Исследование изотерм динамического поверхностного натяжения водных растворов алкилсульфатов натрия
Дипломная
работа
на
тему: Исследование изотерм динамического поверхностного натяжения водных
растворов алкилсульфатов натрия
Введение
Интерес к изучению кинетики адсорбции
поверхностно-активных веществ на границе с газом или жидкостью обусловлен тем,
что многие технологические процессы и поверхностные явления в биологических
системах протекают в условиях, когда равновесие между поверхностным слоем и
объемом раствора не достигается. Велика роль кинетики адсорбции в процессах
смачивания и растекания, имеющих многочисленные приложения в технике: чем ниже
динамическое поверхностное натяжение раствора, тем выше скорость растекания и
смачивания. Эффективность процесса поверхностного разделения веществ также
зависит от скорости адсорбции отделяемого вещества. Скорость адсорбции ПАВ на
границах жидких фаз часто лимитирует скорость химических реакций с их участием.
Очень велика роль кинетики адсорбции белков и других высокомолекулярных
соединений в биологических системах [1].
Настоящая работа - посвящена изучению
динамического поверхностного натяжения водных растворов алкилсульфатов натрия,
особый интерес при этом вызывает эффект появления максимума на изотерме
поверхностного натяжения. Измерение поверхностного натяжения растворов
проводилось методом максимального давления в газовом пузырьке.
1. Уравнение адсорбции Гиббса для
неравновесной поверхности
Неравновесное состояние межфазной области может
характеризоваться либо полным отсутствием термодинамического равновесия, либо
частичным, т.е. нарушением механического, адсорбционно-диффузионного,
поляризационного или других видов равновесий, связанных, например, с
протеканием химических реакций на поверхности. Важную роль в описании
неравновесных поверхностных явлений играет введенный И. Пригожиным принцип локального
равновесия (локальная формулировка второго начала термодинамики), который
гласит, что для неравновесной в целом системы локальная энтропия зависит от
локальных макроскопических переменных: плотности внутренней энергии, тензора
давления, концентрации и т.п.
При применении принципа локального равновесия к
поверхностному слою необходимо учитывать прежде всего два главных момента: 1)
неавтономность поверхностного слоя, 2) возможность возникновения больших
градиентов при изменении термодинамических величин на расстояниях порядка
молекулярных размеров.
Авторами [2] был рассмотрен случай поляризации
поверхностного слоя в отсутствие адсорбционного равновесия вследствие как
ориентации диполей на поверхности , так и образования ионного двойного
электрического слоя. Этот наиболее общий и часто встречающийся на практике
случай будет обсужден здесь.
Рассмотрим многокомпонентную двухфазную систему,
состоящую из молекул (полярных и неполярных) и ионов, с плоским поверхностным
слоем. Обращаясь к многослойной модели, систему разобьем на элементарные слои
(включая объемные фазы: α - жидкость, β
- газ),
в пределах каждого из которых электрохимические (химические) потенциалы можно
считать постоянными. Отметим, что использование многослойной модели в наиболее
полной мере дает возможность учесть две особенности поверхностных слоев, о
которых говорилось выше: ‘неавтономность’ и большие градиенты.
Анализ различных случаев ориентации диполей
(вертикально вверх, вниз, параллельно поверхности и т.д.) показывает, что для
описания ориентации необходимо задать шесть переменных: три значения <cosβ>
и три значения <cos2
β>, где β
- угол
ориентации. Поскольку поляризация при ориентации определяется дипольным
моментом, заменим <cosβ>
и <cos2 β>
на средние значения составляющих дипольного момента <mkix>
и квадрата дипольного момента <mk2ix>
(x=x,
y, z; k
- индекс слоя).
Пусть система находится в механическом и
тепловом равновесии, температура T
и давление p одинаковы всюду,
но адсорбционное и поляризационное равновесие отсутствуют. Используя первое и
второе начала термодинамики, запишем
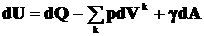 (1)
(1)
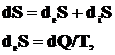 (2)
(2)
где U
- внутренняя энергия системы; Q
- тепло, полученное системой от внешней среды; γ - поверхностное
натяжение; V - объем; А -
площадь поверхности; S
- энтропия; deS - изменение
энтропии, обусловленное взаимодействием системы с внешней средой, а diS
- изменение, связанное с необратимыми процессами внутри системы. Изменение
энтропии diS может быть
записано в виде
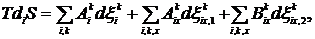 (3)
(3)
где
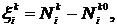
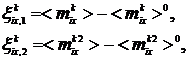 (4)
(4)
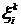 определяет изменение числа молекул N вследствие
переноса, а
определяет изменение числа молекул N вследствие
переноса, а 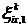 и
и 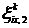 - изменения
- изменения
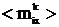 и
и 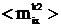 ,
обусловленные ориентацией. Индекс 0 отвечает нулевому времени (начало отсчета).
,
обусловленные ориентацией. Индекс 0 отвечает нулевому времени (начало отсчета).
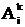 ,
, 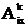 ,
, 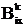 -
соответственно сродство изменения числа молекул в результате переноса и
изменения переменных <mkix> и <mk2ix>
вследствие ориентации.
-
соответственно сродство изменения числа молекул в результате переноса и
изменения переменных <mkix> и <mk2ix>
вследствие ориентации.
Из соотношений (1) - (3) получаем
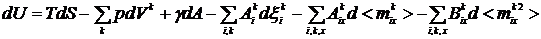
Для свободной энергии F = U - TS
неравновесной двухфазной системы в целом или неравновесного (в отношении
адсорбции и ориентации) плоского поверхностного слоя имеем
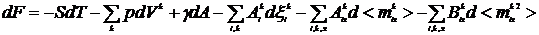 (5)
(5)
Далее определим электрохимический
потенциал 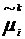
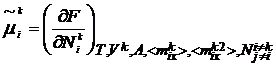 (6)
(6)
частиц сорта i в слое k - для состояния,
в котором ориентация есть постоянная заданная величина.
Определим величину из формулы
(5): 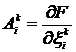 . Из
соотношений (4) и (6) имеем
. Из
соотношений (4) и (6) имеем
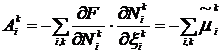 ,
,
и тогда (5) переходит в уравнение
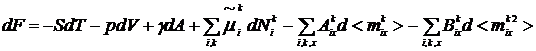 . (7)
. (7)
Вычтем из (7) соответствующие
выражения для объемных фаз
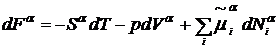 ,
,
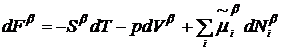
и прибавим и отнимем произведение 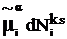 . Тогда
получим
. Тогда
получим
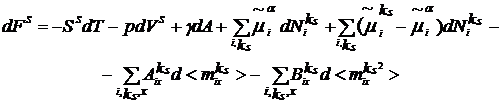 (8)
(8)
Индекс kS означает,
что соответствующая величина относится к переходной зоне между объемными
фазами. В двух последних членах уравнения (8) индекс k заменен на kS, так как
спонтанная ориентация диполей происходит только на поверхности.
Для заданного состояния
неравновесного поверхностного слоя по теореме Эйлера об однородных функциях
имеем
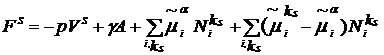 . (9)
. (9)
Продифференцировав (9) и сравнив
результат дифференцирования с (8), получим обобщение уравнения адсорбции
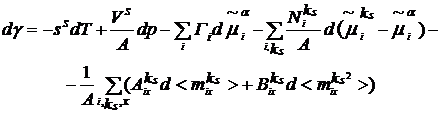 (10)
(10)
где ss=Ss/A,
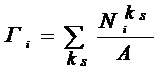
Разность 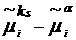 играет роль
адсорбционного сродства для kS-го элементарного слоя на
поверхности, так что два последних члена в (10) отражают наличие неравновесного
процесса на поверхности. В равновесии они исчезают. Уравнение (10) может
использоваться, с одной стороны, как релаксационное уравнение для свежей
поверхности, и, с другой, как уравнение, описывающее зависимость поверхностного
натяжения от параметров состояния, в частности, от концентрации ПАВ.
играет роль
адсорбционного сродства для kS-го элементарного слоя на
поверхности, так что два последних члена в (10) отражают наличие неравновесного
процесса на поверхности. В равновесии они исчезают. Уравнение (10) может
использоваться, с одной стороны, как релаксационное уравнение для свежей
поверхности, и, с другой, как уравнение, описывающее зависимость поверхностного
натяжения от параметров состояния, в частности, от концентрации ПАВ.
Применительно к процессу релаксации
поверхности при заданном составе и состоянии объемной фазы α уравнение
(10) заметно упрощается. Исчезают первые три члена в правой части, поскольку
температура, давление и химические потенциалы фазы α фиксированы.
