Разработка нового метода синтеза алкалоидов азафеналенового ряда
Дипломная работа
Разработка нового метода синтеза
алкалоидов азафеналенового ряда
Содержание
Введение
1. Литературный обзор
1.1 Методы синтеза алкалоидов пергидро- и декагидро [9b]
азафеналенового ряда
1.2 Метатезис как метод создания циклических структур
1.3 Получение AlkLi из AlkI. Присоединение RLi к пиридинам
1.4 Восстановительное транс-α,α'-диаллилирование ароматических азотных
гетероциклов аллилборанами
2. Обсуждения результатов
3. Экспериментальная часть
4. Выводы
Список литературы
Введение
Органический синтез в последние десятилетия характеризуется
все возрастающим вниманием к синтезу природных соединений и их аналогов,
значительным укреплением методической базы (созданием надежных синтетических
методов). Осуществление синтеза сложнейших природных соединений (например,
хлорофилла, витамина В12, биополимеров), создание материалов с необычными
свойствами показывает, что для современного органического синтеза практически
не существует неразрешимых задач.
Данная дипломная работа посвящена синтезу природных
соединений: алкалоидов азафеналенового ряда, которые продуцируются
"божьими коровками". В литературном обзоре представлены работы по
полному синтезу этих алкалоидов. Однако, с развитием органического синтеза,
появлением новых реагентов и методов, открываются все новые возможности для
эффективного синтеза этих природных соединений. Целью работы является
разработка нового метода синтеза соединений пергидро - и декагидро [9b] азафеналенового ряда с
использованием реакций аллилборирования ароматических азагетероциклов и
внутримолекулярного метатезиса.
синтез алкалоид азафеноленовый ряд
1.
Литературный обзор
Кокцинеллиды (Coccinellidae) - одно из крупных семейств
отряда жесткокрылых (Coleoptera), насчитывающее более 5000 видов, из которых около
100 обитает в России.
Сегодня этот вид жуков представляет интерес в качестве
полезных насекомых, так как жуки и личинки кокцинеллид очень прожорливы и,
уничтожая в больших количествах таких опасных вредителей как тли, листоблошки,
червецы, щитовки и клещи, приносят громадную пользу сельскому хозяйству. Самые
блестящие страницы в истории биологического метода борьбы с вредителями
сельского хозяйства вписаны именно при использовании божьих коровок. Достаточно
напомнить о феноменальном успехе, который был получен в результате интродукции
из Австралии божьей коровки родолии (Rodolia cardinalis Muls.) в Калифорнию для
борьбы с австралийским желобчатым червецом-ицерией (Icerya purchasi Mask.). Без
преувеличения можно сказать, что само существование цитрусовых, как культуры,
обязано этому виду божьей коровки. По данным Де Баха (De Bach, 1964) из 225
успешных случаев биологического подавления вредителей в 51 случае результаты
были получены при использовании кокцинеллид [1].
Известно, что божьи коровки крайне редко используются другими
организмами в качестве источника пищи. Это может быть связано с мощной
химической защитой кокцинеллид, которую они демонстрируют яркой окраской и
отпугивающим запахом. Последний свидетельствует о наличии в жуках алкалоидов.
Многие виды кокцинеллид используют эти алкалоиды в защитном механизме,
известном как "reflex bleeding" [2].
На сегодняшний день из кокцинеллид выделено несколько
алкалоидов, они представлены на рисунке 1 [2].
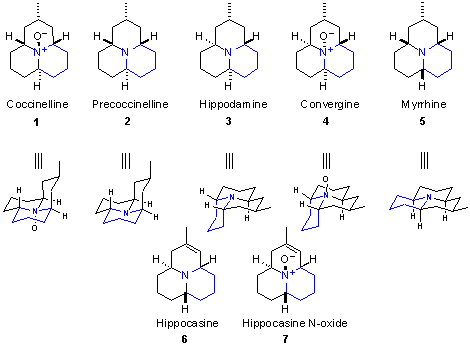
Рис.1. Алкалоиды,
используемые кокцинеллидами.
Алкалоиды coccinelline (1)
и precoccinelline (2) впервые выделил Тарш и его
коллеги хроматографией из метанольного экстакта божьих коровок [3]. Выделенные
вещества согласно масс-спектрометрии высокого разрешения имели молекулярные
формулы C13H23NO и C13H23N
соответственно. Принадлежность coccinelline к алкалоидам азафеналенового ряда
была доказана рентгеноструктурными исследованиями [4]. Кроме того, было
выяснено, что каталитическое гидрирование переводит coccinelline в precoccinelline,
а окисление последнего моноперфталевой кислотой дает снова coccinelline. Это
подтвердило предположение о том, что coccinelline является N-оксидом
precoccinelline.
Алкалоиды hippodamine (3)
и convergine (4) были выделены той же группой
исследователей аналогичным способом. Масс-спектр hippodamine показал
брутто-формулу C13H23N и фрагментацию, почти идентичную
precoccinelline. Convergine было легко превратить в hippodamine восстановлением
литийалюминийгидридом. В начале предположили (ошибочно), что оба алкалоида
представляют собой рацемические смеси, так как оптического вращения не
наблюдалось. Однако, рентгеноструктурный анализ показал, что convergine имеет
хиральную структуру, а так как 4 может быть превращен в 3,
структура hippodamine такая же. Таким образом была установлена абсолютная
кофигурация обоих алкалоидов. [5]
Тарш и сотрудники также смогли выделить и охарактеризовать
алкалоид myrrhine (5) [6]. Снова было показано, что молекулярная формула
C13H23N и фрагментация в масс-спектре почти идентична
precoccinelline и hippodamine. ИК-спектр показал присутствие Больмановских
линий, характерных для аминов с антиперипланарной С-Н связи неподеленной
электронной парой азота. Это позволило предложить, что myrrhine является
ахиральным амином. Данная гипотеза была подтверждена преобразованием
coccinelline в условиях реакции Полоновского (взаимодействие N-окиси третичного амина с ангидридом трифторуксусной
кислоты (рис.2)) [7] до неустойчивого енамина, который при
каталитическом гидрировании дал смесь myrrhine и hippodamine, Стало ясно, что
конфигурации этих двух продуктов отличаются только одним стереоцентром. Таким
образом была установлена структура myrrhine.
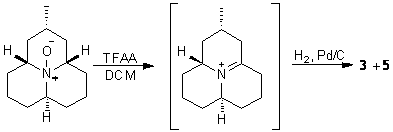
Рис.2. Взаимодействие
coccinelline с ангидридом трифторуксусной кислоты и последующее его
восстановление.
Далее та же исследовательская группа выделила алкалоиды hippocasine (6) и N-оксид hippocasine (7). Была установлена молекулярная формула C13H21N.
По фрагментации в масс-спектре было видно, что соединения содержат двойную
связь или дополнительное кольцо. В 1Н ЯМР-спектре был обнаружен
сигнал одного олефинового протона, что указывает на наличие енаминовой системы
[8]. Однако характерных полос ультрафиолетового поглощения енаминовой системы
при 220-235 нм не было обнаружено. В данном енамине неподеленная пара
электронов азота не параллельна р-орбиталям двойной связи системы, таким
образом, эта структура не должна поглощать в упомянутой области УФ-спектра. Это
послужило аргументом для размещения двойной связи в структурах 6 и 7.
Все из упомянутых алкалоидов в настоящее время синтезированы.
Перейдем к рассмотрению существующих методов синтеза алкалоидов азафеналенового
ряда.
1.1
Методы синтеза алкалоидов пергидро- и декагидро [9b] азафеналенового ряда
Некоторые из методов синтеза основаны на том факте, что
метильная группа в соединениях 1-7 занимает наиболее
термодинамически выгодное эквториальное положение [9, 10 11]. Так Айер и др.
при разработке метода синтеза (±) - dihydrodeoxyepiallocernuine использовали следующий
подход для получения 2-метилпергидро-9b-азафеналеновых алкалоидов myrrhine и hippodamine, (схема 1).
Монолитийпроизводное 2,4,6-коллидина 8 обрабатывали
3-бромопропиональдегиддиметилацеталем с образованием 9. Далее при
добавлении фениллития образуется анион, который взаимодействует с ацетонитрилом
и дает кетон, из которого получили соответствующий ацеталь 10.
Восстановление натрием в изоамиловом спирте дает смесь насыщенных
стереоизомерных аминов 11, которые отхроматографировали, а затем сняли
защитные группы с образованием 12. Нагревание 12 с 2
эквивалентами р-толуолсульфокислоты дает один продукт, кетон 13,
с той же конфигурацией на всех стереогенных центрах, что и у myrrhine (5).
Так как 13 является неустойчивым соединением, его сразу же преобразовали
в тиокеталь 14, который восстанавливали с удалением серы над никелем
Ренея с образованием myrrhine (5). Окислением m-CPBA был получен
соответствующий N-оксид, идентичный природному N-оксиду myrrhine [10].
Интересно, что циклизация в мягких условиях (пирролидин, уксусная кислота)
превращает 12 в смесь двух стереоизомерных кетонов. Получение
соответствующих тиокеталей и последующая их десульфуризация дает смесь myrrhine
(5) и (±) - hippodamine (3), последний был преобразован в N-оксид
(±) - convergine (4). Общий выход 5 и 3 из 12
составил, соответственно, 33% и 23%.

Схема 1. Реагенты:
(I) PhLi; (II) 3-бромопропиональдегиддиметилацеталь; (III) PhLi; (IV) CH3CN, HCl; (V) этиленгликоль, TsOH; (VI) Na/изоамиловый спирт; (VII) HCl; (VIII) TsOH; (IX) TsOH, TEA, этандитиол; (X) никель Ренея
в этаноле; (XI) пирролидин, AcOH; (XII) этандитиол, BF3*Et2O.
Используя подобный подход, Айеру и Фуруичи впервые удалось
синтезировать coccinelline (1) и precoccinelline (2) [11]. Из
2,6-лутидина (15) был получен кеталь 16 с использованием ранее
описанной стратегии (схема 2) [10] Кипячение 19 в пирролидине с
уксусной кислотой дало смесь двух изомерных кетонов 20 и 21.
После разделения стереомерных трициклических кетонов, их обработали
метиллитием, далее дегидратировали и получили myrrhine (5) из 20
и precoccinelline (2) из 21. Precoccinelline был преобразован в
coccinelline реакцией с м-хлорнадбензойной кислотой.

Схема 2.
Реагенты: (I) PhLi; (II) 3-бромопропиональдегиддиметилацеталь; (III) PhLi; (IV) CH3CN, H+; (V) этиленгликоль, TsOH; (VI) Na/изоамиловый спирт; (VII) HCl; (VIII) пирролидин, AcOH; (IX) MeLi, тионилхлорид; (X) H2/Pt.
Стивенс и Ли использовали совершенно другой подход для синтеза
азафеналенов, основанный на методологиии Робинсона-Шопфа [12, 13, 14] (схема
3). Их метод синтеза основан на реакции аминодиальдегида 22 с
диметил ацетондикарбоксилатом, в результате которой получается пергидро-9b-азафенален 23, легко преобразуемый в кетон 21.
Из последнего, в свою очередь, получают precoccinelline и превращают в
соответствующий N-оксид, coccinelline.

Схема 3.
Аминодиальдегид 22 был синтезирован в шесть стадий из
диметилмалоната (схема 4) [15]. На первой стадии малоновый эфир вводили
в реакцию с акролеином, в результате чего был получен альдегид 24,
который затем был защищен ацетальной защитой. Конденсацией Кляйзена из 25
получали кетоэфир 26, который кипятили с хлоридом натрия в мокром DMF, а
затем подвергли восстановительному аминированию с образованием диметилацеталя 27.
Конденсация с диметил ацетондикарбоксилатом (28) и последующий гидролиз
при рН=2 дают только один стереоизомер 23. После декарбоксилирования
кетона 23, был получен precoccinelline (2) обработкой
метилентрифенилфосфином с последующей каталитической гидрогенизацией.
Синтезированный precoccinelline был преобразован в coccinelline (1)
обработкой м-хлорнадбензойной кислотой.

