых устойчиво и в твёрдом
состоянии и в растворе. Примером продукта полного замещения аммиака [Co(NH3)6]3+
на кислотные остатки может служить комплексный анион [Co(NО2)6]3-,
дающий с одновалентными катионами (кроме Li+
и Na+)
малорастворимые в воде кристаллические осадки состава М3[Co(NО2)6].Образование
жёлтой соли калия используется для его открытия [8].
1.2 Области применения молибдена, кобальта и
никеля
Молибден имеет большое значение в современной
технике. Из всего его количества, потребляемого промышленностью, до 80%
используется в черной металлургии для производства жаропрочных, жаростойких,
антикоррозионных, инструментальных, быстрорежущих, магнитных, конструкционных
сталей, жаропрочных и жаростойких чугунов.
Молибден в стали входит в состав, как свободных
выделений карбидов, так и твердого раствора. Молибден повышает способность
стали к цементации. В магнитных сталях и сплавах он увеличивает магнитную
проницаемость. Придает жаропрочность и жаростойкость ряду сплавов на основе
цветных металлов.
В жаропрочных сплавах с цветными металлами
потребляется 4 - 5% вырабатываемого молибдена. 3 - 6,5% Мо выпускают в виде
проволоки, прутков, листа для электро - и радиотехнической промышленности и
других назначений. Для реактивов, красок и других химикатов используется 4 - 5%
Мо. Повышается его применение в сельском хозяйстве.
Молибден вводят в стали в виде сплава с железом
- ферромолибдена. Молибдена в ферросплавах не ниже 50%.
Соединения молибдена применяются как
катализаторы в органическом синтезе и как реактивы в аналитической химии
(парамолибдат аммония и комплексные соединения молибдена), в производстве лаков
и красок для шерсти и шелка. В сельском хозяйстве используются соединения
молибдена в виде слабых растворов: он облегчает усвояемость растениями
питательных веществ из почвы. Но в то же время большие дозы молибдена оказывают
токсическое действие на растительные и животные организмы [9].
Основное количество металлического кобальта идёт
на производство сплавов с особыми физическими свойствами, высококоэрцитивных
магнитов, быстрорежущей и кобальтовой стали, жаропрочных и твёрдых сплавов.
На основе кобальта получены катализаторы для
органического синтеза. Из соединений кобальта изготовляют стойкие эмали и
краски.
Перспективно использование кобальта в
производстве газотурбинных двигателей, постоянных магнитов разных типов, в
устройствах каталитического дожигания выхлопных газов автомобильных двигателей
[10].
2. КАТАЛИЗАТОРЫ
Катализатор - вещество, ускоряющее реакцию, но
не входящее в состав продуктов реакции. Катализаторы подразделяются на гомогенные
и гетерогенные. Гомогенный катализатор находится в одной фазе с
реагирующими веществами, гетерогенный - образует самостоятельную фазу,
отделённую границей раздела от фазы, в которой находятся реагирующие вещества.
Типичными гомогенными катализаторами являются кислоты и основания. В качестве
гетерогенных катализаторов применяются металлы, их оксиды и сульфиды.
Оксид алюминия - основа
катализатора, его получают осаждением из водных растворов, содержащих ионы А13+.
Гелеобразный осадок подвергают старению, фильтруют, промывают и высушивают. В
зависимости от условий осаждения могут быть получены такие модификации
гидрооксида алюминия, как бемит АlO(ОН)
или байерит А1(ОН)3. Прокаливание оксида алюминия до 600 °С
обеспечивает оптимальную кислотность и стабильность структуры. Основными
модификациями для контактов служат h и g-А12О3.
Кислотность их поверхности, в зависимости от степени гидратации, обусловлена
наличием центров Бренстеда (протонодонорные ОН~ группы) и Льюиса
(электроноакцепторные ионы А13+). При хлорировании происходит замена
ОН-групп на ионы С1~, что увеличивает кислотность поверхности за счет смещения
электронов от связи О-Н соседних групп к электроотрицательному иону хлора. Это
важно для практики, так как, изменяя степень галоидирования, можно
контролировать силу поверхностных кислотных центров и, таким образом,
относительные скорости тех реакций, которые ускоряются кислотами.
Удельная поверхность контакта катализатора
должна быть максимально большой, так как лимитирующими стадиями процесса
являются адсорбция - десорбция реагирующих веществ на активных центрах
катализатора, т. е. скорость реакции определяется доступностью активной
поверхности. Наибольшую удельную поверхность имеют контакты на основе h-
А12Оэ. Тем не менее g- А12Оэ
предпочтительнее, так как он более термостабилен и, следовательно, катализатор на
основе g-А12Оэ
способен выдерживать большее число регенераций, чем на основе h-
А12Оэ.
Объем пор и размер зерен влияют на
селективность, стабильность и активность контакта. При быстро проходящей
реакции дегидрирования нафтеновых углеводородов контакты с большим средним
радиусом пор и более широкой кривой распределения по размерам пор позволяют
получать лучшие результаты. Однако, наиболее рационален катализатор со
сферической формой гранул (плотная упаковка, повышенная прочность) с диаметром
1,5-3,0 мм, так как путем уменьшения размеров зерна контакта можно
уменьшить длину пробега молекул реагента, в результате чего скорость протекания
реакции будет определяться не диффузией, а кинетикой химического превращения.
Таким путем удается с максимальной полнотой использовать всю внутреннюю и
наружную поверхность катализатора. Вместе с тем применение очень мелкого
контакта вызывает чрезмерное гидравлическое сопротивление в реакторе.
Механическая прочность должна быть достаточно
высока, чтобы при транспортировке, загрузке в реакторы и эксплуатации (особенно
в системах с непрерывной регенерацией в движущемся слое катализатора) не
образовывались осколки и пыль, которые, накапливаясь в аппаратуре, увеличивают
перепады давления в системе, снижают экономические показатели.
Химический состав должен характеризоваться
ограничением нежелательных примесей (щелочные металлы, железо), ухудшающих
качество контакта, и оптимальным содержанием активных компонентов,
обеспечивающих высокие показатели процесса.
Каталитические свойства должны выражаться в
снижении энергии активации реакций процесса, но не на столько, чтобы
промежуточное активное состояние было устойчивым. Молекулы реагирующих веществ
должны лишь относительно слабо адсорбироваться на катализаторе, не блокировать
его поверхность, что может привести к подавлению каталитических реакций [3].
По данным исследований модельных систем, включая
бинарные Со/ Al2O3
и Мо/ Al2O3,
в составе АКМ катализаторов можно ожидать существование следующих объёмных и
поверхностных соединений: MoO3,
Аl(MoO4)3,
Со3O4,СоAl2O4,
СоMoO4,
СоО, комплексы переменного состава СоМоАl.
При исследовании промышленных катализаторов с
содержанием 2 - 3% СоО и 10 - 12% MoO3
различными методами было установлено, что основной фазой являются СоMoO4
и оксид алюминия. Фаза СоAl2O4
рентгенографически не обнаруживается, по-видимому, потому, что при низком
содержании кобальта эта фаза находится в высокодисперсном состоянии, а её
структура отличается от объёмной шпинели. Однако образование фазы СоAl2O4
обнаружено по линии 1612 см-1 в ИК - спектрах образцов, что
свидетельствует о взаимодействии кобальта с молибденом. Кроме того, показано
стабилизирующее влияние кобальта на молибдат и на его дисперсность. Методом
дериватографии установлено, что в промышленных АКМ катализаторах может
образоваться фаза Аl(MoO4)3.
Состав и количество фаз может
различаться в зависимости от суммарного содержания металлов, атомного отношения
Со/Мо, природы носителя, способа введения металлов и температуры прокаливания.
Например, показано, что даже при одинаковом способе приготовления (пропитка
носителя) порядок введения металлов существенно влияет на его фазовый состав.
Так, катализатор с содержанием 1 - 4% кобальта и 2 - 8% молибдена, полученный
ступенчатым внесением кобальта, имеет синюю окраску. Катализаторы, полученные
однократной пропиткой, изменяют свой цвет от светло-голубого до чёрного при
содержании кобальта >2%, что обусловлено образованием Со3O4,
то есть кобальт присутствует преимущественно в тетраэдрической координации, то
есть не входит в состав СоМо04. При увеличении температуры
прокаливания в интервале 500-750 °С происходит образование фазы СоА12О4.
При ступенчатом введении кобальта в концентрации >2% последний присутствует
первоначально в основном в октаэдрической координации. Однако при повышении
температуры прокаливания с 500 до 750 °С «октаэдрический» кобальт переходит в
«тетраэдрический».
При совместной пропитке
носителя солями Со и Мо создаются условия для образования фазы СоМо04,
которая обнаруживается рентгенографически при концентрации кобальта >2%. В
процессе прокаливания возможно взаимодействие СоМоО4 с А12О3
с образованием фазы СоА12О4[2].
Фаза СоА12О4
- шпинель - минерал класса окислов. По химической конституции соответствуют
двойным окислам типа R2+OR3+O3,
где R2+
- Mg, Fe2+,
Мn2+ и
Zn, иногда Be,
Ni5+
и Co2+;
R3+
- Al, Fe3+,
Cr3+,
иногда Ti4+
и V3+.
В зависимости от преобладающего катиона R3+
различают : алюмошпинели - собственно шпинель MgAl2O4,
ганит ZnAl2O4,
галаксит (Mn, Fe)
Al3O,
и герцинит FeAl.2O4;
ферришпинели - магнезиоферрит MgFe304,
франклинит ZnFe2O4,
якобсит Мn
х
Fe2O4,
магнетит FeFe2O4
и другие; хромошпинели - хромит FeCr2O4,
мангохромит (Mg, Fe)х
Сг2О4,
хромпикотит (Mg, Fе)х
(Сr, А1)2О4
и др.; титанo- и ваиадиошпииели
- ульво-шпинель FeTiO4
и кульсонит FeV3О4.
Сингония кубическая. Структура координационная. По характеру распределения
атомов в тетраэдрических и октаэдрических позициях различают шпинели
нормальные, обращенные и смешанные. В природе наиболее распространены минералы
со структурой нормального типа. Собственно шпинель - распространенный минерал.
Разновидности: благородная шпинель - прозрачная, ювелирного качества, почти
бесцветная пли окрашенная в темно-красный (рубиновая шпинель.), розово-красный
(гиацинтовая шпинель.), синий (сапфировая шпинель) и др. цвета; Химический
состав собственно шпинели (%): MgO
- 28,34; А12О3 - 71,66. Между Mg
и Fe2+ существует
полная изоморфная смесимость, благодаря которой происходит переход к герциниту.
Нередко они частично замещены двухвалентными цинком и марганцем, иногда
кобальтом и никелем. Алюминий замещается трехвалентными железом и хромом с
образованием изоморфных рядов между алюмошпинелями и ферришпинелями,
алюмошпинелями и хромошпинелями (хромошпинелидами). Очень редко отмечаются в
небольших количествах примеси щелочей и кальция. Шпинели кристаллизуется в
гексоктаэдрическом виде симметрии. Встречается в виде кристаллов и сростков,
для герцинита характерны плотные тонкозернистые агрегаты. Искусственные шпинели
характеризуются избытком окиси алюминия. Шпинель образуется также при
нагревании мусковита, глауконита, монтмориллонита и др. слоистых магний-
и алюмосодержащих минералов при нагревании до температуры выше 1000 °С.
Благородную шпинель используют как драгоценный камень. Искусственная шпинель
применяется в качестве огнеупорного материала, в производстве керамики и
стойких керамических красок [10].
В сульфидированных АКМ
катализаторах обнаружены MoS2,
Co9S8
и
в малых концентрациях сульфид алюминия и полимерные продукты S22-
и S32-.
При этом, как показывают данные магнитных измерений, ИК - и УФ-спектроскопии,
сульфидирование оксидных предшественников происходит не полностью и не
образуется дисперсных фаз MoS2
и Co9S8.
Но относительно полноты сульфидирования оксидных АКМ катализаторов в литературе
имеются противоречивые данные. По-видимому, процесс сульфидирования протекает
по-разному в зависимости от температуры, давлении, сульфидирующего агента и
структуры оксидных предшественников. В сульфидированных катализаторах
отмечается очень слабое взаимодействие молибдена с А12О3
в отличие от оксидных систем. Что же касается состояния кобальта в
сульфидированных катализаторах, то обнаружено два типа ионов Со2+.
Установлено, что кобальт, связанный с А12О3 в виде
псевдоалюмината, не сульфидируется. Второй тип ионов Со2+ образует сульфиды,
причем соотношение между ионами молибдена и сульфидирующимися ионами Со2+
достаточно хорошо коррелирует с гидрообессеривающей активностью.
Выявлено, что условия
предварительной обработки (окислительная или восстановительная среда,
температура, длительность, давление, природа сульфидирующего агента) могут
значительно изменять состав сульфидных фаз АКМ катализатора. В общем степень
сульфидирования кобальта зависит от скорости диффузии ионов Со2+ из
объема к поверхности. Часть из них образует Co9S8,
а остальные внедряются в носитель или локализуются между слоями фазы MoS2,
способствуя восстановлению ионов Мо4+ до Мо3+ [2].
2.1 Алюмокобальтмолибденовые (АКМ) и
алюмоникельмолибденовые (АНМ) катализаторы
Среди катализаторов гидрогенизационных процессов
нефтепереработки катализаторы гидрообессеривания нефтепродуктов являются
многотонажной продукцией, поскольку процессы гидроочистки занимают ведущее
место в современных схемах переработки нефти как в нашей стране, так и за
рубежом. Катализаторы гидрообессеривания существуют более 50 лет и за этот
период претерпели несколько этапов эволюции. В настоящее время в мировой
практике широко распространены в основном алюмокобальтмолибденовые (АКМ) и
алюмоникельмолибденовые (АНМ) каталитические системы.
Эффективность катализаторов гидроочистки
определяется как их химическим составом и структурой, так способами
приготовления и активации. Известно, что структура и фазовый состав
катализатора являются одними из определяющих факторов его эффективности. Как
правило, изучают структуру и состав катализаторов гидрообессеривания в оксидной
форме. При этом предполагают, что имеется тесная взаимосвязь между активными
поверхностными фазами катализатора в процессе эксплуатации и его оксидными
предшественниками [2].
Первый промышленный катализатор риформинга
содержал около 9 мас.% оксида молибдена, осажденного на активированном геле
оксида алюминия, и был применен на установке гидроформинга с неподвижным слоем
контакта, а затем и с псевдоожиженным слоем. Другой окисный катализатор (32
мас.% оксида хрома + 68 мас.% оксида алюминия), разработанный фирмой «Socony
Mobil Oil
Co», использовали на
установках «термофор» с подвижным слоем контакта. И, наконец, на единственной
установке «гиперформинг» эксплуатировался молибдат кобальта, нанесенный па
оксид алюминия, стабилизированный оксидом кремния в подвижном слое с
непрерывной регенерацией. Переход от полностью платиновых монометаллических к
би- и полиметаллическим катализаторам - одно из важнейших достижений в области
каталитического риформинга.
Все окисные катализаторы отличаются
устойчивостью к ядам (S,
N), но из-за
относительно низкой активности процесс риформинга протекает в жестких условиях
(Т до 540 °С, Р до 1,0 МПа, v
от 0,7 до 0,05 ч-1, что ведет к усилению реакций расщепления, интенсивному
коксообразованию и необходимости частой или постоянной регенерации контакта
[3].
Механизм повышения стабильности, селективности и
активности катализаторов при модифицировании его добавками различных металлов
пока неясен. Изучение существующих би- и полиметаллических катализаторов
тормозится тем, что общие методы физической характеризации неэффективны из-за
очень высокой дисперсности и низкой концентрации нанесенных металлов в этих
системах. В последнее время появились методы исследования процессов, протекающих
на поверхности контактов, на атомном уровне: метод рассеяния электронов,
дифракция электронов низкой энергии, электронная спектроскопия, ионное
рассеяние и т. п.. Они позволяют определять структуру и химическую связь атомов
и молекул на поверхности, а также константы скорости элементарных стадий
реакций на поверхности катализатора. Однако аппаратурная сложность и высокая
стоимость таких методов затрудняют их широкое распространение. Чтобы лучше
понять природу металлических контактов, ученые используют в качестве модельных
систем массивные сплавы или сплавленные металлы на некислых подложках. Однако
прямая экстраполяция полученных результатов на реальные нанесенные катализаторы
не всегда правомерна [4].
.1.1 Регенерация катализаторов
Срок службы промышленных
катализаторов зависит как от свойств самого катализатора, так и от качества
перерабатываемого сырья и условий проведения процесса.
Катализаторы гидрообессеривания
периодически подвергают регенерации путем окислительного выжига кокса
непосредственно в реакторе при температуре 450 - 550 ºС.
Дезактивирующее влияние на катализаторы гидроочистки кроме отложений кокса и
металлов оказывают также резкое повышение температуры в результате нарушения
теплового режима, забивание пор катализатора пылью, образование SO3
и,
следовательно, сульфатов на поверхности катализаторов.
Процесс регенерации включает в
себя следующие основные стадии:
1. Замена водородосодержащего
газа на инертный газ или водяной пар;
2. Нагрев катализатора до
начала горения кокса;
. Начальный период
выжига кокса;
. Установившийся период
регенерации;
. Окончание регенерации.
Содержание кокса в катализаторе
перед регенерацией составляет 12 - 15% при переработке вакуумного газойля и до
20% при переработке мазута.
В процессе регенерации катализатора
наряду с выжигом кокса происходит окисление сульфидов гидрирующих металлов.
Этот процесс особенно важен для АКМ катализаторов, которые перед использованием
подвергаются сульфидированию.
При температуре ≥ 550 ºС
образуются каталитически неактивные компоненты и наблюдается значительное
спекание, приводящее к уменьшению удельной плотности и объема микропор. Причем
спекание протекает в большей степени у катализаторов с большей исходной
удельной поверхностью и с большим содержание оксидов металлов. При температурах
600 - 760 ºС возгоняется
молибден, а при более высоких температурах (800 - 900 ºС)
изменяется фазовый состав носителя, что ведет к снижению активности и прочности
катализатора.
Таким образом, окислительную
регенерацию катализаторов гидрогенизационных процессов нефтепродуктов следует
проводить при температуре в реакторе не выше 550 - 600 ºС.
Кроме того, следует учитывать, что катализаторы не допускают резкого снижения
или повышения температуры. Поэтому нагрев или охлаждение рекомендуется проводить
с уменьшенными скоростями с целью выравнивания температурного слоя по слою и по
сечению гранул. Так, установлено, что при увеличении скорости изменения
температуры с 30 до 300 ºС/ч
прочность катализатора снижается вдвое. Скорость подъема для промышленных
катализаторов рекомендована до 30 ºС/ч
и, как исключение, не более 50 ºС/ч.
