Химический исследование производственных сточных вод
Содержание
Общие указания
. Метод определения сульфатов
2. Метод определения железа
. Метод определение меди
. Метод определения цинка
. Метод определения хлоридов
. Метод определения нефтепродуктов
. Метод определения взвешенных веществ
Общие указания
О работе в лаборатории
При работе в лаборатории необходимо соблюдать следующие правила:
- соблюдать тишину в лаборатории;
иметь рабочий халат;
перед началом работ изучить сущность используемого метода и устройство
используемой аппаратуры;
подготовить необходимые реактивы и химическую посуду;
рационально строить свою работу
все работы выполнять точно и аккуратно, но без спешки, которая может
привести к порче поставленного опыта;
соблюдать все меры предосторожности при работе с ядовитыми,
взрывоопасными и огнеопасными веществами.
О реактивах и обращении с ними
лаборатория реактив вода вещество
Работающие в лаборатории должны знать основные свойства применяемых ими
реактивов, особенно степень их ядовитости и способность к образованию
взрывоопасных и огнеопасных смесей с другими реактивами.
Приготовлять растворы нужно в таком количестве, которое необходимо для
работы. Необходимо следить, чтобы на всех банках с реактивами обязательно были
этикетки (или надписи восковым карандашом) с обозначением, что находиться в
банке. По своему назначению реактивы делят на две основные группы:
общеупотребительные и специальные. Специальные (наиболее дорогие и редкие)
реактивы хранят отдельно. Все реактивы должны храниться в соответствии с их
свойствами.
О реактивах и обращении с ними надо помнить следующее:
реактивы следует предохранять от загрязнения;
реактивы следует расходовать экономно;
на всех склянках с реактивами всегда должны быть этикетки с указанием
названия реактива и степени их чистоты;
реактивы, изменяющиеся под действием света, следует хранить только в
желтых или темных склянках;
особую осторожность следует соблюдать при обращении с концентрированными
кислотами и щелочами:
с огнеопасными реактивами следует работать вдали от огня и включенных
нагревательных приборах.
Отбор и хранение проб
. Отбор должен обеспечивать представительные пробы, сохранение состава
исследуемой воды до анализа и гарантировать от случайных загрязнений. Объем
отбираемой пробы должен быть достаточным для выполнения анализа, а при
необходимости и повторения его.
Для исключения искажения результатов анализа из-за сорбирования
определяемых компонентов пробы стенками сосудов, пробу следует отбирать в
сосуды, в которых возможно проведение всех последующих операций анализа.
За каждой пробой следует закрепить отдельный сосуд и нанести на нем метки
с целевым обозначением.
Отбор единичных проб для определения нефтепродуктов осуществляют в
отдельные сосуды, так как для анализа следует использовать весь объем каждой
пробы.
. При отборе проб для определения нелетучих растворенных веществ,
пробо-отборное устройство должно быть продуто, если непосредственно перед этим
из него проба не отбиралась. Продувку осуществляют так, чтобы промыть всю
пробоотборную трассу за 15 - 20 мин., после чего устанавливают скорость
истечения жидкости, обеспечивающую запаздывание пробы не более чем на 10 мин.
. При отборе проб для определения веществ, находящихся частично в
нерастворенном состоянии, после продувки устанавливаю скорость входа воды в
отверстие пробозаборного устройства, равную скорость среды в трубопроводе.
Пробоотборная трасса должна быть наиболее короткой для уменьшения запаздывания
пробы.
Недопустимо во время отбора проб менять устанавливающуюся скорость
истечения, прикасаться к запорным органам (вентилям), допускать толчки и удары
по пробоотборной трассе.
. Проба для определения растворенного кислорода должна быть защищена от
контакта с воздухом.
. Отбираемая проба должна быть охлаждена до температуры не превышающей 400С.
. Сроки выполнения анализов определяют требования эксплуатации и
возможность сохранения проб.
Анализы по определению летучих веществ и веществ, находящихся
одновременно в растворенном состоянии и в форме суспензированных частиц,
следует выполнять сразу же после отбора проб.
Допустимые сроки хранения проб для определения:
нефтепродуктов - не более 12 ч.;
других компонентов - не более 2 - 3 ч.
. При определении загрязняющих веществ в производственных в сточных водах
находящихся в очень малых концентрациях, следует использовать дистиллированную
воду очищенную дополнительно. Сущность метода состоит в фильтрации
дистиллированной воды или конденсата через материалы (сорбенты), поглощающие
органические примеси, катионы и анионы, и задерживающие нерастворенные частицы
различной степени дисперсности.
. Очищенную воду следует хранить в полиэтиленовой посуде с плотно
навинчивающейся крышкой. Срок хранения - не более пяти суток.
Построение калибровочных графиков
При массовых фотоколориметрических анализах, определяя концентрацию
испытуемого раствора, не сравнивают каждый раз его светопоглощение со
светопоглощением эталонного раствора, а предварительно строят так называемую калибровочную
кривую. Для этого пользуются серией эталонных растворов различной
концентрации. Имея такую кривую, при определении концентрации испытуемого
раствора достаточно измерить его светопоглощение и по калибровочной кривой
найти величину концентрации, соответствующую найденному светопоглощению.
Для построения калибровочной кривой нужно приготовить серию эталонных
растворов, содержащих различные количества определяемого вещества. Сначала
приготовляют стандартный раствор, содержащий строго определенное количество
исследуемого вещества. С помощью бюретки отбирают в мерные колбы емкостью 100
мл различные, точно измеренные объемы этого стандартного раствора и
соответствующих реактивов, вызывающих окраску анализируемого раствора. Затем
содержимое каждой мерной колбы разбавляют дистиллированной водой, доводя объем
раствора до метки.
С помощью фотоколориметра измеряют оптические плотности приготовленных
эталонных растворов, и результаты измерений записывают в виде таблицы.
Например:
Эталонный раствор………………1 2 3 4 5
Содержание определяемого вещества, мг/100 мл раствора…..1 4 6 8 10
Оптическая плотность …………0,1 0,4 0,6 0,8 1,0
На основании полученных результатов строят кривую зависимости оптической
плотности раствора от его концентрации. Это и есть калибровочная кривая.
I0
Оптическая плотность D = lg I
Концентрация, С
Рис.1 Зависимость оптической плотности раствора от концентрации.
Для ее построения на миллиметровой бумаге откладывают по оси абсцисс
значения концентраций эталонных растворов, а по оси ординат величины их
оптических плотностей. Затем из точек, найденных на осях, восстанавливают
перпендикуляры, и точки их пересечения соединяют одной линией.
Лабораторная работа №1
Метод определения сульфатов
Цель работы: изучить метод гравиметрического (весового) определения
сульфатов
Сущность метода. Метод основан на реакции взаимодействия сульфат-ионов с
ионами бария, сопровождающейся образованием малорастворимого
мелкокристаллического осадка сульфата бария. Осадок сульфата бария
отфильтровывают, промывают, сушат, прокаливают, взвешивают и рассчитывают в нем
содержание SO42- и серы.
