Дисперсные системы
ПЛАН:
1. Ведение…………………………………………………………………..2
2. Основные
типы дисперсных систем…………………………………...2
3. Образование
дисперсных систем………………………………………4
4. Устойчивость
дисперсных систем..........................................................5
5. Классификации
дисперсных систем…………………………………...8
6. Структурообразование
в дисперсных системах и в растворах полимеров……………………………………………………………….16
7. Свойства
дисперсных систем и определение размера частиц……….23
8. Список
использованной литературы. …………………………………24
ВВЕДЕНИЕ
ДИСПЕРСНЫЕ СИСТЕМЫ
- гетерогенные системы из двух или большего числа фаз с сильно развитой
поверхностью раздела между ними. Обычно одна из фаз образует непрерывную
дисперсионную среду, в объеме которой распределена дисперсная фаза (или неск.
дисперсных фаз) в виде мелких кристаллов, твердых аморфных частиц, капель или
пузырьков. Д. с. могут иметь и более сложное строение, напр., представлять
собой двухфазное образование, каждая из фаз которого, будучи непрерывной,
проникает в объем др. фазы. К таким системам относятся твердые тела,
пронизанные разветвленной системой каналов-пор, заполненных газом или
жидкостью, некоторые микрогетерогенные полимерные композиции и др. Нередки
случаи, когда дисперсионная среда "вырождается" до тончайших слоев
(пленок), разделяющих частицы дисперсной фазы.
Основные
типы дисперсных систем.
По дисперсности, т. е. размеру частиц дисперсной
фазы или отношению общей площади межфазной поверхности к объему (или массе)
дисперсной фазы (уд. поверхности), Д. с. условно делят на грубодисперсные и
тонко(высоко)дисперсные. Последние, по традиции, наз. коллоидно-дисперсными или
просто коллоидными системами. В грубодисперсных системах частицы имеют размеры
от 1 мкм и выше (уд. поверхность не более 1 м2/г), в коллоидных - от 1 нм до 1
мкм (уд. поверхность достигает сотен м2/г). Дисперсность оценивают по
усредненному показателю (среднему размеру частиц, уд. поверхности) или
дисперсному составу (см. Дисперсионный анализ). Тонкопористые тела
характеризуют пористостью -понятием, аналогичным дисперсности. В
свободнодисперсных системах сцепление между частицами дисперсной фазы
отсутствует, каждая частица кинетически независима и при достаточно малых
размерах участвует в интенсивном броуновском движении. Для структурированных
(связнодисперсных) систем характерно наличие неупорядоченной пространств. сетки
(каркаса), образованной частицами дисперсной фазы (см. Структурообразование в
дисперсных системах). Особую группу составляют высококонцентрированные Д. с., в
которых частицы находятся в "стесненных" условиях как, напр., в
периодич. коллоидных структурах. Мех. св-ва свободнодисперсных систем
определяются гл. обр. св-вами дисперсионной среды, а связнодисперсных систем -
также св-вами и числом контактов между частицами дисперсной фазы (см.
Реология). По агрегатному состоянию дисперсионной среды и дисперсной фазы
выделяют след. осн. виды Д. с.: 1) аэродисперсные (газодисперсные) системы с
газовой дисперсионной средой: аэрозоли (дымы, пыли, туманы), порошки,
волокнистые материалы типа войлока. 2) Системы с жидкой дисперсионной средой;
дисперсная фаза м. б. твердой (грубодисперсные суспензии и пасты,
высокодисперсные золи и гели), жидкой (грубодисперсные эмульсии,
высокодисперсные микроэмульсии и латексы) или газовой (грубодисперсные газовые
эмульсии и пены). 3) Системы с твердой дисперсионной средой: стеклообразные или
кристаллич. тела с включениями мелких твердых частиц, капель жидкости или
пузырьков газа, напр., рубиновые стекла, минералы типа опала, разнообразные
микропористые материалы. Отдельные группы Д. с. составляют мн. металлич.
сплавы, горные породы, сложные композиционные и др. многофазные системы.
Лиофильные и лиофобные Д. с. с жидкой дисперсионной средой различаются в
зависимости от того, насколько близки или различны по своим св-вам дисперсная
фаза и дисперсионная среда (см. Лиофильность и лиофобность). В лиофильных Д. с.
межмолекулярные взаимод. по обе стороны разделяющей фазы пов-сти различаются
незначительно, поэтому уд. своб. поверхностная энергия (для жидкости -
поверхностное натяжение) чрезвычайно мала (обычно сотые доли мДж/м2), межфазная
граница (поверхностный слой) м. б. размыта и по толщине нередко соизмерима с
размером частиц дисперсной фазы. Лиофильные Д. с. термодинамически равновесны,
они всегда высокодисперсны, образуются самопроизвольно и при сохранении условий
их возникновения могут существовать сколь угодно долго. Типичные лиофильные Д.
с. - микроэмульсии, нек-рые полимер-полимерные смеси, мицеллярные системы ПАВ,
Д. с. с жидкокристаллич. дисперсными фазами. К лиофильным Д. с. часто относят
также набухающие и самопроизвольно диспергирующиеся в водной среде минералы
группы монтмориллонита, напр., бентонитовые глины. Следует отметить, что в
прошлом "лиофильными коллоидами" наз. р-ры полимеров, т. е.
принципиально гомог. системы. Однако в совр. терминологии понятие
"коллоид" относится только к микрогетерогенным системам; по отношению
к гомогенным (однофазным) системам его не употребляют. В лиофобных Д. с.
межмолекулярное взаимод. в дисперсионной среде и в дисперсной фазе существенно
различно; уд. своб. поверхностная энергия (поверхностное натяжение) велика - от
неск. единиц до неск. сотен (и тысяч) мДж/м2; граница фаз выражена достаточно
четко. Лиофобные Д. с. термодинамически неравновесны; большой избыток своб.