В едином процессе релаксации величины 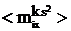 можно считать пропорциональными
можно считать пропорциональными 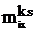 (фактически
имеется лишь одна составляющая
(фактически
имеется лишь одна составляющая 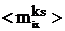 в направлении нормали z к
поверхности, так как и , и
в направлении нормали z к
поверхности, так как и , и 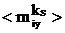 равны нулю
по условиям симметрии). Учитывая также, что каждое ориентационное сродство
пропорционально скорости изменения соответствующей переменной, вытекающее из
(10) релаксационное уравнение можно записать в виде [3]:
равны нулю
по условиям симметрии). Учитывая также, что каждое ориентационное сродство
пропорционально скорости изменения соответствующей переменной, вытекающее из
(10) релаксационное уравнение можно записать в виде [3]:
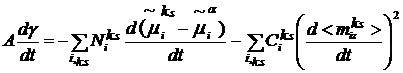 (11)
(11)
где t - время; 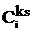 - некоторые
постоянные. В правой части релаксационного уравнения (11) имеется два члена:
диффузионный и ориентационный. Величина
- некоторые
постоянные. В правой части релаксационного уравнения (11) имеется два члена:
диффузионный и ориентационный. Величина 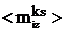 связана с дипольной частью χd
поверхностного потенциала χ соотношением электростатики:
связана с дипольной частью χd
поверхностного потенциала χ соотношением электростатики:
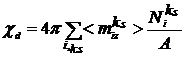 . (12)
. (12)
Поскольку ориентация осуществляется
главным образом в поверхностном монослое, можно считать k = 1.
Упрощая задачу, предположим, что ориентируется только один компонент, т.е. i = 1. Тогда
из уравнения (12) получим:
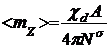 , (13)
, (13)
где 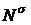 - число молекул в монослое. В этой
модели из уравнения (11) имеем:
- число молекул в монослое. В этой
модели из уравнения (11) имеем:
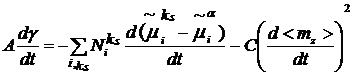 . (14)
. (14)
Здесь С - равна величине Сik
в модели монослоя с одним компонентом.
Величина С положительна, так как поверхностное
натяжение в процессе ориентации молекул уменьшается.
Уравнение (14) можно использовать для получения
соотношения между динамическим поверхностным натяжением и неравновесным
поверхностным электрическим потенциалом. Рассмотрим далее три предельных
случая.
1. Ориентационный
механизм релаксации
Процесс ориентации происходит
намного медленнее диффузии ионов и молекул и практически осуществляется в
условиях диффузионного равновесия 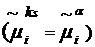 . Тогда число молекул в
поверхностном монослое практически не меняется, и изменение поверхностного
потенциала обусловлено лишь процессом ориентации молекул (dχ ~ d<mZ>). Тогда
из уравнения (14) с учетом (13) следует соотношение:
. Тогда число молекул в
поверхностном монослое практически не меняется, и изменение поверхностного
потенциала обусловлено лишь процессом ориентации молекул (dχ ~ d<mZ>). Тогда
из уравнения (14) с учетом (13) следует соотношение:
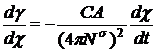 , (15)
, (15)
которое показывает, что 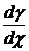 → 0
при t → ∞,
так как
→ 0
при t → ∞,
так как 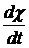 → 0.
→ 0.
Предположим теперь, что процесс
ориентации протекает в условиях диффузионного равновесия всех других компонентов,
но не того, молекулы которого ориентируются на поверхности. Сам акт ориентации
является достаточно быстрым, и скорость формирования дипольного двойного слоя
лимитируется подводом вещества к поверхности. Для этого случая имеем
соотношение:
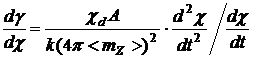 , (16)
, (16)
где k - некоторый
коэффициент. При t → ∞ производные и 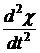 стремятся к
нулю, но их отношение при экспоненциальной зависимости в пределе постоянно, так
что и стремится к
некоторой постоянной величине. 3. Ионно-диффузионный механизм релаксации
стремятся к
нулю, но их отношение при экспоненциальной зависимости в пределе постоянно, так
что и стремится к
некоторой постоянной величине. 3. Ионно-диффузионный механизм релаксации
Диффузия и ориентация диполей
происходят достаточно быстро, а лимитирующей стадией является процесс
формирования ионного двойного слоя. В этом случае из уравнения (14) получаем:
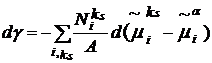 (17)
(17)
где суммирование по i относится к
ионам.
Учитывая стандартное выражение для
электрохимического потенциала, получим:
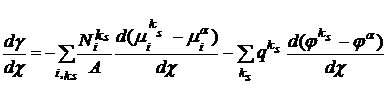
 (18)
(18)
где 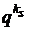 - заряд элементарного слоя kS на единицу
поверхности; φ
- электрический
потенциал в месте нахождения иона.
- заряд элементарного слоя kS на единицу
поверхности; φ
- электрический
потенциал в месте нахождения иона.
В правой части релаксационного
уравнения (18) два члена. Первый описывает влияние адсорбции ионов (в ходе
которой формируется поверхностный потенциал χ) на распределение химических
потенциалов в двойном слое. Второй член уравнения (18) чисто электростатической
природы. Он отличен от нуля даже для электронейтрального поверхностного слоя и
приводит к снижению поверхностного натяжения независимо от знака поверхностного
потенциала.
Проведенный анализ представляет
большой практический интерес, так как может способствовать выяснению механизма
образования электрического поверхностного потенциала. В самом деле, разобранные
случаи говорят о том, что если
) существует диффузионное
равновесие и медленная ориентация диполей, то → 0 при t → ∞,
и в зависимости Δγ
= γ - γ∞
от Δχ
= χ - χ∞
нет линейного члена по Δχ;
) наблюдается быстрая
ориентация диполей и отсутствует диффузионное равновесие этих диполей, то → const при t → ∞,
и в зависимости Δγ
от
Δχ
должен
быть линейный член по Δχ;
) поверхностный потенциал
возникает вследствие образования ионного двойного слоя, то → const при t → ∞,
и в зависимости Δγ
от
Δχ должен быть
линейный член по Δχ.
Следовательно, если в эксперименте наблюдается
линейный член, то мы имеем дело с механизмом образования χ
по типу 2 или 3, если он отсутствует, - то по типу 1. Эффект заряжения
поверхности
Джонсом и Реем обнаружено понижение
поверхностного натяжения водных растворов солей при возрастании концентрации от
0 до 10-3 М, за которым следовало его увеличение.
Работа [4] посвящена исследованию поверхностного
натяжения водных растворов солей в зависимости от возраста поверхности и его
связи с электрическими свойствами поверхности. Было измерено поверхностное
натяжение растворов NaCl
и KCl в зависимости от
концентрации. Измерения проводились на приборе ГАЗП-1 КТ конструкции П.П.
Пугачевича методом максимального давления в газовом пузырьке. Поверхностное
натяжение γ зависит от
концентрации и времени между срывами пузырьков, которые характеризует свежесть
образующейся поверхности. При малом времени (12-17 с) на зависимости Δγ
(разность
поверхностных натяжений раствора и воды) от концентрации с наблюдается заметный
минимум поверхностного натяжения. При дальнейшем увеличении времени минимум
исчезает.
Причиной появления минимума, как указано в [4],
может быть электрическое состояние поверхности. Заряжение поверхности жидкости
происходит в определенном временнОм интервале вследствие нестационарного
электрического потенциала. Известно, например, что свободный заряд на
поверхности жидкости при образовании пузырька составляет 4·10-7 Кл/см2.
Уравнение адсорбции для неравновесного
поверхностного слоя в случае образования свободного заряда q
на поверхности имеет следующий вид:
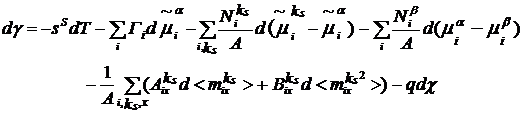 (19)
(19)
где χ
- поверхностный потенциал.
Появление члена qdχ
представляется вероятным в неравновесных условиях образования поверхности при
достаточно свежей поверхности и малой концентрации электролита (когда двойной
электрический слой достаточно протяженный и легко деформируемый).
Обратимся теперь к рассмотрению
баллоэлектрического эффекта [5] - явления образования суммарного электрического
заряда на облаке тумана, получаемого путем распыления жидкости. Можно
сформулировать следующую общую закономерность, справедливую для водных
растворов всех веществ: при низких концентрациях ионов знак баллоэлектрического
эффекта совпадает со знаком заряда присутствующего капиллярно-активного иона,
при высоких концентрациях ионов он противоположен знаку скачка потенциала на
поверхности.