Химический процесс. Сульфат-ион SO42- является анионом сильной серной
кислоты Н2SO4. Из солей серной кислоты
малорастворимы в воде соли бария, стронция, свинца и кальция. Остальные
сульфаты хорошо растворимы. Ион SO42- бесцветен.
Хлорид бария ВаСl2 Ва2+-ион при
взаимодействии с растворами, содержащими SO42- , образует белый осадок ВаSO4. Как малорастворимая соль сильной
кислоты, сульфат бария нерастворим в кислотах. Этим ВаSO4 отличается от солей бария всех других анионов, чем и
пользуются при обнаружении сульфат-ионов:
Ва2+ + SO42- → ВаSO4↓
Таким образом, растворимые соли бария служат реактивом на сульфат-ион.
S2O32- +2H+ →
S↓ + SO2↑ + H2O32- + S2-
+ 6H+ → 3S↓ + 3H2O
Отличить ВаSO4 от серы можно, используя способность
сульфата бария к образованию с KMnO4 смешанных
кристаллов розового цвета.
Весовая форма в этом методе определения идентична форме осаждения. При
обработке осадка при переведении его в весовую форму могут произойти следующие
нежелательные процессы:
а) при озолении фильтра может произойти восстановление сульфата бария
ВаSO4 + 2С → 2СО2 + ВаS
Обратный переход сульфида бария в сульфат происходит в процессе
длительного нагревания осадка на воздухе
ВаS + 2О2 → ВаSO4
б) при прокаливании при слишком высокой температуре наблюдается
термическое разложение сульфата бария
ВаSO4 → ВаО + SO3↑
Экспериментальная часть
Реактив: хлористоводородная кислота, пл. 1,19 г/см3; хлорид
бария, 5%-ный раствор; метиловый оранжевый, 0,1%-ный раствор.
Проведение анализа. Анализируемую воду сначала фильтруют, затем отбирают 25-200
мл, в зависимости от содержания сульфатов, о котором можно судить по
предварительной пробе* (*к небольшому количеству анализируемой воды прибавляют
хлористоводородную кислоту до кислой реакции и хлорид бария до полного
осаждения сульфата бария. Если появится только опалесценция раствора, берут для
количественного определения 200 мл анализируемой воды; если же выпадает осадок,
берут меньшее количество воды. При некотором навыке по виду осадка можно
заключить, какой объем анализируемой воды будет наиболее подходящим для
количественного определения сульфата), Нужный объем пробы переносят в стакан,
подкисляют HC1 по метиловому оранжевому и
выпаривают (или разбавляют) до объема 50 мл.
Если при упаривании образовался осадок, его отфильтровывают через
маленький фильтр и промывают горячей дистиллированной водой, подкисленной хлористоводородной
кислотой. Фильтрат и промывные воды опять упаривают до объема 50 мл, нагревают
до кипения** (** Если анализируемая сточная вода содержит сульфиты или
тиосульфаты, кипятят до полного удаления сернистого газа.) и приливают к ним по
каплям горячий 5%-ный раствор хлорида бария до полного осаждения сульфатов.
Жидкость с образовавшимся осадком оставляют стоять на водяной или песочной бане
в течение 2 ч и затем на холоду - на ночь. Ha следующий день осадок отфильтровывают через плотный фильтр
(синяя лента) *** (*** Осадок сульфата бария рекомендуется переосадить,
особенно в тех случаях, когда анализируемая вода содержит большое количество
солей или окисление при определении «общей серы» проводилось с добавлением
карбоната натрия. Для этого осадок сульфата бария обрабатывают на фильтре
горячим 3-5%-ным раствором ЭДТА, предварительно доведенным до сильнощелочной
реакции аммиаком. K полученному
раствору прибавляют несколько капель 10%-ного раствора хлорида бария и
нейтрализуют раствор хлористоводородной кислотой по метиловому красному),
промывают горячей водой до исчезновения в промывной воде хлоридов (проба с
раствором AgNO3, подкисленным HNO3), сушат, прокаливают 30 мин и взвешивают в виде BaSO4.
Если в сточной воде содержится много кремневой кислоты, необходимо
предварительно ее отделить. При большом содержании в сточной воде железа
рекомендуется предварительно восстановить его до двухвалентного. Минимальное
количество сульфатов, определяемое по этому методу, составляет 2 мг/л.
Обработка измерений
Содержание сульфат-ионов (x) в
мг/л и серы (у) в мг/л вычисляют по формулам:
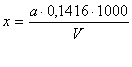
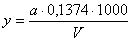
где:
а - масса прокаленного осадка сульфата бария, мг; V - объем взятой
для анализа пробы, мл; 0,4116 - коэффициент пересчета BaSO4 на SO42-; 0,1374 - коэффициент пересчета BaSO4 на серу.
Контрольные
вопросы
1. В чем заключается сущность гравиметрического метода?
2. Какая реакция положена в основу методики определения
сульфат-ионов в растворе?
. Почему именно соли бария служат реактивом на сульфат-ионы?
. Каким образом определяется объем пробы для проведения анализа?
. Каким образом можно отличить выпавшие кристаллы сульфата бария
от кристаллов серы в осадке?
. Какие нежелательные процессы могут возникнуть в процессе
обработки полученного осадка?
. Почему исследуемую пробу необходимо подкислять соляной
кислотой?
. Присутствие каких элементов в исследуемом растворе нежелательно
и почему?
Лабораторная
работа №2
Метод
определения железа
Цель
работы: изучить
метод фотоколориметрического определения железа.
Сущность
метода заключается в
том, что ионы трехвалентного железа образуют с сульфосалицилат-ионами
комплексное соединение, раствор которого в щелочной среде окрашен в желтый
цвет. Двухвалентное железо в условиях анализа переходит в трехвалентное.
Все фотоколориметрические методы определения основаны на одном общем
принципе. Световой поток проходит через кювету наполненную испытуемым
окрашенным раствором. Прошедший через раствор световой поток воспринимается
фотоэлементом, в котором световая энергия превращается в электрическую энергию.
Возникающий при этом электрический ток измеряют при помощи чувствительного
гальванометра. Сила электрического тока, возникающего при действии световой
энергии на фотоэлемент, прямо пропорциональна интенсивности освещения.
Для определения этим методом концентрации исследуемого вещества
измеряют оптическую плотность испытуемого раствора (Dисп) и эталонного раствора, концентрация которого известна (Dэтал). Расчет проводят по формуле:
Сисп = (Dисп/ Dэтал)∙Сэтал
Основными преимуществами фотоколориметрических методов измерения
интенсивности окраски является быстрота и легкость определений при высокой их
точности.
Химический процесс. Железо образует два ряда солей: соли железа (II) и (III). Растворы солей железа (III) содержат трехзарядные катионы Fe3+, а растворы солей железа -
двухзарядные катионы Fe2+. Так как реакции этих ионов
совершенно различны, нужно рассматривать их отдельно.