поверхностной энергии обусловливает протекание в них процессов перехода в более
энергетически выгодное состояние. В изотермич. условиях возможна коагуляция
-сближение и объединение частиц, сохраняющих первоначальные форму и размеры, в
плотные агрегаты, а также укрупнение первичных частиц вследствие коалесценции
-слияния капель или пузырьков газа, собирательной рекристаллизации (в случае
кристаллич. дисперсной фазы) или изотермич. перегонки (мол. переноса) в-ва
дисперсной фазы от мелких частиц к крупным (в случае Д. с. с жидкой дисперсионной
средой - последний процесс наз. переконденсацией). Нестабилизованные и,
следовательно, неустойчивые лиофобные Д. с. непрерывно изменяют свой дисперсный
состав в сторону укрупнения частиц вплоть до полного расслоения на макрофазы.
Однако стабилизованные лиофобные Д. с. могут сохранять дисперсность в течение
длит. времени.
Образование
дисперсных систем.
Возможно двумя путями: диспергационным и
конденсационным. Диспергирование макрофаз с образованием лиофильных Д. с.
происходит самопроизвольно - для этого достаточно энергии теплового движения.
Такой процесс осуществляется при значениях поверхностного натяжения s ниже
нек-рого критич. значения sкр = bkТ/d2, где d - размер частиц дисперсной фазы,
Т - абс. т-ра, k - постоянная Больцмана, b - безразмерный коэф., принимающий
значения примерно 10-30. Образование лиофобных Д. с. путем диспергирования
стабильной макрофазы требует значительных энергетич. затрат, определяемых
суммарной площадью пов-сти частиц дисперсной фазы. В реальных условиях на
образование пов-сти при измельчении твердых тел или при распылении и
эмульгировании жидкостей приходится лишь небольшая часть (доли процента)
подводимой к системе энергии; остальное расходуется на побочные процессы и
рассеивается в окружающем пространстве (см. Диспергирование). Конденсационный
путь образования Д. с. связан с зарождением новой фазы (или новых фаз) в
пересыщенной метастабильной исходной фазе - будущей дисперсионной среде. Для
возникновения высокодисперсной системы необходимо, чтобы число зародышей новой
фазы было достаточно большим, а скорость их роста не слишком велика. Кроме
того, требуется наличие факторов, ограничивающих возможности чрезмерного
разрастания и сцепления частиц дисперсной фазы. Переход первоначально
стабильной гомог. системы в метастабилъное состояние может произойти в
результате изменения термодинамич. параметров состояния (давления, т-ры,
состава). Так образуются, напр., природные и искусственные аэрозоли (туман - из
переохлажденных водяных паров, дымы - из парогазовых смесей, выделяемых при
неполном сгорании топлива), нек-рые полимерные системы - из р-ров при ухудшении
"термодинамич. качества" р-рителя, органозоли металлов путем
конденсации паров металла совместно с парами орг. жидкости или при пропускании
первых через слой орг. жидкости, коллоидно-дисперсные поликристаллич. тела
(металлич. сплавы, нек-рые виды горных пород и искусств. неорг. материалов).
Возможно также образование Д. с. в результате хим. р-ции в гомог. среде, если
продукт р-ции при данных условиях находится в агрегатном состоянии, отличном от
"материнской" фазы, или практически не растворяется в ней. Примерами
подобных систем могут служить аэрозоли с твердыми частицами NH4Cl (образуются
при взаимод. газообразных NH3 и НСl), аэрозоли с капелъно-жидкими частицами H2SO4
(при взаимод. SO3 и водяного пара). В природе и технол. процессах часто
образуются гидрозоли разного состава при гидролизе солей и др. соед.,
неустойчивых к действию воды. Окислит.-восстановит. р-ции используют для
получения золей Аu и Ag, разложение Na2S2O3 разб. серной или соляной к-той -
для получения гидрозоля элементарной серы. Хим. или термохим. разложения
карбонатов, орг. порофоров (порообразователей, вспенивающих агентов) и др.
соед. с выделением газообразных в-в в первоначально жидких средах лежит в основе
пром. произ-ва мн. пеноматериалов.
Устойчивость
дисперсных систем.
Устойчивость дисперсных систем характеризуется
постоянством дисперсности (распределения частиц по размерам) и концентрации
дисперсной фазы (числом частиц в единице объема). Наиб. сложна в теоретич.
аспекте и важна в практич. отношении проблема устойчивости аэрозолей и жидких
лиофобных Д. с. Различают седиментационную устойчивость и устойчивость к
коагуляции (агрегативную устойчивость). Седиментационно устойчивы коллоидные
системы с газовой и жидкой дисперсионной средой, в к-рых броуновское движение
частиц препятствует оседанию; грубодисперсные системы с одинаковой плотностью
составляющих их фаз; системы, скоростью седиментации в к-рых можно пренебречь
из-за высокой вязкости среды. В агрегативно устойчивых Д. с. непосредств.
контакты между частицами не возникают, частицы сохраняют свою индивидуальность.
При нарушении агрегативной устойчивости Д. с. частицы, сближаясь в процессе
броуновского движения, соединяются необратимо или скорость агрегации становится
значительно больше скорости дезагрегации. Между твердыми частицами возникают
непосредственные точечные ("атомные") контакты, к-рые затем могут
превратиться в фазовые (когезионные) контакты, а соприкосновение капель и
пузырьков сопровождается их коалесценцией и быстрым сокращением суммарной
площади межфазной пов-сти. Для таких систем потеря агрегативной устойчивости
означает также потерю седимeнтационной устойчивости. В агрегативно устойчивых
системах дисперсный состав может изменяться вследствие изотермич. перегонки -
мол. переноса в-ва дисперсной фазы от мелких частиц к более крупным. Этот
процесс обусловлен зависимостью давления насыщенного пара (или концентрации
насыщенного р-ра) от кривизны пов-сти раздела фаз (см. Капиллярные явления). Агрегативная
устойчивость и длительное существование лиофобных Д. с. с сохранением их св-в
обеспечивается стабилизацией. Для высокодисперсных систем с жидкой
дисперсионной средой используют введение в-в - стабилизаторов (электролитов,
ПАВ, полимеров). В теории устойчивости Дерягина-Ландау-Фервея-Овербека (теории
ДЛФО) осн. роль отводится ионно-электростатич. фактору стабилизации.