.1.2 Утилизация отработанных
катализаторов
По мере ужесточения экологических норм на
нефтепродукты спрос на катализаторы гидроочистки в мире растет. Увеличение
объемов их потребления делает актуальной проблему утилизации катализаторов,
отработавших свой ресурс и не подлежащих дальнейшей эксплуатации [11].
Отработанные катализаторы по сравнению со
свежими имеют пониженные прочность и удельную поверхность. При этом они
накапливают на своей поверхности вредные соединения натрия и железа при
достаточно высоком содержании кокса. У таких образцов восстанавливать
активность нецелесообразно, поэтому рекомендуется проведение процесса
рекуперации с целью получения металлов в чистом виде или в составе солей.
Вопросы утилизации отработанных
катализаторов, в том числе оксидсодержащих, актуальны, так как их решение
напрямую связано со снижением техногенного загрязнения окружающей среды и
повышением экономической эффективности существующих производств за счет
извлечения ценных продуктов. Для переработки оксидсодержащих отходов используют
также такие эффективные методы, как прокалка, термоокислительное и
термощелочное разложение и окисление, кислотную и кислототермическую
переработку и сочетание указанных методов. Кроме того, отходы отработанных
катализаторов применяют без переработки для получения различных неорганических
материалов, например, абразивных паст на основе металлооксидного катализатора
дегидрирования углеводородов, коагулянтов для очистки воды, строительных
материалов и др.
Для полного извлечения металлов
из катализатора проводят выщелачивание водным раствором винной, гликолевой,
лимонной, янтарной, щавелевой или серной кислотами, или обработку
комплексообразователем с получением раствора, содержащего аммиачные комплексы
типа Co(NH3)5(H2O)3+
или Co(NH3)63+.
Степень извлечения молибдена из катализатора составляет 60-70%, кобальта
примерно 50% [5].
Предложен способ, близкий к промышленной
реализации: гидрометаллургический, основанный на взаимодействии катализатора с
растворами соды и аммиака, при котором достигается достаточно полное извлечение
молибдена [12].
В работе [13] дано описание
технологии, основанной на выщелачивании ценных компонентов из катализаторов
раствором NH4OH
с концентрацией 60 - 80 г/л в присутствии (NH4)2CO3.
Выщелачивание проводилось при температуре 60 - 80 0С, при
соотношении Т:Ж=1:3-1:4 в течении 60 - 90 минут. Степень извлечения Мо
составляет соответственно 95-98%. Раствор после выщелачивания имеет следующий
состав: Мо - 10,5 г/л, Ni
- 1,9 г/л, NH3
- 40
г/л, СО2 - 25 г/л. Далее осадок фильтруется, из раствора отгоняется NH3
методом упарки. Ni
извлекается сорбцией на карбоксильном катионите типа КБ. Сорбированный Ni
десорбируют раствором состава: NН3
- 100г/л, СО2 - 60г/л. Десорбат упаривают до выделения никеля
в виде основного карбоната. Молибденсодержащий раствор после извлечения никеля
упаривают в пять раз, и из этого раствора выделяют Мо в виде парамолибдата
аммония (ПМА).
Предварительные исследования
безобживого варианта, проведённые с катализатором, где Мо содержится 50,4%,
показали, что он достаточно полно разлагается 25% раствором щёлочи NaOH
около 315 г/л при Т:Ж = 1:6, температура 140 0С в течение 6 часов.
Дополнительные исследования показали, что
температуру выщелачивания можно снизить до 90 - 100 0С при
увеличении длительности процесса до 10 - 12 часов и добиться той же степени
извлечения при 15% концентрации щёлочи в растворе; кроме того, вместо последней
можно использовать соду.
Что касается никеля и кобальта, то из
катализаторов аммиачными растворами эти металлы выщелачиваются не более чем на ~
10 - 59%. Дополнительные опыты показали, что введение в выщелачивающие растворы
окислителей (персульфата калия, пероксида водорода) или проведение
выщелачивания при пропускании через смеси катализатор-раствор воздуха не
приводит к заметному изменению степени выщелачивания никеля и кобальта (в
пределах погрешности анализа). Растворы кислот (серной или азотной)
выщелачивают никель несколько полнее, но в условиях проведения данных
экспериментов, не более чем на 22,5%. При этом в кислые растворы (в отличие от
щелочных) переходит заметное количество основы катализатора -
оксида алюминия. Подобная инертность никеля и кобальта может быть обусловлена
только тем, что большая часть этих металлов, находящихся в составе
катализаторов, присутствует в виде никелевой или кобальтовой шпинелей. Та их
небольшая часть, которая присутствует в виде оксидов или молибдатов,
растворяется в аммиачных растворах. Никель и кобальт, находящиеся в форме
шпинелей, химически инертны и не выщелачиваются аммиачными растворами. Там, где
основой катализатора является оксид кремния, никель аммиачным раствором
выщелачивается заметно более полно. Более высокая степень выщелачивания никеля
растворами кислот обусловлена тем, что кислоты растворяют и часть основы -
оксида алюминия, в том числе, находящегося в составе никелевой шпинели. Было
показано, что путем обработки катализаторов растворами кислот, серной или
азотной, относительно полное выщелачивание никеля и кобальта может быть
достигнуто только после растворения большей части носителя -
оксида алюминия. В связи с этим представляется целесообразным ограничиться
избирательным выщелачивание из катализаторов молибдена, а катализаторы после
выделения из них молибдена передавать на специализированные предприятия,
специализирующиеся в области переработки никель-кобальтового сырья.
Что касается состава растворов для
выщелачивания, то, если ограничиться извлечением молибдена, естественно,
следует предпочесть растворы соды [7].
Сода является значительно более эффективным
реагентом. На основе данных выбраны следующие режимы выщелачивания катализатора
(АМК) 10%_ным раствором Na2CO3, Т:Ж = 1 : 4, t =
80 ÷ 95 оС,
продолжительность 1_й стадии 6 ч, 2_й - 4 ч, максимальное извлечение молибдена
в раствор составляет 92,7%. При выщелачивании отработанного катализатора
раствором соды, жидкая фаза содержит 8-11 г/л молибдена. Такой раствор может
быть неоднократно использован для выщелачивания новых порций катализатора, а
далее переработан путем осаждения молибдена в составе молибдата кальция или
направлен после подкисления до pH = 2,0 ÷ 2,5 на
сорбцию Мо анионитами.
В этой работе мы предлагаем
использовать выщелачивание серной кислотой с полным растворением основы АКМ
катализаторов А12О3. Где ценные элементы будут полностью
переходить в раствор. Молибден предположительно в форме Н4Мо8О26,
а кобальт в виде красного СоSO4
в безводном состоянии [8].
В ходе исследований предложен
новый экологически чистый способ извлечения металла из отработанных
катализаторов и получения ценного сырья для различных наукоёмких отраслей
промышленности.
Согласно литературным данным
основа катализаторов - оксид алюминия является трудно растворимым как в
кислотах, так и в щелочах. Процесс растворения основы, в частности, в соляной,
серной и смеси кислот протекает в течение нескольких часов даже при
температурах близких к 100 оС. Скорость процесса растворения можно
увеличить путем повышения концентрации исходных реагентов, но этот вариант
приводит к дополнительным затратам, так как большой избыток как кислоты так и
щелочи теряется безвозвратно в процессе корректировки раствора перед
последующей технологической операцией, связанной с извлечение и
концентрированием ценных компонентов катализатора.
Более целесообразно так
осуществлять выщелачивание, чтобы эффект, обусловленный повышением концентрации
реагента, получать без увеличения его расхода. Анализ изменения концентрации
реагента и скорости по мере протекания процесса показывает, как это сделать. По
мере увеличения степени выщелачивания концентрация реагента линейно убывает и
одновременно уменьшается поверхность твердых частиц, что приводит к
существенному снижению скорости. Увеличить скорость процесса растворения можно
либо за счет увеличения избытка реагентов, либо это можно обеспечить без
повышения общего расхода реагента - переходом от одностадийного процесса к
противоточному двухстадийному, схема которого приведена на рисунке 1[14].
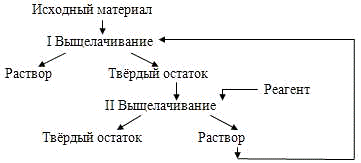
Рисунок 1 - Схема
двухстадийного противоточного процесса выщелачивания
При подаче всего реагента на
вторую стадию выщелачивания уменьшение поверхности твердых частиц
компенсируется повышением концентрации реагента, а в случае обратимой реакции -
также понижением содержания растворимого продукта. Благодаря этому скорость на
заключительном этапе выщелачивания гораздо выше и продолжительность его гораздо
меньше, чем при одностадийном выщелачивании. В результате, хотя скорость
начальной стадии несколько ниже, а ее продолжительность немного больше, чем при
одностадийном процессе, в целом продолжительность полного выщелачивания
значительно сокращается.
При осуществлении выщелачивания
в непрерывном режиме в каскаде реакторов переход от прямоточного к
двухстадийному противоточному варианту позволяет заметно повысить извлечение
ценного компонента в раствор или - при том же извлечении - увеличить
производительность каскада.
Очевидно, что дальнейшее
увеличение числа ступеней противоточного выщелачивания должно сопровождаться
дополнительным сокращением суммарного времени. Однако при осуществлении
процесса в каскаде аппаратов после каждой ступени требуется отделять раствор от
твердого остатка (фильтрацией или отстаиванием и декантацией), что усложняет
процесс. Поэтому обычно ограничиваются двумя стадиями противоточного
выщелачивания [14].
2.2 Способы выделения ценных
компонентов из растворов
2.2.1 Сорбционное извлечение молибдена, никеля,
кобальта
Растворы после утилизации отработанных
катализаторов направляют на дальнейшее выделение из них ценных компонентов.
Соединения могут быть селективно выделены ионным обменом или экстракцией.
Сорбционные методы концентрирования и разделения
неорганических ионов занимают одно из ведущих мест в химическом анализе.
Известные сорбенты можно разделить на две большие группы: неорганические
вещества - алюмосиликаты, оксиды, сульфиды; соединения на основе органических
полимеров, содержащие ионообменные или комплексообразующие группы. На стыке
этих двух групп находятся сорбенты с неорганической основой с нанесённым на
поверхность органическим веществом - модификатором. Интерес к ним вызван
специфическими свойствами последних: неорганический каркас придаёт им такие
свойства, как высокая скорость установления сорбционного равновесия (в случае
широкопористых носителей), ненабухаемость, термическая и радиационная
устойчивость, механическая прочность; органический модификатор обеспечивает
селективность и полноту связывания ионов. Сравнительно давно используются
сорбенты с адсорбционными связями между модификатором и носителем. Слабая фиксация
органического вещества на поверхности носителя и как следствие этого вымывание
его при контакте с раствором являются основным недостатком последних. Эта
проблема решается использованием кремнезёмных сорбентов с ковалентно
закреплёнными лигандами. Однако из-за сложности их синтеза они не получили
широкого распространения. Значительные перспективы для сорбции и
концентрирования ионов различных металлов открывают минерально-органические
материалы на основе природных алюмосиликатов [15].
Процессы ионного обмена самостоятельно стали
рассматриваться в начале 30-х годов. Они относятся к сорбционным процессам, а
точнее к хемосорбции. Это процесс взаимодействия твёрдой фазы с жидкой, причём
в качестве твёрдой фазы (сорбента) применяется ионит - специально полученный
полимер, имеющий форму шариков и характеризуемый наличием электронных связей,
которые определяют свойства ионита. Сорбируемое вещество, содержащееся в
жидкости, путём диффузии проникает в структуру матрицы ионита и удерживается на
ней за счёт образования химической связи. Поэтому процесс называется
хемосорбцией. Тип образуемой химической связи - ионный, отсюда название смолы
[16].
Было установлено [17], что многообразие форм
молибдена(VI)
обусловлено не только общей концентрацией металла и рН раствора, но и его
солевым составом. Для примера: при рН 6,5 - 1 ионное состояние молибдена
определяется концентрацией ионов водорода и не зависит от типа аниона
минеральной кислоты. Подтверждением этому может служить, поглощение
отечественным анионитом АВ-17-10П ионов молибдена из подкисленных различными
минеральными кислотами растворов с рН 2,5.
Обмен молибдена на АВ-17-10П в зависимости от рН
среды можно представить следующими уравнениями:
рН>8 2RCl+
MoO4-2 ↔R2MoO4 +2Cl-
pH 6-3.5 6RCl+
Mo7O24-6↔ R6Mo7O24
+6Cl-
4RCl+ Mo8O26-4↔
R4Mo8O26 +4Cl-
pH<3.5 4RCl+ Mo8O26-4↔
R4Mo8O26 +4Cl-
При изменении концентрации молибдена от 5·10-2
до 10-4 моль/дм³, но
при постоянном рН 5,5 тип сорбируемого аниона изменяется от Mo7O24-6
до MoO4-2.
В растворах с концентрацией молибдена в пределах 5·10-4-10-3 моль/дм³
доминирует
мономерная форма MoO4-2.
2.2.2 Экстракционные методы
Экстракция получила широкое применение в
технологии редких металлов для разделения близких по свойствам элементов. Так
молибден и вольфрам могут разделяться при экстракции ацетофеноном. Экстракция
неорганических веществ обусловлена химическим взаимодействием растворённого
вещества и растворителя.
Экстрагенты могут быть разделены на три большие
группы в зависимости от того, действуют ли они по катионообменному принципу
(например, карбоновые кислоты), анионообменному (например, амины) или за счёт
образования аддуктов (типично для эфиров и нейтральных фосфор органических
соединений) [18].
Молибден, кобальт и никель могут
экстрагироваться разными экстрагентами.
Например, для регенерации кобальта из вторичных
отходов и получения его в форме, пригодной для повторного использования в
производстве твёрдых сплавов, разработаны технологические схемы. Большинство
предприятий твёрдосплавной промышленности применяют солянокислое растворение
вторичных отходов, но Узбекистанский комбинат тугоплавких и жаропрочных
металлов использует технологию разделения металлов в азотнокислых растворах.
При растворении вторичных отходов в соляной кислоте кобальт и все примеси
переходят в раствор. Анионные хлоридные комплексы кобальта, железа, меди,
марганца могут быть извлечены в органическую фазу анионообменными экстрагентами
- солянокислыми солями третичных алкиламинов (ТАА.HCl)
и солями ЧАО. Для азотнокислых растворов необходимы другие экстрагенты.
Предлагается селективная экстракция меди и железа карбоновыми кислотами и в
последующей селективной экстракции кобальта роданидом ЧАО [19].
Рудные концентраты и вторичное сырьё часто
содержат значительные количества молибдена, который попутно извлекается при
получении паравольфрамата аммония.
Эффективным реагентом является перекись
водорода, образующая с вольфрамом и молибденом прочные пероксоанионы и в то же
время не вносящая в растворы никаких примесей, способных перейти в конечную
продукцию.
В содержащих перекись водорода растворах
молибден преимущественно присутствует в составе дипероксоанионов МоО62-,
а вольфрам в составе монопероксида МоО62-. Из таких
растворов нейтральными экстрагентами (например, ЧАО) избирательно
экстрагируется молибден, причём обеспечивается глубокая очистка вольфрама от
молибдена.
При экстракции молибдена и вольфрама ТБФ из
смешанного раствора, содержащего Н2О2 и подкислённого HNO3,
H2SO4
или HCl до
заданного рН, значения DMo
и DW отличаются на
порядок и более (β=
30-50).
Экстракционная способность в отношении молибдена
возрастает в ряду фосфат - фосфонат - фосфиноксид. Так при одинаковых значениях
рН DMo при
экстракции алкилфосфонатом в 3-4 раза выше, чем при экстракции ТБФ. Ещё выше
показатели экстракции ТИАФО. Раствор ТИАФО (2-3%-ный) обеспечивает такие же
коэффициенты распределения молибдена, какие получены при экстракции
неразбавленным ТБФ при значительно меньшей кислотности (рН 1.5), при этом
коэффициент разделения Мо/W
составляет 350. Однако, вследствие низкой концентрации ТИАФО в органической
фазе значительная доля его оказывается связанной даже при сравнительно
небольшом количестве извлекаемого молибдена. Поэтому изотерма экстракции
нелинейна, коэффициент распределения быстро понижается с увеличением
концентрации молибдена и экстракцию ТИАФО рекомендуют для растворов с
содержанием молибдена не более 2-3 г/л.
Причины различий в экстракции молибдена фосфор
органическими экстрагентами из азотнокислых и сернокислых растворов, содержащих
перекись водорода. При экстракции молибдена из азотнокислых растворов
фосфонатом зависимость DMo
- рН проходит через максимум при рН ~ 1.3, тогда как в случае сернокислых
растворов максимума нет, но наблюдается перегиб, после которого DMo
возрастает с увеличением кислотности.
Экстракция молибдена из растворов, содержащих
перекись водорода, протекает по гидратно-сольватному механизму с образованием [H3O.mH2O
nS] HMoO6.
Для ТБФ
и
ДАМФ
n = 3, для
ТИАФО
n = 2. Исходя
из состава экстрагируемого комплекса, запишем реакции:
МоО62-
+ 2Н+
+ 4H2O + nS [
H3O. 3H2O. nS ] HMoO6,
HМоО6
--
+ Н+
+ 4H2O + nS [ H3O. 3H2O.
nS ] HMoO6,
H2МоО6
+ 4H2O + nS [ H3O.
3H2O. nS ] HMoO6.
C целью получения
молибденового продукта с низким содержанием примеси вольфрама можно
использовать селективную экстракцию Д2ЭГФК. Основная часть молибдена (70-80%)
извлекается в органическую фазу. Водный раствор, содержащий вольфрам и
оставшийся молибден, может быть возвращён на разделение экстракцией [20].
3. ЦЕЛИ И ЗАДАЧИ РАБОТЫ
Целью настоящей работы явилось разработка
принципиальной технологии извлечения молибдена и кобальта из дезактивированных
катализаторов гидрообработки нефти, основанной на разложении катализаторов
серной кислотой с последующим извлечением с помощью методов ионного обмена, и
её экспериментальная проверка.