Растворы солей железа (III)
имеют желтую или красно-бурую окраску (в зависимости от образующегося
комплекса). Комплексы образуются в результате соединения между собой
электрически нейтральных молекул более простых по составу солей.
Экспериментальная
часть
Приготовление
растворов
1. Раствор аммония хлористого, 2 моль/дм3.
26,75 г аммония хлористого растворяют дистиллированной водой в мерной
колбе на 250 см3. Объем доводят до метки дистиллированной водой.
2. Аммиак разбавленный 1:1.
В мерную колбу на 250 см3 приливают 125 см3
дистиллированной воды, затем приливают 125 см3 водного аммиака 22%. (раствор
готовят под вытяжкой).
3. Кислота сульфосалициловая, 300 г/дм3.
30 г сульфосалициловой кислоты растворяют дистиллированной водой в мерной
колбе на 100 см3. Объем доводят до метки дистиллированной водой.
4. Кислота соляная 1:1.
В мерную колбу на 100 см3 приливают 50 см3
дистиллированной воды, затем приливают
см3 концентрированной соляной кислоты (раствор готовят под
вытяжкой).
5. Основной стандартный раствор железа,
1мг/см3 ГОСТ 4212-76.
6. Рабочий стандартный раствор, 10мкг/см3.
1 см3 основного стандартного раствора помещают в мерную колбу
вместимостью 100 см3 и доводят объем раствора до метки
дистиллированной водой.
Проведение анализа
Объем исследуемого раствора, содержащий 10-100 мкг железа, помещают в
коническую колбу, добавляют 1 см3 раствора соляной кислоты (1:1) и
упаривают до объема 10-15 см3. Полученный раствор разбавляют
дистиллированной водой и фильтруют, собирая фильтрат в мерную колбу
вместимостью 100 см3, добавляют 2 см3 раствора хлористого
аммония (2 моль/дм3), 2 см3 раствора сульфосалициловой
кислоты (300г/дм3) и 5 см3 аммиака (1:1). Объем раствора
доводят дистиллированной водой до метки и перемешивают. Через 5 мин. делают
замер пробы на КФК-2. В качестве раствора сравнения применяют дистиллированную
воду.
Обработка
измерений
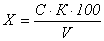
где:
Х
- массовая концентрация железа в исследуемой пробе, мг/дм3;
С
- массовая концентрация железа, найденная по градуировочному графику, мг;
К
- коэффициент перерасчета на мг/дм3;
V - объем пробы взятый для анализа, см3;
Контрольные
вопросы
1. На каком общем принципе основаны методы фотоколориметрического
анализа?
2. Как определяется оптическая плотность раствора?
. В чем заключается сущность фотоколориметрического определения
железа?
. Перечислите основные преимущества этого метода.
. Какое комплексное соединение образуют ионы трехвалентного
железа с
сульфасалицилат-ионом?
. В какой цвет образующийся комплекс окрашивает раствор, и какой при
этом должна быть среда?
7. В чем заключается сущность методики построения калибровочного
графика?
. Какую зависимость отражает калибровочная кривая?
Лабораторная работа № 3
Метод определения меди
Цель работы: изучить метод фотоколориметрического определения меди
Сущность
метода заключается в
том, что исследуемый раствор обрабатывают раствором диэтилдитиокарбамата
натрия, в результате чего образуется желто-коричневый осадок
диэтилдитиокарбамата меди, который экстрагируют хлороформом.
Медь встречается в сточных водах рудообогатительных фабрик, заводов,
производящих электролитную медь, гальванических цехов различных предприятий,
фабрик искусственного волокна, в шахтных водах и т.д.
B
сточных водах медь может присутствовать в виде ионов Cu2+, [Cu(NН3)4]2+
- в сточных водах фабрик искусственного волокна, [Cu(CN)3]2-
- в водах обогатительных фабрик, иногда в водах гальванических цехов, в виде
меднотартратных комплексов и т.д.
Для определения меди в комплексных соединениях часто требуется
предварительное их разрушение. Концентрация меди в сточных водах может быть
самой различной. При больших ее концентрациях рекомендуется проводить
электролитическое или титриметрическое определение; при малых концентрациях
более приемлемы колориметрические методы.
Экспериментальная часть
Приготовление
растворов
1. Диэтилдитиокарбамат натрия, 10г/дм3.
2,5г диэтилдитиокарбамата натрия растворяют в 150-200 см3 дистиллированной
воды. Ставят на мешающее устройство для полного растворения реактива. Раствор
переносят в мерную колбу на 250 см3 и до метки доводят
дистиллированной водой.
Хранят в посуде из темного стекла.
2. Аммиак разбавленный 1:1.
В мерную колбу на 250 см3 приливают 125 см3 дистиллированной
воды, затем приливают 125 см3 водного аммиака 22%. (раствор готовят
под вытяжкой).
3. Хлороформ.
4. Аммоний лимонокислый, 400г/дм3.
100г аммония лимонокислого растворяют в дистиллированной водой в мерной
колбе на 250 см3 .
Объем доводят до метки дистиллированной водой.
5. Трилон Б, 50г/дм3.
12,5г трилона Б растворяют в дистиллированной водой в мерной колбе на 250
см3. Объем доводят до метки дистиллированной водой.
6. Основной стандартный раствор меди,
1мг/см3
ГОСТ 4212-76.
7. Рабочий стандартный раствор, 5мкг/см3.
5 см3 основного стандартного раствора помещают в мерную колбу
вместимостью 1000 см3 и доводят объем раствора до метки
дистиллированной водой.
Мешающее
влияние
Влияние никеля, марганца и железа устраняют добавлением растворов трилона
Б и лимоннокислого аммония.
Для устранения влияния нефтепродуктов проводят предварительную экстракцию
без добавления диэтилдитиокарбамата натрия.
Проведение анализа
Объем исследуемого раствора 50 см3 помещают в делительную
воронку, добавляют 100 см3 дистиллированной воды, 5 см3
раствора лимоннокислого аммония (400г/дм3), 10 см3
раствора трилона Б (50г/дм3), 10 см3 раствора аммиака
(1:1) и 10 см3 хлороформа и встряхивают в течении 2 мин. после
отстаивания слой хлороформа удаляют. Экстракцию продолжают до тех пор, пока
слой хлороформа перестанет окрашиваться. В делительную воронку добавляют 10 см3
раствора диэтилдитиокарбамат натрия (10г/дм3) и экстрагируют
комплексную соль хлороформом 2 раза по 10 см3, энергично встряхивают
раствор в течение 2 мин. после отстаивания слои хлороформа сливают через фильтр
в мерную колбу вместимостью 25 см3. Объем объединенных экстрактов
доводят хлороформом до метки и перемешивают. Оптическую плотность раствора
измеряют на КФК-2. В качестве раствора сравнения используют хлороформ.
Обработка
измерений
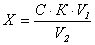
где:
Х
- массовая концентрация меди в исследуемой пробе, мг/дм3;
С
- массовая концентрация меди, найденная по градуировочному графику, мг;
К
- коэффициент перерасчета на мг/дм3;
V1 - объем дистиллированной воды взятый для анализа, V1=100 см3;
V2 - объем пробы взятый для анализа, см3.
Контрольные
вопросы
1. В чем заключается сущность фотоколориметрического определения меди?
2. В каких случаях для определения меди применяют
фотоколориметрические методы, а в каких титриметрические методы анализа?
. В виде каких ионов медь может присутствовать в промышленных
сточных водах?
. Почему перед началом анализа необходимо разрушить комплексные
соединения меди в исследуемой пробе?
. Каким образом устраняют мешающее влияние Ni, Mn, Fe в исследуемой
пробе?
. Каким образом устраняется мешающее влияние нефтепродуктов?
Лабораторная работа № 4
Метод определения цинка
Цинк встречается в сточных водах рудообогатительных фабрик,
гальванических цехов многих предприятий, в сточных водах производства
пергаментной бумаги, минеральных красок, искусственного волокна и других
производств. Концентрация цинка в сточных водах может быть самой различной: от
нескольких сотен миллиграммов до десятых и сотых долей миллиграмма в 1 л.
B
сточных водах обогатительных фабрик и гальванических цехов цинк может
присутствовать в виде комплексного цианоцинката. B этом случае перед его определением надо разрушить
комплексное соединение. B
присутствии больших количеств органических веществ рекомендуется
предварительная обработка.
Больше всех других металлов определению цинка мешает медь. В ее
присутствии, в зависимости от относительного ее количества и выбранного метода
определения цинка, поступают одним из следующих способов: 1) отделяют медь
перед определением цинка осаждением ее внутренним или внешним электролизом или
тиосульфатом натрия; 2) маскируют ее цианидом или тиосульфатом; 3) определяют
суммарное содержание меди и цинка и отдельно - содержание меди; по разности
находят содержание цинка. Указания о возможности применения того или иного
способа даны при изложении каждого метода анализа цинка.
Цель работы: изучить метод титриметрического определения цинка с
применением раствора гексацианоферрата (II) калия (концентрация цинка должна превышать 3 мг/л)
Сущность метода. При добавлении раствора гексацианоферрата (II) калия к раствору соли цинка
выпадает белый осадок K2Zn3[Fe(CN)6]2,
нерастворимый в воде и в разбавленных кислотах:
2K+ + 3Zn2+ + 2[Fe(CN)6]4-
= K2Zn3[Fe(CN)6]2
качестве индикаторов применяют дифениламин -
окислительно-восстановительный индикатор, бесцветный при окислительном
потенциале ниже +0,776 B и
окрашенный в сине-фиолетовый цвет при более высоком окислительном потенциале, и
гексацианоферрат (ІІІ) калия. Последний при отсутствии в растворе
гексацианоферрата (II) калия имеет
окислительный потенциал, превышающий +0,776 B, и, следовательно, вызывает окрашивание дифениламина. Ho когда в растворе имеется
гексацианоферрат (II) калия,
потенциал гексацианоферрата (ІІІ) калия снижается и окраска дифениламина
исчезает.
Рекомендуется обратное титрование. K анализируемому раствopy соли цинка прибавляют гексацианоферрат (III) калия и дифениламин, а затем в
избытке титрованный раствор гексацианоферрата (II) калия. Жидкость остается окрашенной в желто-зеленый цвет.
Затем титруют обратно титрованным раствором соли цинка. Когда весь избыток
гексацианоферрата (II) калия будет
связан цинком в нерастворимое соединение, присутствующий в растворе
гексациапоферрат (III) калия вызовет
окрашивание дифениламина в сине-фиолетовый цвет.
Дифениламин в качестве индикатора рекомендуется заменить
3,3'-диметилнафтидином. При обратном титровании избытка гексацианоферрата (II) солью цинка окраска переходит от
зеленой к пурпурно-красной. Этот переход окраски настолько резок, что
титрование можно проводить даже 0,001 M растворами гексациаиоферрата (II) и соли цинка.
Определению мешают металлы, образующие нерастворимые гексацианоферраты (II): медь, кадмий, кобальт, никель и
марганец. Медь надо предварительно удалить; содержание других; металлов обычно
значительно уступает содержанию цинка, пoэтому ими можно или совсем пренебречь, или ввести
соответствующие поправки, определив их отдельно. Свинец в условиях определения
осаждается в виде сульфата и не мешает. Мешающее влияние железа учтено в ходе
анализа - оно устраняется добавлением пирофосфата.
Реактивы
Гексацианоферрат (II)
калия (желтая кровяная соль). 0,025 М раствор. Взвешивают 10,56 г.
кристаллического гексацианоферрата (II) калия K4[Fe(CN)6]·3H20, прибавляют к навеске 0,2 карбоната натрия,
растворяют в небольшом объеме дистиллированной воды, раствор переносят в мерную
колбу емкостью 1 мл и доводят его объем дистиллированной водой до метки.
Сохраняют в склянке из темного стекла.
Гексацианоферрат (III)
калия (красная кровяная соль). Растворяют 1 г K3[Fe(CN)6] в 100 мл воды.
Сохраняют в темной склянке. Через 5 дней pacтвор становится негодным и его надо заменять
свежеприготовленным.
Сульфат цинка, приблизительно 0,05 M раствор. Растворяют 3,000 г металлического (x.ч.) цинка в разбавленной (1:5)
серной кислоте, взятой в небольшом избытке, и раствор разбавляют
дистиллированной водой до 1 л.
Индикатор. Растворяют 1 г дифениламина в 100 мл концентрированной серной
кислоты.
Пирофосфат натрия. Можно приготовить в лаборатории из двузамещенного
натрия Na2HPO4·12H2O, прокаливая его до тех пор, пока
взятая проба соли, растворенная в воде, не будет давать с нитратом серебра
чисто-белый осадок (не желтоватый). Полученной после прокаливания солью можно
пользоваться непосредственно или растворив ее в воде и выделив десятиводный
кристаллический продукт.
Серная кислота, разбавленная (1:3) раствор.
Перекись водорода, 3%-ный раствор, или персульфат аммония, 5%-ный
раствор.
Сульфат аммония.
Проведение анализа. Отбирают такой объем анализируемой сточной воды, чтобы в нем
содержалось от 2 до 50 мг цинка, подкисляют серной кислотой (раствор должен
стать от 0,5 н. до 1,5 н. по содержанию H2SO4), прибавляют к нему несколько миллилитров
перекиси водорода или раствора персульфата аммония и кипятят для окисления
железа и разложения избытка реактива. Затем прибавляют по 0,7-0,8 г сульфата
аммония на каждые 50 мл раствора, 0,5-1 г пирофосфата натрия (для связывания
железа в комплексное соединение), нагревают до 60 °C и приливают 4 капли раствора гексацианоферрата (III) калия и 2 капли раствора индикатора
(дифениламина). Перемешав жидкость, наливают в нее из бюретки, постепенно и при
непрерывном перемешивании, раствор гексацианоферрата (II) калия сначала до исчезновения сине-фиолетового окрашивания,
а затем приливают 20-25%-ный избыток. Дают постоять 2 мин и потом оттитровывают
избыток гексацианоферрата (II)
калия титрованным раствором соли цинка до перехода окраски из желто-зеленой в
сине-фиолетовую.