Стабилизация обеспечивается электростатич. отталкиванием диффузных частей
двойного электрич. слоя, к-рый образуется при адсорбции ионов электролита на
пов-сти частиц. При нек-ром расстоянии между частицами отталкивание диффузных
слоев обусловливает наличие минимума на потенц. кривой (дальний, или вторичный,
минимум; см. рис.). Хотя этот минимум относительно неглубок, он может препятствовать
дальнейшему сближению частиц, притягиваемых силами межмолекулярного
взаимодействия. Ближний, или первичный, минимум соответствует прочному
сцеплению частиц, при к-ром энергии теплового движения недостаточно для их
разъединения. Сближаясь на расстояние, отвечающее этому минимуму, частицы
объединяются в агрегаты, образование к-рых ведет к потере системой агрегативной
устойчивости. При этом устойчивость системы к коагуляции определяется высотой
энергетич. барьера.
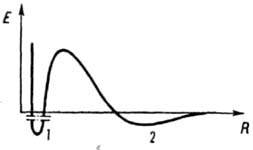
Зависимость энергии взаимодействия Е между частицами
от расстояния R: 1 и 2 - ближний и дальний минимумы соответственно.
При введении в Д. с. в качестве стабилизатора ПАВ
фактором стабилизации м. б. "термодинамич. упругость" пленок среды,
разделяющей частицы. Стабилизация обеспечивается тем, что при сближении частиц,
напр., капель или газовых пузырей, происходит растяжение и утоньшение
разделяющей их прослойки, содержащей ПАВ, и, как следствие, нарушение адсорбц.
равновесия. Восстановление этого равновесия и приводит к повышению устойчивости
прослойки среды, разделяющей частицы. Гидродинамич. сопротивление вытеснению
жидкой дисперсионной среды из прослойки между сближающимися частицами - один из
кинетич. факторов стабилизации Д. с. Он особенно эффективен в системах с
высоковязкой дисперсионной средой, а при застекловывании последней делает
систему неограниченно устойчивой к агрегации частиц и коалесценции.
Структурно-мех. фактор стабилизации, по П. А. Ребиндеру, возникает при
образовании на межфазной границе полимолекулярных защитных слоев из мицеллообразующих
ПАВ, высокомолекулярных соед., а иногда и тонких сплошных или дискретных
фазовых пленок. Межфазный защитный слой должен обладать способностью
сопротивляться деформациям и разрушению, достаточной подвижностью для
"залечивания" возникших в нем дефектов и, что особенно важно, быть
лиофилизованным с внеш. стороны, обращенной в сторону дисперсионной среды. Если
защитный слой недостаточно лиофилен, он, предохраняя частицы от коалесценции,
не сможет предотвратить коагуляции. Структурно-мех. барьер является, по
существу, комплексным фактором стабилизации, к-рый включает термодинамич.,
кинетич. и структурные составляющие. Он универсален и способен обеспечить
высокую агрегативную устойчивость любых Д.с. с жидкой дисперсионной средой, в
т. ч. высококонцентрированных, наиб. важных в практич. отношении. Осн. св-ва Д.
с. определяются поверхностными явлениями: адсорбцией, образованием двойного
электрического слоя и обусловленных им электрокинетических явлений, контактными
взаимодействиями частиц дисперсной фазы. Размер частиц определяет оптич.
(светорассеяние и др.) и молекулярно-кинетич. св-ва (диффузия, термофорез,
осмос и др.). Д. с. повсеместно распространены в природе. Это - горные породы,
грунты, почвы, атм. и гидросферные осадки, растит. и животные ткани. Д. с.
широко используют в технол. процессах; в виде Д. с. выпускается большинство
пром. продуктов и предметов бытового потребления. Высокодисперсные техн.
материалы (наполненные пластики, дисперсноупрочненные композиц. материалы)
отличаются чрезвычайно большой прочностью. На высокоразвитых пов-стях
интенсивно протекают гетерог. и гетерог.-каталитич. хим. процессы. Учение о Д.
с. и поверхностных явлениях в них составляет сущность коллоидной химии.
Самостоят. раздел коллоидной химии - физико-химическая механика - изучаeт
закономерности структурообразования и мех. св-ва структурированных Д. с. и
материалов в их связи с физ.-хим. явлениями на межфазных границах.
Классификации
дисперсных систем.
По степени раздробленности (дисперсности) системы делятся
на следующие классы: грубодисперсные, размер частиц в которых более 10-5
м; тонкодисперсные (микрогетерогенные) с размером частиц от 10-5 до
10-7 м; коллоидно-дисперсные (ультрамикро-гетерогенные) с частицами
размером от 10-7 до 10-9м. Если фиксировать внимание на
двух основных компонентах дисперсных систем, то одному из
них следует приписать роль дисперсионной среды, а другому - роль дисперсной
фазы. В этом случае все дисперсные системы можно
классифицировать по агрегатным состояниям фаз.
Эта классификация была предложена
Оствальдом и широко используется до настоящего времени. Недостатком классификации следует считать
невозможность отнесения дисперсных систем, приготовленных
с твердой или жидкой дисперсной фазой, к какому-либо классу, если размер частиц
составляет несколько нанометров. Пример такой классификации приведен в табл.
1.
Академик П.А. Ребиндер предложил более совершенную классификацию дисперсных систем по агрегатным
состояниям фаз. Он разделил все дисперсные системы на два класса:
свободнодисперсные системы и сплошные (или связнодисперсные) системы (табл. 2 и
3). В свободнодисперсных системах дисперсная фаза не
образует сплошных жестких структур (сеток, ферм или каркасов). Эти системы
называют золями. В сплошных
(связнодисперсных) системах частицы дисперсной фазы образуют жесткие
пространственные структуры (сетки, каркасы, фермы). Такие системы оказывают
сопротивление деформации сдвига. Их
называют гелями.