Закономерность, наблюдающаяся при низких
концентрациях ионов, легко укладывается в рамки теории, связывающей
баллоэлектрический эффект с тангенциальным разрывом двойного электрического
слоя на поверхности, если иметь в виду ионный диффузный двойной слой,
возникающий вследствие различной адсорбируемости анионов и катионов. Для водных
растворов неорганических солей внешняя обкладка ионного двойного слоя на
поверхности раствор/воздух всегда отрицательна. При высокой концентрации ионов
преобладает эффект контактного заряжения [5]. Адсорбция ПАВ
.2 Механизмы адсорбции
Существует два различных подхода [1]. Согласно
первому из них предполагается, что равновесие между поверхностным и
приповерхностным слоями устанавливается очень быстро, а скорость процесса
адсорбции определяется исключительно скоростью диффузии (диффузионный механизм
адсорбции). Второй подход учитывает как скорость диффузии, так и скорость
установления равновесия между поверхностным и приповерхностным слоями
(смешанный механизм адсорбции). Имеется еще один случай, когда скорость диффузии
велика по сравнению со скоростью установления равновесия между поверхностным и
приповерхностным слоями (кинетический или адсорбционный механизм адсорбции).
. Диффузионный механизм адсорбции
Уравнение Уорда и Тордея [6] принимает во
внимание диффузию мономеров из объема раствора на поверхность, а также обратную
диффузию в объем, поскольку поверхность становится более заселенной. Используя
уравнение диффузии Фика, задав начальные и граничные условия (см., например,
[1]), прейдем к уравнению Уорда и Тордея (1946г.):
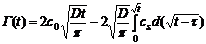 , (20)
, (20)
где с0 объемная концентрация ПАВ, D -
коэффициент диффузии, сS - подповерхностная концентрация, τ - переменная.
Решение уравнения Уорда и Тордея для
t → 0
В начале процесса адсорбции нет
обратной диффузии. Пренебрегая этим членом в уравнении (20), получим:
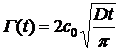 . (21)
. (21)
Поверхностное натяжение раствора при
γ
→ γ0 в случае
идеального поверхностного слоя описывается следующим уравнением: 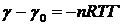 , (22)
, (22)
где n = 1 для
неионных ПАВ и n = 2 для ионных ПАВ. Подставляя
уравнение (22) в уравнение (21), получим:
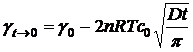 . (23)
. (23)
Решение уравнения Уорда и Тордея для
t → ∞
При t → ∞
подповерхностная концентрация будет приближаться к объемной концентрации, и в
результате получим соотношение:
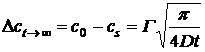 . (24)
. (24)
Принимая во внимание уравнение Гиббса
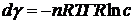 (25)
(25)
при взятии в качестве предела в этом
уравнении Δс → 0,
в итоге имеем:
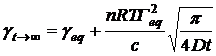 (26)
(26)
. Смешанный механизм адсорбции
Для того чтобы адсорбироваться,
мономер должен совершить работу против возрастающего поверхностного давления π. Когда π велико,
маловероятно, что все молекулы, достигающие подповерхности, будут обладать
достаточной энергией для адсорбции на поверхности. Только молекулы, обладающие
энергией превышающей определенную энергию активации, смогут адсорбироваться.
Рассмотрим модель, предложенную Liggieri и Ravera [7]. Когда
рассматриваются молекулы в подповерхности, только те из них имеющие энергию
большую чем εa
адсорбируются, тогда как среди адсорбированных молекул десорбируются только те,
энергия которых превышает
εd. Параметры εa и εd - энергии
активации адсорбции и десорбции соответственно. Коэффициент диффузии D* связан с
физическим коэффициентом диффузии D
соотношением: 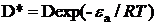 . (27)
. (27)
Когда εa → 0, D* → D и процесс
стремится к диффузионному механизму.
Используя D*, этот
процесс может теперь рассматриваться как диффузионная задача, которая может
быть решена, используя уравнение Фика с новым граничным условием
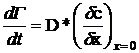 (28)
(28)
Теперь уравнение Уорда и Тордея
имеет вид:
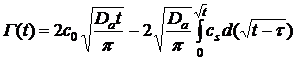 , (29)
, (29)
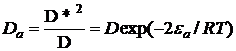 (30)
(30)
2. Роль электростатических
взаимодействий в процессе адсорбции ионогенных ПАВ на межфазной границе раздела
жидкость - газ
Рассмотрим выражение для изотермы
поверхностного давления полученное в работе [8]:
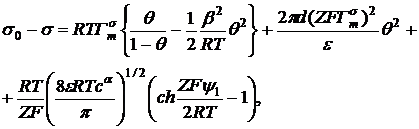 (31)
(31)
где
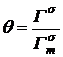 ,
, 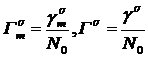 ,
, 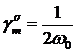
предельная (при 0 К) адсорбция, 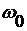 - площадь,
занимаемая молекулой в предельном монослое при ее вертикальной ориентации;
- площадь,
занимаемая молекулой в предельном монослое при ее вертикальной ориентации; 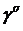 -
поверхностная плотность молекул (ионов) в адсорбционном монослое; N0 - число
Авогадро;
-
поверхностная плотность молекул (ионов) в адсорбционном монослое; N0 - число
Авогадро; 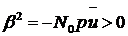 , p - число
ближайших соседей выбранной молекулы (иона) в предельном монослое;
, p - число
ближайших соседей выбранной молекулы (иона) в предельном монослое; 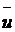 - среднее
значение энергии дисперсионного взаимодействия (притяжения) двух молекул; d -
расстояние между плоскостью адсорбционного монослоя и плоскостью наибольшего
приближения ионов диффузной части ДЭС; Z -
валентность; ψ1
- потенциал
в плоскости наибольшего приближения ионов диффузной части ДЭС к адсорбционному
монослою.
- среднее
значение энергии дисперсионного взаимодействия (притяжения) двух молекул; d -
расстояние между плоскостью адсорбционного монослоя и плоскостью наибольшего
приближения ионов диффузной части ДЭС; Z -
валентность; ψ1
- потенциал
в плоскости наибольшего приближения ионов диффузной части ДЭС к адсорбционному
монослою.
Первые два слагаемых в уравнении
(31) отражают вклад в поверхностное давление дисперсионных сил отталкивания и
притяжения. Последнее слагаемое равно энергии электростатического
взаимодействия в диффузной части ДЭС. Что касается третьего слагаемого, то
нетрудно показать его идентичность электростатической энергии плоского
конденсатора, обкладками которого являются плоскость адсорбционного монослоя и
плоскость наибольшего приближения ионов диффузной части ДЭС.
Таким образом, сумма последних двух
слагаемых представляет собой полную электростатическую энергию ДЭС, которая
входит в изотерму поверхностного давления в виде существенно положительной
величины. Это обстоятельство позволяет сделать вывод о том, что при адсорбции
ИПАВ, в силу электростатического взаимодействия в переходном слое, наряду со
снижением поверхностного натяжения раствора, связанного с собственно адсорбцией
ПАВ (органических ионов) в адсорбционном монослое, имеет место дополнительное
(в сравнении с гипотетическим случаем незаряженной поверхности) снижение
поверхностного натяжения, в точности равное энергии электростатического
взаимодействия.
2.1 Двумерные фазовые переходы в
адсорбционных слоях ПАВ на границе раствор-воздух
В изучаемых нами системах (водные растворы
алкилсульфатов натрия) уже наблюдались фазовые переходы [9, 10, 11], поэтому
здесь будет уместно подробнее освятить этот вопрос.
Монослои дифильных молекул на границе раздела
вода - воздух являются двумерными системами [12]. Различают два типа монослоев:
в первом случае молекулы ПАВ растворяются в водном растворе и адсорбируются на
поверхности с образованием адсорбционного слоя; во втором случае
рассматривается монослой Ленгмюра, который образуется за счет растяжения
длинных цепей нерастворимых в воде ПАВ.
Впервые и наиболее обоснованно возможность
существования двумерных фазовых переходов в адсорбционных слоях растворимых ПАВ
была исследована в работе А.И. Русанова [13], где сформулированы
термодинамические условия для переходов подобного типа в виде неравенства и
показано, что при наличии фазового перехода в поверхностном слое изотерма
поверхностного натяжения должна иметь излом с увеличением наклона изотермы (по
абсолютной величине) с ростом концентрации.
С помощью измерений методом максимального
давления в газовом пузырьке Дмитровской М.В. [9] была получена зависимость
динамического поверхностного натяжения от времени раствора додецилсульфата
натрия при концентрации 1·10-3 моль/л и температуре 20 0С (рис. 1). При
значениях поверхностного натяжения ~ 63 мН/м и 67 мН/м имеются точки излома
(обозначены пунктиром). Резкое изменение характера зависимости поверхностного
натяжения со временем, как это подробно было проанализировано в работе [14],
свидетельствует о возможности существования двумерного фазового перехода.
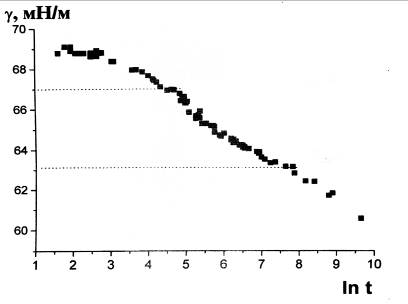
Рис. 1 Фазовый переход в растворе ДДСН.
Рассмотрим теперь влияние температуры и длины
углеводородной цепи на фазовые переходы в монослоях Лэнгмюра [15]. Монослои
Лэнгмюра молекул ПАВ на границе жидкость-газ дают уникальную возможность для
получения представлений о физических свойствах двумерно организованных молекул.