Для достижения данной цели необходимо было
решить следующие задачи:
. Найти оптимальные условия перевода
ценных компонентов из катализаторов на основе оксида алюминия в раствор и
оценить полноту их извлечения;
. Исследовать сорбцию молибдена и кобальта
на ионообменниках различных классов в поисках подходящего ионита для разделения
и максимального извлечения этих элементов;
. Подобрать условия десорбции молибдена и
кобальта из селективных ионитов;
. Разработать принципиальную
технологическую схему процесса переработки молибденкобальтовых катализаторов и
провести лабораторные испытания предлагаемого процесса.
4.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
4.1 Методика проведения
экспериментов
.1.1 Материалы и их подготовка
Образцы используемые в работе
кобальтсодержащих катализаторов были прокалены при температуре 500 °С, т. е.
при температурах, при которых еще не должно было происходить образование
соответственно кобальтовых шпинелей, и не будет возгоняться триоксид молибдена.
С использованием метода эмиссионного спектрального анализа и стандартных
химических методов анализа ранее на кафедре была произведена оценка содержания
ценных и примесных компонентов в образцах катализаторов.
Итак, Мо - Со катализатор,
цилиндрической формы синего цвета, содержал: 7% Мо, < 0.05% Ni,
3.2% Co, < 0.02%
Si, < 0.01% Cu
и < 0.01% Fe, содержание
V не превышало
0.01%.
Важно, что в составе образца
катализатора отсутствовал ванадий, зачастую попадающий в катализаторы из
нефтепродуктов.
В работе использовались
ионообменные смолы зарубежных фирм Purolite,
Dowex, которые представлены в таблице 1.
Таблица 1 - Характеристики
ионитов
|
ионит
|
матрица
|
Функциональные
группы
|
структура
|
|
Purolite А-100
|
Стиролдивинилбензол
|
-N(CH3)2;
-N(CH3)3
|
Макропористый
|
|
Dowex
М-4195
|
Стирольная
|
C6H5N / ─N \ C6H5N
|
Макропористый
|
|
S-930
|
стиролдивинилбензол
|
─N(CH2COOH)2
|
Макропористый
|
|
S-950
|
стиролдивинилбензол
|
─NHC2H4PO3H2
|
Макропористый
|
|
D-5144
|
акрилатная
|
-CONHC2H4NHC2H4NHCH2COOH; -CONHC2H4NCH2COOHC2H4NHCH2COOH; -CONHC2H4NHC2H4N(CH2COOH)2;
-CONHC2H4NCH2COOHC2H4N(CH2COOH)2
|
гелевый
|
|
S-957
|
стиролдивинилбензол
|
─СН(РО3Н2)2
= С(РО3Н2)2 ─SO3H
|
Макропористый
|
В работе использовались
реактивы квалификации ч., чда и тех. Исходные растворы готовились растворением
точно взятых навесок, рабочие растворы - разбавлением исходных растворов.
.1.2 Условия проведения
экспериментов
Эксперименты по изучению
процесса растворения отработанных катализаторов в растворе серной кислоты
проводили в стеклянной колбе, с перемешивающим устройством.
В колбу объемом 250 мл засыпали
навеску катализатора 5 г, который заливали 45,5%-ным раствором серной кислоты в
количестве 21,2 мл по стехиометрии. Пульпу нагревали на водяной бане
(температуру пульпы поддерживали в пределах 90 - 95 °С на протяжении всего
времени растворения). В процессе растворения катализатора пульпа постоянно
перемешивалась.
После окончания процесса
растворения раствор разбавляли дистиллированной водой с таким расчетом, чтобы
концентрация сульфата алюминия в нем была на уровне 200 г/л. Полученные
растворы осветляли преимущественно центрифугированием и анализировали на
содержание ценных компонентов.
Более подробные данные по
условиям проведения опытов приведены в разделе «Результаты и их обсуждение».
Сорбция и десорбция молибдена,
кобальта проводилась в статических и динамических условиях.
Опыты в статических условиях
проводились по ниже следующей методике. Навески сорбента 0.2, 0.4, 0.6 г в
пересчете на сухой сорбент помещали в колбы на 50 мл и заливали 15 мл раствора,
концентрация элементов и кислотность в растворах задавалась заранее, содержимое
колб периодически перемешивали. После установления равновесия проводили анализ
растворов. Сорбенты отделяли от проанализированных растворов, отфильтровывали,
промывали водой и заливали десорбирующими растворами, после чего определяли
концентрацию ценного компонента в десорбатах.
Емкость ионитов по извлекаемым
компонентам рассчитывали по разности исходной и равновесных концентраций
сорбируемого компонента с учетом навески ионита (в пересчете на массу сухого
ионита) и объема раствора.
Значения емкости сорбента
рассчитывали по уравнению:
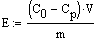 ,
,
где Е - емкость сорбента, мг/г;
Со и Ср - соответственно
начальная и равновесные концентрации сорбируемого компонента, мг/мл;
V-
объем
раствора, мл;
т - навеска
сухого сорбента, г.
Десорбцию сорбированных
компонентов из ионитов проводили следующим образом. Навески сорбентов после
насыщения отделяли от растворов, промывали водой, подсушивали на фильтре,
переносили в сухие колбы и заливали десорбирующими растворами. Десорбцию проводили
так же в течение 5-7 суток (после этого проводили анализ).
При проведении экспериментов в
динамических условиях использовали стеклянные колонны: объёмом 20 мл, в которые
загружали по 20 мл3 набухших ионитов: А100, М - 4195, S
- 930 и D - 5144. Отношение
высоты к диаметру у этих колонок составлял H
: D = 10 : 1.. На
избирательную сорбцию молибдена подавались последовательно модельный раствор с
концентрациями по молибдену 0,041М, по кобальту 0,03М и 200 г/л по Аl2(SO4)3.
Раствор, полученный после сорбции молибдена, имел значение рН = 0,7. В этот
раствор добавляли при перемешивании гидроокись натрия до установления рН = 2,7
и после этого направляли на сорбцию кобальта. Десорбцию молибдена проводили
раствором аммиака (1:1). Десорбцию кобальта проводили одномолярным раствором
серной кислоты. Скорость пропускания растворов при сорбции и десорбции
поддерживали в пределах 0.5 уд. об./ч..
Значения емкости сорбента
рассчитывали по уравнению:
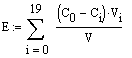 ,
,
где Е - емкость
сорбента, мг/мл;
Со и Сi
- соответственно начальная и на выходе из колонны концентрации сорбируемого
компонента, мг/мл;
Vi
- объем раствора,
мл;. |
V
- объем
сорбента в колонне, мл.
.1.3 Методы анализа
Для определения молибдена и
кобальта в растворе применялись фотоколориметрические методы анализа с
использованием калибровочных графиков.
Методика определения
молибдена[21].
Реактивы:
1) 2.5 мл 8.75 М раствора H2SO4.
2) 0.2 мл 2% раствора CuSO4;
) 2.5 мл раствора
тиомочевины (насыщенный);
) 1.5 мл 4 М раствора NH4CNS;
Ход определения.
В колбу, объёмом 25 мл,
отбирают пробу и разбавляют небольшим количеством воды. Затем при перемешивании
последовательно добавляют все реактивы. Доводят до метки водой, перемешивают и
выдерживают 30 минут. Оптическая плотность измеряется при длине волны λ
= 490 нм
в кюветах толщиной 0.5 см3.
Методика определения кобальта
[22].
Реактивы:
1) 2.5 мл 50% - раствор
ацетата натрия;
2) 1.5 мл нитрозо-R
соль (0,2% - раствор);
) 1.25 мл HNO3
(концентрированная).
Ход определения:
В колбу, объёмом 25 мл, отбирают
пробу и разбавляют небольшим количеством воды. Последовательно добавляют все
реактивы (после каждого добавления реактива колбы перемешивают и кипятят в
течение минуты). Доводят до метки водой, перемешивают и измеряли оптическую
плотность при длине волны λ = 540 нм
в кюветах толщиной 1 см3.
5. РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЯ
.1 Выщелачивания молибдена и кобальта из
катализатора на основе Al2O3
Молибден в отработанных
катализаторах может находиться частично в виде триоксида МоО3,
частично в виде молибдатов никеля NiMoO4
или кобальта СоMoO4
и, возможно, частично в виде молибдата алюминия Al2(MoO4)3,
никель и кобальт - в виде оксидов (NiO,
CoO, Co3O4),
в виде молибдатов и виде шпинелей NiAl2O4
и СоAl2O4
[5,
23]. Кобальтовая шпинель, т.н. тенардова синь, известна как драгоценный камень.
Шпинели представляют собой химически весьма инертные соединения, они трудно
растворимы [24, 25]. Никелевая шпинель образуется при нагревании смеси оксидов
алюминия и никеля при температуре свыше 450 оС, кобальтовая -
650 оС [23, 26-28]. В процессах каталитической гидрообработки
нефтяного сырья и регенерации катализаторов температура может достигать 600 оС
и выше, поэтому в отработанных катализаторах существенная часть никеля и
кобальта может находиться в виде шпинелей. Доля этих металлов, находящихся в
виде шпинелей, будет зависеть от условий эксплуатации и регенерации
катализаторов.
Применение в качестве выщелачивающего реагента
аммиачных растворов солей аммония при переработке катализаторов на основе
оксида алюминия позволяет относительно полно извлечь в растворы только
молибден, но не кобальт и никель. Выщелачивание кобальта и никеля с помощью
аммиачных растворов возможно только из катализаторов на основе кремнезема.
Добиться одновременного перевода в раствор
молибдена и цветных металлов, содержащихся в катализаторах на основе оксида
алюминия, можно только путем полного разложения катализаторов растворами
кислот, предпочтительно, растворами серной кислоты. С учетом цели данной
работы, было принято решение выбрать это направление.
.1.1 Отработка процесса разложения катализаторов
на основе оксида алюминия растворами серной кислоты
Основной задачей этого этапа работ явилась
минимизация расхода серной кислоты при разложении катализаторов растворами
серной кислоты и, тем самым, снижение ее остаточной концентрации в растворах,
что в последующем должно было бы упростить задачу извлечения из полученных
растворов ценных компонентов: молибдена и кобальта. Необходимым представлялось
также рассмотреть кинетику выщелачивания и выяснение необходимости
предварительного измельчения катализатора. Эта часть экспериментов проводилась
преимущественно с использованием молибденокобальтового катализатора 3 как
наиболее «упорного» из всех опробованных образцов.
Реакция разложения основы катализатора -
оксида алюминии описывается уравнением
Al2O3 + 3H2SO4
Û Al2(SO4)3
+ 3H2O, (1)
а реакции перевода в раствор активных
компонентов - упрощенно реакциями
2MoO3 + 2H2SO4
Û H2Mo2O5(SO4)2
+ H2O (2)
и СoO
+ H2SO4 Û
CoSO4 + H2O. (3)
Понятно, что это упрощенная форма записи
протекающих реакций. В действительности кобальт и никель, находящиеся в форме
шпинелей, взаимодействуют с серной кислотой по уравнению
MeAl2O4 + 4H2SO4
Û MeSO4 + Al2(SO4)3
+ 4H2O, (4)
а кобальт, находящийся в форме Co3O4,
по
уравнению
Co3O4 + 3H2SO4
Û 3CoSO4 + 3H2O
+ ½ О2.
(5)
Поскольку основная часть серной кислоты идет на
разложение основы - оксида алюминии, дальнейший расчет
количества серной кислоты, которая идет на разложение катализаторов, с целью
упрощения проводился только с учетом уравнения реакции (1). В соответствии с
этим, стехиометрический расход серной кислоты на разложение катализатора должен
составить 3 моль/моль Al2O3
или ~
30 моль/кг катализатора. Естественно, для полного разложения катализатора
необходим определенный избыток серной кислоты над стехиометрически необходимым
количеством.
Были проведены следующие эксперименты.
Предварительно было проведено одностадийное
выщелачивание молибдена, кобальта растворами серной кислоты с концентрациями
12,7% и 25,4%. Ранее было установлено, что при использовании раствора серной
кислоты с концентрацией 45% для одностадийного растворения
молибден-кобальтового катализатора получаются довольно концентрированные
растворы сульфата алюминия, которые при незначительном охлаждении раствора моментально
кристаллизуются. Этот факт затрудняет использование двухстадийной противоточной
схемы выщелачивания катализатора, которая, как известно, имеет ряд преимуществ
по сравнению с одностадийным выщелачиванием, в частности, она позволяет в более
полной мере использовать концентрацию выщелачивающего агента - серной кислоты,
и тем самым получить конечные растворы сульфата алюминия с более высокими
значениями рН.
Итак, для одностадийного выщелачивания было
проведено два эксперимента. В первом, для растворения 5г катализатора
необходимо 75,9 мл раствора серной кислоты с концентрацией 12,7%. Во втором для
такого же количества катализатора было взято 37,9 мл раствора серной кислоты с
концентрацией 25,4%. Эксперимент проводился при стехиометрически необходимом количестве
раствора серной кислоты. В обоих экспериментах брали одинаковое количество
катализатора и одно и то же количество серной кислоты. Пульпу нагревали на
водяной бане (температура пульпы 95 0С) при постоянном
перемешивании. В течение всего процесса отбирали пробы объемом 0,2 мл,
анализировали на содержание кобальта и рассчитывали степень извлечения кобальта
из катализатора в раствор. Результаты первого эксперимента приведены в таблице
2 и на рисунке 2, а второго, в таблице 3 и на рисунке 2.
Растворение основы катализатора - оксида
алюминия в 12,7% и 25,4% растворах серной кислоты проходило в течении примерно
4,5 часов. По окончании растворения горячие растворы сульфата алюминия
разбавляли водой до получения концентрации по сульфату алюминия 200 г/л, осветляли
центрифугированием. Полученные таким образом растворы имели рН = 0,20 -
0,50, что свидетельствует о небольшом избытке кислоты.
Таблица 2 - Зависимость содержания кобальта в
растворе от времени в процессе растворения катализатора в 12,7%-ном растворе
серной кислоты
|
Время
отбора пробы, мин
|
С,
мг/мл
|
Масса
кобальта, мг
|
Извлечение
Е, %
|
|
5
|
0,58
|
43,5
|
24,7
|
|
15
|
0,62
|
46,5
|
31,5
|
|
25
|
0,80
|
60,0
|
40,7
|
|
35
|
0,96
|
72,0
|
44,8
|
|
45
|
1,00
|
75,0
|
50,8
|
|
65
|
1,20
|
90,0
|
61,0
|
|
85
|
1,32
|
99,0
|
67,1
|
|
115
|
1,40
|
105,0
|
71,2
|
|
145
|
1,51
|
110,6
|
75,0
|
|
205
|
1,56
|
117,0
|
79,3
|
|
265
|
1,71
|
128,3
|
87,0
|
Таблица 3 - Зависимость содержания кобальта в
растворе от времени в процессе растворения катализатора в 25,4%-ном растворе
серной кислоты
|
Время
отбора пробы, минС, мг/млМасса кобальта, мг Извлечение Е, %
|
|
|
|
|
10
|
1,08
|
41,1
|
27,8
|
|
20
|
1,5
|
57,0
|
38,6
|
|
30
|
1,86
|
70,7
|
47,9
|
|
40
|
2,14
|
81,3
|
55,1
|
|
60
|
2,34
|
88,9
|
64,7
|
|
80
|
2,77
|
105,3
|
71,4
|
|
110
|
3,24
|
123,1
|
83,4
|
|
140
|
3,4
|
129,2
|
87,6
|
|
200
|
3,74
|
142,1
|
96,0
|
|
260
|
3,86
|
99,4
|
Таким образом, можно сказать, что с увеличением
концентрации серной кислоты, используемой для растворения, скорость растворения
катализатора увеличивается.
Также было проведено два эксперимента в режиме
противотока с концентрациями по серной кислоте 12,7% и 25,4%. К навеске
катализатора (измельченного) массой 2 г добавляли раствор серной кислоты с
концентрацией 45%. Пульпу нагревали на водяной бане (температура пульпы 95 0С),
при постоянном перемешивании механической мешалкой. Через полчаса 2 г
катализатора растворились, и к полученному раствору добавили ещё 5 г
катализатора. Уже через час после этого рН растворов был равен около 0,50 (для
первого эксперимента) и 0,40 (для второго), что говорит о неполном
израсходовании серной кислоты в растворе.
Дальнейший контакт раствора с избытком
катализатора при перемешивании пульпы в течение часа при температуре 95 0С
приводит к постепенному повышению значения рН раствора до 0,8 - 0,9. Получить
растворы с рН более 0.9 при контакте раствора сульфата алюминия с избытком
катализатора в данном эксперименте нам не удалось.
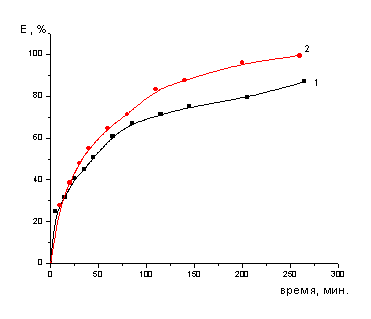
Рисунок 2 - Зависимость степени
извлечения кобальта из катализатора от времени в процессе растворения
катализатора серной кислотой с концентрацией 12,7% (1) и 25,4% (2)
(одностадийное выщелачивание)
Получить растворы с большими
значениями рН нам удалось лишь в случае повторного контакта полученного
раствора сульфата алюминия с рН = 0,5 с большим избытком свежей порции
катализатора (5 г) при нагревании пульпы на водяной бане и при перемешивании в
течение 1 часа. Раствор после выщелачивания отделяли от осадка сначала
декантацией, а затем осветляли его центрифугированием. Результаты экспериментов
приведены в таблицах 4, 5 и на рисунке 3.