Дисперсная система по классификации Ребиндера
обозначается дробью, в которой дисперсная фаза ставится в числителе, а
дисперсионная среда – в знаменателе. Например: Т1/Ж2.
Индекс 1 обозначает дисперсную фазу, а индекс 2 – дисперсионную среду.
Коллоидная химия изучает
свойства как тонко-, так и грубодисперсных систем; как свободно-, так и
связнодисперсных систем.
Включение в одну науку столь большого количества
разнообразных систем, различных как по природе фаз, так и по размерам частиц и
агрегатному состоянию фаз, основано на том, что все они обладают общими
свойствами - гетерогенностью и принципиальной термодинамической
неустойчивостью. Центральное место в коллоидной химии занимают
ультрамикрогетерогенные системы со свободными частицами. Это - так называемые, коллоидные системы.
Таблица 1
Классификация дисперсных систем по агрегатным
состояниям фаз.
|
Дисперсион-ная среда
|
Дисперс-ная фаза
|
Примеры дисперсных систем
|
|
Твердая
|
Твердая
|
Рубиновое стекло; пигментированные
волокна; сплавы; рисунок на ткани, нанесенный
методом пигментной печати
|
|
Твердая
|
Жидкая
|
Жемчуг, вода в граните, вода в бетоне, остаточный мономер в
полимерно-мономерных частицах
|
|
Твердая
|
Газо- образная
|
Газовые включения в различных твердых телах: пенобетоны,
замороженные пены, пемза,
вулканическая лава, полимерные пены, пенополиуретан
|
|
Жидкая
|
Твердая
|
Суспензии, краски, пасты, золи, латексы
|
|
|
|
|
Жидкая
|
Жидкая
|
Эмульсии: молоко, нефть, сливочное масло, маргарин, замасливатели волокон
|
|
Жидкая
|
Газо- образная
|
Пены, в том числе
для пожаротушения и пенных технологий замасливания волокон, беления и
колорирования текстильных материалов
|
|
Газообразная
|
Твердая
|
Дымы, космическая пыль, аэрозоли
|
|
Газообразная
|
Жидкая
|
Туманы, газы в момент сжижения
|
|
Газообразная
|
Газо- образная
|
Коллоидная система не образуется
|
Коллоидные системы необычайно
лабильны, т.е. неустойчивы. Для многих из них достаточно прибавления ничтожного
количества электролита, чтобы вызвать
выпадение осадка. Причина столь легкого изменения состояния коллоидных систем связана с
непостоянством степени их дисперсности. Различают два вида устойчивости любой
раздробленной системы - кинетическую и агрегативную.
Таблица 2
Примеры свободнодисперсных систем
В основу этой классификации положено
агрегатное состояние фаз дисперсной системы.
Понятие агрегативной устойчивости, которое впервые
ввел Н.П. Песков, подразумевает отсутствие агрегирования, т.е. снижения степени
дисперсности коллоидной
системы
при хранении. Для определения кинетической устойчивости необходимо изучать
условия выделения диспергированных частиц в гравитационном или центробежном
поле. Скорость подобного выделения зависит от интенсивности броуновского
движения частиц, т.е. от степени дисперсности системы и разности плотности
дисперсионной среды и дисперсной фазы, а также от вязкости среды.
Таблица 3
Связнодисперсные системы
|
1. Системы с жидкой поверхностью раздела
фаз
|
2. Системы с твердой поверхностью
раздела фаз
|
|
Г1/Ж2 – пены
Ж1/Ж2 –
пенообразные эмульсии
|
Г1/Т2 - пористые
тела, натуральные
волокна,
пемза, губка, древесные угли
Ж1/Т2 – влага в
граните
Т1/Т2 –
взаимопроникающие сетки полимеров
|
Если хотят определить агрегативную устойчивость
системы, то исследуют условия постоянства (или напротив - непостоянства)
степени дисперсности системы. Одно из самых резких и характерных отличий коллоидной системы как от
истинного раствора, так и от
грубодисперсных систем состоит в том, что их степень дисперсности является
чрезвычайно непостоянной величиной и может изменяться в зависимости от самых
разнообразных причин.
В основе этой классификации лежит
агрегатное состояние поверхности раздела фаз.
На основании изложенного выше дадим определение коллоидным системам.
Коллоидными системами называют двух-или многофазные
системы, в которых одна фаза находится в виде отдельных мелких частиц,
распределенных в другой фазе. Такие ультрамикрогетерогенные системы с
определенной (коллоидной) дисперсностью проявляют способность к интенсивному
броуновскому движению и обладают высокой кинетической устойчивостью.
Имея высокоразвитую поверхность раздела фаз и,
следовательно, громадный избыток свободной поверхностной энергии, эти системы
являются принципиально термодинамически неустойчивыми, что выражается в
агрегации частиц, т.е. в отсутствии агрегативной устойчивости. Однако этими
свойствами не исчерпываются все особенности, которыми коллоидные системы отличаются от
других систем. Так, например, на первый взгляд кажется непонятным, почему
коллоидные частицы, совершая энергичные движения и сталкиваясь между собой, не
всегда слипаются в более крупные агрегаты и не выпадают в осадок, как этого
следовало бы ожидать на основании второго закона термодинамики, так как при
этом уменьшалась бы общая поверхность, а с ней и свободная энергия.
Оказывается, во многих случаях устойчивость таких
систем связана с наличием слоя стабилизатора на поверхности
коллоидных частиц. Таким образом, необходимым условием создания устойчивых коллоидных систем является
присутствие третьего компонента - стабилизатора. Стабилизаторами коллоидных систем могут быть электролиты или некоторые
другие вещества, не имеющие
электролитной природы, например высокомолекулярные соединения (ВМС) или
поверхностно-активные вещества (ПАВ). Механизм
стабилизации электролитами и
неэлектролитами существенно различен.