Отдельно определяют отношение концентраций растворов гексацианоферрата (II) калия и соли цинка. Для этого в
другую колбу наливают раствор соли цинка в таком объеме, чтобы в нем находилось
приблизительно столько же цинка, сколько и в анализируемой пробе; разбавляют
водой до такого же объема и продолжают дальше титрование, как при анализе
пробы. Разделив суммарное количество израсходованного раствора соли цинка (на
приготовление исходного раствора и на обратное титрование) на число миллилитров
добавленного раствора гексацианоферрата (II) калия, получают требуемый коэффициент (К).
Обработка измерений. Содержание цинка (x) в мг/л вычисляют по формуле:
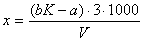
где
а - объем раствора соли цинка, израсходованного на обратное титрование при
анализе пробы, мл; b - объем добавленного раствора гексацианоферрата (II)
калия, мл; K - объем раствора соли цинка, соответствующий 1 мл
раствора гексацианоферрата (II) калия, мл; V - объем анализируемой сточной
воды, мл; 3 - количество цинка, содержащегося в 1 мл раствора сульфата цинка,
мг.
Контрольные
вопросы
1. В чем заключается сущность титриметрического определения цинка?
2. В виде каких ионов цинк может присутствовать в промышленных
сточных водах?
. Почему перед началом анализа необходимо разрушить комплексные
соединения цинка в исследуемой пробе?
. Какая химическая реакция положена в основу методики определения
цинка?
. Какой индикатор применяют в этом методе?
. Каким образом устраняют мешающее влияние Fe в исследуемой пробе?
. Почему в данной методике используется обратное титрование?
Лабораторная работа №5
Метод определения хлоридов
Аргентометрический метод объемного анализа основан на применении в
качестве осадителя стандартного раствора, содержащего ионы серебра AgNO3.
Химический процесс. Определение ионов хлора в растворимых хлоридах основано на
прямом титровании навески анализируемого вещества или его раствора стандартным
раствором азотнокислого серебра в присутствии индикатора - хромата калия:
Ag+ + Cl- → AgCl
Приготовление
растворов
1. Раствор азотнокислого серебра 0,05 моль/дм3.
,4937 г азотнокислого серебра, высушенного при температуре 105ºС, растворяют в 100-200 см3
дистиллированной воды, количественно переносят в мерную колбу на 1дм3
и доводят объем раствора до метки. Если получается мутный раствор, ему дают
отстояться в течение нескольких дней и сифонируют. Раствор хранят в темной
склянке в защищенном от света месте.
Определение точной концентрации азотнокислого серебра
В коническую колбу на 250 см3 отбирают 10 см3
раствора хлористого натрия точной концентрации, приливают 40 см3
дистиллированной воды, 0,5 см3 раствора хромовокислого калия и
титруют раствором азотнокислого серебра. За объем, пошедший на титрование,
принимают среднюю арифметическую величину двух параллельных определений, за
вычетом объема, пошедшего на титрование нулевой пробы.
Нормальность титрованного раствора азотнокислого серебра (N) равна
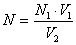
где:
N1 - нормальность раствора хлористого натрия
V1 - объем
раствора хлористого натрия, взятого на титрование, см3
V2 - объем
раствора азотнокислого серебра, пошедшего на титрование, за вычетом результата
нулевой пробы, см3
2. Раствор хромовокислого калия 10%.
100 г хромовокислого калия растворяют в небольшом количестве дистил.
воды, затем для удаления хлоридов добавляют пипеткой по каплям раствор
азотнокислого серебра до начала образования красно-бурого осадка, через 1-2 дня
раствор фильтруют через фильтр «Белая лента» в мерную колбу на 1 дм3
и доводят объем фильтрата дистил. водой до метки.
. Раствор хлористого натрия или хлористого калия 0,05 моль/дм3.
,9221 г хлористого натрия или 3,7277 г хлористого калия, предварительно
высушенного до постоянного веса при 180ºС, растворяют в дистил. воде и
доводят объем раствора в мерной колбе на 1 дм3 до метки.
. Раствор фенолфталеина 0,1%.
,1 г фенолфталеина растворяют в 50 см3 этилового спирта (96%).
Полученный раствор количественно переносят в мерную колбу на 100 см3
и доводят объем раствора до метки дистил. водой
. Раствор гидроокиси натрия.
,0 г гидроокиси натрия растворяют в дистил. воде, не содержащей
углекислоты, объем доводят до 1дм3. раствор хранят в полиэтиленовой
посуде.
. Раствор азотной кислоты.
,5 см3 конц. азотной кислоты приливают пипеткой к 500 см3
дистил. воды, перемешивают и доводят объем до 1 дм3
. Суспензия гидроокиси алюминия.
г квасцов алюмокалиевых или алюмоаммонийных растворяют в 1дм3
дистил. воды, нагревают до 60ºС и постепенно прибавляют 55 см3
концентрированного раствора аммиака при постоянном перемешивании. После
отстаивания в течении 1 часа осадок переносят в стакан, промывают декантацией
дистил. водой до исчезновения свободного аммиака, хлоридов, нитритов и нитратов
Отбор и
консервирование проб
8. Объем пробы воды для определения содержания хлоридов должен быть
не менее 250 см3.
9. Пробы воды, предназначенные для определения хлоридов, не
консервируются, хранят при комнатной температуре. Мутные пробы перед анализом
фильтруют.
Мешающие влияния.
Данным методом, кроме хлоридов, определяются находящиеся в воде ионы
других галогенов (бромиды, иодиды), присутствие которых в обычных условиях не
предполагается. На результаты определения оказывают влияние окрашенные
вещества, кислотность (рН меньше 6,5), присутствие сероводорода и
гидросульфитов.
Окраску прозрачных проб устраняют пропусканием через колонку, наполненную
активированным углем. Первые 150 см3 воды отбрасывают. Оставшуюся
воду используют для определения хлоридов.
При наличии мути и окраски, пробу осветляют суспензией гидроокиси
алюминия (3 см3 на 100 см3 пробы), смесь встряхивают и
фильтруют через фильтр «Белая лента», промывая осадок дистил. водой. Хлориды
определяют во всем объеме фильтрата.
При кислой реакции воды пробу нейтрализуют по фенолфталеину. Для этого
берут 50 см3 пробы, приливают пипеткой 2-3 капли раствора
фенолфталеина (0,1%) и нейтрализуют раствором гидроокиси натрия (0,1 моль/дм3)
до появления розового окрашивания. По объему щелочного раствора, пошедшего на
нейтрализацию 50 см3 пробы, рассчитывают количество раствора
гидроокиси натрия, необходимого для нейтрализации требуемого объема пробы.
При наличии сероводорода и гидросульфитов воду следует подкислить
раствором азотной кислоты (0,1 моль/дм3) по лакмусовой бумаге, и
пропускать в течении нескольких минут ток воздуха до полного удаления
сероводорода.
Обработка измерений
1. Качественное определение хлоридов:
К 5 см3 исследуемой воды пипеткой добавляют 3 капли раствора
азотнокислого серебра с массовой долей 10% и определяют концентрацию хлоридов в
зависимости от интенсивности помутнения
|
Интенсивность помутнения
|
Концентрация хлоридов в
пробе, мг/дм3
|
|
Опалесценция, слабая муть
|
1-10
|
|
Сильная муть
|
10-50
|
|
Хлопья, оседающие не сразу
|
50-100
|
|
Белый объемный осадок
|
свыше 100
|
В зависимости от предполагаемой концентрации хлоридов выбирается объем
пробы для титрования:
|
Концентрация хлоридов в
пробе, мг/дм3
|
Объем пробы для
титрования, см3
|
|
менее 100
|
100
|
|
100-400
|
50
|
|
свыше 400
|
менее 50
|
|
|
2. Количественное определение хлоридов
в коническую колбу на 250 см3 отмеривают пипеткой необходимый
объем исследуемой пробы, добавляют раствор калия хромовокислого из расчета 1 см3
на 100 см3 пробы и титруют по каплям, при непрерывном перемешивании,
раствором азотнокислого серебра.
Конец титрования определяют по неисчезающей при перемешивании
светло-оранжевой окраске, появляющейся от одной капли (избыточной) раствора
азотнокислого серебра.
Параллельно проводят аналогичное определение в нулевой пробе, в качестве
которой используют дистиллированную воду, взятую в эквивалентном определяемой
пробе объеме.
Обработка
измерений
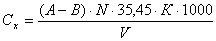
где:
А
- объем раствора азотнокислого серебра, ушедшего на титрование, см3;
В - объем
раствора азотнокислого серебра, ушедшего на титрование нулевой пробы, см3;
N -
нормальность раствора азотнокислого серебра;
К - поправочный коэффициент к титру раствора азотнокислого серебра;
V -
объем анализируемой воды, см3;
,45 - эквивалент хлорид иона;
- независимый
коэффициент пересчета, дм3.
Контрольные вопросы
1. В чем заключается сущность аргентометрического определения хлоридов?
2. Какая химическая реакция положена в основу методики определения
хлоридов?
. Какие мешающие влияния могут быть в исследуемой пробе?
. Как определяют нормальность титрованного раствора?
. Почему по этой методике нужно проводить предварительно
качественный анализ на хлор-ионы?
. Какие еще ионы можно определять данным методом?
Лабораторная работа №6
Метод определения нефтепродуктов
I. Фотоколориметрический способ определения нефтепродуктов
Настоящая методика распространяется на производственные воды тепловой
электростанции и устанавливает методы определения нефтепродуктов в питательной
воде паровых котлов, в конденсатах, возвращаемых от потребителей пара, в
теплофикационных водах.
Сущность метода состоит в образовании окрашенной в коричнево-бурый цвет
жидкости, полученной при взаимодействии примесей нефтепродуктов с серной
кислотой.
Примечание. При отборе проб производственных вод для определения
нефтепродуктов, необходимо учитывать их способность адсорбироваться стенками
сосуда, а также всплывать при значительном содержании или переходить частично в
осадок. Пробу следует отбирать в чистую склянку, по возможности освобожденную
от остатков дистиллированной воды, использованной при промывании склянки.
Чувствительность метода - 0,05 мг нефтепродуктов в пробе.
1. Приготовление основного и стандартного растворов.
Основной раствор концентрации 10 г/кг нефтепродуктов. Если известно,
какими нефтепродуктами могут загрязняться анализируемые воды, то, отвесив 1 г
этих нефтепродуктов в сухую мерную колбу вместимостью 100 см3,
растворяют в гексане (экстрагент), доводят объем раствора этим же растворителем
до метки и хорошо перемешивают. Приготовленный раствор устойчив при его
хранении в хорошо закупоренной колбе.
Стандартный раствор концентрации 0,1 г/кг нефтепродуктов. В мерную колбу
вместимостью 250 см3 отмеривают пипеткой 2,5 см3
основного раствора, доливают принятым экстрагентом до метки и хорошо
перемешивают. Раствор устойчив при хранении его в хорошо закупоренной колбе.
Все отобранное количество воды дважды обрабатывают гексаном. Для этого в
делительную воронку вместимостью 1000 см3 вливают 700 - 800 см3
отобранной воды и 50 см3 гексана. Хорошо взбалтывают, дают
расслоиться , верхний слой гексана сливают в запасную емкость, а воду сливают в
другую делительную воронку такой же вместимостью, в которой повторяют
экстракцию, использую уже 20 см3 экстрагена. После этого воду
отбрасывают и проводят такую же операцию со следующей порцией воды, пока не
отбрасывают все ее количество. Все экстракты сливают во взвешенный и
предварительно высушенный стакан вместимостью 40 - 50 см3, удаляют
экстрагент испарением на водяной бане, остаток высушивают в сушильном шкафу при
60 - 70 0С и взвешивают. После взвешивания все количество
нефтепродуктов растворяют в мерной колбе вместимостью 50 см3 в
принятом растворе, тщательно перемешивают и пользуются в качестве стандартного
раствора. Концентрация этого стандартного раствора определяется массой
выделенных нефтепродуктов.
2. Построение градуированного графика.
В шесть сухих стаканов вместимостью 100 см3 помещают 1, 2, 5,
7, 10 и 15 см3 стандартного раствора нефтепродуктов. На кипящей
водяной бане выпаривают экстрагент, гексан до исчезновения их запаха, затем в
каждый стакан вливают по 10 см3 концентрированной серной кислоты,
тщательно обмывают ею стенки стакана, куда могли попасть капли раствора, и
ставят стаканы на 5 мин в кипящую водяную баню. После этого дают жидкостям
охладиться до комнатной температуры и определяют оптическую плотность (А)
окрашенных растворов на фотоколориметре при светофильтрах областью
светопропускания 440 нм в кюветах с толщиной поглощающего свет слоя 20 мм.
Сравнение проводят относительно 10 см3 концентрированной серной
кислоты, прогретой на водяной бане. По полученным точкам строят градуированный
график, откладывая по оси абсцисс содержание нефтепродуктов в каждом стакане, а
по оси ординат - соответствующие им величины оптической плотности. Если
стандартный раствор приготовлен из навески извлеченных нефтепродуктов, то
учитывают истинную их концентрацию в нем. По методу наименьших квадратов строят
прямую, которой пользуются для вычисления результатов определений. Выполняют не
менее 3 параллельных определений для каждой концентрации. Допускается вычисление
результатов с помощью множителя (К), определяемого по формуле:
m
К = ,
А
где m - масса нефтепродуктов в пробе, мг; А - среднее значение оптической
плотности, соответствующие этой массе.
Проведение анализа.
Отобранный объем анализируемой воды помещают в делительную воронку,
дважды обмывают стенки сосуда, в котором была отобрана проба, гексаном,
используя его по 10 - 15 см3 и сливая в туже воронку. Затем, закрыв
воронку пробкой, сильно взбалтывают ее содержимое, выпуская излишки воздуха
через кран, повернув воронку краном кверху. Взбалтывание проводят 2 - 3 мин,
после чего ставят воронку в штатив и дают жидкости полностью расслоиться. Затем
верхний слой гексана осторожно сливают на сухой бумажный фильтр, полученный
фильтрат собирают в чистый сухой стакан, промывают фильтр гексаном (5 - 10 см3),
собирая промывные растворы в тот же стакан. Для устранения влияния органических
примесей, способных растворяться в гексане, может быть применено фильтрование
экстракта через слой (7 - 10 см) сорбента (активированной окиси алюминия),
загруженного в стеклянную трубку длинной 10 см диаметром 1 см с впаянной в нее
дырчатой стеклянной пластинкой. Верхний конец трубки расширен в виде воронки,
нижний оттянут до диаметра 1 мм. В трубку на стеклянную пластинку помещают слой
стеклянной ваты толщиной 1 см, а сверх него насыпают сорбент толщиной 3 - 5 см.
Сорбент - активированную окись алюминия - прокаливают при 800 0С в
течение 3 - 4 ч, а стеклянную вату прогревают при 250 - 300 0С в
течение 2 - 3 ч. Собранная трубка, т. е. колонка для фильтрования, должна быть
промыта экстрагентом.
Фильтрование через колонку экстракта, полученного извлечением из воды
нефтепродуктов, проводят со скоростью 0,5 - 1,0 см3 в мин, на
фильтрование всей порции экстракта расходуется обычно не более 20 мин. Фильтрат
собирают в чистый сухой стакан; в него же собирают и промывную жидкость, т. е.
5 - 10 см3 экстрагента., пропускаемые через сорбент после окончания
фильтрования экстракта. Фильтрат выпаривают на водяной бане, к оставшимся в
стакане нефтепродуктам приливают 10 см3 концентрированной серной
кислоты и в течение 5 мин нагревают жидкость на кипящей водяной бане. При этом
стремятся смочить серной кислотой все части внутренней поверхности стакана,
куда могли попасть капли экстракта.
Жидкости дают остыть до комнатной температуры и определяют оптическую
плотность ее (Ах) на фотоколориметре со светофильтром областью
светоиспускания 440 нм в кюветах с толщиной поглощающего свет слоя 20 мм. В
качестве раствора сравнения используют 10 см3 концентрированной серной
кислоты, также подвергнутой нагреванию и последующему охлаждению.
При отборе проб производственных вод для определения нефтепродуктов
необходимо учитывать их способность абсорбироваться стенками сосуда, а так же
всплывать при значительном содержанием или переходить частично в осадок.
Обработка результатов.
По градуировочному графику находят массу нефтепродуктов в анализируемой
пробе. Полученную оптическую плотность находят по оси ординат и по ней массу
нефтепродуктов (m) в пробе мг.
Концентрацию нефтепродуктов (С), в мг на кг, вычисляют по формуле:
m
С =
V
где V - объем воды, использованный для определения, см3; m -
масса нефтепродуктов, найденная по градуировочному графику, мг; или по формуле
m = к · Ак. Результат определений округляют до сотых долей.
II.Гравиметрический метод определения нефтепродуктов
Настоящая методика распространяется на производственные сточные воды с
большим содержанием нефтепродуктов.
Сущность метода состоит в излечении нефтепродуктов экстрагентом, который
затем удаляют испарением, остаток сушат и взвешивают. Чувствительность метода 1
мг в пробе.
Проведение анализа.
Весь объем отобранной пробы переливают в делительную воронку
соответствующей вместимости, споласкивают дважды сосуд, в который была отобрана
проба, экстрагентом, используя каждый раз 10 - 15 см3 его и сливая
эти порции, в ту же делительную воронку. Затем сильно встряхивают содержимое
делительной воронки 2 - 3 мин, выпуская воздух через кран при перевернутой
воронке краном вверх. Закончив взбалтывание, ставят воронку в штатив и дают
жидкости расслоиться, экстракт осторожно сливают на сухой беззольный фильтр,
собирая фильтрат в чистый сухой взвешенный бюкс.
Фильтр промывают чистым экстрагентом, используя его 10 - 15 см3
и собирая промывную жидкость в тот же бюкс. Затем ставят бюкс с экстрактом на
кипящую водяную баню и выпаривают экстрагент. Бюкс с нефтепродуктами
выдерживают в сушильном шкафу при 60 - 70 0С в течение 2 ч, после
чего охлаждают его с открытой крышкой в эксикаторе над прокаленным хлористым
кальцием и, закрыв его крышкой, охлаждают.
Анализы с применением экстрагентов необходимо выполнять в вытяжном шкафу.
При оперировании гексаном нельзя пользоваться включенными нагревательными
плитками с открытой спиралью.
Обработка результатов.
Концентрацию нефтепродуктов (С), в миллиграммах, мг/дм3,
вычисляют по формуле:
m
(С) = · 103
V
где V - объем пробы, использованный для определения, см3;
m - масса
высушенного осадка, мг.
Контрольные вопросы
1. В чем заключается сущность определения нефтепродуктов при использовании
серной кислоты?
2. Какой процесс положен в основу гравиметрического метода
определения нефтепродуктов?
. Какой экстрагент применяют в этом методе?
. Почему анализы с применением этого экстрагента необходимо
выполнять в вытяжном шкафу?
. Какие мешающие влияния могут быть в исследуемой пробе?
. Как определяют нормальность титрованного раствора?
. Почему по этой методике нужно проводить предварительно
качественный анализ на хлор-ионы?
. Какие еще ионы можно определять данным методом?
Лабораторная работа №7
Метод определения взвешенных веществ
Методика
гравиметрического определения нерастворимых в воде веществ (взвешенных
веществ). Настоящая методика распространяется на производственные стоки,
производственные воды и устанавливает гравиметрический метод определения
взвешенных веществ в исходной воде, известково-коагулированной, умягченной,
питательной, котловой и теплофикационной водах.
Сущность метода состоит в определении фильтрованием общего содержания
нерастворенных в воде частиц минерального и органического происхождения, с
последующим высушиванием и взвешиванием полученного осадка.
Чувствительность метода определения взвешенных веществ составляет 5
мкг/кг.