Таблица 4 - Зависимость содержания
кобальта в растворе от времени в процессе растворения катализатора в 12,7%-ном
растворе серной кислоты (двухстадийное выщелачивание)
|
Время
отбора пробы, мин
|
С,
г/л
|
Масса
кобальта, мг
|
Извлечение
Е, %
|
|
10
|
0,42
|
31,9
|
19,2
|
|
20
|
0,47
|
35,7
|
24,2
|
|
40
|
0,60
|
45,5
|
30,8
|
|
60
|
0,66
|
50,1
|
34,4
|
|
90
|
0,75
|
56,9
|
38,6
|
|
100
|
0,84
|
63,7
|
43,2
|
|
110
|
1,15
|
87,2
|
59,1
|
|
120
|
1,40
|
106,2
|
72,0
|
|
130
|
1,53
|
116,1
|
78,7
|
|
140
|
1,60
|
121,4
|
82,3
|
|
160
|
1,64
|
124,4
|
84,3
|
|
180
|
1,70
|
129,0
|
87,4
|
|
210
|
1,92
|
145,7
|
98,7
|
|
240
|
1,94
|
147,3
|
99,9
|
Таблица 5 - Зависимость содержания кобальта в
растворе от времени в процессе растворения катализатора в 25,4%-ном растворе
серной кислоты (двухстадийное выщелачивание)
|
Время
отбора пробы, минС, г/лМасса кобальта, мгИзвлечение Е, %
|
|
|
|
|
10
|
0,75
|
28,5
|
19,3
|
|
20
|
0,98
|
37,24
|
25,2
|
|
30
|
1,2
|
45,6
|
30,9
|
|
50
|
1,42
|
53,96
|
36,6
|
|
60
|
2,86
|
108,68
|
73,7
|
|
70
|
3,08
|
117,04
|
79,3
|
|
80
|
3,38
|
128,44
|
87,1
|
|
90
|
3,44
|
130,72
|
88,6
|
|
110
|
3,5
|
132,9
|
90,1
|
|
130
|
3,59
|
136,4
|
92,5
|
|
160
|
3,68
|
139,84
|
94,8
|
Из представленных данных можно сделать вывод,
что скорость извлечения кобальта из катализатора в растворе серной кислоты с
концентрацией 25,4% существенно выше, чем в растворе серной кислоты с
концентрацией 12,7%.
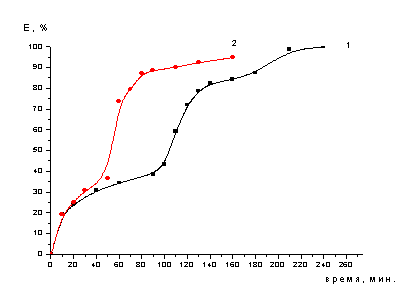
Рисунок 3 - Зависимость степени
извлечения кобальта из катализатора от времени в процессе растворения в серной
кислоте с концентрацией 12,7% (1) и 25,4% (2) (двухстадийное выщелачивание)
При сравнении результатов
экспериментов по растворению молибден-кобальтовых катализаторов в растворах
серной кислоты с разными концентрациями в прямотоке и противотоке становится
очевидным, что при проведении процесса в режиме противотока время растворения
сокращается более чем в 3 раза, при этом расход кислоты на выщелачивание
кобальта остался тем же, что и в режиме прямотока. Кроме того, режим
противоточного выщелачивания обеспечивает более высокие значения рН конечного
раствора сульфата алюминия, содержащего ценные компоненты, такие как молибден и
кобальт.
Был проведен еще один эксперимент.
В стакан с раствором серной
кислоты с концентрацией 45%, нагретым до температуры 95 0С на
водяной бане, засыпали навеску катализатора 5 г. Включалась лопастная мешалка.
Перемешивание производилось при постоянной скорости вращения мешалки, равной 60
об./мин. Через определенные промежутки времени производился отбор проб раствора
(горячей пипеткой) объемом 0,2 мл, которые анализировались на содержание
кобальта. Полностью осуществить процесс выщелачивания не удалось, так как во
время отбора проб раствор очень быстро кристаллизовался, что мешало для
дальнейшего анализа. Экспериментальные данные приведены на рисунке 4.
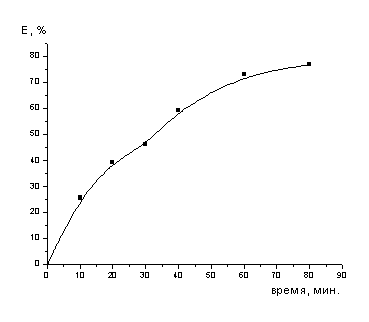
Рисунок 4- Зависимость
степени извлечения в раствор кобальта от времени контакта с 45%-ным раствором
серной кислоты
Из полученных данных следует, что добиться
полного перевода кобальта и молибдена в раствор можно только при полном
разложении основы катализатора - оксида алюминия, а для этого
необходимо брать серную кислоту в избытке по отношению к стехиометрически
необходимому количеству. Однако при этом образуются растворы с относительно
высокой кислотностью или, иными словами, имеющие низкие значения рН, что в
дальнейшем неминуемо вызовет существенные затруднения при решении задачи
выделения ценных компонентов из полученных растворов. Необходимо также иметь в
виду, что образующиеся в результате разложения катализаторов растворы во
избежание кристаллизации сульфата алюминия необходимо разбавлять до
концентрации по Al2(SO4)3
~
200 г/л.
Итак, в результате обработки катализатора 45%
раствором серной кислоты, получается концентрированный раствор по Al2(SO4)3
~
200 г/л, который при небольшом охлаждении кристаллизуется и затрудняет
дальнейший анализ.
5.1.2 Влияние предварительной обработки
катализатора раствором нитрата натрия перед прокаливанием на выщелачивание
кобальта и молибдена
В работах [29, 30] перед выщелачиванием ценных
компонентов из катализаторов предложено вначале обжигать катализаторы для
удаления органических веществ, затем пропитывать 3 моль/л раствором нитрата
натрия с добавкой нитрата железа 1 г/л, затем вновь термообрабатывать
катализаторы при температуре 580 оС, и лишь после этого обрабатывать
катализаторы раствором аммиака. Это, по мнению авторов, позволяет повысить
увеличивает степень извлечения молибдена на 30 - 39%.
Был произведен эксперимент с предварительной
обработкой катализатора 3 М раствором нитрата натрия. Навеску катализатора
помещали в стакан и заливали его 3М раствором нитрата натрия. Через полчаса,
методом декантации, отделили раствор от гранул. Обработанные гранулы высушивали
и затем термообрабатывали при температуре 500 0С в течении двух
часов. Далее проводилось одностадийное растворение катализатора в растворе
серной кислоты, с концентрацией 12,7%.
Навеску катализатора (пропитанного 3моль/л
раствором нитрата натрия) массой 5 г заливали 12,7% раствором серной кислоты
объемом равным 21,2 мл. Пульпу нагревали на водяной бане (температура пульпы 95
0С), при постоянном перемешивании механической мешалкой. В течение
всего процесса отбирали пробы объемом 0,2 мл, анализировали на содержание
кобальта и молибдена, и рассчитывали степень их извлечения из катализатора в
раствор. Результаты эксперимента для молибдена приведены на рисунке 5, а для
кобальта на рисунке 6.
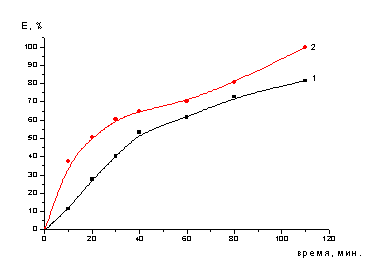
Рисунок 5 - Зависимость степени
извлечения молибдена из катализатора от времени в процессе растворения в 12,7%
-ном растворе серной кислоты (пропитанного NaNO3 (1) и
не пропитанного (2)) (одностадийное выщелачивание)
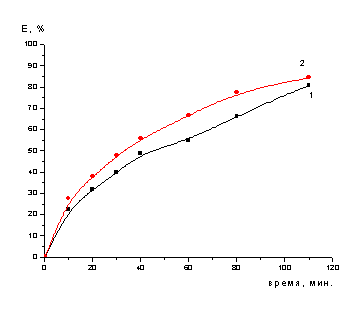
Рисунок 6 - Зависимость степени
извлечения кобальта из катализатора от времени в процессе растворения в 12,7%
-ном растворе серной кислоты (обработанного NaNO3 (1) и
необработанного (2)) (одностадийное выщелачивание)
Полученные в ходе выполнения
предварительных исследований результаты позволяют сделать следующий вывод, что
при термообработке катализатора (обработанного 3моль/л раствором нитрата
натрия) процесс выщелачивания протекает медленнее, чем без обработки.
.1.3 Осветление растворов сульфата
алюминия, полученных при растворении катализаторов в серной кислоте
Вследствие выщелачивания раствор
содержал мелкодисперсные частицы, которые мешали для дальнейшей работы с ним.
Разделение раствора от взвеси проводили при помощи фильтрования. Так как
плотность жидкой фазы пульпы была высокая, она плохо фильтровалась.
Так же был изучен метод расслаивания
пульпы. В пульпу добавляли полиакриламид (желейный раствор) с концентрацией
0,025% и определяли время расслаивания.
Таблица 6 - Определение скорости
расслаивания пульпы
|
Время,
мин
|
Высота
слоя осветленного раствора с ПАА, мм
|
Высота
слоя осветленного раствора без ПАА, мм
|
|
35
|
7
|
1,4
|
|
67
|
12
|
4,5
|
По результатам опыта обнаружено, что с помощью
добавления коагулянта - полиакриламида скорость расслаивания пульпы выше, чем
без его добавления. Однако, несмотря на это скорость осветления раствора
содержащего Al2(SO4)3
с концентрацией 200 г/л является очень низкой и поэтому, с целью увеличения
осветления раствора было использовано центрифугирование. Низкая скорость
расслаивания пульпы может быть связано, по видимому, с высокой плотность и
вязкостью ее водной фазы.
5.2 Сорбция молибдена в статических условиях из
раствора сульфата алюминия при различных рН
Разделение молибдена и кобальта эффективнее
проводить путем сорбции, так как иониты способны избирательно поглощать ионы
металлов из растворов солей. Основными требованиями, предъявляемыми к
сорбентам, являются у одних селективность к молибдену, у других - к кобальту. У
ионитов должны быть высокие емкостные характеристики и возможности
осуществления достаточно полной десорбции из них молибдена растворами аммиака,
а кобальта раствором серной кислоты с концентрацией 1 моль/л.
Известно, что рН растворов сильно влияет на
сорбцию различных элементов. Влияние на сорбцию может оказывать природа ионита,
состав растворов, состояние извлекаемых ионов и другие факторы.
Ранее на кафедре было установлено, что некоторые
иониты селективно способны сорбировать молибден и кобальт. Решено было изучить
сорбцию этих элементов на различных ионитах при различных условиях (различные
значения рН и концентрации сульфата алюминия). Сорбцию проводили в статических
условиях, масса ионита 0,2 г, а объем рабочего раствора составлял 15 мл.
.2.1 Сорбция молибдена из раствора сульфата
алюминия
Растворы, объёмом по 15 мл с содержанием: 0.048
моль/л (4,6 г/л) по Мо, 0.029 моль/л (1,7 г/л) по Со и Al2(SO4)3
с концентрацией 200 г/л и имеющие различные значения рН приводили в
контакт с 0.2 г ионита в пересчёте на сухой сорбент, выдерживали 3-4 суток до
установления равновесия и анализировали. По результатам анализа рассчитывали
емкость ионита при различных равновесных значения рН. Результаты эксперимента
представлены в виде графика на рисунке 7, а численные значения приведены в
таблице 7.
Таблица 7 - Зависимость емкости по молибдену на
анионите А100(Мо) от рН раствора сульфата алюминия
|
рНравн.
|
Сравн.,
г/л
|
Е,
мг/г
|
|
0.67
|
0.39
|
318.3
|
|
0.88
|
0.30
|
324.8
|
|
1.63
|
0.24
|
329.5
|
|
2.38
|
0.43
|
314.8
|
|
2.88
|
0.45
|
313.8
|
.2.2 Десорбция молибдена раствором гидроксида
натрия
После сорбции иониты отфильтровывали, промывали
водой и переносили в колбы. Десорбцию молибдена осуществляли 15 мл 1 моль/л NaOH.
Пробы выдерживали 3-4 суток при периодическом перемешивании. По
результатам анализа десорбатов рассчитывали емкость ионита. Результаты
эксперимента по десорбции молибдена приведены в таблице 8 и представлены в виде
графика на рисунке 7.
Таблица 8 - Зависимость емкости по молибдену на
анионите А100(Мо) от рН растворов
|
рНравн.
(при сорбции Мо)
|
СМо,
г/л (в десорбате)
|
ЕМо,
мг/г
|
|
0.67
|
4,2
|
312
|
|
0.88
|
4,0
|
300
|
|
1.63
|
3,8
|
288
|
|
2.38
|
3,8
|
285
|
|
2.88
|
3,7
|
279
|
Из рисунка 7 видно, что изменение значения рН от
0,7 до 2,9 не значительно сказывается на емкостных характеристиках ионита
А100(Мо), но прослеживается явная тенденция, что с увеличением рН раствора
емкость ионита уменьшается.

Рисунок 7 - Зависимость ёмкости по молибдену(Vl)
на анионите А100 от рН на фоне сульфата алюминия (по результатам сорбции - 1,
по результатам десорбции молибдена - 2)
.3 Сорбция кобальта в статических условиях из
раствора сульфата алюминия при различных значениях рН растворов
5.3.1 Сорбция кобальта в статических условиях из
раствора сульфата алюминия
Изучена сорбция кобальта на фоне сульфата
алюминия. Растворы, объёмом 15 мл, в состав которых входили: 0.048 моль/л (4,6
г/л) по Мо, 0.029 моль/л (1,7 г/л) по Со и Al2(SO4)3
с концентрацией 200 г/л и имеющие различные значения рН приводили в
контакт с 0.2 г ионита в пересчёте на сухой сорбент и направляли на
сорбцию, выдерживали 2 - 3 суток до установления равновесия и анализировали.
Вследствие того, что в растворах, находясь в контакте с ионитами D
- 5041, S - 930, D
- 5144, М - 4195, мало изменялась концентрация по кобальту, нам не
представилось возможным оценить емкостные характеристики этих сорбентов по
результатам сорбции.
Емкостные характеристики ионитов по кобальту
оценивали по результатам анализа десорбатов, полученных после обработке
насыщенных кобальтом ионитов раствором серной кислоты с концентрацией 1 моль/л.
.3.2 Десорбция кобальта в статических условиях
одномолярным раствором серной кислоты
Насыщенные иониты после сорбции отфильтровывали,
промывали водой и переносили в колбы. Десорбцию кобальта осуществляли 15 мл 1
моль/л раствором H2SO4.
Анализ проводили по методике, приведенной выше. Результаты эксперимента
представлены на рисунке 8, численные данные приведены в таблице 9.
Таблица 9 - Результаты по десорбция кобальта
1моль/л раствором серной кислоты
|
рНравн.
при сорбции Со
|
ССо,
г/л (в десорбате)
|
ЕСо,
мг/г (после десорбции)
|
|
D - 5041
|
|
0,72
|
менее
0.003
|
менее
0.1
|
|
0,96
|
менее
0.003
|
менее
0.1
|
|
1,57
|
менее
0.003
|
менее
0.1
|
|
2,06
|
менее
0.003
|
менее
0.1
|
|
2,88
|
менее
0.003
|
менее
0.1
|
|
S - 930
|
|
0,71
|
0.007
|
0.5
|
|
0,98
|
0.011
|
0.8
|
|
1,86
|
0.103
|
7.7
|
|
2,49
|
0.160
|
11.9
|
|
2,97
|
0.203
|
15.2
|
|
D - 5144
|
|
0,71
|
0.004
|
0.3
|
|
1,02
|
0.005
|
0.4
|
|
1,87
|
0.070
|
5.3
|
|
2,57
|
0.160
|
12.0
|
|
2,99
|
0.216
|
16.2
|
|
М
- 4195
|
|
0,66
|
0.013
|
1.0
|
|
0,94
|
0.019
|
1.4
|
|
1,7
|
0.193
|
14.4
|
|
2,25
|
0.587
|
44.0
|
|
2,92
|
0.758
|
56.9
|
Из данных таблицы 9 следует, что десорбция
кобальта прошла не в полной мере, причем ионит S
- 930 уступает D - 5144, М -
4195, D - 5041.
Рисунок 8 - Зависимость емкости ионитов D
- 5041(1), S - 930(2), D
- 5144(3), М - 4195(4) по кобальту от рН растворов
Из полученных данных по сорбции кобальта на
ионитах из растворов Al2(SO4)3,
при различных значениях рН можно сделать следующие выводы:
1) Ионит
М - 4195 лучше всего сорбирует кобальт из растворов с Al2(SO4)3,
чем иониты D - 5041(1), S
- 930(2), D - 5144(3); причем D
- 5041(1) практически не сорбирует кобальт в изученном диапазоне рН.
2) При
увеличении рН увеличивается емкость ионитов, и максимум соответствует рН ~ 2.9
(предельное значение рН для данного состав растворов).
В дальнейшем сорбция кобальта была изучена для
ионитов, S - 930, D
- 5144, М - 4195. Ионит D
- 5041, как бесперспективный, был исключен из дальнейшего исследования.
5.4 Сорбция и десорбция молибдена в статических
условиях
Вообще, ионообменная технология является
наиболее эффективной технологией извлечения молибдена из растворов, получаемых
в результате переработки рудных концентратов молибдена [31-33]. Заключается она
в сорбции молибдена на подходящем ионите с его последующей десорбцией раствором
аммиака и выделением в виде парамолибдата аммония. Ионообменная технология
обеспечивает высокое извлечение молибдена и требуемую чистоту конечного
продукта, поскольку в процессе сорбции происходит очистка молибдена от
посторонних компонентов.
Изотерма сорбции целевого компонента является
важной характеристикой сорбционного процесса. Она характеризует состояние
ионообменного равновесия при постоянной температуре, она связывает между собой
равновесные концентрации обменивающихся ионов в каждой из фаз и позволяет
судить о селективности ионита.
Из изотермы сорбции можно оценить емкость ионита
по целевому компоненту при той или иной концентрации последнего в растворе.
Постановка эксперимента: навески ионитов 0.1,
0.2, 0.3, 0.4, 0.6 и 0.8 г. в пересчёте на сухой сорбент заливали 15 мл
раствора, содержащего 0.094 моль/л (9 г/л) по Мо, 0.03 моль/л (1,7 г/л) по Со и
Al2(SO4)3
с концентрацией 200 г/л, рНисх. = 0,77. Раствор, при
периодическом перемешивании выдерживали неделю до установления равновесия и
анализировали. Результаты эксперимента приведены в таблице 10.
Таблица 10 - Сорбция молибдена на анионите
А100(Мо) в статических условиях
|
рНравн.
|
Сравн.,
г/л
|
Е,
мг/г
|
Е,
мг/см3
|
|
0.77
|
0.08
|
152.6
|
46,2
|
|
0.78
|
0.14
|
202.1
|
61,2
|
|
0.75
|
0.37
|
294.4
|
89,2
|
|
0.74
|
0.87
|
367.5
|
111,4
|
|
0.76
|
2.40
|
436.5
|
132,1
|
|
0.79
|
5.02
|
480.0
|
145,5
|
Ниже на рисунке 9 приведена изотерма сорбции
молибдена на анионите А100(Мо) из рабочего раствора.