Влияние электролитов на устойчивость
коллоидных
систем
носит сложный характер. В одних случаях ничтожные добавки электролита способны
привести к нарушению устойчивости системы. В других - введение электролита способствует
увеличению стабильности.
Образование адсорбционных слоев таких стабилизаторов, как ПАВ,
приобретает особенно большое значение при наличии двухмерных структур,
обладающих повышенными структурно-механическими свойствами. Во многих случаях
стабилизация достигается при покрытии монослоем всего 40-60 % поверхности
коллоидных частиц, когда защитный слой имеет прерывистый характер (в форме
островков). Максимальная устойчивость достигается, естественно, при образовании
полностью насыщенного мономолекулярного слоя.
Структурно-механические свойства адсорбционных слоев в значительной мере
определяют поведение коллоидных
систем.
Эти слои могут быть образованы или изменены небольшими количествами каких-либо
растворенных веществ, поэтому
создается возможность регулирования ряда свойств коллоидных систем, что широко используется
в различных практических приложениях.
Коллоидные системы, состоящие из
частиц диспергированного вещества, способных
свободно перемещаться в жидкой дисперсионной среде совместно с адсорбированными
на их поверхности молекулами или ионами третьего компонента
(стабилизатора), называют лиозолями, а сами частицы, обладающие сложным
строением - мицеллами.
По характеру взаимодействия коллоидных частиц с
дисперсионной средой лиозоли могут быть разделены на лиофильные и лиофобные.
Впервые эта классификация была предложена
немецким ученым-коллоидником Фрейндлихом. Он разделил все системы на два класса
– лиофильные и лиофобные. В соответствии с представлениями, развитыми Фрейндлихом,
лиофобными называют системы, частицы дисперсной фазы которых не взаимодействуют
с дисперсионной средой, не сольватируются и не растворяются в ней. Лиофильные-
это системы, частицы дисперсной фазы которых интенсивно взаимодействуют с
дисперсионной средой.
К лиофобным системам относятся золи драгоценных металлов, золи металлоидов (серы, селена, теллура),
дисперсии полимеров в воде (например, полистирола, фторолона), золи сульфидов мышьяка, сурьмы, кадмия, ртути, золи гидроксидов железа, алюминия и т.д. Эти
системы характеризуются, так называемой, кинетической устойчивостью и
агрегативной неустойчивостью и требуют стабилизации. К лиофильным коллоидным системам Фрейндлих отнес
растворы, образующиеся
при растворении природных или
синтетических ВМС. Таковы растворы белков, крахмала, пектинов, камедей, эфиров целлюлозы и разнообразных
смол, как природных так и синтетических.
Таким образом, растворы ВМС
рассматривались ранее как лиофильные коллоидные системы. Они считались
двухфазными дисперсными
системами
и таким образом сущность классификации Фрейндлиха
сводилась к молекулярным взаимодействиям между дисперсной фазой и дисперсионной
средой. Именно на этом основании проводилось разделение на лиофильные и
лиофобные системы. Лиофильные системы считались двух- или многофазными,
термодинамически неустойчивыми, неподчиняющимися правилу фаз Гиббса. Но такое
представление оказалось неправильным. На самом деле в настоящее время
достоверно установлено, что растворы ВМС - это
истинные растворы, т.е.
однофазные системы, гомогенные, термодинамически устойчивые и подчиняющиеся правилу фаз Гиббса. Считалось, что
обратимость - это характерное свойство лиофильных коллоидных систем, но это не так,
потому что в данном случае растворы ВМС не являются
дисперсными
системами.
В связи с этим академик В.А. Каргин еще в 1948 г.
обратил внимание на то, что классификация Фрейндлиха
совершенно неверна и даже более того - вредна.
Чтобы не менять смысла этих терминов, П.А. Ребиндер
предложил оформить понятия лиофильные и лиофобные коллоидные системы. Дисперсные
много-или двухфазные системы он разделил на два класса, исходя из величины
удельной межфазовой энергии (поверхностного натяжения).
К лиофобным системам были отнесены дисперсные системы с достаточно
высоким межфазовым натяжением (s12), большим некоторого граничного
значения sm:
s12 > sm . (1)
Эти системы характеризуются большой межфазовой
свободной энергией, поэтому граница раздела фаз выражена резко: система
является агрегативно неустойчивой и требует введения стабилизатора. Дисперсность
таких систем является произвольной.
Лиофильные системы – это двухфазные коллоидные системы с низкой, хотя
и положительной межфазовой свободной энергией, меньшей или равной граничному
значению,
s12 ≤ sm . (2)
Это системы с очень малой межфазовой энергией, они
термодинамически устойчивы и образуются самопроизвольно. Дисперсность их вполне
определенна и находится в коллоидной области.
Тот факт, что дисперсные системы классифицируются
по величине свободной поверхностной энергии показывает, что
коллоидные явления тесно связаны со свойствами поверхности раздела фаз.
К лиофильным системам относят:
1) так называемые критические эмульсии, образующиеся в
результате снижения поверхностного натяжения при нагревании
до температуры, близкой к температуре неограниченного
смешения, или в
результате прибавления очень больших количеств ПАВ;
2) ассоциативные коллоидные системы, образуемые в
водной среде веществами типа мыл, некоторых красителей и
дубителей, а в неводной среде некоторыми ПАВ. Такие вещества в разбавленных растворах находятся в
молекулярном состоянии, при увеличении концентрации происходит
агрегация молекул с образованием
частиц коллоидного размера, т.е. образуются мицеллы. Концентрацию вещества в растворе, при которой
происходит переход от истинного раствора к коллоидному,
принято называть критической концентрацией мицеллообразования (ККМ).
Классификацию дисперсных систем можно
проводитьпо удельной поверхности и пористости дисперсной
фазы.
В тех процессах, в которых участвуют две
соприкасающиеся фазы, большое значение имеют свойства поверхности раздела, или
пограничного слоя, отделяющего одну фазу от другой. Молекулы, составляющие
такие слои, обладают особыми свойствами. Если рассматривать монолитную фазу, то
числом молекул, образующих
поверхностный слой, можно пренебречь по сравнению с огромным количеством молекул в объеме тела.
