Патологічна анатомія хромосоми-1
Вступ
Хромосомы - це великі молекулярні
структури, де міститься близько 90 % ДНК клітини. Кожна хромосома складається з
довгого полімеризованого ланцюга ДНК, що містить гени, регуляторні елементи та
проміжні нуклеотидні послідовності. Слово «хромосома» походить від грецьких
слів «хрома» - колір та «сома» - тіло.
Хромосомний набір людини складається
з 23 пар хромосом, в яких зберігається вся спадкова інформація організму. При
пошкодженні спадкової інформації організму відбуваються негативні зміни
(мутації) або хромосомні захворювання.
До хромосомних захворювань вiдносять
форми патологiй, що клiнiчно проявляються множинними вродженими вадами
розвитку, генетичним підгрунтям яких є генетичні вiдхилення вiд нормальної
кiлькостi хромосомного матерiалу в клiтинах органiзму.
Сучасний перiод вiдрiзняється
видiленням цитогенетично i клiнiчно бiльш рiзних синдромiв, що обумовленi
дисбалансом по окремих сегментах практично всiх 22 аутосом i 2 статевих
хромосом. Роль хромосомної патологiї в загибелi ембрiона i плода у людини в
загальнiй кiлькостi множинних вад розвитку дуже велика. В середньому близько 40
% дiагностованих спонтанних абортiв обумовленi хромосомними порушеннями,
близько 6 % всiх мертвонароджених мають змiни хромосомного апарату. На 1000
мертвонароджених немовлят 3-5 мають хромосомнi хвороби. Якщо ж всi випадки
множинних вроджених вад прийняти за 100 %, то 35-40 % будуть припадати на
порушення стану хромосом.
1. Хромосома І, загальні відомості

Мал.1 Ідіограмма першої хромосоми
людини
-а хромосома людини - найбільша.
Складається з 23 людських хромосом, одна з 22 аутосом людини. Хромосома містить
близько 248 млн пар основ, що становить приблизно 8% всього матеріалу ДНК
людської клітини. В даний час вважається, що на 1 -й хромосомі знаходяться 4234
гена, що більше раніше зроблених прогнозів щодо кількості генів.
Згідно цитогенетичної класифікації
хромосома 1 відноситься до хромосом групи А, що означає, що ця хромосома є
метацентриком, тобто має плечі (p - і q -) приблизно рівної довжини. Хромосома
І має велику гетерохроматинову ділянку, яка включає два центромерних бенда 1p11
і 1q11 і прицентромерні бенди 1q12, разом складові займають порядку 5% довжини
хромосоми 1.
Хромосома І містить менше
повторюваних диспергованих послідовностей, ніж у середньому у людському геномі
(32.49 % vs 45%, відповідно). Ця різниця обумовлена меншим вмістом
довгих диспергованих ядерних елементів (LINEs - long interspersed nuclear
elements), яких в хромосомі 1 міститься 11,34 % від всієї довжини хромосоми, у
той час як у середньому людський геном містить близько 20% LINE послідовностей.
Якщо говорити про гени, то хромосома
І має середню щільність генів близько 14.2 генів на 1 млн пар основ, що майже
вдвічі вище, ніж середній показник геному людини (7.8 генів на 1 млн пар основ)
Хромосома І містить кілька кластерів
генів, що кодують функціонально або структурно пов'язані транскрипти. Найбільш
значущим є кластер генів епідермального диференціювання, що займає 2.5 млн пар
основ в бенді 1q21. Цей кластер містить 14 генів, які специфічно експресуються
під час залежного від кальцію диференціювання кератиноцитів, і 13 додаткових
генів, що належать до S100 сімейства кальцій -зв'язуючих білків.
На хромосомі 1 перебувають два
кластери генів канонічних гістонів: у бенді 1q21 знаходиться кластер HIST2, що
містить 6 гістонових генів, а в бенді 1q42 знаходиться кластер HIST3, що
складається з трьох генів.
На q -плече в бендах 1q22 - q23
знаходиться кластер генів, що кодують CD1 антигени (CD1A - CD1E), також на цьому
плечі в бендах 1q23 - q25 знаходиться кластер генів, що кодують флавін -містять
монооксигенази (FM01-FM05)
На p -плече найбільш значущими
кластерами є кластер генів, що кодують амілази (AMY1A - AMY1C, AMY2A, AMY2AB),
який знаходиться в бенді 1p21, кластер генів коннексінов (GJA4, GJA10, GJB3 -
GJB5) в бенді 1p34 - p35.1, а також кластер генів глутатіон -S- трансфераз
(GSTM1-GSTM5) в бенді 1p13.