Схема 4. Реагенты:
(I) MeONa, CH (OMe) 3, H+; (II) TsOH, CH (OMe) 3; (III) NaCl, мокрый DMF, перемешивание; (IV) NaH; (V) NH4OAc, NaCNBH3, молекулярное сито; (VI) H+; (VII) Ph3P=CH2; (VIII) H2, Pd/C; (IX) m-CPBA.
Ланглуа и др. усовершенствовали вышеописанный метод, показав, что
выход альдегида 23 можно повысить, используя вместо диметил
ацетондикарбоксилата соответствующую кислоту [16] (схема 5).

Схема 5.
В серии работ описан еще один подход к синтезу алкалоидов
азафеналенового ряда. Мюллер и его коллеги нашли общий способ синтеза всех
известных алкалоидов кокцинеллид. В качестве исходного реагента они использовали
пергидроборафенален, синтезированный гидроборированием 1,5,9-циклододекатриена
[17, 18]. При синтезе coccinelline и precoccinelline (схема 6), [17, 19]
пергидроборафенален обрабатывали N-хлор-O- (2,4-динитрофенил) гидроксиламином (генерируется in situ) и перекисью водорода с образованием
аминоспирта 30. Окисление в соответствующий кетон дает трициклические
енамин 31 [20, 21]. При добавлении метилфторсульфата к енамину 31
образуется соль 32, которая превращается в аллиламмониевую соль 33
при добавлении к 32 диизопропиламида лития. Аллиламин 34 был
получен путем отщепления метильной группы от 34 этилмеркаптидом лития.
Далее стереоселективное эпоксидирование амина 34 привело к соединению 35,
которое подвергли восстановительному раскрытию эпоксидного цикла с образованием
спирта с гидроксильной группой в аксиальном положении. Спирт, в свою очередь,
окислили до кетона 21, используемого Айером и Фуруичи в синтезе
coccinelline и precoccinelline [11]. После реакции Виттига и изомеризации с
помощью р-толуолсульфоновой кислоты был получен олефин 36,
который при гидрогенизации дал precoccinelline (2). Взаимодействие
полученного продукта с м-хлорнадбензойной кислотой преобразует 2
в coccinelline (1).

Схема 6. Реагенты:
(I) N-хлоро-О- (2,4-динитрофенил) гидроксиламин, Н2О2/NaOH; (II) CrO3/H2SO4; (III) CH3OSO2F; (IV) LDA; (V) LiSEt; (VI) TFA, CF3CO3H; (VII) Li, EDA; (VIII) Ph3P=CH2; (IX) TsOH; (X) H2, Pd/C; (XI) m-CPBA.
Трициклический интермедиат 31 был использован для получения
hippocasine и N-оксида hippocasine (схема
7) [17, 18], а также для нового синтеза hippodamine и convergine.
Гидроборирование 31 с последующим окислением дало два спирта 37 и
38 в соотношении 3:
. При окислении полученной смеси образуются кетоны 39 и 40.
После обработки их мягким основанием соотношение 39: 40 становится
равным 9:
. Во избежание N-алкилирования при внедрении метильной группы в α-положение к карбонильной группе соединения
39, его обрабатывали реагентом Бредерека [22], в результате чего образовался
амин 41. Восстановление "литиевой бронзой" дает нестабильный
аминокетон, который сразу каталитически гидрируют до 42, являющегося
исходным соединением для синтеза hippodamine и hippocasine. Несмотря на
эпимеризацию, произошедшую в результате восстановления 42 по
Кижнеру-Вольфу и давшее hippodamine и его изомер с аксиальным положением
метильной группы в соотношении 2: 1, восстановление 42 литием в
этилендиамине приводит к рацемическому hippodamine, который был преобразован в
его N-оксида посредством обработки м-хлорнадбензойной кислотой. Этот же
кетон был переведен в рацемический hippocasine (6) посредством реакции
Бамфорда-Стивенса. N-оксид Hippocasine был получен окислением 6
перекисью водорода. Сравнение природных и синтезированных алкалоидов показало
их идентичность. [17]

Схема 7. Реагенты: (I) BH3*Me2S,
H2O2/NaOH; (II) CrO3/AcOH/H2SO4;
(III) NaOCH3; (IV) CH3OCH [N (CH3) 2]
2; (V)"литиевая бронза";
(VI) H2, Pd/C; (VII) этандитиол/BH3*Et2O; (VIII) Li/EDA; (IX) m-CPBA;
(X) NH2 NH2, TsCl, t-BuNHLi; (XI) H2O2.
Реакция циклоприсоединения между этилгекса-3,5-диеноатом и
нитроном дает возможность синтеза (±) - epi-hippodamine (64) (схема 8) [23] Нитрон 44,
полученный in situ обработкой 43 гидроксиламином,
взаимодействует с этилгекса-3,5-диеноатом с образованием циклоаддуктов 45,
46 и 47. Восстановлением полученной смеси цинком в уксусной кислоте
и этилендиаминтетрауксусной кислоте получен неустойчивый пиперидин 48,
который подвергается изомеризации, затем внутримолекулярному присоединению по
Митчелу и защите ОН-группы с образованием смеси 53 и 54. После
циклизации по Дикману образуется трициклический амин 56, который
деэтоксикарбонилировали с получением смеси продуктов 57, 58 и 59
в соотношении 80: 14: 6 [23]. Необходимый для дальнейшего синтеза продукт 57
выделяли флэш-хроматографией и вводили в реакцию Виттига с
метилентрифенилфосвином с образованием соответствующего олефина, который в свою
очередь был подвергнут каталитическому гидрированию до удаления силильной защитной
группы с образованием 62. Удаление гидроксильной группы соединения 62
проводили через радикальное восстановление ксантогената 63
трибутилоловогидридом до (±) - epi-hippodamine.

Схема 8.
Реагенты: (I), H2NOH*HC1, NaOAc*3H20,
этилгекса-3,5-диеноат, 1200C; (II) Zn, HOAc, EDTA (aq), перемешивание; (III) DBU (1 eq), C6H6, 200C;
(IV) ButMe2SiC1 (1 eq); (V) LDA, THF, - 780C; (VI) LiCl в DMF, перемешивание; (VII) Ph3P=CH2, Et2O; (VIII) H2, Pd/C, MeOH; (IX) Et4N+F-,
THF; (X) NaH, имидазол, THF; (X) I, CS2,
перемешивание; (XII) Mel; (XIII) Bu3SnH, AIBN, толуол, перемешивание.
Юи и др. предложили еще один способ синтеза precoccinelline, [24]
на основе своей "CN (R, S)" стратегии стереоселективного синтеза цис
- и транс-2,6-диалкилпипиридинов [25, 26] В данной стратегии
используется введение CN-группы в хиральное α-положение к атому азота с образованием R -
или S-конфигураци. Их синтез начинается с (2S, 6R) - 2-циано-6 -
оксазолопипиридина 65, из которого легко получить транс-дизамещенный
пиперидин 66 (Схема 9), а затем кетон 21, используемый
Айером в своем методе синтеза [11].

Схема 9.
Взаимодействие 65 с литийдиизопропиламидом (Схема 10)
и последующее добавление 3-бром-2-метоксипропена дает 66. При реакции
последнего с TBDMSOTf и (1,3-диоксоланил-2-пропил) хлоридом магния образуется
оксазолидинон 68 (механизм реакции показан на рис.3).

Схема 10. Реагенты:
(I) LDA-THF, 3-бром-2-метоксипропен; (II) TBDMSOTf, RMgX.

Рис.3. Предполагаемый
механизм алкилирования с перегруппировкой.
Производное 68, полученное в результате
алкилирования/аза-Коуп перегруппировки, подвергли восстановительному
расщеплению тетрагидрофуранового кольца литийалюминийгидридом с образованием транс-производного
пиперидина 69 (схема 11). Метанолиз и снятие бензильной группы
преобразует 69 в соответствующий ацеталь 71. Гидролизом защитных
групп и внутримолекулярной реакцией Манниха из 71 был получен кетон 21,
из которого синтезировали precoccinelline, используя метод Мюллера [17, 19].

Схема 11. Реагенты:
(I) LAH; (II) муравьиная кислота, метанол; (III) K2CO3, 10% Pd/C; 10%
-ная HCl, CSA.
Еще один стереоселективный метод синтеза (±) - hippodamine и (±) -
epi-hippodamine предложен в [27].
Симметричный спирт 72 был синтезирован по описанной в [28]
методике. Далее гидрокси-группу заместили на фталимидную с образованием
соответствующего диена 73 (схема 12).
Окислительным расщеплением терминального алкена 73
перйодатом натрия в присутствии каталитического количества тетраоксида осмия (2
мольных процента) в растворе ТHF-вода
(соотношение 3:
) был получен диальдегид 74, который по методу
Уодсворта-Эммонса превратили в ациклический диэфир 75, содержащий только
E,E-стереоизомер.

Схема 12. Реагенты:
(I) фталамид, Ph3P, DIAD, THF; (II): OsO4, NaIO4, (III) Et2O (O) PCH2CO2Et, NaH, THF; (IV) (1) NaBH4 (1,4 эквивалента), i-PrOH/H2O (6,2:1), (2) ацетон, AcOH.
Замыкание третьего кольца с образованием пергидро-9b-азафеналенового скелета было осуществлено конденсацией
Дикмана. Соединение 76 обрабатывали t-BuOK в кипящем бензоле с
непрерывным азеотропным удалением этанола с помощью ловушки Дина-Старка с
литием, в результате чего образовалась смесь β-кетоэфира 77 и его соответствующей
енольной формы 78 (схема 13).
Деэтоксикарбонилирование 77 и 78 осуществили
обработкой соляной кислотой в эфире с последующим кипячением в влажном DMF с
LiCl. Поученный таким образом кетон 79 ввели в реакцию Виттига с CH3PPh3Br с образованием олефина 80, из которого получили
мезитиленсульфонат и затем подвергли каталитическому гидрированию. Полученная
смесь состояла из (±) - hippodamine (3) и (±) - epi-hippodamine (81) (соотношение 3: 81 равно
3,5:1)

Схема 13. Реагенты:
(I) t-BuOK, Li, бензол, азеотропное удаление EtOH; (II) (1) HClg, Et2O, (2) LiCl, DMF, перемешивани; (III) CH3PPh3Br, n-BuLi, THF; (IV) Pd/C 10%, H2.
Похожий на предыдущий метод синтеза myrrhine и его N-оксида с
использованием линейного субстрата, описан в [29]. Следует заметить, что N-оксид myrrhine не был
извлечен из кокцинеллид.
N-Boc-аминоалкен (82) был получен
замещением по Мицунобу взаимодействием с NHBocTs, последующим снятием
тозилат-защиты и кросс-метатезисом с этилакрилатом (схема 14).
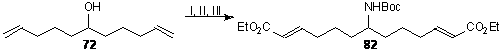
Схема 14. Реагенты:
(I) NHBocTs, DIAD, Ph3P; (II) Mg, MeOH; (III) Hoveyda-Grubbs 2nd gen., CH2=CHCO2Et.
Далее снимали Bос-группу в присутствии кислоты Льюиса, которая
также катализировала конеденсацию по Дикманну с образованием циклических
структур. Обработка соединения 82 трифлатом скандия привела к
бициклическому транс-76 (схема 15), тогда как в
присутствии трифлата олова (II) (0,5 экв.) образуется 83 (с небольшой
примесью цис-76 и транс-76).