Можно считать, что запас энергии системы пропорционален массе, содержащейся в
объеме тела.
При измельчении сплошного тела
число молекул в поверхностном
слое возрастает и достигает максимального значения в коллоидно-дисперсных
системах. Поэтому процессы, протекающие в дисперсных системах, обусловлены
свойствами поверхностных слоев на границе раздела. Образование пен, эмульсий, туманов,
процессы флотации, смачивания и диспергирования, сорбционная
техника и многие-многие другие основаны на свойствах межфазовых поверхностей в дисперсных системах.
Удельной поверхностью называют отношение поверхности
тела к его объему или массе:
Ауд= А/V или Ауд =А/Vr , (3)
где Ауд, А – удельная и суммарная
поверхность, соответственно; r – плотность вещества, V – объем
тела.
Для кубических частиц
Ауд = 6а2/а3= 6а-1
или
Ауд = 6a2/а3r =
6/ar (м2/кг). (4)
Для сферических частиц
Ауд = 4 r2/(4/3 r3)
(м-1),
т.е.
Ауд = 3/r (м-1),
или
Ауд = 3/rr (м2/кг). (5)
Если взять кубик вещества, три его
стороны разделить на 10 частей и провести плоскости в трех направлениях, то
получим более мелкие кубики. Такой процесс можно рассматривать как моделирование процесса диспергирования. Изменение
удельной поверхности в процессе диспергирования показано в табл.
4.
Таблица
4
Зависимость удельной поверхности от дисперсности
|
Длина стороны куба а, см
|
Число
кубов
|
Суммарная
поверхность, м2
|
Удельная поверхность, см-1
|
|
1
10-1
10-2
10-4
10-5
10-6
10-7
|
1
103
106
109
1012
1015
1018
1021
|
6·10-4
6·10-3
6·10-1
6
6·101
6·102
6·103
6·104
|
6
6·101
6·102
6·103
6·104
6·105
6·106
6·107
|
В текстильных коллоидных системах большую роль
играют волокна, нити и пленки. Удельную поверхность таких систем можно
рассчитать по формулам:
для пленки
Ауд = 2l2/l2а =
2/а, (6)
где а- толщина пленки, l- ее ширина и длина;
для цилиндра (волокна, нити)
Ауд= 2lr /r2l = 2/r, (7)
где r - радиус цилиндра, l- его длина.
Связнодисперсные системы - пористые тела - наряду с
внешней удельной поверхностью можно характеризовать размером (радиусом) пор, их
объемом и внутренней удельной поверхностью. Удобную классификацию пор по размерам
предложил М.М. Дубинин. В соответствии с этой классификацией все пористые
тела можно разделить на три класса (в зависимости от адсорбционных свойств): микропористые
тела с радиусом пор 2·10-9 м, мезопористые (переходнопористые) -
(2/50) ·10-9м, макропористые 50·10-9 м.
Микропористые тела в последнее время разделяют на
ультра- и супермикропористые. Такая классификация весьма
приближенно отражает весь спектр возможных размеров пор ( от макропор через
мезопоры и микропоры до субатомных «пор» в виде промежутков между
макрокристаллами в полимерах или точечных дефектов в кристаллах).
В этой связи следует отметить, что любая классификация не может
полностью охватить все многообразие дисперсных систем, существующих в
природе и технологической практике.
Структурообразование
в дисперсных системах и в растворах полимеров.
При повышении концентрации дисперсной фазы
в дисперсных
системах
(или концентрации растворенных
полимеров) возможно образование таких агрегатов частиц (или ассоциатов
макромолекул), которые вызывают отклонение течения таких систем от законов
Ньютона и Пуазейля. Такие жидкости называют
аномально вязкими, а концентрацию, при которой
происходит качественное изменение свойств системы, – критической концентрацией структурообразования. При достижении
критической концентрации дисперсной фазы
в дисперсной
системе
самопроизвольно возникает пространственная структура из взаимодействующих между
собой частиц.
К образованию прочной структуры, называемой
кристаллической, приводит непосредственный контакт между частицами, т.е. такой
контакт, при котором граница раздела фаз между частицами исчезает. Этот процесс
наблюдается при формировании дисперсной системы методом конденсации, когда
отдельные кристаллы срастаются: при
отвердении бетона, при
формировании бумажного полотна или нетканого материала, образовании
пространственных сеток при полимеризации и т.д.
Взаимодействие частиц через тонкую прослойку жидкой фазы приводит к
формированию коагуляционных контактов. После разрушения эти контакты обратимо
восстанавливаются. Это свойство называется «тиксотропия». Такие контакты
возможны в пастах пигментов, в керамических
массах, в растворах и дисперсиях полимеров. На способности
обратимо восстанавливать структуру после снятия нагрузки основаны действие
шлихтующих препаратов и загустителей в печатных красках при
колорировании текстильных материалов, а также склеивание латексом
волокон при получении нетканых материалов, сохранение
формы керамических изделий, удерживание лаков, красок и эмалей на вертикальных
стенках и т.д.
Коагуляционные структуры характеризуются
относительно низкими энергиями взаимодействия и в большинстве случаев возникают
при частичном снижении устойчивости дисперсных систем. В таких
структурах среднее расстояние между частицами соответствует равновесной толщине
пленок жидкости и
характеризуется первым или вторым минимумом на кривых потенциальной энергии
парного взаимодействия частиц.
В соответствии со способом образования
коагуляционных структур частицы могут располагаться на расстояниях Н1»
10-9 м или Н2 » 10-7 м.
Энергия взаимодействия в первом потенциальном
минимуме на два порядка превышает энергию взаимодействия во втором
потенциальном минимуме (потенциальной яме). На практике чаще встречается структурообразование с фиксированием
частиц во втором потенциальном минимуме.