С точки зрения прикладных аспектов монослои Лэнгмюра являются превосходными
модельными системами для мембранной биофизики, так как биологическую мембрану
можно рассматривать как два слабо соединенных монослоя; кроме того они
используются для изучения химических и биохимических реакций, для создания
пленок Лэнгмюра-Блоджетта с целью специального применения в оптических и
электронных приборах, а также для изучения смазывания, трения, смачивания.
В работе [15] исследовались монослои Лэнгмюра
следующих неионогенных ПАВ: СН3(СН2)13ОСН2СН2ОН (С14Е1), СН3(СН2)15ОСН2СН2ОН
(С16Е1), СН3(СН2)17ОСН2СН2ОН (С18Е1).
Для монослоев этих ПАВ авторами были получены
зависимости поверхностного давления от молекулярной площади (π
- А
изотермы) при разных температурах. Уменьшение молекулярной площади путем сжатия
монослоя приводит к ряду двумерных фазовых переходов. При низкой поверхностной
плотности молекулы находятся в состоянии двумерного газа (G).
Увеличение поверхностной плотности путем латерального сжатия ведет к переходу G-фазы
в изотропную двумерную жидко-растянутую фазу (LE).
При дальнейшем сжатии монослой претерпевает переход между LE-фазой
и жидко-конденсированным состоянием повышенной плотности (LC-фазой),
что отражено на π - А изотерме
как излом за которым следует область плато (рис.2).
Монослои также исследовались методом
Брюстеровской угловой микроскопии. После появления точки излома на изотерме
были обнаружены области конденсированной фазы. Для С14Е1 эти области являются
круглыми с полосками внутри. Для С16Е1 и С18Е1 они имеют неправильную форму при
низких температурах, в то время как при высоких температурах они подразделяются
на два сегмента различной яркости.
Кроме уже упомянутой точки излома
соответствующей фазовому переходу LE
- LC, на изотермах для
С14Е1 и С18Е1 имеется еще одна точка излома с последующим коротким плато,
отвечающая второму фазовому переходу между двумя конденсированными фазами
различной сжимаемости и углом наклона молекул к поверхности (рис.2).
Рис. 3 показывает критическое поверхностное
давление (πС) необходимое для LE-LC
фазового перехода в зависимости от температуры для рассматриваемых ПАВ. Рост πС
с ростом температуры связан с тем, что более высокое поверхностное давление
необходимо для фазового перехода при высоких температурах для преодоления
дополнительного напряжения связанного с тепловым движением и гибкостью
углеводородной цепи молекул. Величины dπС/dT
для С14Е1, С16Е1 и С18Е1 равны соответственно 1.224, 0.85 и 0.55 mN/(m
K). Уменьшение величин
dπС/dT
с ростом длины цепи связано с тем, что увеличение числа атомов углерода в
алкильной цепи приводит к усилению Ван-дер-Ваальсовых взаимодействий между
молекулами. Этим же объясняется снижение πС
и увеличение минимальной температуры, необходимой для достижения области
сосуществования LE
и LC фаз, с ростом
числа углеродных атомов в алкильной цепи (рис. 4).
Была рассчитана ΔН
процесса конденсации молекул из LE
в LC фазу с помощью
двумерного уравнения Клапейрона:
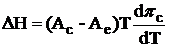 ,
,
где Ае - молекулярная площадь,
соответствующая началу фазового перехода при поверхностном давлении πС, Ас -
площадь конденсированной фазы. Для все ПАВ значения ΔН являются
отрицательными, что говорит об экзотермической природе конденсации. Если мы
сравним величины ΔН для С14Е1
и С16Е1 при 20 0С и ΔН для С16Е1
и С18Е1 при 35 0С, то можно увидеть, что значения ΔН
уменьшаются с ростом длины углеводородной цепи (рис. 5). Отрицательные значения
ΔS, полученные
из соотношения ΔН/Т
означают, что LE - LC фазовый
переход сопровождается потерей вращательного и поступательного движения
молекул.
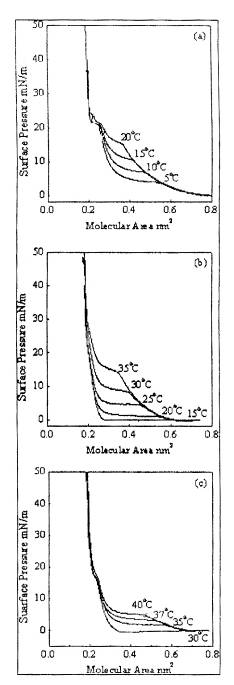
Рис. 2 π - А изотермы
для монослоев С14Е1 (а), С16Е1 (b) и С18Е1 (c).
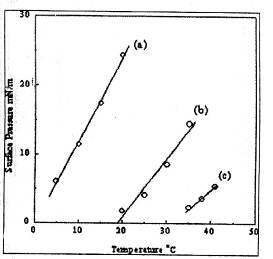
Рис.3
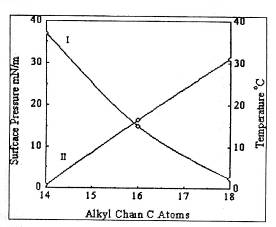
Рис.4 I -
поверхностное давление II - температура

Рис.5
3. Эксперимент
.1 Метод максимального давления в
газовом пузырьке
В работе измерялось динамическое поверхностное
натяжение водных растворов алкилсульфатов натрия методом максимального давления
в газовом пузырьке. Суть метода состоит в следующем. Уровень жидкости в
капилляре, погруженном в раствор при условии, что жидкость смачивает стенки
капилляра и давление внутри и снаружи одинаково, устанавливается несколько выше
уровня жидкости вследствие капиллярного поднятия. Если начать увеличивать
давление внутри капилляра, выдувая из него столбик жидкости, то рост давления с
падением уровня жидкости в капилляре будет линеен вплоть до конца капилляра.
После того, как жидкость покинет капилляр, с ростом размера пузырька начнется
быстрое уменьшение радиуса кривизны мениска жидкости. При этом (если за этот
промежуток времени поверхностное натяжение изменится незначительно) капиллярное
давление, обратно пропорциональное радиусу кривизны поверхности, будет расти.
Когда радиус кривизны растущего пузырька станет равен радиусу капилляра r
(при этом растущий пузырек будет иметь форму, близкую к полусфере) капиллярное
давление достигнет своего максимума. При дальнейшем росте пузырька радиус его
кривизны уже начнет увеличиваться, и давление газа будет больше капиллярного.
Из-за этого происходит отрыв пузырька.
Если процесс формирования пузырька происходит
достаточно медленно, т.е. его можно считать квазистационарным, пузырек должен
отрываться в тот момент, когда его радиус на бесконечно малую величину превысит
радиус капилляра. На практике часто приходится сталкиваться со случаями, когда
подобная квазистационарность не достигается. При этом отрыв пузырька происходит
несколько позже, чем при квазистационарном течении процесса. Получающаяся
разница во времени носит название “мертвого времени”. Оценка показывает, что
реально необходимо учитывать “мертвое время” лишь при очень малых временах
жизни пузырька (порядка нескольких миллисекунд). В этих случаях “мертвое время”
может достигать нескольких процентов от времени жизни пузырька. При больших
временах жизни поверхности процентный вклад “мертвого времени” становится тем
меньшим, чем больше время жизни пузырька, т.е. чем ближе процесс формирования
пузырька к квазистационарному. В данной работе времена жизни пузырька
достаточно велики, что освобождает от необходимости принимать во внимание
“мертвое время”.
Обычно при измерении поверхностного натяжения
методом максимального давления в пузырьке необходимо учитывать гидростатическое
давление столба жидкости над пузырьком. В данной работе для измерения
динамического поверхностного натяжения использовался прибор, предложенный П.П.
Пугачевичем и позволяющий учитывать гидростатическое давление. Его схема
приведена на рис. 6.
Рассмотрим принцип работы газового прибора
Пугачевича. Пусть исследуемая жидкость смачивает стенки капилляра и в начальный
момент давление во всех его частях одинаково и равно Р, рис. 6а.
Манометрическая трубка 1 имеет настолько большой диаметр, что капиллярным
поднятием в ней можно пренебречь. Уровни жидкости в трубках 2 и 4 и в
резервуаре 3 будут находиться на высотах h1,
h2, h3
соответственно (благодаря капиллярному поднятию). С ростом давления в трубках
1, 2 и 4 уровни жидкости в них начнут опускаться, а в резервуаре 3 - подниматься.
На рис.6 б указаны уровни жидкости в приборе при достижении в газовом пузырьке
максимального давления Pmax.
Здесь: h2 - высота,
обусловленная капиллярным поднятием в резервуаре 3; h4
- высота, связанная с вытеснением жидкости формирующимся пузырьком; (h5+h6)
- высота, на которой находился бы уровень жидкости в резервуаре 3, если бы он
имел конечно большой диаметр.
Таким образом, для трубки 1 можно записать:
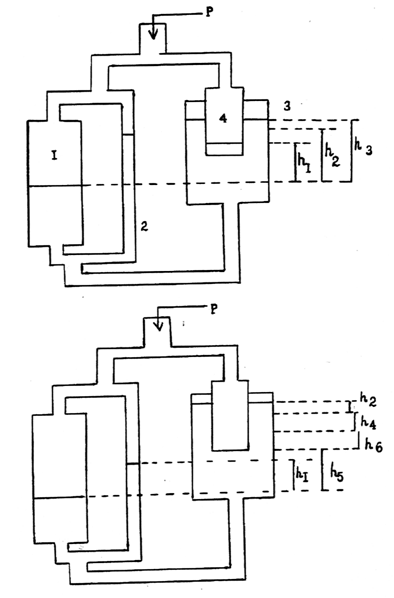
Рис.6а Рис.6б
Рис. 6 Принципиальная схема газового прибора
Пугачевича.
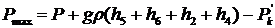 , (32)
, (32)
где g - ускорение
свободного падения; ρ
- плотность
жидкости; 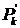 - член,
учитывающий капиллярное поднятие в резервуаре 3.
- член,
учитывающий капиллярное поднятие в резервуаре 3.
Для манометрической трубки 2:
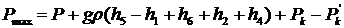 , (33)
, (33)
где Рk - член,
связанный с капиллярным поднятием в трубке 2.
Для максимального давления в газовом
пузырьке можно записать:
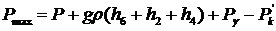 , (34)
, (34)
где Рγ -
капиллярное давление в трубке 4.
При сравнении уравнений (32) и (33)
получается выражение для трубки 1:
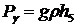 , (35)
, (35)
а для узкой манометрической трубки 2
из уравнений (33) и (34) имеем:
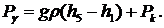 (36)
(36)
Таким образом, применение узких манометрических
трубок вызывает необходимость учитывать капиллярное поднятие в них, поэтому
удобнее применять широкие манометрические трубки и для расчета поверхностного
натяжения пользоваться уравнением (35). Для узких капиллярных трубок
справедливо следующе6е соотношение:
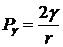 (37)
(37)
Тогда из уравнения (35) можно найти
поверхностное натяжение жидкости по уравнению:
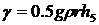 (38)
(38)
В данной работе для вычисления
поверхностного натяжения используется приближение Кантора, согласно которому:
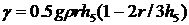 (39)
(39)
Отсюда видно, что при применении
прибора Пугачевича не требуется учитывать глубину погружения капилляра в
жидкость, изменение уровня жидкости в резервуаре (в который погружен капилляр),
обусловленное формированием пузырька и капиллярным поднятием в этом резервуаре.
Для измерения поверхностного натяжения с помощью прибора Пугачевича достаточно
измерить h5, т.е.
расстояние от торца капилляра до уровня жидкости в манометрической трубке в
момент отрыва пузырька. Кроме простоты измерений, такой прибор дает возможность
работать в сложных экспериментальных условиях - при любой температуре,
давлениях, в атмосфере инертных газов. Прибор Пугачевича, использованный в
данной работе, изображен на рис. 7.
В данной работе динамическое
поверхностное натяжение измерялось с помощью установки, изображенной на рис. 8.
С целью уменьшения влияния посторонних вибраций на рабочую часть установки -
газовый прибор ГАЗП-1КТ конструкции П.П. Пугачевича - в капитальную стену 1 были
вмонтированы два металлических уголка 2, на которых помещалась
противовибрационная подвеска, состоящая из резинового буфера (использовались
теннисные шары) 3, и свободно лежащей на ней массивной плиты 4. К плите в
режиме свободной подвески на металлическом стержне 5 крепилась дюралюминиевая
рамка 6, к которой был присоединен газовый прибор Пугачевича 7. Прибор
помещался в термостатированный объем 8, изготовленный из органического стекла.
Контроль температуры осуществлялся двумя термометрами 11. В качестве газа,
создающего нужное давление для формирования пузырька, использовался гелий,
который не растворяется и не адсорбируется на поверхности водного раствора, что
особенно существенно при повышении давления. Газ поступает из баллона 13 под
давлением 40 кг·с/см2. Давление на выходе из баллона контролируется манометром
18. С помощью газового редуктора 14 и контролируемого манометра 19, а также
более точных редуктора 15 и манометра 20, регулирующих подачу газа на вход
газовой магистрали 16, гелий поступает в прибор, откуда после отрыва пузырька,
через отверстие 17 выходит в атмосферу. Для отсчета высоты торца капилляра и
уровня жидкости в манометрической трубке в момент отрыва пузырька использовался
катетометр (КМ - 8) 22, позволяющий отсчитывать расстояние с точностью до 0,02
мм. Для получения четкого изображения, прибор Пугачевича подсвечивался через
прозрачную переднюю панель с помощью лампы 21. адсорбция
поверхностное натяжение изотерма
В процессе освоения установки была
выработана следующая методика измерения поверхностного натяжения жидкостей.
Прибор ГАЗП-1КТ, заполненный исследуемым раствором при помощи специальной
подвески помещался в изолированную камеру. Затем осуществлялось
термостатирование всей установки (по методике необходимо 2 часа). Поверхностное
натяжение исследуемой жидкости определялось при атмосферном давлении.
Максимальное давление, выраженное высотой столба исследуемой жидкости в газовом
приборе, измерялось катетометром. Далее устанавливалось новое значение
давления, затем следующие и, таким образом, операции повторялись.
Поверхностное натяжение
рассчитывалось по формуле Кантора (39).
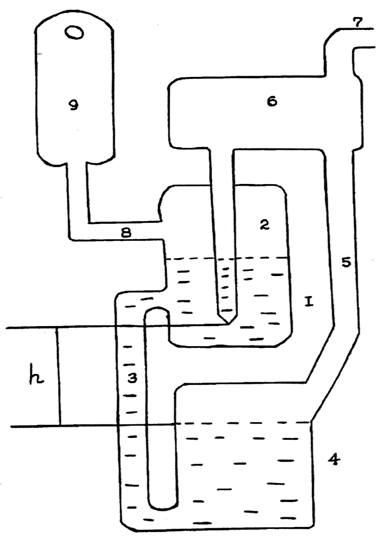
Рис. 7. Газовый прибор Пугачевича,
использованный в данной работе:
1 - капиллярная трубка; 2, 6, 9 - резервуары; 4
- манометрический сосуд; 3, 5, 7,8 - переходные трубки
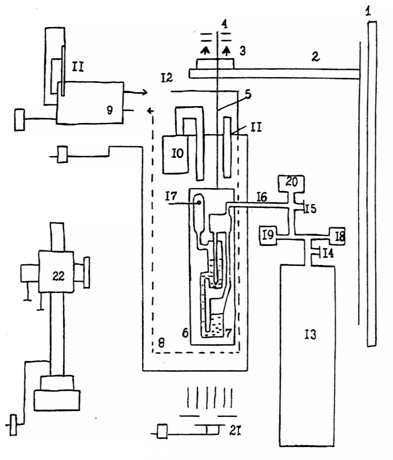
Рис. 8. Схема экспериментальной установки для
измерения динамического поверхностного натяжения методом максимального давления
в пузырке:
- капитальная стена; 2 - металлические уголки; 3
- буфер; 4 - массивная плита; 5 - металлический стержень; 6 - рамка; 7 - прибор
Пугачевича; 8 - термостатированный объем; 9 - водяной термостат; 10 - воздушный
термостат; 11 - термометры; 12 - жидкостная магистраль; 13 - газовый баллон, 14-15
- редукторы; 16 - вход газовой магистрали; 17 - выход газа; 18-20 - манометры;
21 - подсветка; 22 - катетометр КМ-8.
Величина радиуса капилляра была определена ранее
калибровочными опытами с водой и составила 0.04 см.
Указанная методика позволила измерять величину
максимального давления с точностью до 0.02 ч 0.05 мм водяного столба, что в
пересчете на поверхностное натяжение дает 0.05 ч 0.1 мН/м.
Экспериментальная погрешность в определении
величины поверхностного натяжения рассчитывается по формуле:
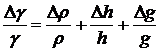 (40)
(40)
Очевидно, что величина погрешности
поверхностного натяжения в основном определяется погрешностью измерения h. По
воспроизводимости результатов была определена ошибка, которая составила для
растворов ПАВ ±0.12 мН/м.
3.2 Объекты исследования
В настоящей работе исследовалось
динамическое поверхностное натяжение трех веществ: децилсульфата натрия (ДСН),
додецилсульфата натрия (ДДСН) и тетрадецилсульфата натрия (ТДСН).
Использовались вещества квалификации “коллоидно-химически чистые”. Исследования
динамического поверхностного натяжения проводились для следующих концентраций
ДСН: 2.5·10-3 м/л (при 20 0С), 4.99·10-3 м/л (при 20 0С и 25 0С), 1.251·10-2
м/л (при 20 0С). Для ДДСН взята концентрация 1.046·10-3 (25 0С). Для ТДСН
исследовались концентрации 2.59·10-5 м/л (при 20 0С) и 8.98·10-5 (20 0С).