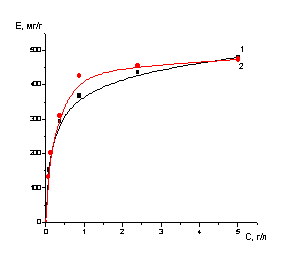
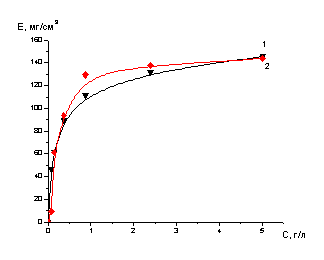
Рисунок 9 - Изотермы сорбции (по результатам
анализов сорбатов - 1, по результатам анализов десорбатов - 2) молибдена на
анионите А100(Мо) в статических условиях
Изотерма сорбции молибдена на анионите А100(Мо)
имеет выпуклую форму, что дает основание рассчитывать на то, что в процессе
сорбции на этом сорбенте будет происходить глубокое извлечение молибдена из
растворов до его низких остаточных концентраций. Емкость анионита по молибдену
достигает 480 г молибдена/г сухого анионита.
Далее в статических условиях были проведены
эксперименты по оценке полноты десорбции молибдена из насыщенного им до разных
значений емкости анионита А100(Мо) раствором аммиака 6 моль/л.
Методика проведения десорбции: навески
насыщенного ионита с массами 0.1, 0.2, 0.3, 0.4, 0.6 и 0.8 г отфильтровывали,
промывали водой, помещали в колбы и заливали раствором аммиака с концентрацией
6 моль/л. Пробы выдерживали в течении 3 - 4 суток при периодическом
перемешивании и анализировали. Молибден из анионита А100(Мо) десорбируется
полно, о чём свидетельствуют данные таблицы 11.
Таблица 11 - Десорбция молибдена из анионита в
статических условиях
|
ЕМо,
мг/г (после сорбции)
|
СМо,
г/л (в десорбате)
|
ЕМо,
мг/г (после десорбции)
|
ЕМо,
мг/см3
|
|
152.6
|
0.08
|
131.3
|
9,5
|
|
202.1
|
0.14
|
202.5
|
61,4
|
|
294.4
|
0.37
|
309.8
|
93,9
|
|
367.5
|
0.87
|
427.0
|
129,4
|
|
436.5
|
2.40
|
454.5
|
137,7
|
|
480
|
5.02
|
474.0
|
143,6
|
Таким образом, вышеприведенные данные
свидетельствуют о том, что анионит А100(Мо), действительно, является весьма
эффективным сорбентом для извлечения молибдена и из растворов с высокой
концентрацией сульфата алюминия.
5.5 Сорбция и десорбция кобальта в статических
условиях
Поскольку кобальт в растворах после
сернокислотного разложения катализаторов находятся в катионной форме (хотя и
возможно образование в растворах ионных пар с сульфат-ионом типа CoSO4),
существенную конкуренцию при сорбции им может составить макрокомпонент -
алюминий, также находящийся в катионной форме. В связи с этим сульфокатиониты,
не проявляющие выраженную избирательность к ионам переходных металлов, к числу
которых принадлежат кобальт и никель, не могут быть использованы для их
извлечения. Для извлечения кобальта из таких растворов, в принципе, могут быть
использованы только комплексообразующие иониты, способные к избирательной
сорбции кобальта и никеля за счет связывания их в комплексные соединения со
своими функциональными группами. Однако функциональные группы большинства
комплексообразующих ионитов проявляют склонность к протонированию, а растворы
после сернокислотного разложения катализаторов имеют сравнительно низкие
значения рН.
При разработке процесса сорбционного извлечения
кобальта из растворов после сернокислотного разложения катализаторов были
опробованы комплексообразующие сорбенты: S
- 930 и S - 950
иминодикарбоксильными и аминофосфоновокислыми функциональными группами
соответственно; катионит
Возможность применения этих ионитов для
извлечения кобальта оценивалась путем снятия изотерм сорбции последних из
раствора с концентрацией по Аl2(SO4)3
200 г/л, имеющему рН 2,90 ± 0,03.
Постановка эксперимента: навески ионитов с
массами 0.2, 0.4, 0.6 г в пересчёте на сухой сорбент заливали 15 мл модельного
раствора, содержащего 0.048 моль/л (4,6 г/л) по Мо, 0.044 моль/л (2,6 г/л) по
Со и сульфат алюминия с концентрацией 200 г/л. Пробы выдерживали в течении 3 -
4 суток при периодическом перемешивании и анализировали. Результаты
эксперимента приведены в таблице 12.
Таблица 12 - Сорбция кобальта на ионитах S-930,
S-950, S-957,
D-5144, M-4195
в статических условиях
|
рНравн.
|
Сравн.,
г/л
|
Е,
мг/г
|
|
S - 930
|
|
2,78
|
0,16
|
10,35
|
|
2,8
|
0,25
|
12,38
|
|
2,83
|
0,45
|
14,22
|
|
2,85
|
1,20
|
18,72
|
|
2,83
|
1,41
|
19,26
|
|
2,93
|
2,03
|
22,36
|
|
S - 950
|
|
3,06
|
2,41
|
9,47
|
|
3,03
|
2,48
|
10,58
|
|
3,07
|
2,54
|
11,41
|
|
S - 957
|
|
2,39
|
2,22
|
6,38
|
|
2,40
|
2,34
|
7,22
|
|
2,53
|
2,43
|
8,01
|
|
D - 5144
|
|
2,86
|
0,32
|
6,50
|
|
2,85
|
0,37
|
8,06
|
|
2,82
|
0,46
|
8,89
|
|
2,80
|
1,03
|
18,15
|
|
2,85
|
1,17
|
21,75
|
|
2,83
|
1,49
|
26,28
|
|
3,01
|
1,77
|
31,13
|
|
2,99
|
2,13
|
35,25
|
|
M - 4195
|
|
2,83
|
0,149
|
40,03
|
|
2,86
|
0,356
|
52,28
|
|
2,8
|
0,464
|
53,40
|
|
2,84
|
0,854
|
65,48
|
|
2,93
|
1,53
|
80,25
|
|
2,94
|
1,99
|
91,50
|
На рисунке 10 представлены изотермы сорбции
кобальта на сорбентах S-930,
S-950, S-957,
D-5155, M-4195.
Вид кривых на рисунке 10 говорит о насыщении
ионообменных сорбентов кобальтом. Ёмкость по кобальту для ионита М - 4195 в два
раза больше, чем у остальных анионитов.
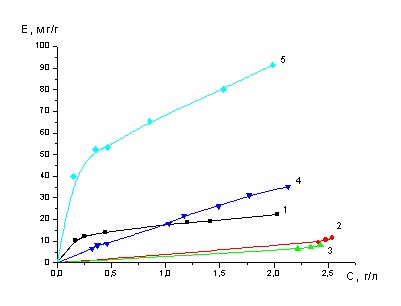
Рисунок 10 - Изотермы сорбции кобальта на
сорбентах S-930 (1), S-950
(2), S-957 (3), D-5144
(4), M-4195(5)
(представлены данные по результатам анализа сорбатов)
Далее, иониты, насыщенные кобальтом
отфильтровывали, промывали водой и десорбировали кобальт 15 мл 1моль/л
раствором серной кислоты. Пробы выдерживали в течении 3-4 суток при
периодическом перемешивании и анализировали. Данные по десорбции приведены в
таблице 13 и представлены на рисунке 11.
Таблица 13 - Десорбция кобальта в статических
условиях 1моль/л Н2SO4
|
ССо,
г/л
|
ЕСо,
мг/г
|
ЕСо,
мг/см3
|
|
S - 930
|
|
0,4
|
6,90
|
2,16
|
|
0,53
|
8,74
|
2,73
|
|
1,48
|
13,60
|
4,25
|
|
1,98
|
14,60
|
4,56
|
|
2,37
|
14,62
|
4,57
|
|
2,39
|
14,80
|
4,63
|
|
S - 950
|
|
2,55
|
1,19
|
0,39
|
|
2,57
|
1,20
|
0,40
|
|
2,58
|
1,29
|
0,43
|
|
S - 957
|
|
2,57
|
0,80
|
0,22
|
|
2,59
|
0,87
|
0,24
|
|
2,59
|
1,05
|
0,29
|
|
D - 5144
|
|
0,46
|
5,80
|
2,15
|
|
0,53
|
7,23
|
2,68
|
|
1,47
|
14,20
|
5,26
|
|
1,55
|
15,02
|
5,56
|
|
1,91
|
17,23
|
6,38
|
|
2,12
|
18,91
|
7,00
|
|
2,28
|
19,80
|
7,33
|
|
2,34
|
20,22
|
7,49
|
|
M - 4195
|
|
0,80
|
37,80
|
15,12
|
|
0,99
|
44,95
|
17,98
|
|
1,07
|
49,93
|
19,97
|
|
1,24
|
54,27
|
21,71
|
|
1,78
|
74,42
|
29,77
|
|
1,90
|
76,39
|
30,56
|
Из таблицы 13 видно, что десорбция кобальта с
ионита М - 4195 протекает лучше, чем c
остальных ионитов.
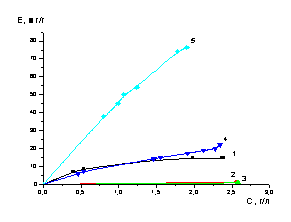
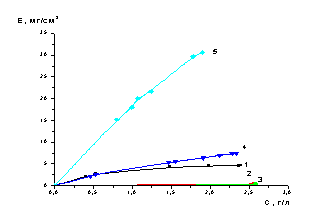
Рисунок 11 - Изотермы сорбции кобальта на
сорбентах S-930 (1), S-950
(2), S-957 (3), D-5144
(4), M-4195(5)
(полученные по результатам анализа десорбатов кобальта)
Результаты представленные в таблицах 12, 13 и на
рисунках 10, 11 показывают, что полученные данные по сорбции и десорбции
различаются. Изотермы, полученные по результатам сорбции кобальта показывают
более высокие значения емкости для всех ионитов, по сравнению значений емкостей
полученных по результатам десорбции. Этот факт, вероятно, связан с тем, что при
промывки водой насыщенного кобальтом ионита при соответствующей равновесной
концентрации происходит частичная десорбция.
Известно [34], что из всех синтезированных к
настоящему времени и выпускаемых в промышленном масштабе ионитов только ионит с
бис-пиколиламинными функциональными группами проявляет способность к сорбции
кобальта из слабокислых растворов. Нами было установлено, что этот ионит
эффективно сорбирует ионы кобальта и из растворов, содержащих сульфат алюминия,
по крайне мере до его концентрации 200 г/л.
Аниониты S-930
(1), D-5144 (4) также
сорбируют кобальт из растворов, содержащих Аl2(SO4)3,
но имеют емкость примерно в два раза меньше, чем М - 4195, однако их можно
использовать для извлечения кобальта. Учитывая высокую стоимость анионита Dowex
XFS 4195,
использование для извлечения кобальта ионитов S-930,
D-5144 может
оказаться экономически более выгодным.
.6 Сорбция и десорбция молибдена в динамических
условиях
Далее для оценки достигаемой степени извлечения
молибдена, степени его концентрирования и соотношения значений полной обменной
емкости и емкости до проскока были проведены эксперименты по сорбции и
десорбции молибдена и кобальта в динамических условиях на выбранных ионитах.
Для этого использовались одинаковые стеклянные сорбционные колонны: объёмом 20
мл, в которые загружали по 20 мл3 набухших ионитов: А100(Мо), М -
4195, S - 930 и D
- 5144. Отношение высоты к диаметру у этих колонок составлял H
: D = 10 : 1. На
избирательную сорбцию молибдена подавались последовательно модельный раствор с
концентрациями по молибдену 0,041 моль/л (3,98 г/л), по кобальту 0,03 моль/л
(1,76 г/л) и 200 г/л по Аl2(SO4)3
при рН 0.7. Растворы подавались снизу вверх со скоростью 0.5 уд. об./ч. При
сорбционном извлечении молибдена из растворов проводился отбор проб на выходе
из колонки, которые анализировали на содержание молибдена и кобальта. Рабочие
растворы пропускали до полного насыщения анионита, о чём свидетельствует
выравнивание концентраций молибдена на входе и выходе сорбционной колонны.
Выходная кривая сорбции молибдена на анионите А100(Мо) в динамических условиях
представлены на рисунке 12, а численные данные приведены в таблице 14. В
процессе сорбции молибдена(Vl)
концентрация кобальта в сорбатах была постоянной и практически соответствовала
исходной концентрации 0,03 моль/л..
Таблица 14 - Сорбция молибдена(Vl)
на анионите А100(Мо) в динамических условиях
|
V, мл об. пробы
|
∑V,
мл
об. пробы
|
V, кол. об.
|
Сравн.,
г/л
|
|
57
|
57,0
|
2,85
|
0
|
|
40
|
97,3
|
4,87
|
0
|
|
151
|
248,3
|
12,42
|
0
|
|
346
|
594,3
|
29,72
|
0,01
|
|
10,3
|
604,6
|
30,23
|
0,01
|
|
12
|
616,6
|
30,83
|
0,08
|
|
21,6
|
638,2
|
31,91
|
0,11
|
|
10
|
648,2
|
32,91
|
0,16
|
|
30
|
678,2
|
33,91
|
0,23
|
|
20
|
698,2
|
34,91
|
0,33
|
|
11
|
709,2
|
35,46
|
0,38
|
|
76
|
785,2
|
39,29
|
0,93
|
|
10
|
795,2
|
39,76
|
1,06
|
|
20
|
815,2
|
40,76
|
1,51
|
|
21
|
836,2
|
41,81
|
1,85
|
|
22
|
858,2
|
42,91
|
2,16
|
|
20
|
878,2
|
43,91
|
2,37
|
|
20
|
898,2
|
44,91
|
2,66
|
|
10
|
909,2
|
45,46
|
2,82
|
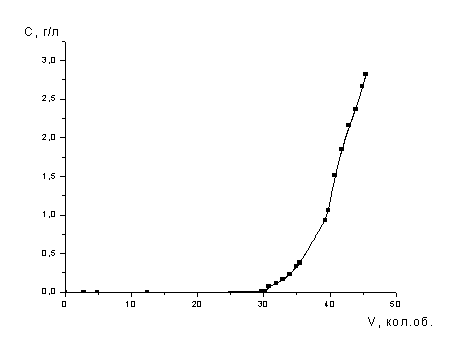
Рисунок 12 - Выходная кривая
сорбции молибдена на ионите А100(Мо)
Рисунок 12 демонстрирует высокую эффективность
сорбционного способа извлечения молибдена из растворов сложного состава при
использовании в качестве сорбента анионита А100(Мо). Следует отметить, что
кобальт практически не сорбируется, о чем свидетельствует изменение его
концентрации в первых объемах сорбата от 0 до исходной концентрации. Результат
обработки выходной кривой сорбции молибдена показал, что динамическая обменная
ёмкость для ионита соответствует 119,4 мг Мо/мл набухшего ионита, а полная
динамическая обменная емкость соответствует 165,4 мг Мо/мл набухшего ионита.
По окончании процесса сорбции молибдена(Vl)
колонку промывали водой (объем = 71мл). Отмывка сорбента проводилась до
практически полного удаления из фазы сорбента сульфата алюминия. Содержание
алюминия в промывных водах контролировали визуально по образованию осадка
гидроокиси алюминия. После чего проводили десорбцию. Десорбция молибдена проводилась
раствором аммиака с концентрацией = 6 моль/л. Скорость пропускания растворов
при десорбции составляла 0,5 уд. об./ч. На выходе из колонны контролировали
содержание молибдена. Полученные данные представлены в виде выходной кривой
десорбции молибдена на рисунке 13, численные данные приведены в таблице 15.
Таблица 15 - Десорбция молибдена с анионита
А100(Мо) раствором аммиака (6 моль/л) в динамических условиях
|
V, мл об. пробы∑V,
мл
об. пробыV, кол.
об.Сравн., г/л
|
|
|
|
|
5,5
|
5,5
|
0,275
|
0,056
|
|
5
|
10,5
|
0,53
|
19,28
|
|
5
|
15,5
|
0,775
|
59
|
|
5.5
|
21
|
1,05
|
130,5
|
|
5
|
26
|
1,3
|
120
|
|
5
|
31
|
1,55
|
98
|
|
6
|
37
|
1,85
|
70,75
|
|
5
|
42
|
2,1
|
57,77
|
|
5
|
47
|
2,35
|
44,75
|
|
5
|
52
|
2,6
|
36,4
|
|
5
|
57
|
2,85
|
25,4
|
|
5
|
62
|
3,1
|
17,6
|
|
5,5
|
67,5
|
3,375
|
13,3
|
|
5
|
72,5
|
3,625
|
9,45
|
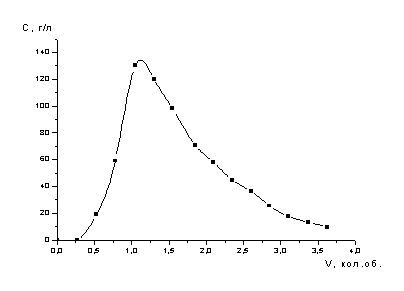
Рисунок 13 - Выходная кривая
десорбции молибдена(Vl)
Из данных представленных на рисунке
видно, что десорбция основного количества молибдена происходит 0,5 - 3,5
колоночными объёмами раствора аммиака, причём максимальная концентрация
молибдена составляет 130,5 г/л, что более чем в 30 раз больше по сравнению с
исходной концентрацией. Десорбат практически не содержит кобальт и алюминий.
Результат обработки выходной кривой сорбции молибдена показал, что из одного мл
насыщенного сорбента было десорбировано 181,2 мг Мо.
Проведенный эксперимент по сорбции и
десорбции молибдена показал, что полная динамическая обменная емкость анионита
А100 (Мо) по данным сорбции соответствует 165,4 мг/мл, а по данным десорбции -
181,2 мг/мл. Такое расхождение значений ПДОЕ, по-видимому, связано с
погрешностью эксперимента.
.7 Сорбция и десорбция кобальта в динамических
условиях
Раствор, полученный после сорбции молибдена,
имел значение рН = 0,7. В этот раствор добавляли при перемешивании гидроокись
натрия до установления рН = 2,7 и после этого направляли на сорбцию кобальта.
Раствор подавался снизу вверх со скоростью 0.5 уд. об./ч. При сорбционном
извлечении кобальта из растворов проводился отбор проб на выходе из колонки,
которые анализировали на содержание кобальта. Рабочий раствор пропускали до
полного насыщения ионита. Результаты экспериментов по сорбции кобальта в
динамических условиях на ионитах М-4195, S-930, D-5144
представлены ниже.
Выходная кривая сорбции кобальта на ионите М -
4915 в динамических условиях представлены на рисунке 14, численные значения в
таблице 16.
Таблица 16 - Сорбция кобальта на анионите М -
4915 в динамических условиях
|
V, мл об. пробы∑V,
мл
об. пробыV, кол.
об.Сравн., г/л
|
|
|
|
|
22
|
22
|
1,1
|
0
|
|
15
|
37
|
1,9
|
0
|
|
16
|
2,7
|
0
|
|
60
|
113
|
5,7
|
0,23
|
|
20
|
133
|
6,7
|
0,42
|
|
20
|
153
|
7,7
|
0,72
|
|
23
|
176
|
8,8
|
0,84
|
|
23
|
199
|
10,0
|
0,94
|
|
20
|
219
|
11,0
|
1,01
|
|
20
|
239
|
12,0
|
1,06
|
|
21
|
260
|
13,0
|
1,11
|
|
20
|
280
|
14,0
|
1,15
|
|
20
|
300
|
15,0
|
1,21
|
|
20
|
320
|
16,0
|
1,26
|
|
22
|
342
|
17,1
|
1,33
|
|
20
|
362
|
18,1
|
1,42
|
|
20
|
382
|
19,1
|
1,50
|
|
13
|
395
|
19,8
|
1,64
|
Результат обработки выходной кривой сорбции
кобальта показал, что динамическая обменная ёмкость для ионита соответствует
4,7 мг Со/мл набухшего ионита, а полная динамическая обменная емкость
соответствует 19,6 мг Со/мл набухшего ионита.
По изотерме сорбции кобальта на анионите М-4195,
равновесной концентрации кобальта 1,78 г/л соответствует емкость по кобальту
29,8 мг/мл (таблица 13 и рисунок 11).
Десорбция кобальта проводилась одномолярным
раствором серной кислоты. Скорость пропускания растворов при десорбции
колебалась в пределах 0,5 уд. об./ч.
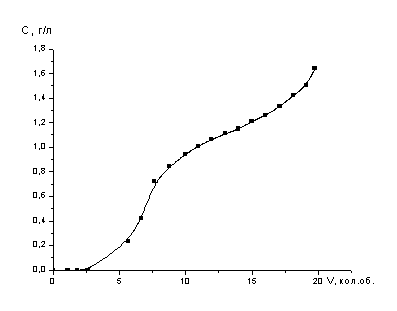
Рисунок 14 - Выходная кривая
сорбции кобальта на ионите M
- 4915
На выходе из колонны контролировали содержание
кобальта. Результаты экспериментов по десорбции кобальта в динамических
условиях на ионитах М-4195, S-930, D-5144
представлены ниже.
Полученные данные представлены в виде выходной
кривой десорбции кобальта с анионита М - 4195 на рисунке 15, численные данные
приведены в таблице 17.
Таблица 17 - Десорбция кобальта с анионита М -
4195 1моль/л раствором серной кислоты в динамике
|
V, мл об. пробы
|
∑V,
мл
об. пробы
|
V, кол.об.
|
Сравн.,
г/л
|
|
6,0
|
6
|
0,3
|
0,36
|
|
5,5
|
11,5
|
0,58
|
2,49
|
|
5,2
|
16,7
|
0,84
|
9,70
|
|
3,0
|
19,7
|
0,95
|
13,15
|
|
3,0
|
22,7
|
1,14
|
14,99
|
|
3,2
|
25,9
|
1,30
|
15,00
|
|
3,2
|
29,1
|
1,46
|
12,10
|
|
3,0
|
32,1
|
1,61
|
10,16
|
|
4,0
|
36,1
|
1,81
|
8,21
|
|
4,5
|
40,6
|
2,03
|
6,00
|
|
5,5
|
46,1
|
2,31
|
3,70
|
|
5,4
|
51,5
|
2,58
|
2,22
|
|
3,3
|
54,8
|
2,74
|
1,84
|
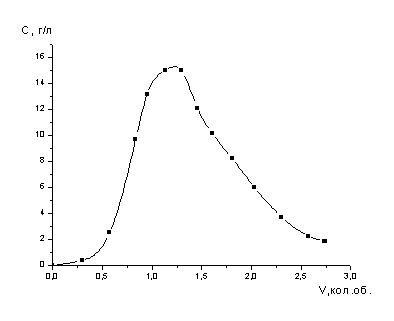
Рисунок 15 - Выходная кривая
десорбции кобальта с анионита М-4195
Из данных представленных на рисунке
15 видно, что десорбция основного количества кобальта происходит 0,5 - 2,5
колоночными объёмами раствора серной кислоты, причём максимальная концентрация
кобальта составляет 15 г/л, что в 8 раз больше по сравнению с исходной
концентрацией. Результат обработки выходной кривой сорбции кобальта показал,
что из одного мл насыщенного сорбента было десорбировано 18,1 мг Со.
Проведенный эксперимент по сорбции и
десорбции кобальта показал, что полная динамическая обменная емкость ионита М -
4195 по данным сорбции соответствует 19,6 мг/мл, а по данным десорбции - 18,1
мг/мл. Такое расхождение значений ПДОЕ, по-видимому, связано с погрешностью
эксперимента.
В аналогичных условиях была изучена
сорбция кобальта в динамических условиях на ионите 930.
Выходная кривая сорбции кобальта на
ионите S - 930 в
динамических условиях представлены на рисунке 16, численные значения в таблице
18.
Таблица 18 - Сорбция кобальта на
анионите S - 930 в
динамике
|
V, мл об. пробы
|
∑V,
мл
об. пробы
|
V, кол. об.
|
Сравн.,
г/л
|
|
20,0
|
20,0
|
1
|
0,22
|
|
11,6
|
31,6
|
1,58
|
1,13
|
|
5,5
|
37,1
|
1,86
|
1,30
|
|
6,0
|
43,1
|
2,16
|
1,40
|
|
6,0
|
49,1
|
2,46
|
1,47
|
|
4,1
|
53,2
|
2,66
|
1,51
|
|
9,1
|
62,3
|
3,12
|
1,56
|
|
10,0
|
72,3
|
3,62
|
1,59
|
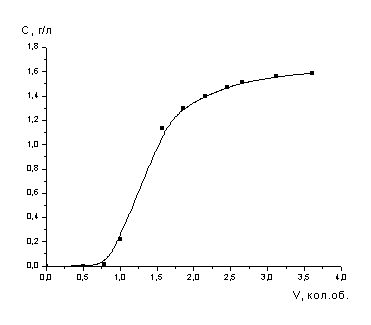
Рисунок 16 - Выходная кривая
сорбции кобальта на ионите S
- 930
Результат обработки выходной кривой сорбции
кобальта показал, что динамическая обменная ёмкость для ионита соответствует
2,98 мг Со/мл набухшего ионита, а полная динамическая обменная емкость
соответствует 1,76 мг Со/мл набухшего ионита.
По изотерме сорбции кобальта на ионите S
- 930, равновесной концентрации кобальта 1,78 г/л соответствует емкость по
кобальту 4,3 мг/мл (таблица 13 и рисунок 11).
В аналогичных условиях была изучена десорбция
кобальта в динамических условиях на ионите S
- 930.
Выходная кривая десорбции кобальта с ионита S
- 930 в динамических условиях представлены на рисунке 17, а численные значения
в таблице 11.
Таблица 19 - Десорбция кобальта с анионита S
- 930 1 моль/л раствором серной кислоты в динамических условиях
|
V, мл об. пробы∑V,
мл
об. пробыV, кол.
об.Сравн., г/л
|
|
|
|
|
10
|
10
|
0,5
|
0,04
|
|
8,2
|
18,2
|
0,91
|
1,77
|
|
5
|
23,2
|
1,16
|
2,9
|
|
4
|
27,2
|
1,36
|
0,77
|
|
3,3
|
30,5
|
1,53
|
0,21
|
|
3
|
33,5
|
1,68
|
0,13
|
|
3,5
|
37
|
1,85
|
0,09
|
|
5
|
42
|
2,1
|
0,05
|
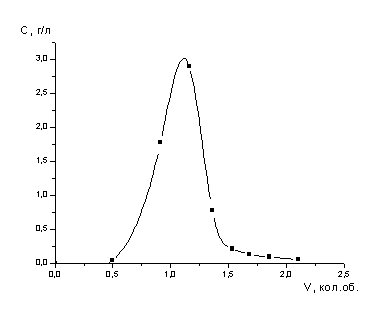
Рисунок 17 - Выходная кривая
десорбции кобальта с анионита S - 930
Из данных представленных на рисунке
17 видно, что десорбция основного количества кобальта происходит 0,5 - 1,5
колоночными объёмами раствора серной кислоты, причём максимальная концентрация
кобальта составляет 2,9 г/л, что в 1,5 раз больше по сравнению с исходной
концентрацией. Результат обработки выходной кривой сорбции кобальта показал,
что из одного мл насыщенного сорбента было десорбировано 1,5 мг Со.
Проведенный эксперимент по сорбции и
десорбции кобальта показал, что полная динамическая обменная емкость ионита S - 930 по
данным сорбции соответствует 1,8 мг/мл, а по данным десорбции - 1,5 мг/мл.
Данные по сорбции и десорбии кобальта практически совпадают.
В аналогичных условиях была изучена
сорбция кобальта в динамических условиях на ионите D - 5144.
Выходная кривая сорбции кобальта на
ионите D - 5144 в
динамических условиях представлены на рисунке 18, численные значения в таблице
20.
Таблица 20 - Сорбция кобальта на
анионите D - 5144 в
динамических условиях
|
V, мл об. пробы∑V,
мл
об. пробыV, кол.
об.Сравн., г/л
|
|
|
|
|
10
|
10,0
|
0,50
|
0
|
|
20
|
30,0
|
1,50
|
0,18
|
|
10,5
|
40,5
|
2,03
|
0,39
|
|
10
|
50,5
|
2,53
|
0,67
|
|
8,5
|
59,0
|
2,95
|
0,81
|
|
11
|
70,0
|
3,50
|
1,01
|
|
10
|
80,0
|
4,00
|
1,15
|
|
10,5
|
90,5
|
4,53
|
1,20
|
|
10
|
100,5
|
5,03
|
1,24
|
|
9
|
109,5
|
5,48
|
1,26
|
|
11
|
120,5
|
6,03
|
1,28
|
|
10
|
130,5
|
6,53
|
1,32
|
|
11,5
|
142,0
|
7,10
|
1,37
|
|
10
|
152,0
|
7,60
|
1,42
|
|
10
|
162,0
|
8,10
|
1,44
|
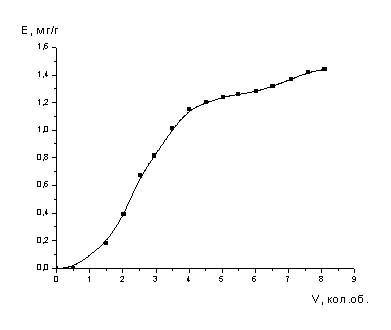
Рисунок 18 - Выходная кривая
сорбции кобальта на ионите D
- 5144
Результат обработки выходной кривой сорбции
кобальта показал, что динамическая обменная ёмкость для ионита соответствует
0,9 мг Со/мл набухшего ионита, а полная динамическая обменная емкость
соответствует 7,1 мг Со/мл набухшего ионита.
По изотерме сорбции кобальта на анионите D
- 5144, равновесной концентрации кобальта 1,78 г/л соответствует емкость по
кобальту 6,3 мг/мл (таблица 13 и рисунок 11).
В аналогичных условиях была изучена десорбция
кобальта в динамических условиях на ионите D
- 5144.
Выходная кривая десорбции кобальта с ионита D
- 5144 в динамических условиях представлены на рисунке 19, а численные значения
в таблице 21.
Таблица 21 - Десорбция кобальта с анионита D
- 5144 1М раствором серной кислоты в динамических условиях
|
V, мл об. пробы
|
∑V,
мл
об. пробы
|
V, кол. об.
|
Сравн.,
г/л
|
|
4,2
|
4,2
|
0,21
|
0,14
|
|
6,4
|
10,6
|
0,53
|
0,17
|
|
7,8
|
18,4
|
0,92
|
2,09
|
|
5,1
|
23,5
|
1,18
|
4,87
|
|
5,0
|
28,5
|
1,43
|
6,47
|
|
5,5
|
34,0
|
1,70
|
6,07
|
|
5,0
|
39,0
|
1,95
|
3,80
|
|
5,5
|
44,5
|
2,23
|
1,50
|
|
5,5
|
50,0
|
2,50
|
0,56
|
|
6,0
|
56,0
|
2,80
|
0,05
|
|
5,8
|
61,8
|
3,09
|
0,01
|
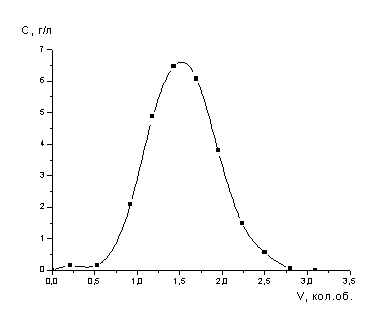
Рисунок 19 - Выходная кривая
десорбции кобальта с анионита D - 5144
Из данных представленных на рисунке
19 видно, что десорбция основного количества кобальта происходит 0,5 - 2,5
колоночными объёмами раствора серной кислоты, причём максимальная концентрация
кобальта составляет 6,5 г/л, что в 5 раз больше по сравнению с исходной
концентрацией. Результат обработки выходной кривой сорбции кобальта показал,
что из одного мл насыщенного сорбента было десорбировано 6,9 мг Со.
Проведенный эксперимент по сорбции и
десорбции кобальта показал, что полная динамическая обменная емкость ионита D - 5144 по
данным сорбции соответствует 7,1 мг/мл, а по данным десорбции - 6,9 мг/мл.
Таким образом, последовательное пропускание
растворов, полученных в результате разложения отработанных катализаторов
растворами серной кислоты вначале через анионит А100(Мо), затем через иониты М
- 4195, S - 930, D
- 5144 обеспечивает раздельное и практически полное извлечение молибдена и
кобальта.
Из представленных данных можно сделать вывод,
что иониты М - 4195, D-5144
сорбируют кобальт из растворов, содержащих сульфат алюминия, гораздо лучше, чем
ионит S-930. Ионит D-5144
можно рекомендовать для извлечения кобальта наряду с ионитом М-4195.
.8 Принципиальная технологическая схема
извлечение молибдена и кобальта из катализатора, основанная на выщелачивании
серной кислотой
Результаты проведенных исследования позволяют
предложить принципиальную технологическую схему извлечения молибдена и кобальта
из отработанных катализаторов, с предварительным растворением их в растворе
серной кислоты.
Схема, представленная на рисунке 20, включает
проведение следующих операций. В начале проводится двухстадийное противоточное
выщелачивание молибдена и кобальта из катализатора раствором серной кислоты,
далее раствор направляется на сорбцию молибдена на анионите А100(Мо), после
сорбции молибдена из сорбатов проводится сорбционное извлечение кобальта ни
ионите М - 4195 или ионите D-5144.
Десорбция молибдена осуществляется 6 моль/л раствором аммиака, а десорбция
кобальта 1 моль/л раствором серной кислоты.
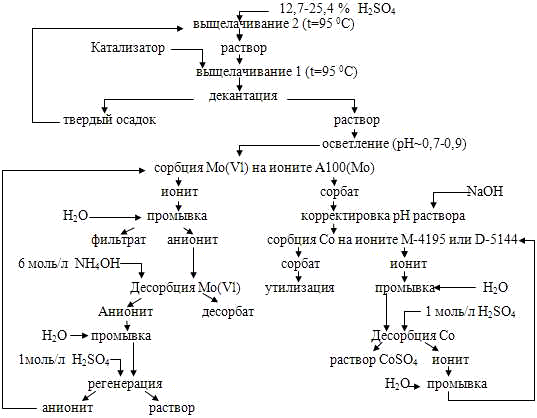
Рисунок 20 - Принципиальная технологическая
схема процесса извлечения молибдена и кобальта из раствора сульфата алюминия
ЗАКЛЮЧЕНИЯ И ВЫВОДЫ
катализатор
алюминий молибден кобальт
1 Установлено, что оптимальными
условиями разложения катализатора являются концентрация серной кислоты 12,7 -
25,4%, температура процесса 95 - 98 0С и режим противоточного
двухстадийного разложения. Показано, что при данных условиях скорость
разложения ускоряется в 3-4 раза по сравнению с одностадийным разложением
катализатора.
2. Установлено, что для
извлечения молибдена из раствора сульфата алюминия может быть рекомендован
макропористый слабоосновный анионит Purolite А100(Мо), а для извлечения
кобальта ─ комплексообразующий ионит Dowex XF 4195 с би-спикалинаминными
функциональными группами и D
- 5144 содержащий одновременно полиэтиленполииминные и карбоксильные
функциональные группы.
3. Предложена принципиальная схема переработки
молибденкобальтовых катализаторов с получением растворов индивидуальных ценных
компонентов.
СПИСОК ИСПОЛЬЗОВАННЫХ ИСТОЧНИКОВ
1. Гаджиев,
Р.Б. Получение формиата никеля из отработанных каталитических комплексов / Р.Б.
Гаджиев, С.М. Москвичев, И.П. Петрюк. - Химическая промышленность - 3изд., 2006
г.
3. Сеньков,
Г.М. Промышленные катализаторы риформинга / Г.М. Сеньков, Н.С. Козлов. - Наука
и техника,1986.- 264 с.
. Некрасов,
Б.В. Основы общей химии / Б.В Некрасов. - М.: Химия, 1965. - Т.1 - 519с.
. Нефедов,
Б.К. Катализаторы процессов глубокой переработки нефти / Б.К. Нефедов, Е.Д.
Радченко, Р.Р. Алиев. - М.: Химия, 1992. - 272 с.
. Блохин,
А.А. Извлечение молибдена из отработанных катализаторов гидроочистки нефтяного
сырья. Каталитические процессы и катализаторы / А.А. Блохин, Ю.В. Мурашкин,
О.В. Полянская, С.С. Никитин. - СПб.: СПбГТИ(ТУ), 2006 г.
. Блохин,
А.А. Ионообменное извлечение рения при переработке дезактивированных
платинорениевых катализаторов / А.А. Блохин, Ю.В. Мурашкин, М.А. Плешков и др.
Цветные металлы, 2006.
. Зеликман,
А.Н. Металлургия редких металлов / А.Н. Зеликман. - М.: Металлургия, 1980. -
328с.
. Большаков,
К.А. Химия и технология редких и рассеянных элементов. Высшая школа/ К.А.
Большаков. - М.: 1976. - Т3 - 320с.
10. Браун,
М.П. Энциклопедия неорганических материалов / М.П. Браун, М.В. Волощенко, В.Н.
Гриднёв и др.; под ред. И.М. Федорченко. - В 2 т. - Т.2 - Киев: Главная
редакция украинской советской энциклопедии,1977. - 812 с.
. Абросимов,
А.А. Экологические аспекты производства и применения нефтепродуктов/
А.А. Абросимов. - М.: БАРС, 1999. - 733 с.
12. Евграфова,
А.К. Цветные металлы / А.К. Евграфова, Л.А. Павлинова, Л.Ш. Цемехман и
др. - 1995. - №5. - 41-43 с.
. Некрасов,
Б.В. Основы общей химии / Б.В Некрасов. - М.: Химия, 1965. - Т.3 - 519с.
. Вольдман,
Г.М. Теория гидрометаллургических процессов: Учеб. Пособие для вузов/ Г.М.
Вольдман, А.Н. Зеликман. - 4-е изд. перераб. и доп. - М.: Интермет Инжинеринг,
2003. - 145 с.
. Михнев,
А.Д. Извлечение Mo
и Ni из отработанных
катализаторов. Цветные металлы / А.Д. Михнев, Г.Л. Пашков, С.В. Дроздов и др. -
2000. - №11-12. - 90-93с.
. Теоретические
аспекты использования сорбционных и хромоторафических процессов в металлургии и
химической технологии: тезисы докладов международной конференции. -
Екатеринбург: ГОУВПОУГТУ-УПИ. - 2006. - 103с.
. Акимбаева,
А. М. Сорбция ионов Сu
(II) органоминеральным
катионитом на основе бентонита. Цветные металлы / А.М. Акимбаева, Е.Е. Ергожин,
А. Д. Товасаров. - 2006. - №3.
. Хансон,
К. Последние достижения в области жидкостной экстракции/ Под ред. К.
Хансона.-М.: Химия, 1974.-448с.
. Красников,
Г.П. Экстракционные технологические схемы получения кобальта высокой чистоты.
Химическая экстракция/ Г.П. Красников, М.Л. Навтанович. - Новосибирск:
Наука,1984.
. Зеликман,
А.Н. Закономерности экстракции и разделения Мо и W
фосфорорганическими соединениями. Химическая экстракция/ А.Н. Зеликман, Г.М.
Вольдман, Ю.М. Конюхов.- Новосибирск: Наука,1984г.
. Марченко,
З. Фотометрическое определение элементов/ З. Марченко. - М.: Мир, 1972.-502с.
. Шарло,
Г. Методы аналитической химии/ Г. Шарло.-М.: Химия, 1969.-1184с.
23. Радченко,
Е.Д. Промышленные катализаторы гидрогенизационных процессов нефтепереработки /
Е.Д. Радченко, Б.К. Нефедов, Р.Р. Алиев. - М.: Химия, 1987.-224 с.
. Федорченко,
И.М. Энциклопедия неорганических материалов: В 2 т. Т.1 //Под ред. И.М.
Федорченко. Киев: Главная редакция украинской советской энциклопедии,1977.-
840с.
. Рипан,
Р. Неорганическая химия. В 2 т. Т. 2./ Р. Рипан, И.М. Четяну. - Мир, 1971. -
869с.
. Агиевский,
Д.А. Сульфидные катализаторы гидрогенизации на основе молибдена и вольфрама и
пути их совершенствования. /Д.А. Агиевский, М.В. Ланда, М.А. Кипнис, Л.И.
Павлова. - М.: ЦНИИТЭнефтехим, 1982.- 78с.
27. Е.Д.Радченко,
Е.Д. Повышение качества реактивных топлив и регулирование их углеродного
состава / Е.Д. Радченко, М.В. Ландау, В.Я. Кругликов и др. - М.:
ЦНИИТЭнефтехим, 1976. - 75 с.
. Дорогочинский,
А.З. Гидрирование ароматических углеводородов на металлоцеолитных катализаторах
/ А.З. Дорогочинский, Е.А. Есипко.-М.: ЦНИИТЭнефтехим, 1980.- 48 с.
. Сухарев,
С.Б. Синтез, свойства оксигидратов металлов и их применение в сорбционных и
каталитических процессах. Автореферат к.т.н. Екатеринбург, 2008. - 24 с.
. Зеленин,
В.И. Переработка молибденсодержащих сорбентов и катализаторов / В.И.Зеленин,
В.Н.Рычков, С.Б.Сухарев, А.И. Никитин // Вестник УГПИ-УПИ. №15(67). Актуальные
проблемы физической химии твердого тела/ Сб. науч. трудов. - Екатеринбург: ГОУ
ВПО УГПИ-УПИ, 2005. - 175-179 с.
. Гедгагов,
Э.И. Пористые аниониты в технологии производства молибдена /Э.И.Гедгагов,
А.Г.Маурина, А.М.Затворницкий и др. // Цвет. мет. - 1979. №3. 43-44
с.
. Холмогоров,
А.Г. Модифицированные иониты в технологии молибдена и вольфрама/ А.Г.
Холмогоров, М.В. Мохосоев, Э.Л. Зонхоева.-Новосибирск: Наука, 1985. - 181 с.
. Гедгагов,
Э.И. Современное состояние и перспективы развития вольфрамо-молибденовой
подотрасли в странах СНГ //Цвет. мет. / Э.И. Гедгагов, А.Д. Бессер. - 1998,
№3,49-56с.
34. Grinstead
R.R. Selective absorption of copper, nickel, cobalt and other transition metals
ions from sulfuric acid solutions with the chelating ion exchange resin XFS
4195 //Hydrometallurgy. 1984. V.
12, N 3. P.
387-400.
. ГОСТ
12.4.021-75 CCБТ Системы
вентиляционные. Общие требования.
. Правила
эксплуатации электроустановок потребителей (ПЭЭП-92) м: Энергоатом
издат,1992.-228 с.
. Инструкция
по безопасности проектирования ИБП 105-95. Определение категорий помещений и
зданий по взрывопожарной и пожарной опасности.-М.: Стройиздат, 1995.-26 с.
. НБП-05-95
Определение категорий помещений и зданий по взрывопожарной и пожарной
опасности.
. Вредные
вещества в промышленности: Справ. / Под. ред. Н.В. Лазарева.-Т.2-Неорганические
и металлорганические вещества.- Химия, 1971.-624с.
. Бандман,
А.Л. Вредные химические вещества. Неорганические соединения элементов I-IV
групп: Справ. Изд./ А.Л. Бандман, Г.А. Гудзовский, Л.С. Дубейковская и др.; Под
ред. В.А. Филова и др. - Л.: Химия, 1988-512с.
. ГОСТ
12.1.005-88 ССБТ Воздух рабочей зоны. Общие санитарно-гигиенические требования.
. Пряников,
В.И. Техника безопасности и промышленная санитария: Справ. Для работников химической
промышленности. - Т2 / В.И. Пряников, А.И. Родионова. - М.: Химия, 1979-270с.
. СТП
СПбГТИ 017-97 Виды учебных занятий. Положение о выпускной работе
дипломированного специалиста. Общие требования / 21 с.
. Р01-2007
Библиографическое описание документа. Примеры оформления. - Введ. 2008-01-01. -
М.: Госстандарт России: Изд-во стандартов, 2008.-11с.
. СТП
СПбГТИ 006 - 2005 Подготовка и оформление авторских текстовых оригиналов для
издания / 2005. - 32с.
. ГОСТ
8.417-2002 ГСИ. Единицы величин. - взамен ГОСТ 8.417-81; введен. 2003-09-01.-
Минск: Межгос. Совет по стандартизации, метрологии и сертификации; М.: Изд-во
стандартов, 2003.- 27 с.- (Государственная система обеспечения единства
измерений)
ПРИЛОЖЕНИЕ А
(обязательное)
Патентный поиск
Целью патентного поиска являлось выявление
аналогов и прототипов разрабатываемого технологического процесса. В ходе работы
были изучены следующие патенты и авторские свидетельства, посвященные проблеме
переработки молибдено-кобальтовых катализаторов на основе оксида алюминия, с
целью извлечения молибдена, никеля и кобальта.
Таблица А.1-Перечень отобранных в процессе
поиска аналогов
|
Страна
|
№заявки
или Охранного Документа (А.с. или патента)
|
Название
изобретения
|
Дата
публикации
|
|
Франция
|
0204563
|
Способ
гидроформирования с участием катализатора на основе кобальта в ионной
неводной жидкости с возвращением в процессе улучшения катализатора
|
15.10.2003
|
|
США
|
6713519
|
Получение
и применение катализатора, содержащего углеродные нанотрубки
|
30.03.2004
|
|
Испания
|
230101328
|
Извлечение
металлов из летучей фракции золы, образующейся на тепловых электростанциях
|
14.04.2004
|
|
США
|
6841686
|
Катализаторы
окисления на основе молибдена
|
11.01.2005
|
|
Австралия
|
200110840
|
Ионообменный
способ извлечения никеля и кобальта из окисленной руды
|
27.01.2005
|
|
США
|
5687163
|
Способ
получения осадка и катализатора на основе молибдена
|
15.03.05
|
|
Россия
|
2003112243
|
Селективное
извлечение Мо (VI)
|
27.12.2004
|
|
Россия
|
2006133630
|
Способ
извлечения молибдена
|
27.01.2006
|
|
Россия
|
2335499
|
Способ
извлечения молибдена
|
27.03.2008
|
ПРИЛОЖЕНИЕ Б
(обязательное)
Охрана труда и окружающей среды
Охрана окружающей среды и здоровья людей -
первоочередная задача любого государства, которое хочет иметь свое будущее.
Поэтому необходимо повышать эффективность мер по охране природы, шире внедрять
малоотходные и, по возможности, безотходные технологические процессы, развивать
комбинированные производства, обеспечивающие полное и комплексное использование
природных ресурсов, сырья и материалов, исключающие или существенно снижающие
вредное воздействие на окружающую среду.
Экспериментальная часть настоящей дипломной
работы была выполнена в лаборатории №3 кафедры технологии редких и рассеянных
элементов, Санкт - Петербургского Государственного Технологического института.
Характеристика использованных в работе веществ и материалов представлена в
таблицах Б.1 - Б.4.
Технологические операции, проводившиеся в
работе, рассмотрены в таблице Б.4.
Характеристика основных производственных отходов
сведена в таблице Б.5.
Анализ вредных и производственных факторов
Основные опасные и вредные производственные
факторы по ГОСТ 12.0.003-74 [35] классифицируются как химические и физические.
Данные представлены в таблицах Б.1 - Б.2.
Основными источниками тепловой энергии в
лаборатории, представляющими потенциальную пожарную опасность являются: газовая
горелка, муфельная печь, электроплитка, а также прочие электроприборы в случае
их неисправности.
Лаборатория характеризуется следующими
условиями, создающими опасность поражения электрическим током:
) Возможность одновременного прикосновения
человека к имеющим соединения с землей металлоконструкциям зданий,
технологическим аппаратам, механизмам и т.п., с одной стороны, и к
металлическим корпусам электрооборудования - с другой;
) Химически активная или органическая среда
(разрушающая изоляцию и токоведущие части электрооборудования);
Согласно [36] лаборатория относится к классу
помещений с повышенной опасностью поражения людей электрическим током.
В лаборатории проводятся работы с жидкостями с
температурой вспышки паров выше 61 0С, с твердыми сгораемыми
веществами и материалами. Таким образом, по НПБ 105-95 [37] данное помещение
относится к категории "пожароопасное В - 4". В лаборатории при
нормальной эксплуатации взрывоопасные смеси горючих газов или паров ЛВЖ не образуются,
а возможны только в результате аварий и неисправностей. ЛВЖ имеются в небольших
количествах, недостаточных для создания взрывоопасной смеси в зоне, превышающей
5% свободного объема помещения. По НПБ 105-95 [38] данное помещение относится к
взрывоопасным класса В - 1б.
На основании данных таблиц Б.1 - Б.2 можно
сделать следующие выводы:
1) В
лабораторных установках отсутствуют вещества, нагретые выше температуры их
самовоспламенения;
2) Вещества,
воспламеняющиеся при соприкосновении с водой, а также способные самовозгораться
на воздухе, в процессе работы не образуются.
Химически нестойкие вещества в работе не
применяются.
Таблица Б.1-Характеристика физико-химических
свойств сырья
|
Физико-химические
свойства
|
|
Вещества
|
Агрегатное
состояние
|
Температура
плавлен.,°С
|
Температура
кипения, °С
|
Плотность
кг/м3
|
|
Азотная
кислота
|
жидк.
|
-41.6
|
83
|
1513
|
|
Cерная кислота
|
жидк.
|
10.3
|
330
|
1,834
|
|
Аммиак
|
жидк.
|
-77.7
|
-33.3
|
6.81
|
|
Гидроксид
натрия
|
жидк
|
328
|
1388
|
2130
|
|
Тиомочевина
|
тв
|
182
|
|
1.405
|
Таблица Б.2 - Характеристика токсических свойств
сырья [39-41]
|
Токсические
свойства
|
|
Вещества
|
Характеристика
действия на организм человека
|
Класс
опасности
|
Предельно-допустимая
концентрация в воздухе рабочей зоны (ПДК), мг/м3
|
|
Серная
кислота
|
Тяжелые
ожоги, раздражает слизистые, токсична
|
I
|
1
|
|
Азотная
кислота
|
Токсична,
вызывает пожелтение кожи, хим. ожоги, язвы
|
II
|
5,0
|
|
Аммиак
|
Ядовит,
раздражает слизистые оболочки, вызывает воспаление легких, отдышку.
|
III
|
0.02
|
|
Гидроксид
натрия
|
Тяжелые
ожоги кожи и глаз.
|
II
|
0.5
(аэрозоль)
|
Таблица Б.3 - Анализ технологических операций с
точки зрения опасностей и вредностей при их проведении [42]
|
Наименование
технологических операций
|
Оборудование
для проведения операции
|
Реактивы
для проведения операции
|
Возможные
опасности и вредности при проведении операции
|
|
Приготовление
растворов
|
Мерная
термостойкая посуда, высокоточные весы, плитка
|
Концентрированные
кислоты, сульфаты металлов, дистиллированная вода
|
1)
Химический ожог; 2) Раздражение верхних дыхательных путей и слизистых
оболочек глаз, носа и рта
|
|
Проведение
выщелачивания Мо, Со
|
Термостойкий
стеклянный стакан, плитка, мешалка
|
Концентрированные
кислоты, дистиллированная вода
|
1)
Химический ожог; 2) Раздражение верхних дыхательных путей и слизистых
оболочек глаз, носа и рта
|
|
Приготовление
растворов для сорбции, сорбция
|
Мерная
посуда, стеклянная пипетка, резиновая груша
|
Концентрированные
кислоты, сульфаты металлов, аммиак, дистиллированная вода, сорбент
|
1)Химический
ожог; 2)Порезы
|
Таблица Б.4 - Характеристика производственных
отходов
|
Наименование
отходов
|
Агрегатное
состояние
|
Характеристика
отходов
|
Место
сброса
|
Количество
отходов
|
|
1)
Водные растворы после анализа
|
жидк.
|
ПАВ,
реактивы
|
Хранение
и дальнейшая утилизация после нейтрализации
|
100
л
|
|
2)
Битое стекло
|
тв.
|
-
|
Сборник
твердых отходов
|
0,05
кг
|
|
3)
Вентиляционные выбросы
|
газ
|
Пары
реактивов
|
Атмосфера
|
0.03
м3
|
Обеспечение безопасности работы в лаборатории
Для устранения указанных опасностей были
предприняты следующие меры:
перед работой был проведен инструктаж по ТБ,
согласно [42] и разработанным на кафедре инструкциям;
все виды работ проводились в халате;
все работы с едкими жидкостями, и радиоактивными
веществами проводились на подносе под тягой;
отработанные кислотные растворы сливались в
специальные емкости и после нейтрализации сливались в канализационную сеть;
при работе соблюдались правила
электробезопасности (все приборы имели заземление, перед и во время работы
производилась проверка состояния изоляции, качества заземления);
при возникновении неисправностей приборы
отключались от сети, и об этом немедленно сообщалось руководителю работ.
Аптечка и ее содержание
В препараторской на кафедре находится аптечка,
укомплектованная в соответствии с ТУ 9398-001-46930922-97.
Содержимое аптечки представлено в таблице Б.5.
Таблица Б.5 - Содержимое аптечки на кафедре ТРРЭ
|
название
препарата
|
количество,
шт.
|
|
корвалол
|
2
|
|
настойка
пустырника
|
2
|
|
настойка
боярышника
|
2
|
|
настойка
валерианы
|
2
|
|
перекись
водорода 3% раствор
|
2
|
|
аммиак
водный 10% раствор
|
1
|
|
парацетамол
|
2
|
|
цитрамон
|
2
|
|
настойка
йода
|
1
|
|
бинт
стерильный 5Х10
|
3
|
|
вата
250гХ60
|
1
|
|
кордиамин
|
1
|
|
анальгин
|
2
|
|
андипал
|
2
|
|
уголь
активированный
|
3
|
|
валидол
|
2
|
|
нитоглицерин
|
1
|
Меры первой медицинской помощи пострадавшим [42]
) При химических ожогах кислотами или щелочами
пораженные участки кожи промывают струей холодной воды в течение не менее 15
минут. Затем накладывают примочки из 2-х процентного бикарбоната натрия, а при
ожогах щелочами - 2-х процентный раствор уксусной кислоты.
) При отравлениях необходимо вывести
пострадавшего на свежий воздух или в проветриваемое помещение. Пострадавшего необходимо
уложить, создать ему полный покой, укрыть и ждать врача.
) При порезах необходимо наложить марлевую
повязку, предварительно обработав рану йодом и удалив осколки стекла под струей
холодной воды.
4 При
поражении электрическим током первая помощь состоит из двух этапов:
освобождение пострадавшего от действия электрического тока и оказания ему
первой медицинской помощи (создать пострадавшему полный покой, проверить пульс,
дыхание; если он плохо дышит (резко, судорожно) или вообще не дышит, делать искусственное
дыхание и наружный массаж сердца). Необходимо срочно вызвать врача, даже если
кажется, что пострадавший вне опасности.
Средства тушения пожара:
вода;
ящик с песком;
асбестовое одеяло.
огнетушитель ОХВП - 10.
Санитария и гигиена
Освещенность в лаборатории организована по
IV-разрядной норме - 200лк.
Вентиляция состоит из приточно-вытяжной
вентиляции с местной вентиляцией (Кв=3,5-3,8) ч-1 [38].
Скорость движения воздуха в рабочем проеме - 1,6 м/с соответствует классу
опасности веществ [41].
ПРИЛОЖЕНИЕ В
(обязательное)
Маркетинговое исследование
Настоящая дипломная работа носит поисковый
характер. Целью НИР является разработка новой промышленной технологии
извлечения молибдена, кобальта и никеля из отработанных катализаторов на основе
оксида алюминия, а также улучшение разделения этих элементов путем сорбции.
Потенциальными потребителями данной идеи является промышленное предприятие по
производству молибдена, кобальта, никеля и их концентратов.
В настоящее время потенциальным потребителем
результатов проведенных исследований могут быть промышленные предприятия России
(Приокский завод цветных металлов в Рязанской области г. Касимов), стран СНГ
(Узбекистан), дальнего зарубежья (Китай).
Предлагаемая к промышленному производству идея
имеет некоторые преимущества:
) сравнительно недорогие реагенты;
) извлечение молибдена, кобальта и никеля из
отработанных катализаторов на 70 - 100%;
) простое технологическое оборудование.
Данное производство может быть экономически
выгодным. Однако, оно требует больших капиталовложений. Оно может позволить
перерабатывать исходное сырьё (отходы производств) и на 70 - 100% извлекать
оттуда ценные элементы, необходимые для разных отраслей промышленности. Ведь в
России недостаточное количество месторождений молибдена, и при современных
ценах на этот металл производство может принести с годами ощутимые прибыли.
Кобальт и никель - дорогие цветные металлы, извлечение их также говорит о
выгоде данного промышленного производства.
ПРИЛОЖЕНИЕ Г
(обязательное)
Технико-экономическая оценка результатов
исследования
Важным показателем проводимых исследований
является договорная цена на научно-исследовательскую работу (НИР) Цн определяется
по формуле Г.1:
Цн = Зн ∙(1+  )
∙ K, (Г.1)
)
∙ K, (Г.1)
где Зн - затраты на выполнение
исследования, предусмотренного планом дипломной работы, руб.;
Р - уровень рентабельности исследования, %;
К ─ коэффициент, учитывающий поощрительную
надбавку за качество разработки.
Для определения договорной цены рассчитаны
текущие затраты на выполнение НИР, к ним относятся:
сырьё, материалы, реактивы;
энергетические затраты;
оплата труда персонала с обязательными
начислениями;
амортизация оборудования и приборов для
научно-экспериментальных работ.
Затраты на сырьё, материалы и реактивы (Зм),
израсходованные на проведение исследования, определены расчётом по формуле
(Г.2), результаты которого сведены в таблице Г.1.
Зм = ∑ Рi
∙
Цi,
(Г.2)
i
где Рi
- расход i-го вида
материальных ресурсов, нат. ед.;
Цi
- планово-заготовительная цена i-го
вида материальны ресурсов, руб.
Таблица Г.1 - Расчёт затрат на сырьё, материалы,
реактивы
|
Наименование
|
Техническая
характеристика
|
Единица
измерения
|
Израсходовано
материала
|
Цена
по каталогу, руб.
|
Сумма,
руб.
|
|
Гидроксид
натрия
|
ЧДА
|
Кг
|
0.5
|
54
|
27.00
|
|
Серная
кислота
|
ЧДА
|
Кг
|
2
|
52
|
104.00
|
|
Аммиак,
вод.
|
ЧДА
|
Кг
|
1
|
42
|
42.00
|
|
Азотная
кислота, разб. 56%
|
ХЧ
|
Кг
|
0.3
|
47.90
|
14.37
|
|
Ацетат
натрия
|
ЧДА
|
Кг
|
0.3
|
72
|
21.6
|
|
Нитрозо-р-соль
|
Ч
|
Кг
|
0.02
|
980
|
19.6
|
|
Тиомочевина
|
Ч
|
Кг
|
0.5
|
139
|
69.50
|
|
Медь
(II)
сернокислая 5/в
|
Ч
|
Кг
|
0.002
|
118
|
0.236
|
|
Роданид
аммония
|
Ч
|
Кг
|
0.7
|
127
|
88.9
|
|
Алюминий
сернокислый 18/в
|
Кг
|
0.6
|
64
|
38.4
|
|
Кобальт
(II)
сернокислый 7/в
|
Ч
|
Кг
|
0.01
|
590
|
5.9
|
|
Персульфат
калия
|
Ч
|
Кг
|
0.01
|
102
|
1.02
|
|
Молибденовая
кислота
|
Ч
|
Кг
|
0.1
|
487
|
48.7
|
|
Отработанный
катализатор
|
|
Кг
|
0.07
|
1600
|
112
|
|
Аскорбиновая
кислота
|
Ч
|
Кг
|
0.00015
|
665
|
0.1
|
|
Итого
|
|
593.3
|
В состав энергетических затрат включены затраты
на электроэнергию (Зэ), горячую и холодную воду.
Затраты на электроэнергию определены следующим
образом по формуле Г.3:
Зэ = М ∙ Ки ∙
Т ∙ Ц, (Г.3)
Где М - паспортная мощность электроприборов,
использованных при проведении исследования, кВт;
Ки - коэффициент использования
мощности электрооборудования;
Т - время эксплуатации электрооборудования за
весь период выполнения исследования, ч;
Ц - цена 1 кВт∙ч электроэнергии, руб., (Ц
= 1.70 руб/кВт∙ч).
Результаты расчёта представлены в таблице Г.2.
Таблица Г.2 - Расчёт затрат на электроэнергию
|
Прибор
|
Паспортная
мощность, кВт
|
Коэффициент
использования мощности
|
Время
работы, ч
|
Расход
энергии, кВт∙ч
|
|
Дистиллятор
|
7.2
|
0.9
|
100
|
648
|
|
Плитка
электрическая
|
3.0
|
0.85
|
400
|
1020
|
|
Сушильный
шкаф
|
6.8
|
0.9
|
10
|
61.2
|
|
Вытяжной
шкаф
|
1.5
|
0.7
|
520
|
546
|
|
Весы
аналитические
|
0.006
|
0.8
|
10
|
0.048
|
|
Фотоколориметр
|
0.2
|
0.7
|
400
|
56
|
|
Центрифуга
|
8.2
|
0.9
|
5
|
36.9
|
|
рН-метр
|
0.6
|
0.7
|
50
|
21
|
|
Итого
|
|
2389.1
|
Затраты на электроэнергию составили:
Зэ = 2389.1 ∙ 1.70 = 4061.6
руб.
Затраты на воду (Зв) определены,
исходя из её ориентировочного расхода и цены для условий института (30 руб./м3
с учётом канализации). Источником горячей и дистиллированной воды в лаборатории
является дистиллятор, поэтому расход холодной воды представляет собой сумму
расходов на лабораторные нужды, на охлаждение дистиллятора и на получение
дистиллированной воды.
Для данной работы суммарный расход составил м3,
а затраты:
Зв = 28 ∙ 30 = 840 руб.
Затраты на основную заработную плату исполнителя
(Зи) исследования определены умножением размера месячной стипендии
на число месяцев, отводимых на выполнение дипломной работы:
Зи = 2200 ∙ 4 = 8800 руб.
Основная заработная плата руководителей работы
(Зрук) и консультантов (Зконс) определена, исходя из
суммарной нормы затрат их рабочего времени на одну дипломную работу и
среднечасовой заработной платы
Зрук = 33 ∙ 300 = 9900 руб.
Зконс = 2 ∙ 235 = 470 руб.
Дополнительная заработная плата (Здоп)
определена в размере 20% к сумме затрат по статье «основная заработная плата»:
Здоп = (8800+9900+470) ∙0.2 =
3834 руб.
Отчисления на социальное страхование (Зсс)
принимаются в размере 26% от суммы основной и дополнительной заработной платы:
Зсс = (8800+9900+470+3834) ∙0.26
= 5981.04 руб.
Затраты на стеклянные приборы и посуду
определены в зависимости от вида и емкости стеклянных изделий, использованных
для выполнения исследовательской работы и цен на них по данным кафедры. Текущие
затраты определены, как 50% от суммарной стоимости этих изделий, приведенной в
таблице Г.3.
Таблица Г.3 -Затраты на стеклянные приборы и
посуду
|
Наименование
|
Объём,
см3
|
Количество,
шт.
|
Цена,
руб.
|
Сумма,
руб.
|
|
Пипетка
|
1
|
2
|
48.75
|
97.5
|
|
2
|
2
|
48.75
|
97.5
|
|
5
|
3
|
42.5
|
127.5
|
|
10
|
2
|
48.75
|
97.5
|
|
15
|
1
|
56.25
|
56.25
|
|
Колба
мерная
|
10
|
1
|
42.5
|
42.5
|
|
25
|
5
|
43.75
|
218.72
|
|
100
|
1
|
46.25
|
46.25
|
|
Цилиндр
мерный
|
25
|
1
|
23.75
|
23.75
|
|
100
|
1
|
43.75
|
43.75
|
|
Воронка
|
50
|
1
|
31.25
|
31.25
|
|
Стакан
|
1000
|
1
|
127.5
|
127.5
|
|
Колба
коническая
|
50
|
35
|
40
|
1400
|
|
Стакан
химический
|
2000
|
1
|
176.4
|
176.4
|
|
Бюкс
|
32х50
|
1
|
61.25
|
61.25
|
|
Колба
Бунзена
|
250
|
1
|
268.75
|
268.75
|
|
Воронка
Бюхнера
|
50
|
1
|
87.5
|
87.5
|
|
Палочка
стеклянная
|
|
1
|
4
|
4
|
|
Колонка
сорбционная
|
|
2
|
575
|
1150
|
|
Итого
|
|
4157.9
|
Сумма амортизационных отчислений на реновацию А
определена, исходя из стоимости по данным кафедры использованных для выполнения
исследовательской работы оборудования и приборов Ф, годовых норм их амортизации
на реновацию Нар и времени их использования (мес.) для данного
исследования Т по формуле (Г.4):
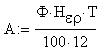 (Г.4)
(Г.4)
Результаты представлены в таблице Г.4
Таблица Г.4 - Расчёт амортизационных отчислений
на реновацию
|
Прибор
|
Стоимость,
руб.
|
Годовые
нормы амортизации на реновацию, %
|
Время
работы, мес.
|
Сумма
амортизационных Отчислений, руб.
|
|
Дистиллятор
|
15120
|
8
|
0.6
|
60.5
|
|
Плитка
электрическая
|
350
|
8
|
2.3
|
5.4
|
|
Сушильный
шкаф
|
14580
|
14
|
0.06
|
10.2
|
|
Вытяжной
шкаф
|
20009
|
9
|
3
|
450.2
|
|
Весы
аналитические
|
21600
|
10
|
0.06
|
10.8
|
|
Фотоколориметр
|
36500
|
8
|
2.3
|
560
|
|
Центрифуга
|
9500
|
12
|
0.03
|
2.9
|
|
рН-метр
|
5880
|
8
|
0.3
|
11.8
|
|
Итого
|
|
1111.8
|
Накладные расходы приняты в размере 50% от суммы
прямых затрат.
Для определения общей суммы текущих затрат в
таблице Г.6 составлена смета, представляющая собой свод всех текущих затрат Зн
на выполнение исследовательской работы за весь период её выполнения.
Уровень рентабельности принимается равным 25%.
Для вычисления коэффициента, учитывающего
поощрительную надбавку за качество разработки, необходимо рассчитать величину
научно-технического эффекта (Энт), который определяется по формуле:
Энт = ∑ уi
∙ki
(Г.5)
i
где уi
─
величина частного показателя, балл;
ki
─
весовой коэффициент i-го частного
показателя степени новизны, научно-технический уровень, глубина воздействия на
процесс.
Величина частного показателя и весовой
коэффициент представлены в Таблице Г.5.
Таблица Г.5 - Расчёт рассматриваемых параметров
|
i
|
уi
|
ki
|
|
1
|
70
|
0.25
|
|
2
|
70
|
0.25
|
|
3
|
60
|
0.5
|
Энт =
70 ∙ 0.25
+ 70 ∙
0.25 + 60 ∙ 0.5 =
65 балла
Таблица Г.6 - Смета затрат на проведение
научно-исследовательской работы
|
Наименование
статей затрат
|
Сумма,
руб.
|
Доля
статей в общей сумме затрат, %
|
|
1.Сырьё,
материалы, реактивы
|
593.3
|
0.01
|
|
2.
Энергетические затраты
|
|
|
|
а)
электроэнергия
|
4061.5
|
6.8
|
|
б)
вода
|
840
|
1.4
|
|
Итого
|
4901.5
|
8.2
|
|
3.Основная
заработная плата
|
|
|
|
а)
исполнителя
|
8800
|
14.8
|
|
б)
руководителя и консультантов
|
10370
|
17.4
|
|
Итого
|
19170
|
32.2
|
|
4.
Дополнительная заработная плата
|
3834
|
6.4
|
|
5.
Отчисления на социальное страхование
|
5981.04
|
10.0
|
|
6.
Стеклянные приборы и посуда
|
4157.9
|
7.0
|
|
7.
Амортизационные отчисления
|
1111.8
|
1.9
|
|
Итого
прямых затрат
|
39749.5
|
66.66
|
|
8.
Накладные расходы
|
19874.8
|
33.33
|
|
Всего
затрат
|
59624.3
|
100
|
На основании величины научно-технического
эффекта коэффициент, учитывающего поощрительную надбавку за качество
разработки, равен 1.3.
Итак с учётом всех затрат договорная цена на НИР
ЦН равна:
ЦН = 59624.3 ∙ (1+25/100) ∙1.3
= 96889.5 руб.
Выводы по технико-экономической оценке
результатов НИР
При выполнении данной работы ставилась задача
усовершенствовать метод извлечения и разделения молибдена и кобальта из
отработанных катализаторов на основе оксида алюминия путём сорбции. Поскольку
работа посвящена в основном изучению влияния различных факторов на процесс
сорбции металлов, а также нахождение оптимальных условий ионного обмена, то на
данной стадии выполнения работы не представляется возможным проведение
экономического анализа. Данная работа носит характер начальных разработок и
поиска оптимальных условий проведения процесса. Она будет дальше продолжена на
кафедре редких и рассеянных элементов. В результате исследований, выяснилось:
1. В работе используются не дорогие
реагенты (0.01% от общих затрат) ;
2. Стеклянные приборы и посуда вполне
доступны (7,0%);
. Энергетические затраты сравнительно не
высоки (8.2%);
. Метод можно использовать в промышленном
масштабе;
5. Основными статьями расходов являются накладные
расходы и основная заработная плата исполнителя;
Приложение Д
(обязательное)
Стандартизация
При выполнении настоящей дипломной работы
учитывались требования СТП ЛТИ 03-85 КС УКДВ. Порядок
введения и контроля за соблюдением стандартов в институте.
Состав работы и содержание ее основных частей
соответствует СТП СПбГТИ 017-97. Виды учебных занятий. Положение о выпускной
квалификационной работе дипломированного специалиста (инженера). Общие
требования.
Библиографическое описание соответствует рекомендациям
Р 01-97. Библиографическое описание.
Оформление пояснительной записки выполнено
согласно требованиям СТП СПБГТИ 006-2005. Подготовка и оформление авторских
текстовых оригиналов для издания.
Порядок проведения научно-исследовательских
работ соответствовал ГОСТ 7.32-2001 СИБИД. Отчет о
научно-исследовательской работе. Общие требования и правила оформления.
При составлении библиографического описания
сокращения слов и словосочетаний руководствовались ГОСТ 7.12-77
СИБИД. Сокращение русских слов и словосочетаний в библиографическом описании.
Единицы физических величин приведены в
соответствии с ГОСТ 8.417-2002. Единицы величин.
При выполнении экспериментальной части дипломной
работы соблюдались санитарные нормы и правила согласно:
1) ГОСТ
12.0.003-74
ССБТ. Опасные и вредные производственные факторы. Классификация.
2) ГОСТ
12.1.005-88
ССБТ. Воздух рабочей зоны. Общие санитарно-гигиенические требования.
3) СНиП
23-05-95
Естественное и искусственное освещение производственных помещений.
4) ГОСТ
12.4.021-75
ССБТ. Системы вентиляционные. Общие требования.
Использованные в работе сырье и материалы
соответствовали:
|
Гидроксид
натрия
|
ГОСТ
4328-77
|
ГОСТ
4204-77
|
|
Карбонат
натрия
|
ГОСТ
83-79
|
|
Тиомочевина
|
ГОСТ
6344-73
|
|
Роданид
аммония
|
ГОСТ
27067-86
|
|
Соляная
кислота
|
ГОСТ
3118-77
|
|
Гидроксид
аммония
|
ГОСТ
3760-79
|
|
Сульфат
меди(II)
|
ГОСТ
4165-78
|
|
Карбонат
аммония
|
ГОСТ
3770-75
|
|
Хлорид
аммония
|
ГОСТ
1773-72
|
|
Сульфат
натрия
|
ГОСТ
4166-76
|
|
Гидроксид
алюминия
|
ТУ
6-09-1473-77
|
|
Гидрокарбонат
аммония
|
ГОСТ
3762-69
|
|
Молибдат
натрия
|
ГОСТ
18289-78
|
При постановке экспериментов и обработке их
результатов использовался: СТП ЛТИ 2.075.008-81 КС УКДВ. Методы
обработки результатов наблюдений. Прямые измерения с многократными
наблюдениями.
ПРИЛОЖЕНИЕ Е
(обязательное)
Виды и объемы, выполненных с использованием ЭВМ
При выполнении работ настоящей дипломной работы
широко использовалась электронно-вычислительная техника. Как указано в разделе
« Технико-экономическая оценка результатов исследования», общие затраты
машинного времени составили 50 часов.
Наиболее активно вычислительная техника
применялась для построения графиков, таблиц и общего оформления дипломной
работы.
Все работы были проведены в лаборатории
вычислительной техники и инструментальных методов анализа кафедры технологии
редких и рассеянных элементов на персональной ЭВМ IBM
PS/AT
Pentium.
При помощи ЭВМ были реализованы:
обработка экспериментальных данных
построение графиков, таблиц и общее оформление
дипломной работы.
Для обработки экспериментальных данных и
построения графиков, применялись следующие готовые программные средства: Microcal
Origin (версия 6.1), Microsoft
Word (версия Office
2007). В результате применения электронно-вычислительной техники была
значительно увеличена скорость обработки экспериментальных данных, а также
повышена точность её результатов.