Объемная доля дисперсной фазы, при которой
происходит образование коагуляционной структуры, зависит от формы частиц.
Асимметричные частицы могут образовывать структуру при значительно меньшей концентрации, чем
сферические. Асимметричная форма частиц характерна для гидроксидов железа и алюминия, для глины и некоторых пигментов. Прочность структуры
характеризуют напряжением, необходимым для разрушения пространственной
структуры.
Структурированные жидкости не подчиняются
законам течения Ньютона и Пуазейля. Различают два типа структурированной жидкости: с
жидкообразной и с твердообразной структурой.
Жидкости с жидкообразной
структурой характеризуются реологическими кривыми течения, у которых отсутствует
критическое напряжение сдвига, а присутствуют два линейных участка
псевдоньютоновского течения.
Твердообразные структуры должны быть разрушены
прежде, чем начинается течение. Иными словами такая структура до разрушения
обладает свойствами твердого тела.
Область коллоидной химии, занимающаяся
изучением закономерностей образования и разрушения структуры в дисперсных системах и в растворах полимеров, называется
«реологией». В реологии оперируют
такими понятиями, как деформация, т.е.
относительное смещение части системы без нарушения ее целостности. Деформация может быть
упругой и остаточной. При упругой деформации форма тела
восстанавливается после снятия напряжения.
На рис. 2.30 показана схема однородного сдвига куба
с длиной ребра l, условно выделенного из изучаемой системы, под действием
касательного напряжения Р. Мерой сдвига служит отношение смещения х к
первоначальной длине ребра куба l, т.е. высота, на которой происходит смещение
x / l = tga = g , (2.4.52)
где a – угол смещения элемента структуры.
Мерой скорости деформации служит градиент
скорости смещения:
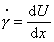
 . (2.4.53)
. (2.4.53)
Реология оперирует тремя
идеализированными зависимостями между Р и g(или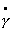 ) для описания
трех структурных свойств (упругости, вязкости и пластичности)
и использует комбинации этих зависимостей для описания более сложных процессов,
протекающих в структурированных дисперсных системах.
) для описания
трех структурных свойств (упругости, вязкости и пластичности)
и использует комбинации этих зависимостей для описания более сложных процессов,
протекающих в структурированных дисперсных системах.
Упругая деформация (или упругость)
пропорциональна напряжению сдвига:
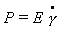 , (2.4.54)
, (2.4.54)
где Е – модуль Юнга.
Уравнение (2.4.54) носит название закона Гука. Зависимость,
которая описывается уравнением (2.4.54) для идеального упругого тела, показана
на рис. 2.31, а. Физическую модель идеального упругого тела Гука изображают
обычно в виде спиральной пружины, закрепленной за один из концов и
растягиевамой за другой.
Мерой упругости служит модуль Юнга, определяемый как
ctga зависимости, приведенной на рис. 2.31, а. Эта зависимость для идеального
тела линейна. Физический смысл упругой деформации заключается в
изменении межатомных расстояний при создании напряжения и стремлении тела
вернуть атомы в исходное
равновесное состояние, характеризуемое минимумом свободной энергии. В этой
связи идеальное упругое тело восстанавливает свою форму и размеры практически
мгновенно после снятия напряжения. Для восстановления первоначальных
размеров и формы в реальных упругих телах требуется некоторое незначительное
время.
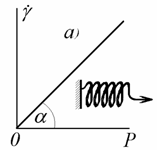
Вязкое течение описывают уравнением Ньютона (2.4.1,
а) в форме 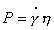 . Схема модели
вязкого течения и зависимость градиента скорости смещения от напряжения
приведены на рис. 2.31, б. Вязкость жидкости определяется
как сtgb. В качестве механической модели идеальной вязкой ньютоновской жидкости служит поршень
в цилиндре, между которыми возможно перетекание.
. Схема модели
вязкого течения и зависимость градиента скорости смещения от напряжения
приведены на рис. 2.31, б. Вязкость жидкости определяется
как сtgb. В качестве механической модели идеальной вязкой ньютоновской жидкости служит поршень
в цилиндре, между которыми возможно перетекание.
|
|
|
|
|
Рис. 2.31. Модель и
зависимость деформации от
напряжения: а – идеально упругого тела (Гука); б – идеально вязкой жидкости (Ньютона); в
– идеально пластического тела (Кулона)
|
Физическая модель вязкого течения связана с
термически активируемым процессом перестройки взаимодействующих друг с
другом молекул. Естественно,
что при действии напряжения одни связи между молекулами жидкости разрываются, а
другие – образуются вновь. В истинно вязкой ньютоновской жидкости коэффициент вязкости остается
постоянным от очень малых нагрузок вплоть до напряжений, при которых ламинарный
режим течения переходит в турбулентный. В ряде случаев при изучении вязкого
течения используют величину, обратную вязкости, которую
называют текучестью.
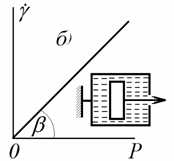
Пластичность, или
пластическое течение, не является линейной функцией напряжения. В качестве
модели пластической деформации используют твердое тело, лежащее на
плоскости (рис. 2.31, в) и удерживаемое на месте силами сухого трения вплоть до
некоторого напряжения, способного преодолеть это сухое (кулоновское) трение. Такое течение
возможно, например, в пастах пигментов, когда
происходит последовательное разрушение-восстановление контактов между
частицами, которые фиксируются в пространстве через некоторую прослойку жидкой
фазы. В том случае, если в системе образуется кристаллическая структура при
непосредственном контакте между частицами, течение начнется только после
необратимого разрушения таких контактов и критическое напряжение будет
соответствовать их прочности.
Конечно, в практическом приложении структурообразования
и разрушения (например, при разрушении структуры в
загущенных полимерами печатных красках при перемешивании и в процессе ее
нанесения на ткань и при восстановлении структуры в том
рисунке, который нанесен на ткань, или при нанесении раствора полимера - шлихтующего
препарата - на нити), одновременно могут проявляться и различные виды деформаций: упругая деформация, затем вязкое
или пластическое течение и последующее структурирование.