4. Результаты эксперимента
На рис. 9 -16 представлены зависимости
динамического поверхностного натяжения от логарифма времени, измеряемого в
секундах. Пунктирная линия соответствует равновесному поверхностному натяжению.
На всех графиках можно выделить три участка. При
малых временах наблюдается рост поверхностного натяжения со временем. Далее
следует максимум - это область ожидания адсорбции. Время, отвечающее
максимальному поверхностному натяжению, колеблется для разных изотерм от 50
секунд до 2 минут. Подобные максимумы на изотермах динамического поверхностного
натяжения наблюдались и ранее (см. [16]). И, наконец, третий участок
соответствует области интенсивной адсорбции.
Рост поверхностного натяжения на начальном этапе
и как следствие появление максимума на изотермах может быть связано с
электрическим состоянием поверхности. Ранее в разделе 2.2 нами был рассмотрен
эффект заряжения поверхности жидкости. В нашем случае как раз может иметь место
такое явление. Вследствие локального нарушения электронейтральности двойного
электрического слоя на поверхности образуется свободный заряд q
и в уравнении адсорбции для неравновесного поверхностного слоя (19) появляется
член qdχ,
чем и обусловлены низкие значения поверхностного натяжения при малых временах и
возникновение максимума на изотермах. Появление члена qdχ
представляется вероятным в неравновесных условиях образования поверхности при
достаточно свежей поверхности (малые времена) и низкой концентрации электролита
(когда двойной электрический слой достаточно протяженный и легко
деформируемый). В нашем случае также для самой маленькой концентрации ДСН -
2,5·10-3 М (см. рис. 9) наблюдается наиболее сильный рост поверхностного
натяжения на начальном этапе. Для концентрации ДСН 1,251·10-2 М (рис. 12)
наблюдается уже меньший рост поверхностного натяжения (максимум более пологий).
Дело в том, что чем ниже концентрация электролита тем более протяженным
является диффузный слой, легче происходит его тангенциальный разрыв и тем
больший образуется заряд на поверхности (тем существеннее вклад члена qdχ
в
динамическое поверхностное натяжение). Увеличение поверхностного натяжения Δγ
при
малых временах до величины максимума на наших изотермах составляет несколько
десятых мН/м, оценки показывают, что это вполне соответствует величине вклада в
поверхностное натяжение, который может давать член qdχ
(если
воспользоваться литературными значениями q
= 4·10-7 Кл/см2 и Δχ
= 0.1 В).
Для раствора ДСН концентрации 4.99·10-3 м/л при
20 0С на изотерме динамического поверхностного натяжения наблюдается излом
(обозначен пунктиром) предположительно связанный с наличием двумерного фазового
перехода в адсорбционном слое (рис. 16). Были рассчитаны скорости процесса
релаксации vγ = -dγ/dlnt.
Для участка 2 (более пологого) vγ
= 0.0712 мН/м, для третьего участка, соответствующего резкому снижению γ,
vγ = 0.3011 мН/м. Это
говорит о том, что процесс релаксации на участке 3 протекает быстрее, чем на
участке 2.
Влияние концентрации на поверхностное натяжение.
Для растворов ДСН изотермы соответствующие большим концентрациям лежат в целом
ниже изотерм соответствующих меньшим концентрациям. То же наблюдается и для
двух растворов ТДСН. Т.е. поверхностное натяжение уменьшается с ростом
концентрации ПАВ.
Влияние температуры на поверхностное натяжение.
Для раствора ДСН концентрации 4,99·10-3 М были получены две изотермы для двух
температур 20 и 25 0С (рис. 10, 11). Анализируя полученные зависимости можно
отметить, что поверхностное натяжение уменьшается с ростом температуры.
Определение механизма адсорбции. Были построены
зависимости γ-t1/2
для трех растворов: ДСН (С = 4,99·10-3 М), ДДСН (С = 1,046·10-3 М) и ТДСН (С =
8,98·10-5 М) (рис. 17 - 19). С этой целью выбирались значения t
соответствующие условию t
→
0, условно, в соответствии с [17], мы принимаем область при t
→
0 как область начала интенсивной адсорбции. Линейный характер полученных
зависимостей говорит о том, что для исследованных участков кинетика адсорбции
диффузионно-контролируемая, при этом угловой коэффициент для этих зависимостей
определяется по уравнению (23) и равен:
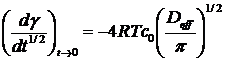 . (41)
. (41)
С помощью уравнения (41) были
рассчитаны эффективные коэффициенты диффузии для указанных выше концентраций
ПАВ. Для ДСН Deff = 4.8·10-19
м2/с (20 0С), для ДДСН Deff = 2.6·10-16 м2/с (25 0С),
для ТДСН Deff = 1.7·10-14
м2/с (20 0С). Отсюда можно видеть, что коэффициент диффузии увеличивается с
ростом длины углеводородной цепи молекул ПАВ. Эту зависимость можно объяснить с
помощью представлений о гидрофобном эффекте. Известно, что с ростом длины
углеводородной цепи уменьшается энергия агрегации молекул ПАВ. Для наших
веществ были рассчитаны энергии Гиббса мицеллообразования по формуле [18]:
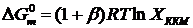 , (42)
, (42)
где XККМ - ККМ,
выражаемая молярной долей, β - степень связывания противоионов, β ≈ -
1/2k -1, k - tg угла
наклона зависимости lg СККМ(nc - число
атомов углерода в цепи). В нашем случае k = - 0.306,
а β = 0.63.
Энергии Гиббса мицеллообразования для ДСН (при 20 0С), ДДСН (при 20 0С) и ТДСН
(при 40 0С) равны соответственно -28.9, -34.8 и -43.4 (кДж/моль).
Т.е. процесс выталкивания молекул
ПАВ из воды с ростом длины цепи становится более энергетически выгодным. Это
означает ускорение процесса адсорбции и увеличение коэффициентов диффузии
молекул ПАВ с ростом длины цепи.
Расчет адсорбции. Используя данные
по динамическому поверхностному натяжению, для изученных ПАВ была рассчитана
адсорбция (таблицы 1 - 9). Расчет проводился по формуле [3]:
 . (43)
. (43)
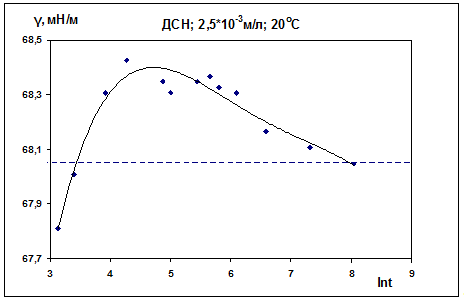
Рис. 9
Таблица 1. ДСН, С = 2,5·10-3 М, 20
0С
|
t,
сек
|
lnt
|
h,
мм
|
γ, мН/м
|
Г,
моль/м2*105
|
|
|
|
|
|
|
23
|
3,14
|
433,37
|
67,81
|
0,101
|
|
30
|
3,40
|
433,27
|
68,01
|
0,097
|
|
50
|
3,91
|
433,12
|
68,31
|
0,091
|
|
72
|
4,28
|
433,06
|
68,43
|
0,089
|
|
130
|
4,87
|
433,1
|
68,35
|
0,090
|
|
150
|
5,01
|
433,12
|
68,31
|
0,091
|
|
230
|
5,44
|
433,1
|
68,35
|
0,090
|
|
285
|
5,65
|
433,09
|
68,37
|
0,090
|
|
330
|
5,80
|
433,11
|
68,33
|
0,091
|
|
445
|
6,10
|
433,12
|
68,31
|
0,091
|
|
720
|
6,58
|
433,19
|
68,17
|
0,094
|
|
1500
|
7,31
|
433,22
|
68,11
|
0,095
|
|
3120
|
8,05
|
433,25
|
68,05
|
0,096
|
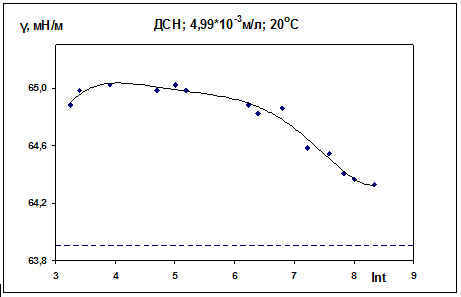
Рис. 10
Таблица 2. ДСН, С = 4,99·10-3 М, 20 0С
|
t,
сек
|
lnt
|
h,
мм
|
γ, мН/м
|
Г,
моль/м2*105
|
|
|
|
|
|
|
26
|
3,26
|
435,89
|
64,88
|
0,161
|
|
30
|
3,40
|
435,84
|
64,98
|
0,159
|
|
50
|
3,91
|
435,82
|
65,02
|
0,159
|
|
110
|
4,70
|
435,84
|
64,98
|
0,159
|
|
150
|
5,01
|
435,82
|
65,02
|
0,159
|
|
180
|
5,19
|
435,84
|
64,98
|
0,159
|
|
510
|
6,23
|
435,89
|
64,88
|
0,161
|
|
600
|
6,40
|
435,92
|
64,82
|
0,163
|
|
900
|
6,80
|
435,9
|
64,86
|
0,162
|
|
1380
|
7,23
|
436,04
|
64,58
|
0,168
|
|
1980
|
7,59
|
436,06
|
64,54
|
0,168
|
|
2520
|
7,83
|
436,13
|
64,41
|
0,171
|
|
3000
|
8,01
|
436,15
|
64,37
|
0,172
|
|
4200
|
8,34
|
436,17
|
0,173
|
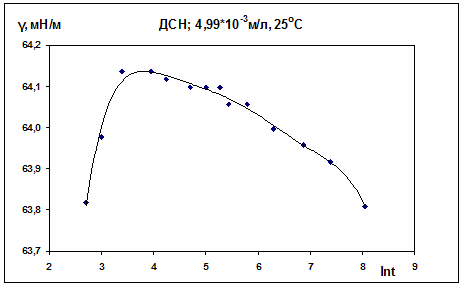
Рис. 11
Таблица 3. ДСН, С = 4,99·10-3 М, 25 0С
|
t,
сек
|
lnt
|
h,
мм
|
γ, мН/м
|
Г,
моль/м2*105
|
|
|
|
|
|
|
15
|
2,71
|
435,4
|
63,82
|
0,165
|
|
20
|
3,00
|
435,32
|
63,98
|
0,161
|
|
30
|
3,40
|
435,24
|
64,14
|
0,158
|
|
52
|
3,95
|
435,24
|
64,14
|
0,158
|
|
70
|
4,25
|
435,25
|
64,12
|
0,158
|
|
110
|
4,70
|
435,26
|
64,10
|
0,159
|
|
150
|
5,01
|
435,26
|
64,10
|
0,159
|
|
195
|
5,27
|
435,26
|
64,10
|
0,159
|
|
230
|
5,44
|
435,28
|
64,06
|
0,160
|
|
330
|
5,80
|
435,28
|
64,06
|
0,160
|
|
540
|
6,29
|
435,31
|
64,00
|
0,161
|
|
960
|
6,87
|
435,33
|
63,96
|
0,162
|
|
1620
|
7,39
|
435,35
|
63,92
|
0,163
|
|
3120
|
8,05
|
435,405
|
63,81
|
0,165
|
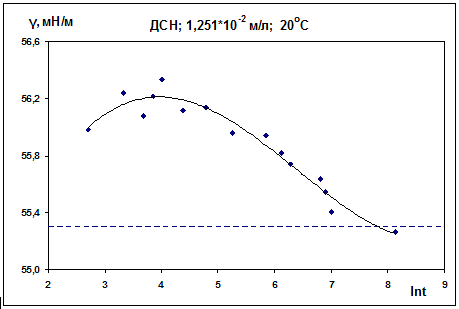
Рис. 12
Таблица 4. ДСН, С = 1,251·10-2 М, 20 0С
|
t,
сек
|
lnt
|
h,
мм
|
γ, мН/м
|
Г,
моль/м2*105
|
|
|
|
|
|
|
15
|
2,71
|
439,8
|
55,98
|
0,344
|
|
28
|
3,33
|
439,67
|
56,24
|
0,339
|
|
40
|
3,69
|
439,75
|
56,08
|
0,342
|
|
47
|
3,85
|
439,68
|
56,22
|
0,339
|
|
55
|
4,01
|
439,62
|
56,34
|
0,337
|
|
80
|
4,38
|
439,73
|
56,12
|
0,341
|
|
120
|
4,79
|
439,72
|
56,14
|
0,341
|
|
190
|
5,25
|
439,81
|
55,96
|
0,345
|
|
345
|
5,84
|
439,82
|
55,94
|
0,345
|
|
450
|
6,11
|
439,88
|
55,82
|
0,347
|
|
540
|
6,29
|
439,92
|
55,74
|
0,349
|
|
900
|
6,80
|
439,975
|
55,63
|
0,351
|
|
990
|
6,90
|
440,02
|
55,54
|
0,353
|
|
1100
|
7,00
|
440,09
|
55,40
|
0,356
|
|
3420
|
8,14
|
440,16
|
55,26
|
0,359
|
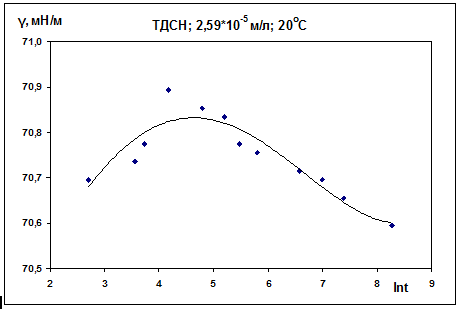
Рис. 13
Таблица 5. ТДСН, С = 2,59·10-5 М, 20 0С
|
t,
сек
|
lnt
|
h,
мм
|
γ, мН/м
|
Г,
моль/м2*105
|
|
|
|
|
|
|
15
|
2,71
|
432,21
|
70,70
|
0,042
|
|
35
|
3,56
|
432,19
|
70,74
|
0,041
|
|
42
|
3,74
|
432,17
|
70,77
|
0,041
|
|
65
|
4,17
|
432,11
|
70,89
|
0,038
|
|
120
|
4,79
|
432,13
|
70,85
|
0,039
|
|
180
|
5,19
|
432,14
|
70,83
|
0,039
|
|
240
|
5,48
|
432,17
|
70,77
|
0,041
|
|
330
|
5,80
|
432,18
|
70,76
|
0,041
|
|
720
|
6,58
|
432,2
|
70,72
|
0,042
|
|
1080
|
6,98
|
432,21
|
70,70
|
0,042
|
|
1620
|
7,39
|
432,23
|
70,66
|
0,043
|
|
3900
|
8,27
|
432,26
|
70,60
|
0,044
|
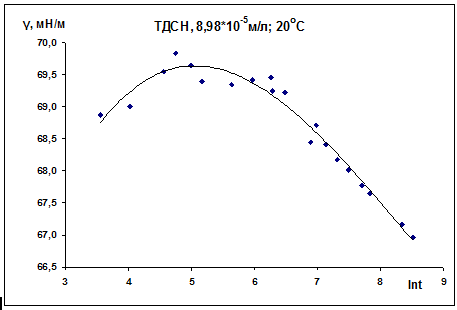
Рис. 14
Таблица 6. ТДСН, С = 8,98·10-5 М, 20 0С
|
t,сек
|
lnt
|
h,
мм
|
γ, мН/м
|
Г,
моль/м2*105
|
|
|
|
|
|
|
35
|
3,56
|
433,25
|
68,87
|
0,080
|
|
56
|
4,03
|
433,175
|
69,02
|
0,077
|
|
96
|
4,56
|
432,905
|
69,55
|
0,066
|
|
115
|
4,74
|
432,76
|
69,84
|
0,060
|
|
147
|
4,99
|
432,86
|
69,64
|
0,064
|
|
176
|
5,17
|
432,98
|
69,41
|
0,069
|
|
282
|
5,64
|
433,01
|
69,35
|
0,070
|
|
392
|
5,97
|
432,97
|
69,43
|
0,068
|
|
526
|
6,27
|
432,95
|
69,47
|
0,067
|
|
540
|
6,29
|
433,06
|
69,25
|
0,072
|
|
660
|
6,49
|
433,07
|
69,23
|
0,072
|
|
990
|
6,90
|
433,46
|
68,45
|
0,088
|
|
1080
|
6,98
|
433,33
|
68,71
|
0,083
|
|
1260
|
7,14
|
433,48
|
68,41
|
0,089
|
|
1500
|
7,31
|
433,6
|
68,17
|
0,094
|
|
1800
|
7,50
|
433,68
|
68,01
|
0,097
|
|
2220
|
7,71
|
433,8
|
67,78
|
0,102
|
|
2520
|
7,83
|
433,86
|
67,66
|
0,105
|
|
4200
|
8,34
|
434,11
|
67,16
|
0,115
|
|
4980
|
8,51
|
434,21
|
66,96
|
0,119
|
Таблица 7. ДДСН, С = 1,046·10-3 М, 25 0С
|
t,сек
|
lnt
|
h,
мм
|
γ, мН/м
|
Г,
моль/м2*105
|
|
|
|
|
|
|
17
|
2,83
|
433,44
|
66,14
|
0,118
|
|
14
|
433,44
|
66,14
|
0,118
|
|
16
|
2,77
|
433,44
|
66,14
|
0,118
|
|
17
|
2,83
|
433,44
|
66,14
|
0,118
|
|
19
|
2,94
|
433,41
|
66,20
|
0,116
|
|
15
|
2,71
|
433,41
|
66,20
|
0,116
|
|
18
|
2,89
|
433,41
|
66,20
|
0,116
|
|
21,5
|
3,07
|
433,41
|
66,20
|
0,116
|
|
25
|
3,22
|
433,37
|
66,28
|
0,115
|
|
31
|
3,43
|
433,37
|
66,28
|
0,115
|
|
27
|
3,30
|
433,37
|
66,28
|
0,115
|
|
32,5
|
3,48
|
433,37
|
66,28
|
0,115
|
|
45
|
3,81
|
433,37
|
66,28
|
0,115
|
|
40
|
3,69
|
433,37
|
66,28
|
0,115
|
|
25
|
3,22
|
433,35
|
66,32
|
0,114
|
|
36
|
3,58
|
433,34
|
66,34
|
0,114
|
|
61,5
|
4,12
|
433,34
|
66,34
|
0,114
|
|
51
|
3,93
|
433,34
|
66,34
|
0,114
|
|
49
|
3,89
|
433,32
|
66,38
|
0,113
|
|
74,5
|
4,31
|
433,3
|
66,42
|
0,112
|
|
90
|
4,50
|
433,35
|
66,32
|
0,114
|
|
107
|
4,67
|
433,28
|
66,46
|
0,111
|
|
133
|
4,89
|
433,36
|
66,30
|
0,114
|
|
115
|
4,74
|
433,34
|
66,34
|
0,114
|
|
165
|
5,11
|
433,42
|
66,18
|
0,117
|
|
196
|
5,28
|
433,43
|
66,16
|
0,117
|
|
290
|
5,67
|
433,44
|
66,14
|
0,118
|
|
290
|
5,67
|
433,51
|
66,00
|
0,121
|
|
364
|
5,90
|
433,48
|
66,06
|
0,119
|
|
500
|
6,21
|
433,67
|
65,68
|
0,127
|
|
675
|
6,51
|
433,72
|
65,58
|
0,129
|
|
900
|
6,80
|
433,94
|
65,14
|
0,138
|
|
1774
|
7,48
|
434,58
|
63,87
|
0,163
|
Таблица 8. ДСН, С = 4,99·10-3 М, 20 0С.
|
t,
сек
|
t1/2,
с1/2
|
γ,
Н/м
|
|
|
|
|
900
|
30,00
|
0,0649
|
|
1380
|
37,15
|
0,0646
|
|
1980
|
44,50
|
0,0645
|
|
2520
|
50,20
|
0,0644
|
|
3000
|
54,77
|
0,0644
|
Таблица 9. ДДСН, С = 1,046·10-3 М, 25 0С.
|
t,
сек
|
t1/2,
с1/2
|
γ,
Н/м
|
|
|
|
|
364
|
19,08
|
0,0661
|
|
500
|
22,36
|
0,0657
|
|
675
|
25,98
|
0,0656
|
|
900
|
30,00
|
0,0651
|
|
1774
|
42,12
|
0,0639
|
Таблица 10. ТДСН, С = 8,98·10-5 М, 20 0С.
|
t,сек
|
t1/2
|
γ, Н/м
|
|
|
|
|
526
|
22,93
|
0,0695
|
|
540
|
23,24
|
0,0692
|
|
660
|
25,69
|
0,0692
|
|
990
|
31,46
|
0,0685
|
|
1080
|
32,86
|
0,0687
|
|
1260
|
35,50
|
0,0684
|
|
1500
|
38,73
|
0,0682
|
|
1800
|
42,43
|
0,0680
|
|
2220
|
47,12
|
0,0678
|
|
2520
|
50,20
|
0,0677
|
Выводы
. Методом максимального давления в газовом
пузырьке измерено динамическое поверхностное натяжение водных растворов
алкилсульфатов натрия. Использовались ПАВ квалификации “коллоидно-химически
чистые”. Вещества были синтезированы и очищены во ВНИИ ПАВ г. Шебекино, что
позволило получить надежные данные по свойствам этих растворов.
. Поверхностное натяжение растворов ПАВ
традиционно уменьшается с ростом температуры и ростом концентрации ПАВ, что
согласуется с полученными результатами.
. Расчетные величины адсорбции имеют традиционно
величину ~ 10-6 моль/м2.
. Анализ полученных изотерм показал, что в
области малых концентраций и малых времен жизни поверхности растворов
нарушается электронейтральность поверхности, что приводит к увеличению
поверхностного натяжения со временем. Это явление подтверждается ранее открытым
эффектом Джонса-Рея для неорганических электролитов и исследованиями
поверхностной электризации, опубликованными в работе [4].
. Установлено, что в области “интенсивной
адсорбции” наблюдается диффузионный характер адсорбции. Причем, с ростом длины
углеводородной цепи молекул ПАВ происходит ускорение процесса адсорбции, что
связано с усилением действия гидрофобного эффекта.
. В адсорбционном слое децилсульфата натрия
предположительно обнаружен двумерный фазовый переход от “жидко-растянутого” к
“жидко-конденсированному” состоянию ПАВ.
Литература
1. Мырзахметова Н.О. Динамическое
поверхностное натяжение водных растворов: адсорбционный и электризационный
эффекты // Дис. … канд. хим. наук. - Санкт-Петербург, СПбГУ. - 1993. - 150с.
. Кочурова Н.Н., Русанов А.И.
Термодинамика неравновесного поверхностного натяжения жидкостей //В кн.:
Вопросы термодинамики гетерогенных систем и теории поверхностных явлений. - Л.:
Изд-во ЛГУ, 1988. - Вып. 8. - С. 109 - 122.
. Кочурова Н.Н., Русанов А.И.
Релаксация поверхностных свойств водных растворов поверхностно-активных веществ
и механизм адсорбции // Успехи химии. - 1993. - Т. 62. - № 12. - С.1150 - 1162.
. Кочурова Н.Н., Русанов А.И.,
Мырзахметова Н.О. Эффект Джонса-Рэя и поверхностная электризация // Докл. АН
СССР. - 1991. - Т. 316. - № 6. - С. 1425 - 1427.
.Натансон Г.Л. К вопросу о механизме
баллоэлектрических явлений // Докл. АН СССР.
- 1950. - Т. 73 - № 5. - С.
975 - 978.
. Eastoe J., Dalton J.S.
Dynamic surface tension and adsorption mechanisms of surfactants at the
air-water interface // Advances in Colloid and Interface Science. 2000. V. 85.
Issues 2-3. P.103 - 144.
. Ravera F., Liggieri L.,
Steinchen A. Sorption kinetics considered as a renormalized diffusion process
// Journal of Colloid and Interface Science. 1993. V.156.
Issue 1. p.
109.
. Побережный В.Я., Сотскова Т.З.,
Кульский Л.А. Роль электростатических взаимодействий в процессе адсорбции
ионогенных ПАВ на межфазной грантце раздела жидкость - газ // Доклады АН УССР.
Сер. Б. Геол., хим. и биол. науки. - 1989. - №5. - С. 41 - 45.
. Дмитровская М.В., Кочурова Н.Н.
Возникновение двумерных фазовых переходов в адсорбционных слоях
поверхностно-активных веществ (ПАВ) и их смесей с полимерами на границе раствор
- воздух // Годовой научный отчет за 2006 год по гранту Президента Российской
Федерации МК-5943.2006.3. - С.1 - 12.
. Свитова Т.Ф., Смирнова Ю.П.,
Чураев Н.В., Русанов А.И. Динамика поверхностного натяжения и двумерные фазовые
переходы в монослоях растворимых ПАВ на поверхности раздела вода/воздух //
Коллоидный журнал. 1994. Т.56.
№3. С.441
- 445.
.Nicolov A., Martynov G.,
Exerova D. Associative interactions and surface tension in ionic surfactant
solutions at concentrations much lower than the CMC // Journal of Colloid and
Interface Science. 1981. V.81. № 1. P. 116 - 124.
. Vollhardt D., Phase transition
in adsorption layers at the air-water interface // Advances in Colloid and
Interface Science. 1999. V.79.
P. 19 - 57
. Русанов А.И. Фазовые равновесия и
поверхностные явления. - Л.: Химия, 1967. - 388 с.
. Дмитровская М.В., Кочурова Н.Н., Petzold
G. Исследование
поверхностного натяжения водных растворов хлорида
додециламидоэтилдиметилбензиламмония // Коллоидный журнал. 2004.
Т.66.
№5. С.
1 - 5
. Islam N., Kato T. Influence
of temperature and alkyl chain length on phase behavior in Langmuir monolayers
of some oxyethylenated nonionic surfactants // Journal of Colloid and Interface
Science. 2006. V.
294. Issue 2. P.
288 - 294.
. Пчелинцева М.Н., Кочурова Н.Н.,
Петцольд Г., Лунквиц К. Динамическое поверхностное натяжение водных растворов
полиэлектролитных комплексов // Коллоид. журн. - 2000. - Т. 62. - № 5. - С. 672
- 677.
. Дмитровская М.В. Динамическое
поверхностное натяжение водных растворов хлорида
додециламидоэтилдиметилбензиламмония и его смесей с полиэлектролитами // Дис. …
канд. хим. наук. - Санкт-Петербург, СПбГУ. - 2005. C.
97. 18. Русанов А.И. Мицеллообразование в растворах поверхностно-активных
веществ. - СПб.: Химия, 1992. С.100.