I. Метод с использованием бумажных фильтров или мембранных
фильтров (при определении взвешенных веществ массовой концентрации до 30 мг/дм3)/
Аппаратура
1. Весы аналитические.
2. Шкаф сушильный.
. Бюксы с крышками.
. Эксикатор
. Воронки лабораторные d=50-100мм.
. Стаканы химические на 500-700 см3.
. Фильтры ФОС по ГОСТ 12026-76.
. Колбы химические.
Отбор проб.
Пробу отбирают и хранят в стеклянных или полиэтиленовых бутылях.
|
Предполагаемое содержание
взвешенных веществ, мг/кг
|
менее 10
|
0 - 50
|
0 - 100
|
100 - 500
|
более 500
|
|
Объем пробы, дм3
|
1,5
|
1,0
|
0,5
|
0,25
|
0,1
|
Перед отбором порции воды для анализа, пробу следует тщательно взболтать
для равномерного распределения взвешенных веществ по всей жидкости и быстро, не
давая осесть взвеси, отбирать необходимый объем воды.
При подготовке можно использовать метод фильтрования через мембранные
фильтры.
Проведение анализа
1. Отобранный для анализа объем жидкости фильтруют через бумажный фильтр.
Первые порции фильтрата возвращают в фильтруемую пробу, так как в них могут
содержаться волокна фильтра. При фильтровании весь осадок должен быть перенесен
на фильтр. Для этого используют порции фильтрата. После окончания фильтрования
влажный фильтр с осадком помещают в тот же бюкс и вновь высушивают до
постоянного веса в сушильном шкафу при температуре 105-1100С.
Повторяют высушивание до получения веса, отличающегося от предыдущего не более,
чем на 0,01 г. После последнего взвешивания фильтр с осадком осторожно
переносят во взвешенный фарфоровый тигель, в котором тигель озоляют, а остаток
прокаливают при температуре 800-8500С до полного сгорания углистых
частиц. После охлаждения в эксикаторе, тигель с осадком взвешивают.
2. Подготовленный мембранный фильтр, высушенный в сушильном шкафу при
температуре 1050С до постоянного веса, взвешивают на аналитических весах
и помеченный карандашом накладывают матовой стороной вверх на прокладку из
фильтровальной бумаги, диаметр которой равен диаметру нижней поверхности
аппарата для фильтрования. Фильтр с прокладкой помещают в нижнюю часть
аппарата, смачивают дистиллированной водой и включают отсос. Плотно укрепляют
фильтр, прикручивая шайбой верхнюю часть аппарата.
В зависимости от количества взвешенных веществ, пробу объемом 100-500 см3
фильтруют, применяя вакуум. Частички, приставшие к стенкам аппарата Зейтца,
смывают дистиллированной водой на мембранный фильтр. По окончании фильтрования
фильтр с осадком, не снимая с прокладки, подсушивают на воздухе, затем сушат
при t= 1050С в сушильном шкафу
до постоянного веса, охлаждают в эксикаторе и взвешивают.
Фильтрование через мембранные фильтры невозможно для маслянистых или
жирных веществ.
Обработка результатов.
Общее содержание взвешенных веществ (Хобщ.), в мг/кг вычисляют
по формуле
( а - б )
Хобщ =
· 106 , где
V
а - масса бюкса с фильтром или мембранного фильтра с осадком взвешенных
веществ после высушивания, г;
б - масса бюкса с сухим фильтром или вес мембранного фильтра, г;
V -
объем пробы, взятой для определения, см3.
Концентрацию прокаленных взвешенных веществ (Х прок), в мг/кг
вычисляют по формуле
А - Б
Х прок =
· 106 , где
V
А - масса тигеля с прокаленным осадком, г;
Б - масса пустого тигеля, г;
V -
объем пробы, взятой для определения, см3.
II. Метод с использованием бумажных фильтров (при определении
взвешенных веществ массовой концентрации от 30 мг/дм3 и более)
Сущность
метода. Нерастворимые
в воде вещества отделяют путем фильтрования исследуемого раствора, промывают
дистиллированной водой и определяют их массу.
Подготовка
к анализу
Два беззольных фильтра (белая лента), свернутых в форме воронки,
предварительно помещают в бюксы и высушивают с открытыми крышками в течении
1,5-2,0 ч при 105ºС. Затем охлаждают бюксы в эксикаторе с закрытыми
крышками до температуры 20-25ºС, взвешивают на аналитических весах
с точностью до третьего десятичного знака.
Операцию высушивания в сушильном шкафу, охлаждение в эксикаторе и
взвешивание повторяют не менее трех раз до достижения постоянной массы бюкса с
фильтрами.
Выполнение измерений
Объем исследуемого раствора, содержащий 20-1000 мг нерастворимых в воде веществ,
помещают в стакан и фильтруют декантацией через двойной фильтр, доведенный до
постоянной массы, как указано выше.
Первые две-три порции фильтрата в объеме 50-100 см3 повторно
фильтруют через тот же двойной фильтр. Осадок на фильтре трижды промывают
горячей дистиллированной водой, затем фильтр с осадком помещают в стаканчик для
взвешивания (бюкс) и высушивают с открытыми крышками в течении 1,5-2,0 ч при
105ºС. Затем охлаждают бюксы в эксикаторе
с закрытыми крышками до температуры 20-25ºС, взвешивают на аналитических весах
с точностью до третьего десятичного знака.
Операцию высушивания в сушильном шкафу, охлаждение в эксикаторе и
взвешивание повторяют не менее трех раз до достижения постоянной массы бюкса с
фильтрами
Обработка
измерений
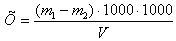
где:
Х
- массовая концентрация взвешенных веществ в мг/дм3;
m1 - масса бюкса с фильтром и высушенным осадком, мг;
m2 - масса бюкса с чистым фильтром, мг;
V - объем пробы, взятой для анализа, см3.
Контрольные
вопросы
1. В чем заключается сущность гравиметрического метода определения
взвешенных веществ?
2. Почему при отборе пробы на определение взвешенных веществ воде
могут быть взяты различные по объему пробы?
. Какое мешающее влияние ограничивает область применения
мембранных фильтров?
. Какие способы фильтрования растворов Вы знаете?
Учебно-методическая литература
1. Алексеев В.Н. Курс качественного химического
полумикроанализа М., Химия, 1988-584 с
. Крешков А.П. Основы аналитической химии (в 4-х томах). М.,
Химия, 1970 - 1928 с
. Лурье Ю.Ю., Рыбникова А.И. Химический анализ
производственных сточных вод. М.,Химия, 1974.-336 с.
. Методика технологического контроля работы очистных
сооружений городской канализации.// М-во жил.-коммун. хоз-ва. М., Стройиздат,
1977. - 299 с.
5. Вредные вещества в промышленности: в 3-х т. справочник для
химиков, инженеров и врачей/ под общ. Ред. Н.В.Лазарева, Э.Н.Левиной.- изд. 7-е
перераб. и доп.-Л: Химия 1,2,3 том. 1976 - 1770 с.