2. Загальна характеристика
хромосомних аномалій
Нормальне диплоїдне число хромосом у
людини дорiвнює 46. Через недосконалiсть цитологiчної технiки загальне число
хромосом у людини довго (з 1912 до 1956 року) вважали рiвним 48. I лише в 1956
роцi шведськi цитологи Н.Tjio i A. Levan застосовували удосконалену цитологiчну
методику, довели, що модельна кiлькiсть хромосом у людини дорiвнює 46. Цi данi
були пiдтвердженi в тому ж роцi англiйськими вченими С.Е. Ford i J.L. Hamerton.
З цього часу почався бурхливий розвиток цитогенетики.
Хромосомний комплекс людини
складається з 23 пар. З них 22 пари -аутосоми, а 23-тя пара - статевi
хромосоми, основна роль яких полягає у визначеннi статi. Так, жiноча стать
представлена двома Х-хромосомами - ХХ, а чоловiча - ХY-хромосомами. Слiд
вiдмiтити, що багато спадкових захворювань як генних, так i хромосомних,
пов'язанi iз змiнами в статевих хромосомах (Х i Y).
хромосом мiстять соматичнi клiтини.
Статевi клiтини мають хромосомний набiр вдвiчi менший - 23 хромосоми. Аутосоми
однаковi як у чоловiкiв, так i у жiнок.
А - комплекс ознак, що дозволяють
запiдозрити хромосомну аномалiю. Це загальнi ознаки: фiзичний недорозвиток, ряд
дизморфiй мозкового i лицьового черепа, клишоногiсть, ряд аномалiй внутрiшнiх
органiв (серця, легень, нирок).
В - ознаки, якi зустрiчаються в
основному при певних хромосомних захворюваннях. Їх комбiнацiя дозволяє в
бiльшостi випадкiв дiагностувати хромосомну аномалiю. Зокрема, при трисомiї
18-ї хромосоми (хвороба Едвардса) - це долiхоцефалiя (89,6 %), флексорне
положення кистей (96,1 %), "стопа-качалка" (76,2 %), короткий i
широкий палець стопи (70,6 %). При трисомiї по 13-й хромосомi (хвороба Патау) -
це щiлина верхньої губи i пiднебiння (68,7 %), флексорне положення кистей (44,4
%), косоокiсть (31,4 %), дефект скальпа (30,5 %).
С - ознаки, характернi лише для
окремих хромосомних аномалiй.
Вважають, що 0,5 % всiх
новонароджених мають хромосомнi аномалiї. Приблизно 1:400 хлопчикiв i 1:600
дiвчаток мають порушення статевих хромосом. У недоношених дiтей хромосомнi
аномалiї зустрiчаються в 4 рази частiше, нiж у доношених. Ще бiльше хромосомних
захворювань в матерiалi абортiв - в середньому 40 %.
. Патанатомія хромосоми 1
Хромосома 1 - це найважливіша
хромосома в генетичному наборі людини. Порушення її будови може призвести до
серйозних наслідків. Звісно, при мутаціях у першій хромосомі не можна говорити
про повноцінне життя людини. Виникнення тієї чи іншої хвороби обумовлено
пошкодженням певних локусів, де розташовуються гени 1 хромосоми. На малюнку
нижче приведено розташування цих локусів на хромосомі.
хромосома патологічна аномалія
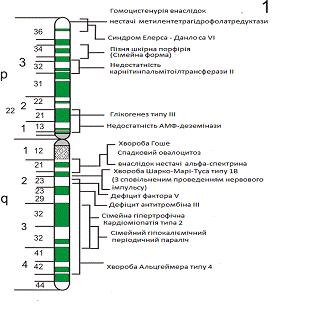
Мал.4 Локуси захворювань на
хромосомі 1
Майже всi хромосомнi захворювання
супроводжуються численними ураженнями скелету, порушеннями психiки, вродженими
вадами зовнiшнiх i внутрiшнiх органiв, затримкою росту, ураженням нервової,
ендокринної та iнших систем, зниженою регенераторною функцiєю, пiдвищеною
захворюванiстю i смертнiстю.
Розглянемо декілька прикладів
захворювань, спричинених мутаціями у будові хромосоми 1, а також шляхи їх
лікування.
Гомоцистенурія
Вперше гомоцистинурія описана в 1962
р. Карсен і Нейлом. До теперішнього часу описано більше 100 хворих. В основі
захворювання лежить відсутність або зниження активності ферменту
цистатіонінсинтетази, йому в якості кофактора потрібен вітамін В12, а в якості
субстрату - фолієва кислота.
Гомоцистеїн є проміжним продуктом
розпаду метіоніну і в нормі не міститься в плазмі та сечі, але дефекти на трьох
різних етапах ферментації можуть призвести до гомоцистинемії і гомоцистинурії.
Існує класична гомоцистинурія, пірідоксинчутлива і пірідоксинрезістентна.
Захворювання є аутосомно - рецесивним, частота захворювання при народжені
становить 1:200000населення.
Клінічні прояви гомоцистинурії
різні, однак найбільш характерні наступні: розумова відсталість, ектопія
кришталиків, кісткові деформації, тромбоемболії і серцево - судинна патологія.
Такі діти при народженні виглядають_здоровими.
Симптоми: Хворобу встановлюють у
віці 3 - х років, коли виявляють підвивих кришталика, але в більшості випадків
яскрава клініка розвивається до 10 років. Батьки помічають, що під час швидкого
руху головою райдужки дитини тремтять. Потім проявляються й інші очні симптоми:
міопія, астигматизм, глаукома, катаракта, відшарування сітківки, атрофія
зорового нерва. Аномалії скелета у таких дітей проявляються особливо часто.
Перш за все, відзначається диспропорція статури у вигляді укорочення тулуба,
подовження кінцівок, помірно виражений остеопороз кісток, сколіоз, викривлення
гомілки, деформації грудної клітки, високе піднебіння, порожниста стопа. У цих
дітей блакитні очі і специфічна еритема у формі метелика (гарячі вилиці) і
еритематозна плямистість кінцівок. Часто прогресує розумова відсталість, проте
у деяких хворих інтелект не порушений. Близько 10-15 % хворих страждають від
судом.
Наступним ускладненням цього
захворювання є тромбоемболія судин різного діаметру, зокрема головного мозку,
яка може проявлятися в будь-якому_віці.
Діагностика захворювання полягає у
виявленні гомоцистину в сечі. Проводять пробу з нітропрусидом, а також паперову
хроматографію амінокислот. Визначають вміст амінокислот в плазмі.
Лікування: великі дози вітаміну В6 і
фолієвої кислоти, дієтотерапію - продукти з низьким вмістом метіоніну.
Допустимий вміст метіоніну - 29-45 мг на 1 кг маси 1 раз на добу. Замість
коров'ячого молока можна використовувати козяче, яке містить менше метіоніну, а
також кобиляче молоко.
Показником ефективності лікування
гомоцистинурии є рівень метіоніну в крові до 0,01 г / л і відсутність
гомоцистину в крові. Враховуючи те, що наркоз підвищує ризик внутрішньосудинної
коагуляції, важливо мати на увазі, що планові операції хворим гомоцистинурія
під загальним наркозом протипоказані, як і ангіографії.
Синдром Елерса-Данлоса - дуже
рідкісне спадкове захворювання сполучної тканини, яке характеризується
патологічною рухливістю в суглобах, надлишкової еластичністю шкіри і крихкістю
тканин.
Цей синдром має декілька різновидів,
які визначаються аномаліями в різних генах, що контролюють вироблення сполучної
тканини. У багатьох дітей виявляється патологічна рухливість у суглобах
(доброякісна гіпермобільність) без інших симптомів; з часом вона зазвичай
зменшується.
Симптоми: Шкіру хворого можна
розтягнути на кілька сантиметрів, але вона повертається в нормальне положення,
якщо її відпустити. Характерна патологічна рухливість у суглобах. Часто вздовж
кісткових утворень, особливо ліктів, колін і гомілок, формуються широкі рубці.
Під шкірою можуть розвиватися невеликі тверді круглі вузли, які помітні на
рентгенограмі.
Незначні пошкодження можуть
супроводжуватися утворенням широких зяючих ран, причому кровотеча зазвичай
невелика. У невеликої частини хворих з синдромом Елерса- Данлоса
спостерігається тенденція до сильної кровоточивості при пошкодженні покрівних
тканин. Накладення швів на рану ускладнено, оскільки шви прорізують неміцні
тканини. Органи тіла легко пошкоджуються, що часто викликає ускладнення під час
операцій. Характерні розтягнення і вивихи. Приблизно у 25% дітей розвиваються
горб і викривлення хребта (кіфосколіоз), 90 % мають плоскостопість. Часто
утворюються грижі і патологічні випинання стінок кишечника (дивертикули). У
рідкісних випадках виникають кровотечі або розриви (перфорації) кишечника.
Для вагітних жінок з цим синдромом
характерні передчасні пологи, оскільки тканини у таких жінок легко
розтягуються. Якщо синдром Елерса- Данлоса наявний у плоду, це може призвести
до раннього розриву плодових оболонок. Проведення вагітній жінці операції для
полегшення пологів, наприклад Кесаревого розтину або розрізу поблизу
вагінального отвори (епізіотомії), може становити небезпеку через неміцність
тканин. До, під час або після пологів можливе важке кровотеча.
Незважаючи на можливість різних
ускладнень, пацієнти з синдромом Елерса- Данлоса мають нормальну тривалість
життя, однак розвиток ускладнення (наприклад, розрив кровоносної судини) може
привести до летального результату.
Причини: Синдром Елерса-Данлоса - це
патологія генів, що кодують білок колаген - компонент сполучної тканини. При
цій патології колаген структурно і функціонально неповноцінний, що і призводить
до розвитку симптомів.
Діагностика: При підозрі на синдром
Елерса-Данлоса, яке виникає виходячи з клінічної симптоматики, можливе
проведення молекулярно-генетичного дослідження.
Лікування: Лікування не розроблено.
Через неміцність тканин хворим потрібно уникати травм. Якщо хворі з синдромом
Елерса-Данлоса хочуть мати дітей, їм рекомендують порадитись з лікарем, яке
дозволяє оцінити ризик успадкування синдрому їх дітьми.
Хвороба Альцгеймера
Хвороба Альцгеймера - це невиліковне
дегенеративне захворювання центральної нервової системи, одна з форм деменції,
характеризується поступовою втратою розумових здібностей (пам'ять, мова,
логічне мислення).
Прийнято виділяти два типи хвороби
Альцгеймера:
з раннім початком: перші симптоми
з'являються у віці до 60 років. Цей тип хвороби зустрічається рідше, проте він
швидше прогресує,
з пізнім початком: цей тип хвороби,
як правило, зустрічається частіше. Перші симптоми хвороби проявляються у віці
після 60 років.
Симптоми: Симптоми хвороби
Альцгеймера можна розділити на ранні та пізні. Ранні симптоми хвороби
Альцгеймера включають в себе:
труднощі з вирішенням завдань, які
вимагають розумового процесу;
втрата можливості знайти дорогу при
звичному маршруті;
проблеми з мовленням, пов'язані з
неможливістю підібрати назви звичних речей;
втрата колишніх інтересів і
захоплень;
проблеми з поверненням речей на їх
звичайні місця;
З прогресуванням хвороби симптоми
стають більш великими:
порушення режиму дня і сну, часті
пробудження вночі;
депресія, відчуття тривоги;
труднощі з виконанням звичних
завдань: приготуванням їжі, вибором одягу по сезону, водінням;
труднощі з читанням і письмом;
забудькуватість деталей недавніх
подій;
забудькуватість деталей власного
життя і власного «я», галюцинації;
втрата здатності розпізнавати
небезпеку;
плутання промови, нездатність
правильного формулювання пропозиції і розповіді;
проблеми з підтримкою соціальних
контактів.
На останніх стадіях хвороби люди
більше не здатні:
сприймати людську мову;
пізнавати близьких людей;
виконувати повсякденні справи, такі
як прийом їжі, одягання і дотримання особистої гігієни.
Підсумок
Отже хвороби, спричинені мутацією
структури 1 хромосоми, можуть призвести до найгірших наслідків. Для уникнення,
або зменшення захворюваності генетичними хворобами, треба як можна активніше
проводити пропаганду здорового способу життя, і спрямовувати найбільшу роботу
на жінок. Оскільки дуже часто генетична інформація плоду порушується внаслідок
того, що мати вела неправильний спосіб життя, вживала алкоголь та різні
наркотичні засоби або шкідливі препарати. Також дуже важливим є спосіб життя
батька, оскільки і його генетичний матеріал бере участь у формуванні нового
організму.
Звісно, що не кожна хвороба,
спричинена мутацією генів хромосоми 1 залежить від способу життя, який вела
мати або батько, багато хвороб можуть передаватися від батьків внаслідок вже
існуючої мутації у їхньому спадковому матеріалі. Наприклад хвороба Альцгеймера
або хвороба Гоше. Все це визначає необхiднiсть знання акушерами, педiатрами,
дитячими ендокринологами, сiмейними лiкарями, психоневрологами, ортопедами,
патологоанатомами i iншими спецiалiстами етiологiї, клiнiчної картини,
принципiв дiагностики, лiкування i профiлактики цiєї великої групи патологiчних
станiв.
Список використаної літератури
1.Медична біологія\За ред. В.П. Пішака, Ю.І. Бажори, Підручник.-
Вінниця: НОВА КНИГА, 2004р.
.http://www.zdorovieinfo.ru
.http://vse-pro-geny.com
. http://uk.wikipedia.org