Если в системе внешнее напряжение расходуется на
преодоление упругой деформации и вязкого
течения, то используют модель, предложенную Максвеллом, из последовательно
соединенных элементов моделей Гука и Ньютона (рис. 2.32, а). В таких системах
типично проявление релаксации напряжения,
описываемого уравнением
P0(t) = P0exp( t/tp),
(2.4.55)
где P0= E0g0 –
начальное напряжение; tр= h/Е – время релаксации.
При t < tp система ведет себя как твердое тело. При t>>tp
модель Максвелла соответствует жидкоподобному течению. Явление релаксации связано с тем, что
для перестройки структуры при относительно невысоком напряжении требуется
определенное время. Поэтому при кратковременном (мгновенном) приложении
напряжения в системе возникают постепенно снижающиеся внутренние напряжения.
Возможно, что снятие внутреннего напряжения будет реализовано при t®¥. Для жидкости, описываемой
моделью Максвелла, характерна необратимость деформации.
Таким образом, свойства системы (твердое тело или
жидкость) зависят от времени релаксации, определяемого
по пересечению касательной к начальному участку деформационной кривой с осью
абсцисс (см.рис. 2.32, а).
Если в системе наблюдается нарастание деформации во времени при
постоянном напряжении и полный спад деформации в течение
определенного времени после снятия нагрузки, то такие системы описываются
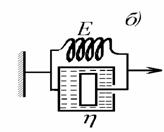
моделью Кельвина-Фойгта, состоящей из соединенных параллельно элементов моделей
Гука и Ньютона (рис. 2.33). Эта модель характерна для механически обратимого
твердообразного структурированного тела. Для такой структуры обычно используют
уравнение при Р = соnst
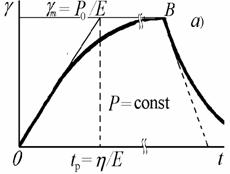
g(t) = P0[1 – exp (– t/tp
)] / E . (2.4.56)
|
|
|
|
Рис. 2.33. Зависимость
напряжения от времени (а) и модель вязкоупругого течения (б) Кельвина –
Фойгта
|
Это уравнение описывает восходящую ветвь кривой на
рис 2.33. Нисходящая ветвь (при Р = 0) описывается уравнением
g = gmexp(– t/tp) . (2.4.57)
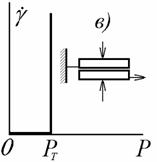
Наиболее точной для описания поведения реальных
систем следует считать модель из соединенных параллельно элементов моделей
Ньтона и Кулона, предложенную Бингамом. Схема модели и деформационная кривая
показаны на рис. 2.34.
|
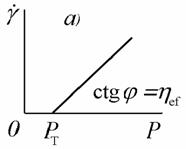 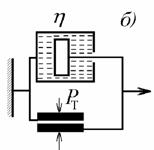
Рис. 2.34. Зависимость деформации от напряжения
(а) и модель вязкопластического течения (б) Бингама
|
При напряжениях, меньших напряжения текучести Рт,
система обладает упругими свойствами. После достижения этого напряжения
начинается пластическое течение, для описания которого Бингам предложил
уравнение
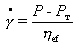 . (2.4.58)
. (2.4.58)
|
Рис. 2.35. Потенциальная
энергия взаимодействия сферических частиц
|
Такое вязкопластическое течение характерно для
многих коагуляционных структур – пигментированных расплавов и растворов полимеров, печатных красок, глинистых растворов,
концентрированных эмульсий и т.д. Часто
увеличение напряжения приводит к дополнительному разрушению структуры. В таких
системах следует говорить об «эффективной» вязкости hef,
уменьшающейся при увеличении напряжения до некоторого предельного значения,
соответствующего полному разрушению структуры в системе.
Свойства
дисперсных систем и определение размера частиц.
Раздел «Свойства коллоидных систем» включает
рассмотрение диффузии,
броуновского движения, осмоса, седиментации, рассеяния света
и его поглощения, рассматриваются также общие принципы определения наиболее
важной характеристики систем - среднего размера частиц. Частицы в дисперсных
системах обычно имеют распределение
по размерам, поэтому знание студентами способов определения параметров этих
распределений позволит им правильно понимать, что свойства коллоидных
систем являются функцией не только
степени раздробленности (дисперсности) измельченной (дисперсной) фазы, но и ее
распределения по размерам частиц.
Этот факт проявляется в тех производственных дисперсных
системах, которые применяются в
производстве и облагораживании текстильных материалов, например, при
использовании дисперсных и сернистых красителей или
дисперсий пигментов
при печатании рисунков на тканях и окрашивании волокон в массе. В процессе хранения
в дисперсных системах (красках) на основе пигментов или в
колорированной массе волокнообразующего полимера происходит выделение
грубодисперсной фракции или неравномерное распределение частиц в массе полимера, что может изменить
оттенок или даже цвет колорированных волокон, так как интенсивность отражения
света и его рассеяние зависят от размера частиц.
Определение размера частиц или капель эмульсии важно также и для
создания эффективного процесса эмульсирования натуральных волокон в
процессе их переработки или при авиважной обработке синтетических волокон.
Взаимодействие волокон с частицами, например, полимера латексов,
применяемых для склеивания
волокон в нетканых материалах или при аппретировании тканей, зависит от их размера, поэтому при рассмотрении
теоретических аспектов прилипания частиц к волокнам, коагуляции (агрегирования частиц) и гетерокоагуляции (осаждения
частиц на волокнах), умение определять размеры частиц несомненно должно
представлять собой один из важнейших навыков, которые выработают студенты,
изучившие этот раздел учебника.
Оценка дисперсности гетерогенных
систем является сложной задачей в
связи с разнообразием формы их частиц, полидисперсностью, возможным
агрегированием первичных частиц. Поэтому определяют обычно некоторую среднюю
величину и погрешность в 10-20 % считается допустимой.
Список
использованной литературы:
1. