Реакции окисления в органической химии
Санкт-Петербургский
Государственный Университет
Реакции
окисления в органической химии
Воронаев Иван Геннадьевич
1. Введение
Окисление органических соединений занимает
важное место в органическом синтезе. Помимо этого, реакции окисления находят
широкое применение в промышленном органическом и нефтехимическом синтезе.
Окисляют парафиновые углеводороды, нафтены, арены, олефины, диены, спирты,
альдегиды, меркаптаны, амины и другие органические соединения.
Окислительно-восстановительные реакции в
органической химии представляют наибольший интерес, т.к. селективность перехода
из одной степени окисления в другую сильно зависит от правильного выбора
реагента и условий проведения реакций.
Под окислительным процессом понимают
превращение, протекающее с увеличением степени окисления атома. Их подразделяют
на окислительное замещение (замена одного или нескольких атомов молекулы на
более электроотрицательные атомы), окислительное присоединение (разрыв кратных
связей и образование простых связей с более электроотрицательным элементом) и
окислительное элиминирование (реакции дегидрирования).
Следует отметить, что термины «окислительный
процесс» и «реакции окисления» несколько различны. Под реакциями окисления
понимают реакции, сопровождающиеся введением в молекулу атомов кислорода, или
формальное отщепление молекулы водорода. Окислительные же процессы
подразумевают более широкий круг превращений. Процессы окисления по разным
признакам можно классифицировать следующим образом:
парциальное окисление;
глубокое окисление (обычно до СО2 и Н2О);
гомогенное газофазное окисление - обычно
радикально-цепное автоокисление или инициированное окисление, процессы горения;
гомогенное гетерофазное (жидкофазное) окисление
(делится на радикально-цепное автоокисление (или инициированное окисление) и
каталитическое окисление);
гетерогенно-каталитическое окисление.
Понятия «восстановительные процессы» и «реакции
восстановления» практически тождественны и означают формальное присоединение
водорода или замену атома на водород.
2. Основные окислители
Для окисления органических веществ обычно
используют соединения переходных металлов, кислород, озон, перекиси и
соединения серы, селена, йода, азота и другие.
Из окислителей на основе переходных металлов
преимущественно применяют соединения хрома и марганца.
Наиболее распространенные соединения хрома (VI)
- это раствор бихромата калия K2Cr2O7
в серной кислоте, раствор триоксида хрома CrO3
в разбавленной серной кислоте (реактив Джонса), комплекс триоксида хрома с
пиридином (реактив Коллинза) и реактив Кори - комплекс CrO3
с пиридином и HCl
(пиридинийхлорхромат).
Также для окисления метильной группы в
ароматических соединениях до альдегидной (реакция Этара) используют
хромилхлорид (CrO2Cl2).
При окислении органических веществ хром (VI)
в любой среде восстанавливается до хрома (III),
однако, окисление в щелочной среде в органической химии не находит
практического применения.
Перманганат калия (KMnO4)
в разных средах проявляет различные окислительные свойства, при этом сила
окислителя увеличивается в кислой среде.
Манганат калия K2MnO4
и оксид марганца (IV) MnO2
проявляют окислительные свойства только в кислой среде. Диоксид марганца -
селективный окислитель, применяется для дегидрирования гетероциклов,
ароматизации циклоалкенов и селективного окисления первичных и вторичных
спиртов до альдегидов и кетонов. Селективно окисляет аллильный гидроксил в
присутствии других ОН-групп.
Гидроксид меди (II)
обычно используется для окисления альдегидов. Реакция проводится при
нагревании, при этом голубой гидроксид меди (II)
превращается сначала в гидроксид меди (I)
желтого цвета, который затем разлагается до красного оксида меди (I).
В качестве окислителя альдегидов также применяют аммиачный раствор гидроксида
серебра (реакция серебряного зеркала).
Следует обратить внимание на то, что уравнения
полуреакций восстановления для указанных соединений хрома, марганца, меди и
серебра не зависят от окисляемого органического вещества, а определяются только
кислотностью среды:
 (кислая среда)
(кислая среда)
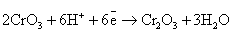 (нейтральная среда)
(нейтральная среда)
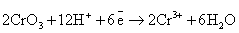 (кислая среда)
(кислая среда)
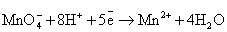 (кислая среда)
(кислая среда)
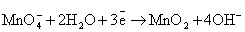 (нейтральная среда)
(нейтральная среда)
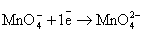 (основная среда)
(основная среда)
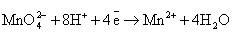
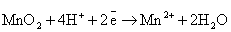
2 Cu(OH)2 + 2ē ® CuO2 +
2OH¯ + H2O
[Ag(NH3)2]+ +
1ē ® Ag + 2NH3
Большинство окисляющих агентов,
используемых в органическом синтезе, относятся к различным классам
неорганических соединений (простые вещества, оксиды металлов, перекисные
соединения, соли, кислоты, содержащие элемент в высокой степени окисления), и
лишь малую их часть составляют органические окислители (пероксикислоты, хиноны
и др.).
3. Окисление различных классов органических
соединений
.1 Окислительная деструкция алканов
Как уже говорилось, окисление органического
вещества - введение в его состав кислорода и (или) отщепление водорода.
Восстановление - обратный процесс (введение водорода и отщепление кислорода).
Учитывая состав алканов (СnH2n+2), можно сделать вывод об их неспособности
вступать в реакции восстановления, но возможности участвовать в реакциях
окисления.
Алканы - соединения с низкими степенями
окисления углерода, и в зависимости от условий реакции они могут окисляться с
образованием различных соединений.
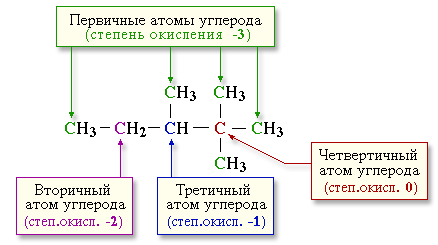
При обычной температуре алканы не вступают в
реакции даже с сильными окислителями (Н2Cr2O7, KMnO4 и т.п.). При внесении в
открытое пламя алканы горят. При этом в избытке кислорода происходит их полное
окисление до СО2, где углерод имеет высшую степень окисления +4, и воды.
Горение углеводородов приводит к разрыву всех связей С-С и С-Н и сопровождается
выделением большого количества тепла (экзотермическая реакция).
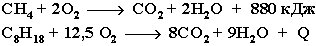
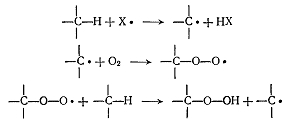
Общепринято, что механизм окисления алканов
включает радикальный цепной процесс, поскольку сам по себе кислород
малореакционноспособен, чтобы оторвать атом водорода от алкана нужна частица,
которая будет инициировать возникновение алкильного радикала, который будет
реагировать с кислородом, давая пероксирадикал. Затем пероксирадикал может
оторвать атом водорода от другой молекулы алкана с образованием
алкилгидропероксида и радикала.
Возможно окисление алканов кислородом воздуха
при 100-150оС в присутствии катализатора - ацетата марганца, данную реакцию
применяют в промышленности. Окисление происходит при продувании тока воздуха
через расплавленный парафин, содержащий соль марганца.
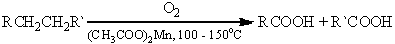
Т.к. в результате реакции образуется
смесь кислот, то их отделяют от непрореагировавшего парафина растворением в
водной щелочи, а затем нейтрализуют минеральной кислотой.
Непосредственно в промышленности
этот метод применяется для получения уксусной кислоты из н-бутана:
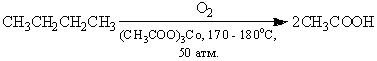
.2 Окисление алкенов
Реакции окисления алкенов
подразделяют на две группы: 1) реакции, в которых сохраняется углеродный
скелет, 2) реакции окислительной деструкции углеродного скелета молекулы по
двойной связи.
.2.1 Реакции окисления алкенов с
сохранением углеродного скелета
. Эпоксидирование (реакция
Прилежаева)
Ациклические и циклические алкены
при взаимодействии с перкислотами в неполярной среде образуют эпоксиды
(оксираны).
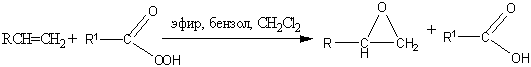
Также оксираны можно получить
окислением алкенов гидропероксидами в присутствии молибден-, вольфрам-,
ванадийсодержащих катализаторов:
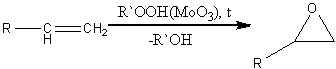
Простейший оксиран - окись этилена -
получают в промышленности окислением этилена кислородом в присутствии серебра
или оксида серебра как катализатора.
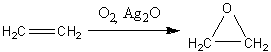
. анти-Гидроксилирование (гидролиз
эпоксидов)
Кислотный (или щелочной) гидролиз
эпоксидов приводит к раскрытию оксидного цикла с образованием трансдиолов.
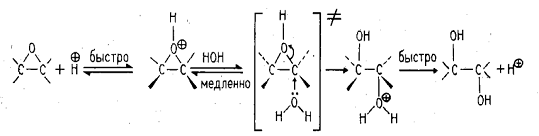
В первой стадии происходит
протонирование атома кислорода эпоксида с образованием циклического оксониевого
катиона, который раскрывается в результате нуклеофильной атаки молекулы воды.
Раскрытие эпоксидного кольца,
катализируемого основанием, также приводит к образованию транс-гликолей.

. син-Гидроксилирование
Одним из старейших методов окисления
алкенов является реакция Вагнера (окисление перманганатом калия). Первоначально
при окислении образуется циклический эфир марганцевой кислоты, который
гидролизуется до вицинального диола:
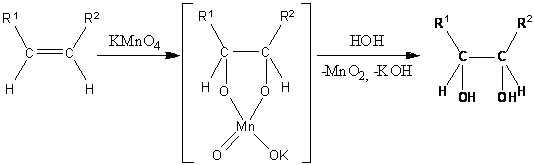
Помимо реакции Вагнера существует
другой метод син-гидроксилирования алкенов под действием оксида осмия (VIII), который
был предложен Криге. При действии тетраоксида осмия на алкен в эфире или
диоксане образуется черный осадок циклического эфира осмиевой кислоты - осмат.
Однако присоединение OsO4 к кратной связи заметно
ускоряется в пиридине. Полученный черный осадок осмата легко разлагают
действием водного раствора гидросульфита натрия:

.2.2 Окислительное расщепление
алкенов
К окислительному расщеплению алкенов
относятся реакции взаимодействия их с перманганатом калия в щелочной или в
серной кислоте, а также окисление раствором триоксида хрома в уксусной кислоте
или дихроматом калия и серной кислотой. Конечным результатом таких превращений
является расщепление углеродного скелета по месту двойной связи и образование
карбоновых кислот или кетонов.
Однозамещенные алкены с концевой
двойной связью расщепляются до карбоновой кислоты и углекислого газа:
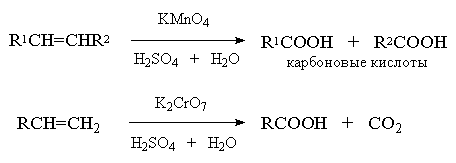
Если оба атома углерода при двойной
связи содержат только по одной алкильной группе, то образуется смесь карбоновых
кислот:
А вот если тетразамещенный при
двойной связи алкен - кетон:
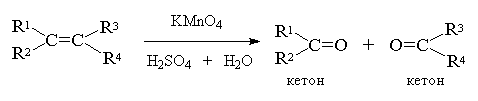
Гораздо большее препаративное
значение приорела реакция озонолиза алкенов. В течение многих десятилетий эта
реакция служила основным методом определения строения исходного алкена. Данная
реакция проводится пропусканием тока раствора озона в кислороде раствор алкена
в хлористом метилене или этилацетате при -80 … -100оС. Механизм данной реакции
установлен Криге:
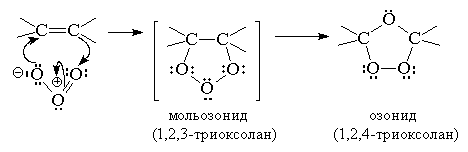
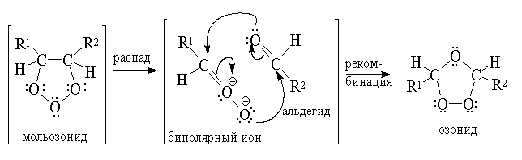
Озониды - нестабильные соединения, разлагающиеся
со взрывом. Существует два способа разложения озонидов - окислительное и
восстановительное.
При гидролизе озониды расщепляются на
карбонильные соединения и перекись водорода. Перекись водорода окисляет
альдегиды до карбоновых кислот - это есть окислительное разложение:
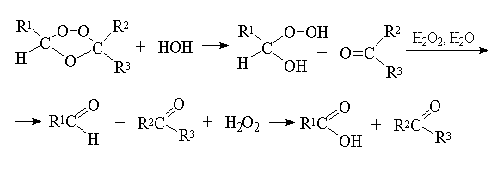
Гораздо более важное значение имеет
восстановительное расщепление озонидов. В качестве продуктов озонолиза
оказываются альдегиды или кетоны в зависимости от строения исходного алкена:
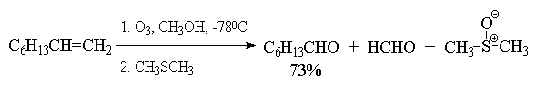
Помимо приведенных выше методов существует еще
один метод, предложенный в 1955 г. Лемье:

В методе Лемье не возникает трудоемких процедур
по отделению диоксида марганца, т.к. диоксид и манганат вновь окисляются
перйодатом до перманганат-иона. Это позволяет использовать только
каталитические количесвта перманганата калия.
.3 Окисление алкинов
Алкины окисляются менее легко, чем алкены,
однако при контролируемом окислении можно сохранить С-С связь и получить в
качестве продуктов реакции карбонильные соединения:
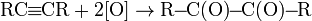
В качестве окислителя выбирают перманганат калия
в нейтральной среде при pH
7-8 и 0oC:
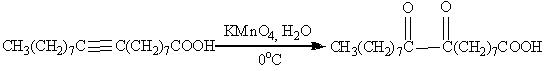
Ацетилен
<#"555928.files/image030.gif">(глиоксаль <#"555928.files/image031.gif">(глиоксалевая кислота
<#"555928.files/image032.gif">(щавелевая кислота
<#"555928.files/image033.gif">
Реакции алкинов с озоном также
протекают значительно медленнее, чем с алкенами. Так например в углеводороде
содержащем двойную и тройную связь, озонолиз преимущественно затрагивает
двойную связь. Полное же расщепление тройной связи в общем случае приводит к
образованию карбоновых кислот, однако частичное превращение дает
1,2-дикарбонильное соединение:
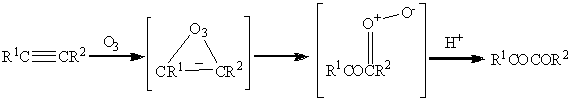
Окисление алкинов также возможно тетраоксидами
осмия и рутения такие реакции приводят к получению дикарбонильных соединений.
.4 Окисление диенов
Реакции окисления диенов во многом похожи на
окисление алкенов. Так, например, кумулированные диены чрезвычайно легко
окисляются под действием тех же реагентов, что и алкены. Водный раствор
перманганата калия в нейтральной или слабоосновной среде вызывает расщепление
алленов до карбонильных соединений:
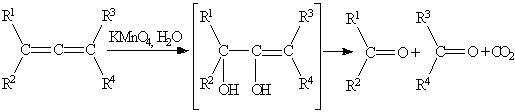
Окисление сопряженных диенов в
зависимости от используемого окислителя и условий проведения реакции может
протекать с образованием различных кислородсодержащих соединений.
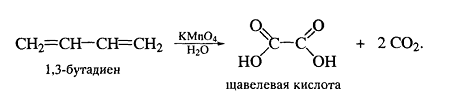
органический синтез
окисление циклический
Следует отметить, что в данных
условиях щавелевая кислота может легко окислиться до углекислого газа и воды.
Озонолиз для алкадиенов протекает по
тому-же механизму, что и для алкенов:
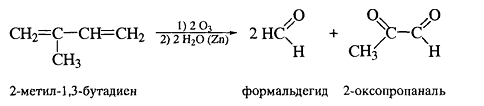
Озон также расщепляет аллены до карбонильных
соединений и углекислого газа, и этот тест может быть использован для
определения кумуленов.
.5 Окисление циклических соединений
Рассмотрим, как ведет себя циклопропан с
окислителями, оказывается, что кольцо циклопропана очень устойчиво по отношению
к действию самых разнообразных окислителей: озона, перманганата и дихромата
калия, что резко отличает циклопропан и его производные от алкенов. Так,
например, алкилциклопропаны реагируют только по α-положению
без раскрытия цикла. Аналогично окислительный озонолиз
транс-1,2-дифенилциклопропана дает транс-циклопропан-1,2-дикарбоновую кислоту:
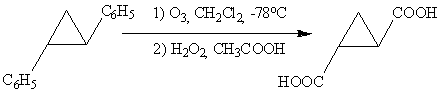
.6 Окисление аренов
Бензольное кольцо устойчиво к
действию окислителей, что резко отличает бензол от других углеводородов.
Известны двухстадийные процессы
окисления бензола в фенол, которые используются в промышленности. Например,
метод Рашига:
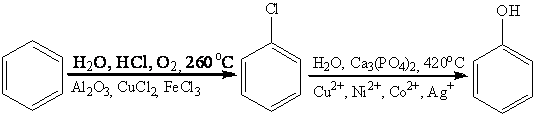
и множество других методов, через
хлорирование, сульфирование; кумольный метод, гетерогенное окисление бензола на
V2O5 в
малеиновый ангидрид, окисление реактивом Фентона (водный раствор FeSO4 и H2O2) или
реактивом Уденфринда (ион Fe2+, кислород, аскорбиновая
кислота и ЭДТА), но все эти методы позволяют окислить бензол в фенол.
Более богата реакциями окисления
химия производных бензола, поскольку алкильные группы, присоединенные к
бензольному кольцу, легко окисляются под действием окислительных агентов.
Наиболее часто для окисления боковых
цепей используют соединения хрома (VI) и
перманганат калия.
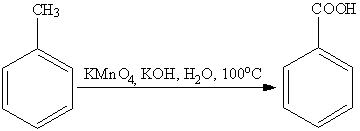
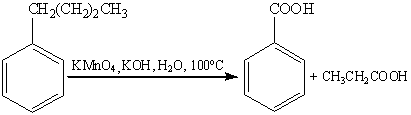
Однако, детальный механизм данных
реакций неизвестен до сих пор. Предполагается, что в качестве интермедиата
образуется бензильный катион, который затем превращается в эфир хромовой или
марганцевой кислоты. Гидролиз эфира и последующее окисление бензилового спирта
приводят к карбоновой кислоте.
Интересным является факт, что если
алкильная группа не содержит атомов водорода в α-положении по
отношению к бензильному кольцу, такая трет-алкильная боковая группа не
окисляется под действием Na2Cr2O7 или KMnO4. Так,
например, трет-бутилбензол окисляется в жестких условиях перманганатом калия до
триметилуксусной кислоты, т.е. окисляется само бензольное кольцо:
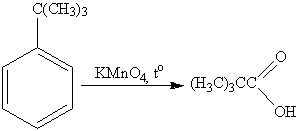
Оксид хрома (VI) в уксусном
ангидриде является превосходным реагентом для окисления метильной группы аренов
до альдегидной. Дальнейшему окислению в кислоту препятсвует образование
диацетата, который устойчив в этих условиях. Катализируемый кислотой гидролиз в
водном спирте приводит к образованию ароматического альдегида:
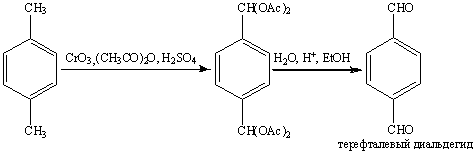
Уникальным является озонолиз аренов,
уникальность заключается в том, что этот метод позволяет расщеплять
ароматический углеводород в соответствии с его каноническими структурами,
составляющими резонансный гибрид.
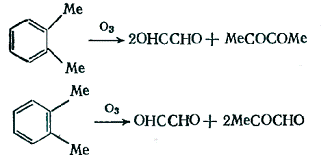
Бензольное кольцо в реакции с озоном значительно
менее реакционноспособно, чем, например, алкеновая двойная связь, а среди
простейших ароматических углеводородов реакционная способность возрастает при
переходе от бензола к нафталину и далее к фенантрену, т.к. увеличивается
энергия локализации связей.
Важнейшее промышленное значение имеют реакции
прямого окисления орто- и пара- ксилолов кислородом воздуха до фталевой и
терефталевой кислот соответственно в присутствии ацетата кобальта (II)
в уксуснокислом растворе:
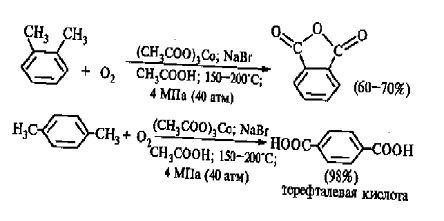

Окисление конденсированных ароматических
соединений приводит к различным продуктам в зависимости от используемого
реагента и условий проведения реакции. Реагенты на основе Cr
(VI) окисляют в кислой
среде нафталин и алкилнафталины до нафтохинонов, а вот дихромат натрия в водной
растворе окисляет только алкильные группы. Окисление нафталина перманганатом
калия в щелочной среде сопровождается разрушением одного ароматического кольца
с образованием моноциклических дикарбоновых кислот:

Также практическое значение имеет реакция
окисления нафталина кислородом над V2O5:
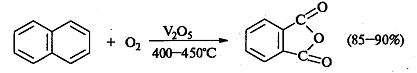
.7 Окисление спиртов
Окисление спиртов - это классический и хорошо
разработанный метод получения карбонильных соединений. Строение соединения,
образующегося при окислении спирта, зависит от того, сколько атомов водорода
связано с углеродом, несущим ОН-группу, т.е. от того, является ли спирт
первичным, вторичным или третичным.
Имеет место упомянуть, что первичные спирты
окисляются в альдегиды, а вторичные в кетоны. Также считается, что третичные
спирты в обычных условиях не окисляются, а в очень жестких их окисление
сопровождается деструкцией углеродного скелета.
Известно четыре основных пути превращения
первичных спиртов в альдегиды, а вторичных - в кетоны.
. Действие сильных окислителей.
Чаще всего, однако, для окисления вторичных
спиртов до кетонов используют реагент Джонса - раствор CrO3
(или Na2Cr2O7)
в водной серной кислоте (или водном ацетоне с добавкой H2SO4).
Титрование реагентом Джонса вторичных спиртов приводит к быстрому их окислению
до кетонов с высоким выходом, причем при этом не затрагиваются двойные или
тройные связи, присутствующие в молекуле субстрата.
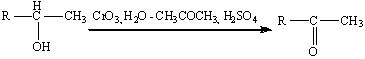
Окисление вторичных спиртов может
протекать с деструкцией углеродного скелета, т.е. полученные кетоны можно
окислить до карбоновых кислот в более жестких условиях. Классическим примером
служит промышленное окисление циклогексанола 50%-ой азотной кислотой до
адипиновой кислоты:
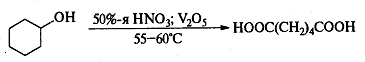
Самостоятельную и наиболее сложную
проблему при окислении первичных спиртов до альдегидов составляет дальнейшее
окисление альдегидов до карбоновых кислот. Так, например, реагент Джонса
окисляет первичные спирты до карбоновых кислот, альдегиды, несомненно,
образуются в качестве промежуточных продуктов, но быстро окисляются под
действием этого окислителя.
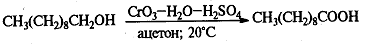
Для предотвращения этой реакции в
качестве окислителя используют комплексы хромового ангидрида с третичными
аминами, которые уменьшают окислительную способность хромового ангидрида и
делают окисление более селективным. В настоящее время лучшими реагентами
являются комплекс CrO3 с двумя молями пиридина: CrO3*2C5H5N (реагент
Саррета-Коллинза) и хлорхромат пиридиния CrO3Cl-C5H5NH+ (реагент
Кори) в хлористом метилене, последний не затрагивает кратную связь при
окислении.
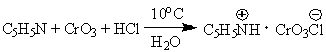
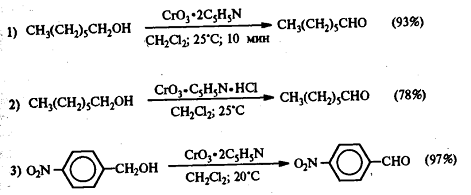
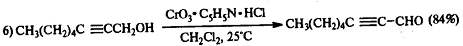
Для получения α,β-непредельных
альдегидов хорошо зарекомендовал себя активный MnO2
(свежеприготовленный из MnSO4 и KMnO4) в
инертных растворителях.
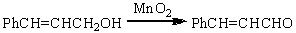
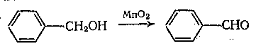
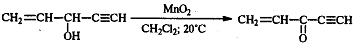
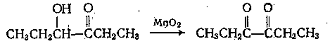
Перманганат калия редко применяется
для окисления спиртов в кетоны и альдегиды. Этот реагент менее селективен, чем
хром (VI); здесь
серьезную проблему представляет переокисление.
Мягким окислителем первичных спиртов
в альдегиды оказался карбонат серебра на целите. Этот дорогостоящий реагент
употребляется при стереоселективном мягком окислении лабильных циклопропил- и
циклобутилкарбинолов:

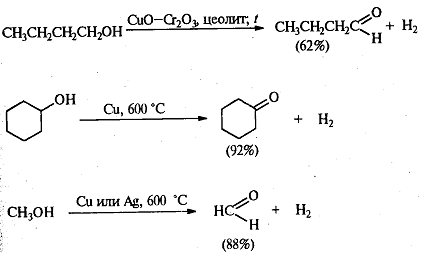
. Каталитическое дегидрирование.
При окислении первичных спиртов в
альдегиды каталитическое дегидрирование обладает преимуществом перед действием
сильных окислителей, а именно: предотвращает более глубокое окисление до
кислоты. Чаще всего применяют хромит меди, но используют и другие катализаторы
(серебро и медь). По этой реакции синтезированы и многие кетоны. Этим методом в
промышленных условиях получают формальдегид из метанола, масляный альдегид из
н-бутилового спирта и циклогексанон из циклогексанола.
. Окисление по Оппенауэру.
Если в качестве окислителя
используется кетон в присутствии основания (кетон при этом восстанавливается до
вторичного спирта), реакцию называют окислением по Оппенауэру. Наиболее широко
в качестве кетона применяется ацетон, бутанон и циклогексанон, а в качестве
основания - трет-бутилат алюминия. Главное достоинство этого метода состоит в
его высокой селективности. Может использоваться для окисления первичных
спиртов, но наибольшее значение метод имеет для синтеза кетонов из вторичных
спиртов (сохраняет С=С связь).
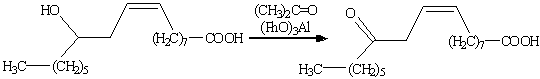
. Окисление по Сверну.
При окислении по Сверну используют
реагент, получающийся при взаимодейтвии ДМСО с оксалилхлоридом в дихлорметане
при -78оС - диметилхлорсульфонийхлорид.
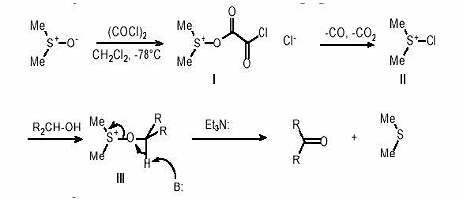
Образование аддукта I с оксалил хлоридом при
низкой температуре, который разлагается до диметилхлорсульфоний хлорида II.
Дальнейшие взаимодействие иметилхлорсульфоний хлорида со спиртами ведет
промежуточному аддукту III, который под действием основания превращается в
карбонильное соединение и диметилсульфид.
Помимо приведенных, существуют и другие методы
окисления (окисление по Кори-Киму, Дессу-Мартину), которые являются дорогой, но
зато хорошей альтернативой окисления спиртов.
Окислительное расщепление двухатомных спиртов
(1,2-диолов)
Существуют два классических метода
окислительного расщепления диолов и полиолов с помощью парайодной кислоты H5IO6
и ее солей (реакция Малапрада), а также тетраацетатом свинца Pb(OOCCH3)4
(реакция Криге):
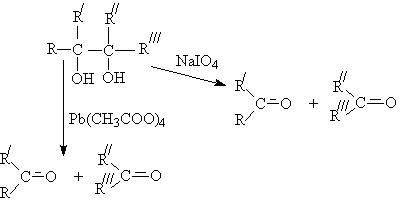
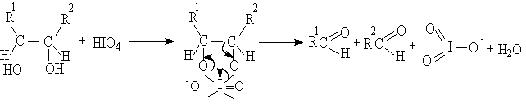
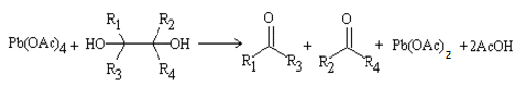
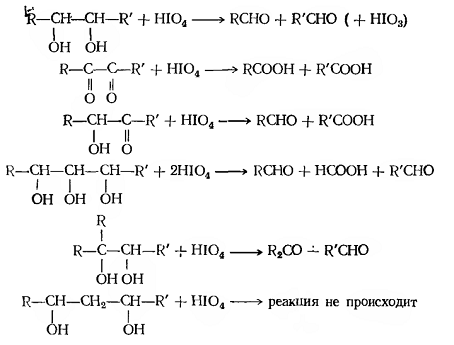
В общем случае процесс окисления диолов
протекает по сложной схеме, включающей последовательные и параллельные реакции,
в общем виде их можно изобразить так:
При этом, как и в случае одноатомных спиртов, по
продуктам окисления можно судить о структуре диола.
.8 Окисление фенолов
Окисление фенолов относится к числу сложных,
многостадийных процессов, механизм которых мало изучен. Очевидно лишь то, что
механизм окисления может сильно меняться в зависимости от природы окислителя и
включает стадию образования стабилизированных резонансом ароксильных радикалов:
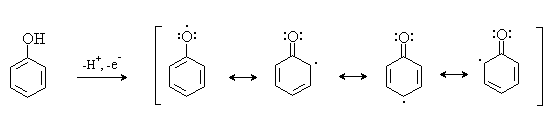
Направление дальнейших превращений зависит от
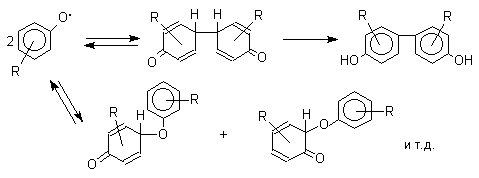
характера заместителей в ароматическом кольце. При окислении большинства
фенолов образуется несколько различных форм димеров в результате образования
новых связей С-С между орто-орто, орто-пара- и пара-пара-положениями
ароксильных радикалов, а также новых С-О связей между атомом кислорода одного
радикала и орто- или пара-положением другой радикальной частицы. Всего, таким
образом, образуется потенциально не менее пяти различных типов димеров, которые
находится в равновесии с исходным ароксильным радикалом. Например, для
монозамещенного фенола:
Сам фенол при окислении дихроматом натрия или
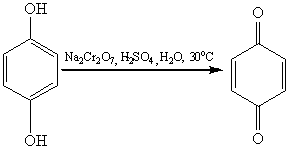
диоксидом марганца в серной кислоте образует пара-хинон:
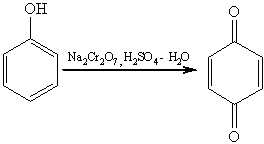
Универсальным окислителем фенолов
является соль Фреми - нитрозодисульфонат калия. Такое окисление идет в очень
мягких условиях по радикальному механизму и приводит к пара-хинонам:
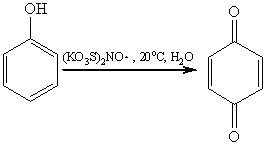
Двухатомные фенолы окисляются
подобно одноатомному фенолу, так например окисление гидрохинона (в качестве
окислителей можно использовать самые разнообразные реагенты: дихромат натрия,
гексацианоферрат (III) калия, перкислоты, оксид серебра,
диоксид свинца и пр.):
.9 Окисление альдегидов
.9.1 Окисление альдегидов
неорганическими окислителями
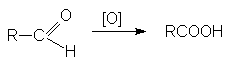
Альдегиды легко окисляются до
карбоновых кислот при действии самых разнообразных окислителей, среди которых
наиболее часто используются перманганат калия или реагент Джонса. Группа С=О
очень легко окисляется, что часто приводит к образованию карбоновых кислот под
действием кислорода воздуха просто при их хранении.
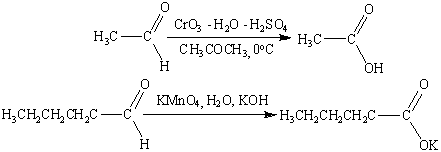
К сожалению, реагент Джонса не
обладает высокой избирательностью по отношению к другим функциональным группам,
во всех такого рода случаях идеальным селективным окислителем оказывается
водно-спиртовой аммиачный раствор оксида серебра (реагент Толленса, не
затрагивает С-С связь, двойную или тройную, гидроксильную группу спиртов и
др.).
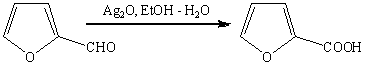
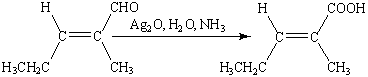
Эта реакция широко используется для
идентификации альдегидов (реакция «серебряного зеркала»).
В водном растворе аммиака
<#"555928.files/image077.gif">
при добавлении к которому альдегида
<#"555928.files/image078.gif">
Если реакция проводится в сосуде с
чистыми и гладкими стенками, то серебро выпадает в виде тонкой плёнки, образуя
зеркальную поверхность. При наличии малейших загрязнений, серебро выделяется в
виде серого рыхлого осадка.
Также в аналитической практике имеет
место быть реакция окисления альдегидов гидроксидом меди (II), при этом
можно выделить три формы его приготовления:
а) в виде свежеприготовленного
осадка Cu(OH)2 при нагревании,
б) в форме комплекса с аммиаком
[Cu(NH3)4](OH)2,
в) в составе комплекса с солью
винной кислоты (тартрат калия-натрия) (реактив Фелинга).
При этом образуется красно-кирпичный
осадок оксида меди (I) или металлическая медь (реакция «медного зеркала», более
характерная для формальдегида):
CH=О + 2Cu(OH)2  RCOOH +
Cu2O↓ + H2ОC=О + Cu(OH)2 HCOOH + Cu↓ + H2О
RCOOH +
Cu2O↓ + H2ОC=О + Cu(OH)2 HCOOH + Cu↓ + H2О
R-CH=O +
2[Cu(NH3)4](OH)2 RCOOH +
Cu2O↓ + 4NH3 + 2H2OCH=O + 2Cu(OH)2/ (CuSO4+ NaKC4H4O6·4H2O+NaOH) RCOOH +
Cu2O↓ + 2H2O
3.9.2 Автоокисление альдегидов
Как уже упоминалось, альдегиды
медленно окисляются в нейтральной среде на воздухе при комнатной температуре до
карбоновых кислот. Эта реакция ускоряется при облучении или при добавлении Fe (II) и
представляет собой типичный цепной радикальный процесс. Обычно цепь зарождается
в результате образования ацильного радикала за счет фотохимического
диспропорционирования:
Ацильный радикал возникает и при
отрыве атома водорода альдегидной группы с помощью инициатора - перекисного
радикала или молекулярного кислорода:
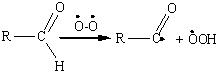
Ключевой стадией развития цепи
является образование перкислоты RCO3H, которая
далее окисляет альдегид до карбоновой кислоты:
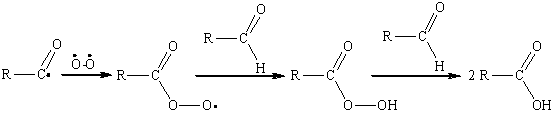
Следует отметить, что ароматические
альдегиды подвергаются окислению кислородом гораздо легче, чем алифатические,
вследствие более высокой стабильности бензоил-радикала  .
.
.9.3 Окислении по Байеру-Виллигеру
Важное практическое значение имеет
реакция окисления ароматических альдегидов перкислотами. Продуктами этой
реакции оказываются либо карбоновые кислоты, либо сложные эфиры фенолов
(окисление по Байеру-Виллигеру).
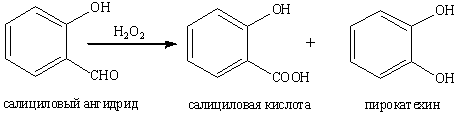
В сущности окисление ароматических
гидроксиальдегидов перкислотами является дальнейшим развитием реакции Дэкина,
заключающейся в превращении о- и п-гидроксибензальдегидов в пирокатехин и
гидрохинон при окислении водным раствором перекиси водорода.
.9.4 Окислительно-восстановительные
реакции альдегидов
Альдегиды при нагревании в
водно-спиртовом растворе щелочи могут диспропорционировать с образованием
равных количеств первичного спирта и карбоновой кислоты (реакция Канниццаро).
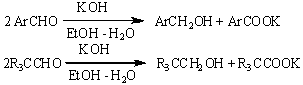
Под действием алкоголятов алюминия
альдегиды также склонны к диспропорционированию с образованием сложных эфиров
(реакция Тищенко):
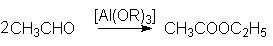
.10 Окисление кетонов
Кетоны инертны по отношению к
большинству окисляющих агентов, но медленно расщепляются горячим раствором
перманганата в щелочной среде или азотной кислоте с деструкцией углеродного
скелета.
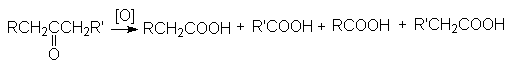
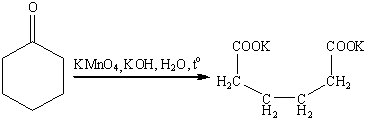
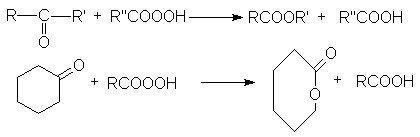
Под действием перкислот кетоны, как
и альдегиды, вступают в реакцию Байера-Виллигера, в результате которой в связь
ОС-С формально внедряется атом кислорода. Из ациклических кетонов образуются
сложные эфиры, а из циклических - лактоны.
.11 Окисление аминов
Все амины сравнительно легко
окисляются из-за своей основной природы. Легче всего окисляются до оксидов
третичные амины. В качестве окислителей используют перекись водорода в воде,
органические перкислоты (трифторперуксусная), перманганат калия.
Перекись водорода и надкислоты
окисляют третичные амины до N-оксидов.
R3N + HOOH →
R3N+-O- + H2O
В случае первичных и вторичных
аминов первоначально образующиеся N-оксиды перегруппировываются в производные
гидроксиламина.
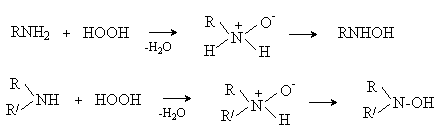
Такое окисление протекает сложно,
так как гидроксиламины сами легко окисляются. В случае первичных аминов
конечными продуктами окисления являются нитросоединения, например:
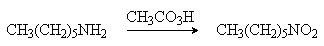
Первичные амины, в которых аминогруппа соединена
с третичным атомом углерода, окисляются в нитросоединения перманганатом калия в
водном ацетоне.
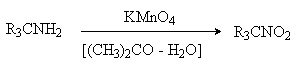
Амины, содержащие атомы водорода в α
-положении, при действии сильных окислителей (KMnO4)
дают смесь веществ, в которой преобладают карбонильные соединения. Процесс
протекает через промежуточное образование иминов, которые при гидролизе дают
альдегиды или кетоны.
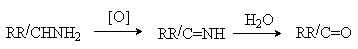
(R=Alk; R/=H, Alk)
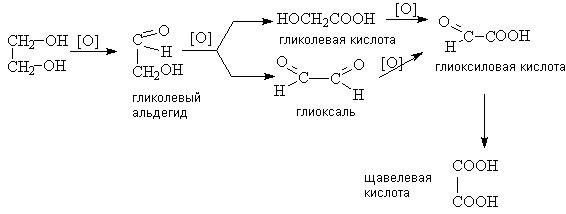
.12 Окисление углеводов
Окисление углеводов - важнейший процесс во
всяком живом организме. В суточном рационе человека и животных преобладают
углеводы, организмы животных не способны синтезировать углеводы из
неорганических веществ. Они получают их от растений с пищей и используют в
качестве главного источника энергии, получаемой в процессе окисления:
(H2O)y + xO2 → xCO2 + yH2O + энергия.
3.12.1 Окисление моносахаридов
Самыми простыми углеводами являются моносахариды
(альдозы, пример глюкоза; кетозы, пример фруктоза). Существует четыре важных
метода окисления альдозы: а) реактивом Фелинга или Толленса, б) бромной водой,
в) азотной кислотой, г) надйодной кислотой.
Альдозы и кетозы восстанавливают реактив
Толленса до альдегидов. Они так же восстанавливают реактив Фелинга. Однако эти
реакции не очень полезны, т.к. при помощи них нельзя отличить альдозы от кетоз.
Также окисление реактивом Фелинга или Толленса нельзя применить для получения
глюконовых кислот из альдоз. Оба эти реактива являются щелочными, а обработка
сахаров щелочами может привести к изомеризации или разрыву цепи.
Бромная вода окисляет альдозы, но не кетозы, это
кислый реагент и он вызывает изомеризацию молекулы. Поэтому с помощью бромной
воды можно отличить альдозы от кетоз, и ее используют для синтеза глюконовой
кислоты из альдозы:

Обработка альдозы азотной кислотой вызывает
окисление СНО-группы и СН2ОН - группы и приводит к образованию дикарбоновой
(сахарной) кислоты.
Подобно другим соединениям, содержащим группы ОН
или =О при соседних атомах углерода, углеводы подвергаются окислительному
расщеплению йодной кислотой (реакция Малапрада).
.12.2 Окисление дисахаридов
Дисахариды также подвергаются реакциям
окисления. При осторожном окислении дисахаридов альдегидная группа восстанавливающего
остатка моносахарида превращается в карбоксильную и получаются кислоты, которые
называются альдобионовыми. Например, при окислении лактозы бромной водой
образуется лактобионовая кислота:
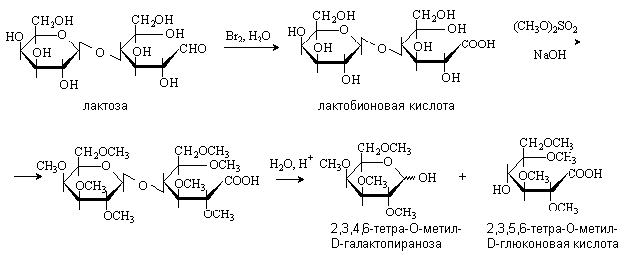
Окисление дает возможность определить, остаток
какого моносахарида находится на восстанавливающем конце.
.12.3 Окисление полисахаридов
Окисление полисахаридов в водных растворах
хлора, как это происходит при отбелке целлюлозы, вызывает расщепление
гликозидных связей по радикальному механизму. Образуется главным образом
D-глюконовая кислота. Однако в обработанной хлором целлюлозе были
идентифицированы и многочисленные другие монокарбоновые кислоты.
Существует метод расщепления
полисахаридных цепей, называемый дегидратация по Смиту. Он основан на окислении
полисахарида солями йодной кислоты - перйодатами. При этой реакции
<http://www.xumuk.ru/bse/2325.html>происходит разрыв С-С-связи в гликолях
<http://www.xumuk.ru/encyklopedia/1089.html> с образованием диальдегида.
Строго обязательным условием для периодатного окисления
<http://www.xumuk.ru/encyklopedia/2/3013.html> является наличие двух
гидроксильных групп <http://www.xumuk.ru/encyklopedia/1039.html> у
соседних углеродных атомов <http://www.xumuk.ru/encyklopedia/401.html>,
между которыми и разрывается С-С-связь. Данный метод позволяет узнать строение
полисахаридов.
4. Список литературы
1.
Реутов О.А., Курц А.Л., Бутин К.П. Органическая химия. В 4-х частях (часть
1-3). Москва. Бином. Лаборатория
знаний,
2004
.
Barton Sir Derek, Ollis W. David. Comprehensive Organic Chemistry. Пер.
с англ. Торгова В.И., Цветкова Ю.Е. Общая органическая химия (том 1-3). Москва,
«Химия», 1983
.
Кери Ф., Санберг Р. Углубленный курс органической химии (книга 2). Москва,
«Химия», 1981
.
Хейнс А. Методы окисления органических соединений. Москва, «Мир», 1988
.
Моррисон Р., Бойд Р. Органическая химия. Москва, «Мир», 1974
.
Травень В.Ф. Органическая химия (том 1). Москва, «Академкнига», 2004
.
Костиков Р.Р., Кузнецов М.А., Новиков М.С., Соколов В.В., Хлебников А.Ф.
Практикум по органическому синтезу, Санкт-Петербург, 2009
.
Вейганд-Хильгетаг. Методы эксперимента в органической химии, Москва, «Химия»,
1968
.
Марч Дж. Органическая химия (в 4 томах). Москва, «Мир», 1988
.
Беккер Г., Бергер В., и др. Органикум. Практикум по органической химии (том
1,2). Москва, «Мир», 1979
.
Гауптман З., Грефе Ю., Ремане Х. Органическая химия. Москва, «Химия», 1979
.
Титце Л., Айхер Т. Препаративная органическая химия. Москва, «Мир», 1999