Схема 16. Реагенты:
(I) Sc (OTf) 3 (3 экв.), PhMe, Δ; (II) Sn (OTf) 2 (2 экв.), PhMe, Δ.
Интересно, что использование больших количеств трифлата олова (II)
приводит к образованию исключительно транс-76, без примесей цис-76
или 83.
Трицикл 83 был использован в качестве исходного соединения
для получения myrrhine и N-оксида myrrhine. Гидролиз эфира, декарбоксилирование
и реакция Виттига с илидом CH2=PPh3 дали экзоциклической алкен 84 (схема
16). Диастереоселективное восстановление двойной связи осуществили
протонированием атома азота р-толуолсульфоновой кислотой с последующим
гидрированием в присутствии никеля Ренея. Была получена смесь myrrhine и epi-myrrhine с соотношением изомеров 10: 1
соответственно. N-оксид myrrhine 85
был синтезирован обработкой myrrhine м-CPBА.
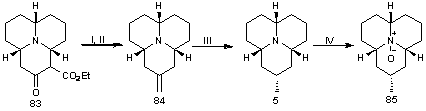
Схема 17. Реагенты:
(I) Na2CO3, EtOH, H2O, Δ; (II) CH2PPh3, THF, 600C; (III) Ni Ренея, H2, p-TsOH, MeOH, H2O; (IV) m-CPBA, CH2Cl2, 00C.
Алкалоиды "божьих коровок" заинтересовали не только
биологов, но и стимулировали развитие органической химии, поставив ряд
аналитических, синтетических и биосинтетических проблем. Из всех видов
кокцинеллид, только небольшой их процент был изучен химически. Продолжение
исследований по химии и биологии этой интересной группы жуков поможет по-новому
взглянуть на химические аспекты жизни насекомых.
1.2
Метатезис как метод создания циклических структур
Метатезис является одним из самых мощных и надежных методов
создания циклических систем, поэтому в настоящее время он часто используется в
многоступенчатом целевой синтезе.
Вообще метатезисом назвается дисмутация алкенов путем
формального обмена алкилиденовых групп [31, c.63]:
RCH=CHR + R1CH=CHR1 →
2RCH=CHR1
Для осуществления этой реакции имеется ряд каталитических
систем, как гомогенных, так и гетерогенных. Наиболее активные гомогенные
катализаторы обычно получают из соединении молибдена, вольфрама, сильного
восстанавливающего и (или) алкилирующего агента, например алкильного
производного алюминия или олова, и добавки для ингибирования побочных реакций и
улучшения селективности. В качестве таких добавок использовали пиридин,
ацетоннтрил, этанол и трифенилфосфин.
Типичными катализаторами метатезиса являются системы WCl6 - EtAlCl2, WCl6 - SnMe4, Мо
(РРh3) 2 (NО) 2С12
- А12Мe3С13, PhWCl3 - AlCl3 и WCl6 - BuLi [31, c.63].
Катализаторы реакций метатезиса (катализаторы Граббса) на
основе рутения из-за своей высокой стабильности и активности нашли широкое
применение в органическом синтезе, некоторые из них представлены на рисунке 4
[32]; в качестве растворителя обычно используют хлористый метилен и толуол.

Рис.4. Некоторые
катализаторы метатезиса.
Метатезис алкенов обычно является обратимым термодинамически
контролируемым процессом. В случае простых алкенов энтальпия реакции
практически равна нулю (образуются и разрываются подобные двойные
углерод-углеродные связи), поэтому распределение продуктов определяется
энтропийными факторами и алкилиденовые группы распределяются почти
статистически.
В течение последних десятилетий большие усилия были направлены на
выяснение механизма метатезиса алкенов. Было предложено несколько различных
схем, однако почти все имеющиеся данные согласуются с цепным металл-карбеновым
механизмом (схема 18) [33].
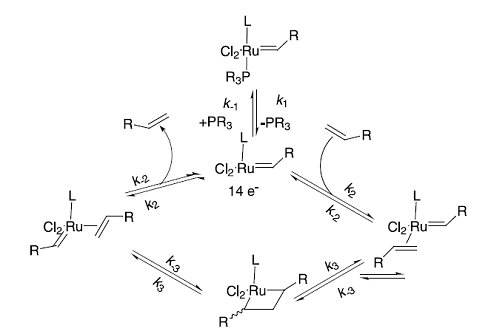
Схема 18.
Например, из таких легкодоступных алкенов как октадецена-9 (4
моль) и гексадецена-2 (1 моль) в присутствии катализатора, полученного из WC16, EtAlCl3 и этанола в соотношении
1: 3: 5, за 5 мин с выходом 10% получен трикозен-9 цис-изомер которого,
является феромоном комнатной мухи. Несмотря на то, что выход желаемого алкена
невысок, использование относительно дешевых реагентов и короткое время реакции
составляют существенные преимущества этого пути, причем мольное соотношение
алкен - вольфрам составляло примерно 1000.
Другим примером применения метатезиса является его использование в
синтезе антибиотика эверниномицина (схема 19) [32].

Схема 19. Реагенты:
(I) 87 (15 моль %), СH2Cl2, 400C.
1.3
Получение AlkLi из AlkI. Присоединение RLi к пиридинам
Успех обмена лития на галоген с образованием первичного
алкиллитийпроизводного (схема 20) в решающей степени зависит от выбора
галоида, металлирующего агента и растворителя. Предпочтительнее использовать
алкилйодиды, а не бромиды, так как последние могут инициировать побочные
радикальные процессы [34].
Наилучшим металлирующим агентом является t-BuLi. Использование 2
мольных эквивалентов реагента делают процесс необратимым, поскольку второй
эквивалент t-BuLi выводит из реакции образующийся t-BuI
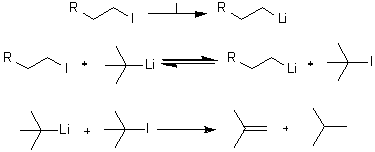
Схема 20.
Реагенты: (I) t-BuLi (2 экв), nC5H12-Et2O, - 780C.
Наилучшим растворителем при литий-йод обмене является система из
углеводородного растворителя и некоторго количества простого эфира, например, Et2O. В такой системе t-BuLi
сольватирован и является димером. Данная реакция экзотермическая, поэтому, как
правило, ее проводят при низкой температуре, чтобы избежать побочных процессов.
Эффективность литий-йод обмена сильно зависит от соотношения
углеводородного растворителя и эфира [34]. Было выяснено, что наилучшие выходы
литийорганических производных получаются в смеси гептан-tBuOСН3 (объемное соотношение 19:
), при этом побочных продуктов почти не образуется.
Для синтеза цис - и транс-несимметрично
2,6-замещенных 3-пиперидинов разработан удобный метод, который основан на
комбинации хорошо известного 1,2-присоединения RLi к пиридину и последующего транс-аллилборирования (схема
21) [35].
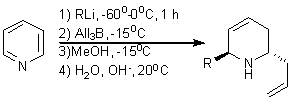
Схема 21.
1.4
Восстановительное транс-α,α'-диаллилирование
ароматических азотных гетероциклов аллилборанами
Аллилбораны обладают повышенной по сравнению с другими
классами органических производных бора химической активностью, что обусловлено
спецификой их строения, а именно наличием двойной связи в β,γ-положении относительно бора. В отличие от алкил - и
арилборанов аллильные аналоги в определенном отношении проявляют свойства
классических металлоорганических соединений (Mg, Zn и Li), однако их
использование во многих случаях имеет ряд преимуществ. Так, они легко присоединяются
к карбонильным соединениям и иминам; реализовано много хиральных вариантов этих
реакций (с применением оптически активных аллилборанов). Уже более трех
десятилетий аллилбораны используют в синтезе различных классов органических
соединений, в том числе многих природных веществ и их аналогов [36].
В начале 90-х годов в лаборатории алифатических
борорганических соединений ИНЭОС РАН была открыта серия реакций транс-α,α'-диаллилирования ароматических азотных гетероциклов
аллилборанами (схема 22). Сущность этих реакций состоит в присоединении
бораллильных фрагментов по иминной связи гетероцикла в присутствии воды или
спирта с образованием транс-диаллилированных тетрагидропроизводных
гетероциклов. Так, при обработке комплекса пиридина с триаллилбораном спиртом,
водой или R2NH происходит его полная перестройка и получается (с
выходом до 97%) транс-2,6-диаллил-1,2,5,6-тетрагидропиридин [35, 36].
Существенно, что транс-2,6-диаллилированные
тетрагидропиридины гладко изомеризуются в соответствующие цис-изомеры
при нагревании (125-130°С) с эквивалентным количеством триаллилборана (схема
22)
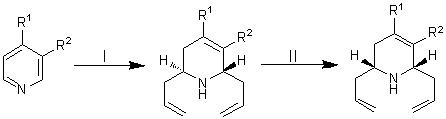
Схема 22. Реагенты: (I) 1) All3B,
) ROH,
) NaOH; (II) 1) All3B, 1300C,
) ROH,
3) NaOH
Стоит заметить, что спирт в реакции аллилирования является
реагентом, а не растворителем, на что указывает образование 5-дейтерированного
3-пиперидеина при обработке комплекса дейтерометанолом (рис.6). Из
комплекса триметаллилборана 2,6-бисметаллильный гетероцикл получается при
комнатной температуре. На примере реакции с участием трикротилборана показано,
что присоединение аллильных групп к пиридиновому кольцу происходит с аллильной
перегруппировкой (рис.5)
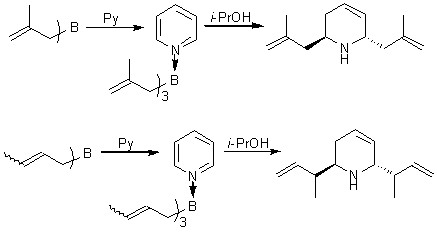
Рис.5.
Присоединение аллильных групп к пиридиновому кольцу с аллильной
перегруппировкой.
Возможный механизм восстановительного транс-2,6-диаллилирования
пиридина приведен на рисунке 6. На первом этапе протекает внутримолекулярное
1,2-аллилборирование пиридинового кольца и образуется диаллил (амино) боран
(этот процесс равновесный). Роль спирта заключается в расщеплении ковалентной
связи B-N, протекающем с перегруппировкой аллильного типа. Образующийся в
результате азометин (вернее, его комплекс с A112BOR) сразу же
подвергается аллилборированию по связи C=N через шестицентровое переходное
состояние. Существенно, что присоединение второй аллильной группы
осуществляется стереоселективно - в транс-положение относительно первой.
Образовавшийся аминоборан деборируется спиртом, взятом в избытке (4 экв.), в
результате чего образуются аллилборонат и диаллилированный продукт.
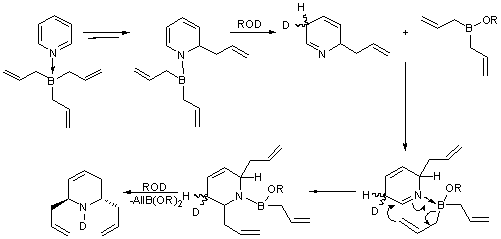
Рис.6. Возможный
механизм восстановительного транс-2,6-диаллилирования.
Как раз с использованием реакций аллилборирования ароматических
азагетероциклов нами была предпринята попытка синтеза соединений пергидро - и
декагидро [9b] азафеналенового ряда.
2.
Обсуждения результатов
Не сложно заметить, что у 2,6-диаллилированных
тетрагидропиридинов не хватает одного атома углерода для достройки углеродного
скелета алкалоидов азафеналенового ряда (схема 23). Наиболее простым и
прямым способом такой достройки углеродного скелета является введение остатка
угольной кислоты в виде карбамата и последующее его двойное внутримолекулярное
алкилирование соответствующими магнийорганическими производными (схема 23).
С другой стороны возможными недостатками такой схемы являются: а) сложность
эффективного получения металлорганического производного; б) реализация двух
путей ухода групп - OR и - NR2 из бициклического продукта первой стадии алкилирования.
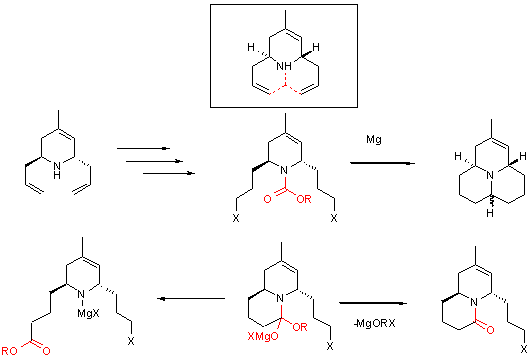
Схема 23.
На первом этапе работы был получен по стандартной методике транс-2,6-диаллил-4-метил-1,2,3,6-тетрагидропиридин
95 взаимодействием 4-пиколина с триаллилбораном и последующей обработкой
изопропиловым спиртом и NaOH (схема
24). По литературным данным выход должен быть 75% [37], однако у нас выход
составил 37%. Из-за этого несоответствия предположили, что выход реакции в
случае пиридинов с электронодонорными заместителями сильно зависит от условий
нагревания. Потому что даже в литературной методике использовался 2-кратный
избыток 4-пиколина, чтобы увеличит выход реакции. Снижение выхода продукта
аллилборирования связано с протолизом комплекса 4-пиколина с триаллилбораном,
который идет тем легче, чем выше температура. Поэтому было решено после
прибавления изопропилового спирта не кипятить реакционную смесь при 800С,
как предлагается в оригинальной методике, а выдержать ее при той температуре,
выше которой уже начинается видимый протолиз (выделение пропилена), а именно
при 500С. В результате такого изменения температурного режима
синтеза выход соединения 95 составил 91%.

Схема 24. Реагенты:
(I) All3B; (II) изопропиловый спирт; (III) NaOH.
По измененной методике мы попробовали синтезировать
диаллилпроизводное из 4-метоксипиколина, которое не удавалась получить ранее (схема
25). Выходы диаллилпроизводных по литературной и измененной методикам
указаны в таблице 1.
Таблица 1. Выходы диаллилпроизводных по литературной и
измененной методикам.
|
R
|
t0С
|
|
-Me
|
80
|
37 (75)
|
|
-Me
|
50
|
91
|
|
-OMe
|
80
|
0
|
|
-OMe
|
50
|
59
|

Схема 25. Реагенты:
(I) All3B; (II) изопропиловый спирт; (III) NaOH.
Далее соединения 95 и 98 были превращены в цис-измеры
нагреванием с триаллилбораном и последующей обработкой метанолом и водной
щелочью (схема 26).

Схема 26. Реагенты:
(I) All3B; (II) (1) изопропиловый спирт, (2) NaOH.
О цис-2,6-диаллил-4-метокси-1,2,3,6-тетрагидропиридине
стоит упомянуть отдельно. Данный изомер диаллилпроизводного 4-метоксипиридина с
помощью реакции метатезиса можно перевести в соответствующий бицикл, содержащий
фрагмент енольного типа, который обладает высокой реакционной способностью к
всевозможным реакциям электрофильного присоединения. Это открывает новые
возможности для синтеза различных биологически активных соединений, например,
аналогов кокакина (рис 7).
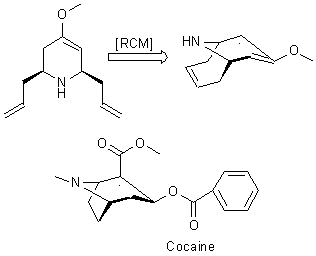
Рис.7. Возможное
применение 100 и его аналогов для синтеза природных соединений.
Чтобы получить исходное соединение для синтеза алкалоида по схеме
23 соединение 95 гидроборировали тексилбораном, а затем окислили
пероксидом водорода в щелочной среде с образованием аминоспирта 102 (схема
27). Использование тексилборана позволяет провести гидроборирование
селективно по терминальным атомам углерода. Стоит так же заметить, что
необходимо брать два эквивалента тексилборана. Один эквивалент идет на защиту
аминогруппы, а другой, собственно, на гидроборирование.
Полученный аминоспирт 102 защитили Boc-группой по методике, описанной в [38], где к раствору амина в системе
THF/вода предлагалось сначала добавить K2CO3, а затем
Boc2O. В полученном таким образом соединении 103 гидроксильные
группы заместили на хлор с образованием 104.
Так как Boc-группа в
сильнокислой среде расщепляется, обычное замещение с помощью хлороводорода
провести нельзя, поэтому было решено использовать трифенилфосфин в
тетрахлорметане.

Схема 27. Реагенты:
(I) H2O2/OH-; (II) Boc2O, THF; (III) Ph3P/CCl4.
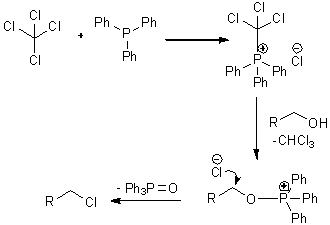
Схема 28. Механизм
образования катализатора.
Так как дихлорид 104 является малоактивным, для получения
магнийорганического производного использовали высокоактивный магний Райка,
полученный восстановлением смеси безводных бромида и иодида магния
металлическим калием в ТГФ (схема 29).
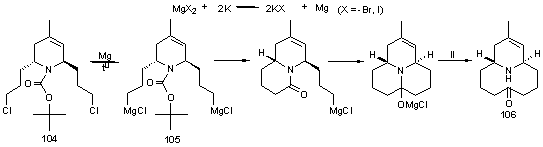
Схема 29. Реагенты:
(II) K2CO3, H2O.
После внутримолекулярного нуклеофильного присоединения
образовавшегося реактива Гриньяра к карбонильному атому углерода Boc-группы и обработки реакционной массы
водным раствором карбоната калия удалось выделить смесь продуктов 106 и 107
(рис.6). Согласно данным ЯМР в смеси преобладала енольная форма 107.
Масс-спектр высокого разрешения подтвердил наличие моно-, и дипротонированных
молекулярных ионов аминокетона с массой 208.1696 и 209.1729.

Рис.6.
Продукты, выделенные после реакции дихлорида с магнием Райка.
Судя по всему, выделенные соединения подвергаются олигомеризации,
кроме того был низкий выход продукта. Поэтому было решено провести еще один
синтез в соответствии со схемой 23, но использовать менее стерически
нагруженную этоксикарбонильную группу. Кроме того, нужно было выяснить
реакционную способность обоих транс - и цис-изомеров.
Цис-изомер
так же был переведен в соответствующий спирт 110. Оба изомерных
аминоспирта обрабатывали сначала HCl, и затем SOCl2. Полученные гидрохлориды дихлорпроизводных не выделяли, а сразу
ацилировали этилхлорформиатом в присутствии диизопропилэтиламина (схема 30).

Схема 30. Реагенты:
(I) HCl; (II) SOCl2;
(III) EtOC (O) Cl; (IV) (i-Pr) 2NЕt.
Дихлориды 109 и 112 как и в предыдущем случае были
введены в реакцию с магнием Райка, но на последней стадии реакционную смесь еще
обработали триацетоксиборгидридом или борогидридом натрия (схема 31).
Однако выделить из реакционной массы ничего, кроме исходных соединений, не
удалось.

Схема 31. Реагенты
(II) K2CO3; (III) H2O; (IV) NaBH (OAc) 3.
Исходя из новых данных, была предложена другая схема синтеза
алкалоида божьей коровки, представленная на рис.8. Для замыкания
третьего цикла предполагается использование реакции метатезиса.
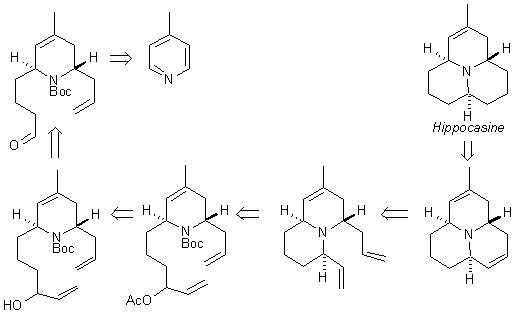
Рис.8. Ретросинтетическая
схема синтеза алколоида азафеналенового ряда.
Для введения алкилкарбонильной группы на первой стадии нам
требовалось получить 4,4-диэтоксибутил-1-ена. Для этого мы разработали новый и
эффективный способ аллилирования триэтилформиата триаллилбораном в присутствии
каталитических количеств кислот Льюиса (схема 32).
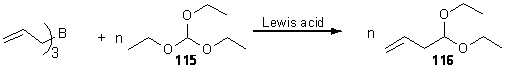
Схема 32.
В первую очередь нами был опробован в качестве кислоты Льюиса BF3*Et2O. Однако реакция не идет до конца, а продукт был загрязнен
полимерами и исходным соединением (температуры кипения у триэтилортоформиата и
4,4-диэтоксибутил-1-ена близки), поэтому решено было использовать другие
кислоты Льюиса, а именно ZnCl2, InCl3 и Zn (OTf) 2.
Наилучшие результаты были получены с трифлатом цинка (таблица 2). Кроме
того, реакция с данными катализаторами шла при комнатной температуре, в отличие
от BF3, где было необходимо охлаждение.
Таблица 2. Катализаторы и соответствующие им выходы
4,4-диэтоксибутил-1-ена.
|
Lewis
acid
|
Yield,
%
|
|
BF3*Et2O
|
56
|
|
ZnCl2
|
62
|
|
InCl3
|
50
|
|
Zn (OTf) 2
|
77
|
Отработав получение 116 в препаративных количествах
можно было продолжить синтез. Гидроборирование 116
борандиметилсульфидным комплексом с последующим расщеплением борпроизводного I2/EtOK в этаноле был получен
иодид 117. Присоединения литийпроизводного, полученного из 117 и t-BuLi, к 4-пиколину и
последующей обработкой литий амида триаллилбораном дает соединение 120 (схема
33) исключительно с транс-конфигурацией заместителей, что является
хорошо установленным фактом [35].

Схема 33. Реагенты: (I) BH3*Me2S;
(II) I2, KOEt; (III) t-BuLi, - 780C; (IV) All3B;
(V) MeOH; (VI) NaOH.
Для проведения дальнейшего синтеза необходимо было защитить
аминогруппу соединения 120, что было сделано по уже упомянутой выше методике
(схема 34). После расщепления ацеталя 121 нагреванием в смеси
THF, H2O, AcOH, полученный альдегид 122 ввели в реакцию с
винилмагнийбромидом при охлаждении до - 780 С. В результате, был
получен спирт аллильного типа 123 в виде единственного диастереомера,
строение которого мы не смогли установить.
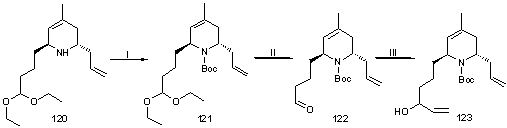
Схема 34. Реагенты:
(I) Boc2O, THF; (II) THF/H2O/AcOH (4/3/5); (III) 1) винилмагнийбромид, THF, - 780C;2) H2O.
Для синтеза бициклического диена была использована следующая
стратегия (схема 35). Из аллилового спирта 123 взаимодействием с
уксусным ангидридом в растворе пиридина был получен ацетат 124,
Вос-защиту с аминогруппы которого сняли взаимодействием с трифторуксусной
кислотой. Каталитическое внутримолекулярное аллильное аминирование под
действием аллильного комплекса палладия приводит к образованию двух изомерных
бициклов 126 в соотношении ~3: 1 (катализатор получали in situ взаимодействием All2Pd2Cl2 с Ph3P):
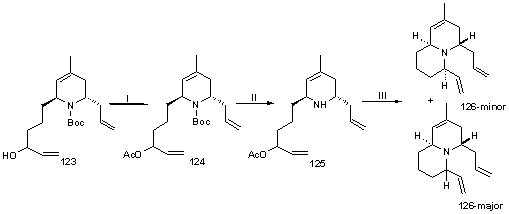
Схема 35. Реагенты: (I) Ac2O, Py; (II)
CF3CO2H; (III) All2Pd2Cl2,
Ph3P.
Относительная конфигурация, вновь образованного стереоцентра
в основном изомере 126-major, доказывалась с помощью ЯМР: вначале проводилось
полное отнесение сигналов в протонном спектре соединения (эксперимент COSY 1H-1H), а затем определялись
пространственно взаимодействующие группы (NOESY).
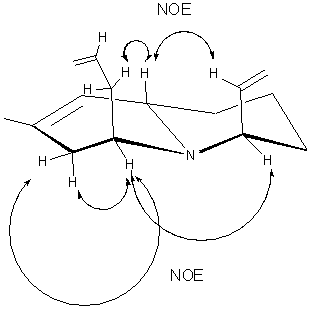
На основании полученных данных установлена относительная 4R*-конфигурация в основном изомере и соответственно 4S* - в другом изомере. Из этого следует, что конфигурации
стереоцентров минорного изомера соответствуют центрам в алкалоиде hippocasine. Поэтому далее была проведена реакция
внутримолекулярного метатезиса 126-minor с использованием катализатора Граббса второго поколения (Схема
36).
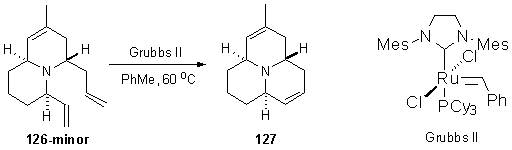
Схема 36.
Реакция метатезиса успешно прошла и было выделено
соответствующее производное азафеналена, содержащее две различные связи С=С,
строение которого подтверждено методом масс-спектрометрии и ЯМР спектроскопии.
Поскольку реакционная способность ди - и тризамещенных С=С связей при
каталитическом гидрировании сильно отличается, существует возможность
селективного восстановления С=С связи возникающей после реакции метатезиса, что
позволит синтезировать требуемый алкалоид.
Таким образом, цель работы - разработка нового способа
синтеза соединений азафеналенового ряда с использованием реакций
аллилборирования и метатезиса была достигнута.
3.
Экспериментальная часть
Все операции с борорганическими соединениями выполняли в
атмосфере сухого аргона. Растворители абсолютировали согласно стандартным
методикам. Гексан (ч.), этилацетат (х. ч.) использовались без дополнительной очистки.
Спектры ЯМР регистрировали на спектрометрах Bruker, Avance-300, 400 и 600 МГц.
Химические сдвиги d (ppm) в спектрах 1Н и 13С
даны относительно остаточных сигналов растворителя в CDCl3; Масс
спектры регистрировали на приборе Finnigan Polaris Q Ion Trap. Для тонкослойной
хроматографии использовали пластины "Silicagel 60 F254", для колоночной
хроматографии использовали силикагель (размер 60-230 меш) фирмы Merck (Германия). Обнаружение
веществ на хроматограммах проводили обработкой пластин парами йода, а также
раствором бихромата калия в серной кислоте.
Синтез
транс-2,6-диаллил-4-метил-1,2,3,6-тетрагидропиридина (95).
4-пиколин (20,5 г, 21,4 мл, 0,22 моль) добавляли по каплям в
атмосфере аргона к охлажденному до - 300С триаллилборану (13,4 г,
16,8 мл, 0,1 моль). Когда температура поднялась до 00С, добавили
пропан-2-ол (24,0 г, 30,6 мл, 0,4 моль) одной порцией в реакционную смеси и
нагревали в течении 2 часов при 500С, затем добавляли с
перемешиванием раствор NaOH (5 N, 40 мл, 0,2 моль). Органическую фазу разбавляли
гексаном, отделяли, сушили над K2CO3, упарили на роторном испарителе, вещество
перегнали под вакуумом. Поучено 32,04 г (91%). ЯМР 1H (300 МГц, DMSO - [D6]): d = 5.90-5.76 (м, 2H);
5.38 (м, 1H); 5.10-5.02 (м, 4H); 3.27 (уш. с, 1H); 2.88 (дт, J = 5.5,
7.3, 1H); 2.17-2.09 (м, 4H); 1.85 (дд, J = 4.1, 17.1, 1H); 1.70 (уш. с,
2H); 1.65 (с, 3H) ppm.
Синтез смеси цис - и
транс-2,6-диаллил-4-метокси-1,2,3,6-тетрагидропиридина (98).
4-метоксипиридин (6 г, 5,6 мл, 0,055 моль) добавляли по
каплям в атмосфере аргона к охлажденному до - 300С триаллилборану
(3,685 г, 4,8 мл, 0,0275 моль). Когда температура поднялась до 00С,
добавили пропан-2-ол (6,6 г, 8,4 мл, 0,11 моль) одной порцией в реакционную
смеси и нагревали в течении 2 часов при 500С, затем добавляли с
перемешиванием раствор NaOH (5 N, 8 мл, 0,05 моль). Органическую фазу разбавляли
гексаном, отделяли, сушили над K2CO3, упарили на роторном испарителе, вещество
перегнали под вакуумом. Получено 6,24 г (59% считая на триаллилборан). Согласно
спектру ПМР соотношение транс/цис = 2.2/1
Синтез
цис-2,6-диаллил-4-метил-1,2,3,6-тетрагидропиридина (99).
транс-2,6-Диаллил-4-метил-1,2,3,6-тетрагидропиридин
(14 г, 13,87 мл, 79,1 ммоль) по каплям добавляли триаллилборан (11,92 г, 15,2
мл, 89 ммоль) при перемешивании в атмосфере аргона при температуре 130-1350С
в течении 6 часов. Затем смесь обрабатывали МеОН и 20% -ным NaOH, разбавляли гексаном,
отделяли органический слой, сушили K2CO3, упарили на роторном испарителе, перегоняли под
вакуумом. Получено 10,78 г (77%).1H ЯМР (400 МГц, dmso - [D6]): d = 5.81 (м, 1H); 5.26 (с,
1H); 5.07-5.00 (м, 4H); 3.27 (уш. с, 1H); 2.69 (квартет т, J = 4.0; 6.2;
6.5, 1H); 2.11 (т, J = 6.8, 2H); 2.07 (т, J = 6.6, 2H); 1.75 (дт,
J = 3.1; 16.8, 1H); 1.70 (уш. д, J = 10.3, 1H); 1.60 (с, 3H) ppm.13C
ЯМР (100 МГц, dmso - [D6]): d = 137.14 CH; 137.00 CH; 133.42 C; 125.46 CH;
117.86 2CH2; 55.14 CH; 53.55 CH; 41.88 CH2; 41.85 CH2;
37.80 CH2; 24.16 CH3 ppm.
Синтез
цис-2,6-диаллил-4-метокси-1,2,3,6-тетрагидропиридина (100).
транс-2,6-Диаллил-4-метокси-1,2,3,6-тетрагидропиридин
(3,93 г,20 ммоль) по каплям добавляли триаллилборан (2,95 г, 3,8 мл, 22 ммоль)
при перемешивании в атмосфере аргона при температуре 130-1350С в
течении 6 часов. Затем смесь обрабатывали 1,8 мл МеОН и 5,2 мл 20% -ного NaOH, разбавляли гексаном,
отделяли органический слой, сушили K2CO3, упарили на роторном испарителе, перегоняли под
вакуумом. Получено 3,36 г (86%).1H ЯМР (300 MHz, CDCl3): δ = 5.96-5.73 (м, 2H), 5.22-5.12 (м, 4H), 4.58 (уш. с, 1H), 3.57
(с, 3H), 3.53-3.47 (м, 1H), 2.95-2.86 (м, 1H), 2.43 - 2.16 (м, 4H), 2.06-2.03
(м, 2H) 2.00 (уш. с, 1H, NH) ppm.
Синтез транс-3,3’-
(4-метил-1,2,3,6-тетрагидропиридин-2,6-диил) дипропан-1-ола (102).
К BH3·Me3S (6,916 г, 8,8 мл, 91 ммлоль) в атмосфере аргона при 0-50С
прибавляли тетраметилэтилен (7,31 г, 10,5 мл, 87 ммоль), перемешивали 30 минут
при постепенном повышении температуры. Затем смесь упарили на водоструе и
разбавили в 12,3 мл ТГФ, прибавляли ее к раствору транс-2,6-диаллил-4-метил-1,2,3,6-тетрагидропиридина
в 41 мл ТГФ при 00С, перемешивали 4 часа при этой температуре. Затем
добавили 16,5 мл MeOH и окисляли при 0-50С смесью 16,5 мл 30% -ой Н2О2,
84,4 мл 20% -ного NaOH и 62 мл воды. Далее перемешивали 30 минут при температуре 400С,
час кипятили при перемешивании. Затем к реакционной смеси прибавили смесь
этилацетата и эфира (15 мл×2 р), экстрагировали.
Экстракты промыли концентрированным раствором NaCl, сушили K2CO3, упарили под вакуумом.
Получено 6,257 г (74%). Методом ЯМР контролировали полноту гидроборирования по
исчезновению сигналов олефиновых протонов. Соединение 102 использовали
далее без дополнительной очистки.
Синтез транс-3,3’ - [1-Boc-
(4-метил-1,2,3,6-тетрагидропиридин-2,6-диил)] дипропан-1-ола (103).
К 1,266 г (5,54 ммоль) транс-3,3’-
(4-метил-1,2,3,6-тетрагидропиридин-2,6-диил) дипропан-1-ола в растворе 11,3 мл
ТГФ и 11,3 мл H2O прибавили K2CO3 (1,0615 г, 7,72 ммоль), затем прибавляли Boc2O (2,7 мл, 11,89 ммоль)
при 00С. Перемешивали 5 часов, упарили на роторном испарителе. К
полученной смеси добавили 20% -ный KHSO4, экстрагировали этилацетатом, выделили
хроматографически (элюент: этилацетат/гексан=1/1) Получено 1,54 г (83%).1H ЯМР (400 МГц, CDCl3): δ = 5.51 (c, 1H); 4.09 (c, 1H); 3.90 (c, 1H); 3.65-3.62 (m, 4H); 2.60 (уш. с, 2H, 2OH); 2.25 (д, J = 15.1, 1H); 1.95 (дд, J = 2.6, 16.0, 1H); 1.75-1.72 (m, 1H); 1.73 (c, 3H); 1.64-1.50 (m, 7H); 1.45 (c, 9H) ppm.13C ЯМР (75 МГц,, CDCl3): δ = 155.70; 132.05; 122.87; 79.74; 62.55; 62.32; 52.13; 51.28;
32.32; 31.90; 29.92; 29.83; 28.50; 23.85 ppm.
Синтез транс-1-Boc-2,6-бис (3-хлорпропил) -
4-метил-3,6-дигидропиридина (104).
К раствору транс-3,3’ - [1-Boc-
(4-метил-1,2,3,6-тетрагидропиридин-2,6-диил)] дипропан-1-ола (2,86 г, 8,17
ммоль) в 53 мл ССl4 прибавили Ph3P (4,28 г, 16,34 ммоль) и кипятили. Полноту
прохождения реакции проверяли по ТСХ (элюент: этилацетат/гексан=1/2). Выделяли
хроматографически. Получено 1,677 г (59%).1H ЯМР (300 МГц, CDCl3): δ = 5.46-5.44 (m, 1H); 4.12-4.10 (m, 1H); 3.90-3.86 (m, 1H); 3.54-3.50 (m, 4H); 2.24 (дм, J = 16.3, 1H); 1.94 (дд, J = 3.6, 16.3, 1H); 1.75 (c, 3H); 1.87-1.58 (m, 8H); 1.47 (c, 9H) ppm.13C ЯМР (75 МГц, CDCl3): δ = 155.42; 132.37; 122.74; 79.72; 52.24; 51.29; 45.22; 44.99;
32.74; 32.60; 31.10; 30.42; 28.60; 28.49 3C; 23.72 ppm.
Синтез смеси 106 и 107.
При перемешивании в атмосфере аргона добавили йод (1,52 г, 2
ммоль) к магнию (0,15 г, 6,17 ммоль) в 35 мл THF, затем 1,2-дибромэтан
(0,772 г, 0,35мл, 4,1 ммоль). Нагревали до полного растворения магния. Далее
добавили металичесский калий и кипятили 3 часа. К полученному коллоидному
магнию Райки по каплям добавили дихлорид 104 (0,282 г, 0,8 ммоль) в 3 мл
THF. Полноту прохождения
реакции проверяли по ТСХ (элюент: этилацетат/МеОН/NH3=90/1/0,2). Далее
обработали смесь раствором K2CO3 (4 г, 29 ммоль), осадок отфильтровали,
экстрагировали гексаном, экстракты сушили K2CO3, упарили на роторном
испарителе, выделяли хроматографически.
Синтез этил транс-2,6-бис (3-хлорпропил) -
4-метил-3,6-дигидропиридин-1 (2H) - карбоксилата (109).
К раствору аминодиола 102 (8.5 гр, 40 ммоль) в CH2Cl2 (50 мл) добавили раствор
HCl 4M в диоксане (10 мл, 40
ммоль), затем прибавили по каплям SOCl2 (23.8 гр, 14.4 мл, 0.2 моля) при перемешивании.
Полученный раствор кипятили 1 час, затем удалили растворители при пониженном
давлении, разбавили CCl4 (15 мл) и вновь упарили. Остаток растворили в CH2Cl2 (50 мл), добавили
этилхлорформиат (6.51 гр, 5.73 мл, 60 ммоль) остудили до 0 0С в бане
со льдом и прибавили i-Pr2NEt (12.9 гр, 17 мл, 0.1 моль). Перемешивали в течение ночи. Промыли
водой, сушили поташем, упаривали, а остаток перегоняли в вакууме т.к.156-158 0С
(0.1 ммHg). Получено: 4 гр (31%) 109 в виде желтоватого масла.1H ЯМР (400 МГц, CDCl3): δ = 5.47 (c, 1H);
.20-4.09 (m, 3H); 3.91-3.89 (m, 1H); 3.58-3.47 (m, 4H); 2.75 (дм, J = 16.5, 1H); 1.98 (дд, J = 3.5, 16.2, 1H); 1.81-1.71 (m, 8H); 1.76 (c, 3H); 1.28 (т, J = 7.0, 3H) ppm.13C ЯМР (100 МГц,, CDCl3): δ = 156.90; 133.19 br.; 123.22; 61.69; 53.11; 52.02; 45.72; 45.53;
33.49; 32.90; 31.65; 30.92; 29.17; 24.33; 15.28 ppm.
цис-2,6-бис (3-хлорпропил) - 4-метил-3,6-дигидропиридин-1
(2H) - карбоксилат (112) получали аналогично транс-изомеру
109. Сигналы в протонном спектре уширены вследствие затрудненного
вращения в амидной группе 1H ЯМР (300 МГц, CDCl3): δ = 5.41 (c, 1H); 4.50 (уш. с, 1H); 4.30 (уш. c, 1H);
.20-4.18 (m, 2H); 3.62-3.58 (m, 4H); 2.38 (дм, J = 15.4, 1H); 1.97-1.77 (m, 10H); 1.66-1.56 (m, 2H); 1.31 (т, J = 7.0, 3H) ppm.13C ЯМР (75 МГц,, CDCl3): δ =155.89; 129.91; 119.38; 61.28; 51.07; 47.77; 44.86; 44.77;
34.01; 33.78; 31.75; 30.03; 29.71; 23.77; 14.66 ppm.
Синтез 4,4-диэтоксибутил-1-ена (116).
Метод А. Колбу заполнить аргоном, поместили в нее
триэтилортоформиат (20,55 г, 23,1 мл, 0,1389 моль) и BF3*Et2O (5 мол %, 0,979 г, 0,87
мл, 6,9 ммоль). При охлаждении и перемешивании (6-70С) по каплям
прибавили триаллилборан (8,46 г, 10,9 мл, 0,063 моль). Перемешивали при этой
температуре 20 часов. Полученную смесь по каплям прибавили к 96 мл 10% -ого
NaOH. Экстрагировали эфиром, сушили K2CO3, перегнали в вакууме (tкип=690С
при 45 мм рт ст). Получено 11,181 г (56%).
Метод B. Колбу заполнили аргоном, поместили в нее триэтилортоформиат
(1,78 г, 2 мл, 12,03 моль) и ZnCl2 (6 мол %, 0,98 г, 0,722 ммоль). При охлаждении и
перемешивании (00С) по каплям прибавили триаллилборан (0,733 г, 0,95
мл, 5,5 моль). Перемешивали при комнатной температуре 2 часа. Полученную смесь
по каплям прибавили к 8,4 мл 10% -ого NaOH. Экстрагировали эфиром, сушили K2CO3, перегнали в вакууме (tкип=690С
при 45 мм рт ст). Получено 1,08 г (62%)
Метод С. Колбу заполнили аргоном, поместили в нее
триэтилортоформиат (1,78 г, 2 мл, 12,03 моль) и InCl3 (5 мол %, 0,113 г,
0,6015 ммоль). При охлаждении и перемешивании (00С) по каплям
прибавили триаллилборан (0,733 г, 0,95 мл, 5,5 моль). Перемешивали при
комнатной температуре сутки. Полученную смесь по каплям прибавилиь к 8,4 мл 10%
-ого NaOH. Экстрагировали эфиром, сушилиь K2CO3, перегнали в вакууме (tкип=690С
при 45 мм рт ст). Получено 0,87 г (50%).
Метод D. Колбу заполнили аргоном, поместили в нее триэтилортоформиат
(1,78 г, 2 мл, 12,03 моль) и Zn (OTf) 2 (5 мол %, 0,22 г, 0,6015 ммоль).
При комнатной температуре по каплям прибавили триаллилборан (0,733 г, 0,95 мл,
5,5 моль). Перемешивали при комнатной температуре сутки. Полученную смесь по
каплям прибавили к 8,4 мл 10% -ого NaOH. Экстрагировали эфиром, сушили K2CO3, перегнали в вакууме (tкип=690С
при 45 мм рт ст). Получено 1,34 г (77%).1H ЯМР (300 МГц, CDCl3): δ = 5.86 (ddt, J = 17.2, 10.2, 7.0, 1H); 5.21-5.10 (m, 2H);
4.57 (t, J = 5.8, 1H); 3.73 (m, 2H); 3.55 (m, 2H); 2.45 (tt, J = 1.0, 5.8, 2H); 1.26 (t, J =
7.1, 6H) ppm.13C ЯМР (75 МГц, DMSO - [D6]): δ = 133.76 CH=; 116.94 CH2=; 101.55 CHO2; 60.44 2CH2O; 38.07 CH2; 15.12 CH3 ppm.
Синтез 1,1-диэтокси-4-йодбутана (117).
К раствору 116 (16,584 г, 0,1052 моль) в 60 мл THF при 00С
прибавили BMS (2,985 г, 3,73 мл, 39,3 ммоль) за 10 минут, перемешивали еще 10
минут, затем убрали охлаждение и перемешивали еще час при комнатной
температуре. Далее прибавили 1,2 мл этанола для удаления избытка гидрида.
Охладили смесь до - 50С и прибавили йод одной порцией, затем по
каплям раствор t-BuOK (13,85 г, 0,1236 моль) в EtOH (25 мл) за 10 минут.
Перемешивали сутки, затем прибавили к смеси Na2SO3 (7,3 г, 58 ммоль).
Экстрагировали пентаном, сушили K2CO3, перегнали под вакуумом.1H ЯМР (300 МГц, CDCl3): δ = 4.54 (t, J = 5.6, 1H); 3.73-3.63 (m, 2H); 3.58-3.47 (m, 2H); 3.25 (t, J = 6.9, 2H); 2.00-1.90 (m, 2H); 1.79-1.72 (m, 2H); 1.24 (t, J = 7.1, 6H) ppm.13C ЯМР (75 МГц,, CDCl3): δ = 101.87; 61.19 2C; 34.46; 28.82; 15.32 2C; 6.74 ppm.
Синтез транс-2-аллил-6- (4,4-диэтоксил) -
4-метил-1,2,3,6-тетрагидропиридина (120).
В атмосфере аргона к раствору йодида 117 (4,3 г, 16,7
ммоль) в смеси пентан/эфир (78 мл/45 мл) при - 780С довольно быстро
прибавили t-BuLi (22,2 мл 1,5 М раствора, 33 ммоль). Перемешивали полтора часа при
- 200С, затем прибавили 4-пиколин (1,55 г, 1,62 мл, 16,7 ммоль),
перемешивали час при комнатной температуре. Далее смесь охладили до - 100С
и по каплям прибавили триаллилборан (2,57 г, 3,3 мл,19 ммоль), смесь нагрели до
00С, прибавили 3 мл МеОН, затем 6,1 мл 20% -ого NaOH. Экстрагировали
гексаном, сушили K2CO3, упарили на роторном испарителе, выделяли хроматографически
(элюент: этилацетат/метанол/ NH3=100/2/0,42). Получено 2,93 г (64%).1H ЯМР (300 МГц, CDCl3): δ = 5.87-5.73 (m, 1H); 5.40 (c, 1H); 5.15-5.08 (m, 2H); 4.49 (т, J = 5.5, 1H); 3.70-3.60 (m, 2H); 3.54-3.44 (m, 2H); 3.32 (уш. с, 1H); 3.01-2.93 (m, 1H); 2.75 (уш. с, 1H, NH); 2.21-2.17 (m, 2H); 1.96 (дд, J = 3.5, 17.3, 1H); 1.78 (дд, J = 8.1, 17.4, 1H); 1.68 (c, 3H); 1.67-1.62 (m, 2H); 1.45 (уш. с, 4H); 1.21 (т, J = 7.0, 6H) ppm.13C ЯМР (75 MHz, CDCl3): δ = 135.31; 131.79; 123.44; 117.21; 102.58; 60.78; 60.75; 51.89;
46.94; 39.91; 35.99; 35.09; 33.43; 28.29; 23.30; 21.51; 15.18 ppm. МС (70 eV, EI): m/z 382/381 (M+, 0,08/0,28); 336 (12);
236 (30); 194 (43); 190 (15); 180 (40); 151 (10); 150 (100); 148 (46); 138
(17); 107 (10); 94 (59); 93 (21); 88 (9); 57 (10). Найдено (%): С, 72.46; H,
11.18; N,
4.79. С17Н31NO2. Вычислено (%): С, 72.55; Н, 11.10; N, 4.98.
Синтез трет-бутил транс-2-аллил-6-
(4,4-диэтоксибутил) - 4-метил-3,6-дигидро-1 (2H) - пиридинкарбоксилата
(121).
В колбу поместили раствор 121 (3,4 г, 12,36
ммоль) в 10 мл THF, затем Boc2O (3,67 г, 16,8 ммоль). Полноту прохождения
реакции проверяли по ТСХ (элюент: этилацетат/гексан=1/15). Упарили, выделяли
хроматографически. Получено 2,45 г (53%).1H ЯМР (300 МГц, CDCl3): δ = 5.84-5.70 (m, 1H); 5.56 (уш. с, 1H); 5.04-5.00 (m, 2H); 4.48 (т, J = 5.6, 1H); 4.05-4.01 (m, 2H); 3.70-3.60 (m, 2H); 3.55-3.45 (m, 2H); 2.37-2.23 (m, 2H); 2.16 (дд, J = 9.0, 13.3, 1H); 1.98 (д, J = 15.1, 1H); 1.76 (c, 3H); 1.74-1.60 (m, 4H); 1.49 (c, 9H); 1.39-1.28 (m, 2H); 1.22 (т, J =
7.0, 6H) ppm.13C ЯМР (75 MHz, CDCl3): δ = 155.17; 136.29;
131.71; 122.94; 116.71; 102.87; 79.22; 60.91; 60.84; 52.32; 51.47; 38.23 br.; 35.64 br.; 33.67; 31.34; 29.68;
28.49 3C;
23.67; 20.66; 15.32 ppm. МС (70 eV, EI): m/z 337/336 ([M-EtO] +, 3/11);
237 (5); 236 (32); 194 (50); 190 (15); 180 (36); 151 (14); 150 (100); 148 (48);
138 (16); 136 (6); 122 (6); 107 (9); 94 (46); 93 (23); 88 (12); 80 (7); 57 (8);
41 (10). Найдено (%): С, 70.06; H, 10.38; N, 3.57. С22Н39NO4. Вычислено (%): С,
69.25; Н, 10.30; N, 3.67.
Синтез трет-бутил транс-2-аллил-4-метил-6-
(4-оксобутил) - 3,6-дигидро-1 (2H) - пиридинкарбоксилата (122).
Раствор 121 (3,103 г, 8,24 ммоль) в смеси 4 мл THF, 3 мл H2O и 5 мл AcOH нагревали 14 часов при
температуре 500С. Полноту прохождения реакции проверяли по ТСХ
(элюент: этилацетат/гексан=1/4). Экстрагировали гексаном, сушили K2CO3, упарили на роторном
испарители, выделяли хроматографически. Получено 1,78 г (72%).1H ЯМР (400 МГц, CDCl3): δ = 9.74 (т, J = 1.6, 1H); 5.73 (дддд, J = 6.1, 8.6, 11.4, 17.7,
1H); 5.52 (м, 1H); 5.00-4.97 (м, 2H); 4.06 (m, 1H); 3.99 (m, 1H); 2.42 (тт, J = 1.2, 7.5, 2H); 2.29-2.20 (m, 2H); 2.15 (дт, J = 8.8, 13.4, 2H); 1.96 (дд, J = 1.6, 15.9, 1H); 1.73 (s, 3H); 1.72-1.68 (m, 2H); 1.64-1.53 (m, 2H); 1.46 (c, 9H) ppm.13C NMR (100 MHz, CDCl3): δ = 202.61; 155.22; 136.17; 132.19; 122.52; 116.83; 79.45; 52.02;
51.49; 43.90; 38.21 br.; 35.00 br.; 31.44; 28.48 3C; 23.67; 17.86 ppm. МС (70 eV, EI): m/z 308 (MH+, 5,8); 252 (5); 190 (8);
180 (25); 166 (100); 148 (38); 138 (17); 131 (15); 122 (22); 94 (54); 93 (32);
91 (17); 79 (10); 41 (9). Найдено (%): С, 70.26; H, 9.48; N, 4.60. С18Н29NO3. Вычислено (%): С,
70.32; Н, 9.51; N, 4.56.
Синтез трет-бутил транс-2-аллил-6-
(4-гидрокси-5-гексенил) - 4-метил-3,6-дигидро-1 (2H) - пиридинкарбоксилата
(123).
К охлажденному до - 780С альдегиду 122 в
атмосфере аргона по каплям прибавили раствор винилмагнийбромида. Прохождение
реакции контролировали по ТСХ (элюент: этилацетат/гексан=1/2). Далее разбавили
водой, экстрагировали гексаном, сушили K2CO3, упарили на роторном
испарителе, выделяли хроматографически. Получено 0,422 г (27%).1H ЯМР (400 МГц, CDCl3): δ = 5.87 (дддд, J = 2.3, 6.2, 10.4, 16.7, 1H); 5.80-5.70 (m, 1H); 5.55 (c, 1H); 5.24 (ддд, J = 1.5, 3.1, 17.2, 1H); 5.10 (дт, J = 1.4, 1.9, 10.4, 1H); 5.02-4.98 (m, 2H); 4.16-4.06 (m, 2H); 4.02-3.95 (m, 1H); 2.33 (дм, J = 13.3, 1H); 2.26 (дм, J = 15.9; 1H); 2.16 (дт, J = 8.9, 13.3, 1H); 1.97 (дд, J = 2.3, 15.9, 1H); 1.74 (c, 3H); 1.73 (м, 1H); 1.69 (уш. с, 1H); 1.56-1.50 (м, 2H); 1.48 (с, 9H); 1.39-1.30 (м, 2H) ppm.13C ЯМР (100 MHz, CDCl3): δ = 155.27; 141.28 и 141.20; 136.31; 131.18; 122.96; 116.76;
114.49; 79.35; 73.21 и 73.05; 52.26; 51.53; 38.26; 37.02 и 36.95; 35.64 br.; 31.41 и 31.38; 28.53 3C; 23.71; 21.09 ppm. МС (70 eV, EI): m/z 336/335 (M+, 0.5/2.2); 243 (4); 236
(4); 195 (8); 194 (76); 192 (12); 180 (51); 177 (9); 176 (66); 174 (47); 138
(29); 136 (11); 134 (16); 120 (18); 107 (12); 96 (20); 95 (17); 94 (100); 93
(40); 88 (16); 80 (17); 78 (6); 77 (10); 41 (10). Найдено (%): С, 71.65; H,
9.85; N,
4.16. С20Н33NO3. Вычислено (%): С, 71.60; Н, 9.91; N, 4.18.
Синтез трет-бутил транс-6 - [4- (ацетилокси) -
5-гексенил] - 2-аллил-4-метил-3,6-дигидро-1 (2H) - пиридинкарбоксилата (124).
Спирт 123 (0,422 г, 1,28 ммоль) смешали с пиридином
(0,64 мл) и Ac2O (0,39 г, 0,37 мл, 3,84 ммоль) и грели на водяной бане.
Прохождение реакции контролировали по ТСХ (элюент: этилацетат/гексан=1/2).
Экстрагировали гексаном, экстракты промыли раствором водой и раствором соды,
сушили K2CO3, упарили на роторном испарителе, выделяли хроматографически.
Получено 0,478 г (99%) 1H ЯМР (400 МГц, CDCl3): δ = 5.84-5.70 (m, 2H); 5.55 (уш. с, 1H); 5.34-5.16 (m, 3H); 5.04-4.99 (m, 2H); 4.06-4.00 (m, 2H); 2.37-2.12 (m, 3H); 2.08 (c, 3H); 1.98 (дд, J = 1.4, 15.9, 1H); 1.71-1.60 (m, 4H); 1.50 (c, 9H); 1.36-1.24 (m, 2H) ppm.13C ЯМР (100 MHz, CDCl3): δ = 170.33, 155.16, 136.52 и 136.47; 136.25; 131.92; 122.84;
116.76; 116.61 и 116.50; 79.29; 74.83 и 74.75; 52.18; 51.47; 38.22; 35.45;
34.20 и 34.16; 31.37; 28.49 3C; 23.67; 21.20 и 20.97 ppm. МС (70 eV, EI): m/z 379/378 (MH+, 0.2/1.5); 266 (5); 237
(10); 236 (63); 180 (38); 177 (14); 176 (100); 174 (47); 147 (5); 146 (5); 138
(20); 134 (11); 120 (12); 108 (8); 96 (17); 95 (12); 94 (75); 93 (31); 91 (11);
88 (13); 80 (12); 44 (6). Найдено (%): С, 70.15; H, 9.45; N, 3.66. С22Н35NO4. Вычислено (%): С,
69.99; Н, 9.34; N, 3.71.
Синтез 1-{3 - [ (2R*,6R*) -
6-аллил-4-метил-1,2,5,6-тетрагидропиридин-2-ил] пропил}проп-2-ен-1-ил ацетата
(125).
К 124 (0,478 г, 1,448 ммоль) прибавили трифторуксусную
кислоту (2 мл), перемешивали. Прохождение реакции контролировали по ТСХ
(элюент: этилацетат/гексан=1/2). Кислоту отогнали на роторе, разбавили CH2Cl2, промыли раствором K2CO3, сушили K2CO3, упарили на роторе,
выделялил хроматографически (элюент: этилацетат/метанол=100/1). Получено 0,28 г
(70%). Использовали далее без дополнительной очистки.1H ЯМР (400 МГц, CDCl3): δ = 5.86-5.74 (m, 2H); 5.45 (c, 1H); 5.32-5.19 (m, 5H); 3.65 (m, 1H); 3.33 (m, 1H); 2.63-2.59 (m, 1H); 2.38 (дт, J = 7.9, 13.7, 1H); 2.26 (дд, J = 3.3, 17.9, 1H); 2.15 (c, 3H); 2.09-2.05 (m, 1H); 1.87-1.75 (m, 2H); 1.78 (c, 3H); 1.73-1.60 (m, 2H); 1.53-1.45 (m, 2H) ppm. МС (70 eV, EI): m/z 279/278 (MH+, 0.12/0.71); 266 (2);
236 (26); 214 (14); 176 (47); 174 (35); 149 (10); 147 (10); 136 (20); 134 (10);
132 (9); 131 (9); 120 (15); 107 (14); 105 (10); 96 (15); 95 (14); 94 (100); 91
(11); 77 (12); 43 (12); 41 (12).
Синтез (4R*,6S*,9aS*) - и (4S*,6S*,9aS*) -
6-Аллил-8-метил-4-винил-1,3,4,6,7,9a-гексагидро-2H-хинолизина (126-minor) и (126-major).
К раствору трифенилфосфина (32 мг, 0,122 ммоль) в CH2Cl2 при перемешивании в
атмосфере аргона прибавили All2Pd2Cl2 (10 моль/%, 23 мг, 0,06 ммоль), затем раствор 125
в CH2Cl2 и порошок K2CO3 (для удаления
образующейся AcOH). Прохождение реакции контролировали по ТСХ (элюент:
этилацетат/метанол=100/1). Добавили K2CO3, разбавили CH2Cl2, промыли водой, сушили K2CO3, упарили на роторном
испарителе. Выделяли хроматографически два изомера.
(4R*,6S*,9aS*) - 126-minor Rf max 1H NMR (600 MHz, CDCl3): δ = 5.80-5.75 (br. m, 1H); 5.68 (dddd, J = 5.8, 8.5, 10.7, 16.2, 1H); 5.23-5.17 (m, 2H); 5.09 (br. d, J = 9.5, 1H); 5.00-4.97 (m, 2H); 3.24 (br. s, 1H); 3.24 (br. s, 1H); 3.04 (br. s, 1H); 2.83 (br. s, 1H); 2.35 (dd, J = 5.4, 13.0, 1H); 2.31 (br. s, 1H); 1.95 (dd, J = 10.6, 20.9, 1H); 1.87 (d, J = 17.4, 1H); 1.79-1.72 (m, 2H); 1.68 (s, 3H); 1.66-1.58 (m, 3H); 1.46-1.38 (m, 1H) ppm.13C NMR (100 MHz, CDCl3): δ = 141.08 br; 136.81 br; 130.74; 123.68 br; 116.50; 115.77 br; 63.82; 54.65; 53.16;
33.55 br; 32.90 br; 32.43 br; 27.37; 24.26; 23.18 ppm. MS (70 eV,
EI): m/z 217 (M+,
); 216 (5); 199 (5); 183 (16); 177 (13); 176
(96); 175 (16); 174 (100); 172 (7); 163 (5); 152 (6); 149 (24); 146 (14); 134
(17); 132 (18); 120 (17); 107 (18); 94 (27); 83 (31); 67 (11); 57 (8); 41 (10).
(4S*,6S*,9aS*) -
126-major Rf min 1H NMR (600 MHz, CDCl3): δ = 5.90-5.84 (m, 1H, CH=vinyl); 5.80
(dddd, J = 6.0, 7.9, 10.2, 16.9, 1H, CH=allyl);
.20-5.16 (m, 2H, CH2=vinyl);
5.05 (d, J = 11.1, 1H, CH=); 5.04-5.00 (m, 2H, CH2=allyl);
3.63 (br. s, 1H, CHNCH=); 3.18-3.15 (m, 1H, CHNAll); 3.05 (td, J = 1.9, 9.2,
1H, CHNVinyl); 2.39 (br. s, 1H, CHAHB allyl); 2.17 (dt, J
= 9.0, 13.5, 1H, CHAHB allyl); 2.07 (dm, J = 19.0, 1H, CHAHBCCH3);
1.80-1.74 (m, 1H, CHAHBCHNCH=); 1.70 (s, 3H, CH3);
1.65-1.62 (m, 1H, CHAHBCHNCH=); 1.58-1.48 (m, 4H, CHAHBCCH3,
CH2CHNVinyl, CHAHB); 1.42-1.34 (m, 1H, CHAHB)
ppm.13C NMR (150 MHz, CDCl3): δ = 142.21; 136.64; 132.48; 122.52; 116.20;
115.25; 59.62; 53.81; 48.49; 37.49; 33.16; 30.64; 26.43; 23.53; 19.84 ppm.
Синтез (3aS*,6aS*,9aR*) - 5-Метил-2,3,3a,6,6a,7,9a,9b-октагидро-9b-азафеналена (127).
К дегазированному раствору диену 126-minor (40 мг, 0.18 ммоля) в
толуоле (5 мл) добавили катализатор Граббса второго поколения (6 мг) и
полученный раствор нагревали при 60 0C в течение 20 минут. За
ходом реакции следили с помощью метода ТСХ (n-C6H14: EtOAc = 3:
). По окончании реакции раствор пропустили через колонку с
силикагелем, продукт элюировали смесью EtOAc: Et3N = 60: 1, что дает 29 мг
(84%) азафеналена 127.1H NMR (300 MHz, CDCl3): δ = 5.78-5.67 (m, 2H, CH=CH); 5.47-5.45 (m, 1H, CH=); 3.52-3.43 (br. m, 2H, 2CHNCH=); 3.43-3.33 (m, 1H, CHN (CH2) 2); 2.39 (dt, J = 17.7, 5.0, 1H, CHAHBCH=); 2.25 (dd, J = 5.0, 17.5, 1H, CHAHBCCH3); 2.08 (td, J = 1.8, 9.4, 1H, CHAHBCH=); 2.00 (dd, J = 9.4, 17.7, 1H, CHAHBCCH3); 1.86-1.75 (m, 1H, CH2CHAHBCH2); 1.71 (s, 3H, CH3); 1.56-1.36 (m, 5H, CH2CHAHBCH2, 2CH2) ppm.13C NMR (100 MHz, CDCl3): δ = 129.80 CH=CH; 129.40 br. C=; 123.63 br.
CH=C; 122.38 CH=CH; 57.12 CHNCH=; 56.82 CHNCH=; 41.83 CHN (CH2) 2;
38.89 CH2CCH3; 34.27 CH2CH=; 27.75 CH2;
27.69 CH2; 24.48 CH2CH2CH2; 22.76
CH3 ppm. MS (70 eV, EI): m/z 189 (M+, 30); 188
(100); 186 (14); 184 (7); 174 (31); 160 (15); 149 (12); 146 (21); 144 (16); 132
(12); 131 (15); 120 (10); 94 (25); 81 (10); 67 (10); 57 (9); 41 (12).
4. Выводы
1. Разработан подход к синтезу соединений пергидро- и
декагидро [9b]
азафеналенового ряда.
2. Усовершенствована методика получения транс-диаллилированных
тетрагидропроизводных гетероциклов.
. Разработан новый метод получения
1,1-диэтоксибутил-1-ена.
Список
литературы
1. Яблоков-Хнзорян
С.М. Обзор семейства жуков-кокцинеллид фауны СССР. // Зоологический сборник.
Институт зоологии АН Армянской ССР. с.94-161.
2. King
A. G., Meinwald J. Review of the Defensive Chemistry of Coccinellids. // Chem.
Rev. 1996, Vol.96, No.3, p.1105-1122.
. Tursch
B.; Daloze D.; Dupont M.; Pasteels J. M.; Tricot M. - C. A defense alkaloid in
a Carnivorous Beetle. // Experientia 1971, Vol.27, p.1380-1381.
. Karlsson
R.; Losman D. J. The crystal structure of the hemihydrochloride of coccinellin,
the defensive N-oxide alkaloid of the beetle Coccinella Septempunctata, a case
of symmetrical hydrogen bonding. // Chem. Soc., Chem.commun. 1972, p.626-627.
. Tursch
B.; Daloze D.; Pasteels J. M.; Cravador A.; Braekman J. C.; Hootele C.;
Zimmermann D. Two Novel Alkaloids from the American Ladybug Hippodamia
Convergens (Coleoptera, Coccinellidae) // Bull. Soc. Chim. Belg. 1972, Vol.81,
p.649-650.
. Tursch,
B.; Daloze, D.; Braekman, J. C.; Hootele, C.; Pasteels, J. M. Chemical ecology
of arthropods - X. The structure of myrrhine and the biosynthesis of
coccinelline. // Tetrahedron 1975, Vol.31, p.1541-1543.
. Lalonde
R. T.; Auer E.; Wong C. F.; Muralidharan P. J. Polonovski transformation of (+)
- nupharidine. Stereochemistry and utility in synthesis. // J. Am. Chem. Soc.
1971, Vol.93, p.2501-2506.
. Ayer,
W. A.; Jenkins, J. K.; Valerde-Lopez, S.; Burnell, R. H. Can. The alkaloids of
Lycopodium cernuum L.I. The structures of cernuine and lycocernuine. // J.
Chem. Soc. 1967, Vol.45, p.433-443.
. Mueller
R. H.; Thompson M. E. Synthesis of the ladybug alkaloids (±) - propyleine and (±)
- isopropyleine. Modification of the published structure of propyleine. //
Tetrahedron Lett. 1980, Vol.21, p.1097-1100.
. Ayer
W. A.; Dawe R.; Eisner R. A.; Furuichi K. A total synthesis of myrrhine, (±) -
hippodamine, and (±) - convergine. // Can. J. Chem. 1976, Vol.54, p.473-481.
. Ayer
W. A.; Furuichi K. The total synthesis of coccinelline and precoccinelline. //
Can. J. Chem. 1976, Vol.54, p.1494-1495.
. Menzies
R. C.; Robinson R. J. A synthesis of ψ-pelletierine. // Chem. Soc. 1924,
Vol.125, p.2163-2168.
14. Schopf
V. C.; Lehmann G. Die Aldolkondensation zwischen Aldehyden und β-Ketosäuren und ihre Bedeutung für die Biogenese einiger
Naturstoffe // Justus Liebigs Ann. Chem. 1935, Vol.518, p.127-155.
15. Stevens
R. V.; Lee A. W. M. J. Stereochemistry of the Robinson-Schoepf reaction. A
stereospecific total synthesis of the ladybug defense alkaloids precoccinelline
and coccinelline. // J. Am. Chem. Soc. 1979, Vol.101, p.7032-7035.
. Langlois
M.; Yang D.; Soulier J. - L.; Florac C. Synthesis of Derivatives of 1, 2,
6-Trisubstttuted-4-Piperidones. // Synth.commun. 1992, Vol.22, p.3115-3127.
. Mueller
R. H.; Thompson M. E. Synthesis of the ladybug defensive agents (±) -
hippodamine, (±) - convergine, (±) - hippocasine and (±) hippocasine oxide //
Tetrahedron Lett. 1980, 21, 1093-1096.
. Mueller
R. H.; Thompson M. E.; DiPardo R. M. Stereo - and regioselective total
synthesis of the hydropyrido [2,1,6-de] quinolizine ladybug defensive alkaloids.
// J.org. Chem. 1984, Vol.49, p.2217-2231.
. Mueller
R. H.; Thompson M. E. Stereoselective total synthesis of the ladybug defensive
agents coccinellin and precoccinellin. // Tetrahedron Lett. 1979, p. 1991-1994.
. Rotermund
G. W.; Koste, R. Isomere des 9b-Bora-perhydrophenalens. // Liebigs Ann. Chem.
1965, Vol.686, p.153-166.
. Brown,
H. C.; Dickason, W. C. Synthesis of the cis,cis,cis-perhydro-9b-phenalenol, a
very highly strained system, via the carbonylation reaction. High rate of
solvolysis of the corresponding p-nitrobenzoate. // J. Am. Chem. Soc. 1969,
Vol.91, p.1226-1228.
. Bredereck
H.; Effenberger F.; Simchen G. Säureamid-Reaktionen, XLVI: Synthese von N. N.
N′-Trimethyl-formamidin und Bis-dimethylamino-methoxy-methan
(Aminalester) // Chem. Ber. 1965, Vol.98, p.1078-1080.
. Adams,
D. R.; Carruthers, W.; Crowley, P. J. Chem. Soc., Chem.commun. 1991, p.261-263.
. Yue
C.; Nicolay J. - F.; Royer J.; Husson H. - P. A new stereoselective synthesis
of ladybug defense alkaloid precoccinelline. // Tetrahedron 1994, Vol.50,
p.3139-3148.
. Bonin
M.; Grierson D. S.; Royer J.; Husson H. - P. A stable chiral
1,4-dihydropyridine equivalent for the asymmetric synthesis of substituted
piperidines: 2-cyano-6-phenyloxazolopiperidine. // Org. Synth. 1992, Vol.70, p.54-59.
. Yue
C.; Royer J.; Husson H. - P. Asymmetric synthesis 26. An expeditious
enantioselective synthesis of the defense alkaloids (-) - euphococcinine and
(-) - adaline via the CN (R,S) method. // J.org. Chem. 1992, Vol.57,
p.4211-4214.
. Rejzek
M.,, Stockman R. A., Hughesb D. L.combining two-directional synthesis and
tandem reactions: an efficient strategy for the total syntheses of (±) -
hippodamine and (±) - epi-hippodamine. // Org. Biomol. Chem., 2005, Vol.3,
p.73-83.
. Kitching
W., Lewis J. A., Perkins M. V., Drew R.,. Moore C. J, Schurig V., Konig W. A.,
Francke W. Chemistry of fruit flies.composition of the rectal gland secretion
of (male) Dacus cucumis (cucumber fly) and Dacus halfordiae. Characterization
of (Z,Z) - 2,8-dimethyl-1,7-dioxaspiro [5.5] undecane. // J.org. Chem., 1989,
Vol.54, p.3893-3902.
29. Dıaz-Gavila M.,´ Galloway W. R. J. D.,
O’Connell K. M. G., Hodkingson J. T., Spring D. R. Diversity-oriented synthesis
of bicyclic and tricyclic alkaloids. // Chem.commun., 2010, Vol.46, p.776-778.
. Boyer
F. D.; Hanna I. Substituent effects in tandem ring-closing metathesis reactions
of dienynes // Eur. J.org. Chem. 2006, Vol.2, p.471-482.
31. Колхаун
Х.М., Холтон Д., Томпсон Д., Твигг М. Новые пути органического синтеза.
Практическое использование переходных металлов. (Пер. с англ. М.С. Ермоленко,
В.Г. Киселева; Под ред. М.Н. Пастушенко). М.: Химия, 1989, 400 с.
32. Nicolaou
K. C., Bulger P. G., Sarlah D. Metathesis Reactions in Total Synthesis. //
Angew. Chem. Int. Ed. 2005, Vol.44, p.4490-4527.
. Harmon
R. E., Parsons J. L., Cooke D W., Gupta S. K., Schoolenberg J. Homogeneous
catalytic hydrogenation of unsaturated organic compounds. // J.org. Chem.,
1969, Vol.34, p.3684-3685.
. William
F. Bailey, Jason D. Brubaker, Kevin P. Jordan Effect of solvent and temperature
on the lithium-iodine exchange of primary alkyl iodides: reaction of t-butyllithium
with 1-iodooctane in heptanes-ether mixtures. // J.organometallic Chem., 2003,
Vol.681, p.210-214.
35. Бубнов
Ю.Н., Климкина Е.В., Игнатенко А.В. Получение транс - и цис-2-аллил-6-алкил
(арил) - 1,2,3,6-тетрагидропиридинов на основе восстановительного
транс-2,6-диалкилирования пиридина. Синтез (±) - эпидигидропиридина и (±) -
дигидропиридина. // Изв. АН. Серия хим., 1998, № 3, с.467-474.
. Бубнов
Ю.Н. Аллилбораны. Принципы реагирования и применение в органическом синтезе. //
Вестн. Моск. Ун-та, Сер.2, Химия, 2005, т.46, с.140-154.
37. Kuznetsov
N. Yu., Khrustalev V. N., Godovikov I. A., Bubnov Yu. N. Synthesis of Bridged
Azabicycles from Pyridines and Pyrrole by a Diallylboration - Ring Closing
Metathesis Sequence. // Eur. J.org. Chem., 2006, p.113-120.
. Sanchez-Sancho
F., Herradon B. Short syntheses of (S) - pipecolic acid, R-coniine, and (S) - δ-coniceine using
biocatalytically-generated chiral building blocks. // Tetrahedron: Asymmetry.
1998, Vol.9, p. 1951-1965.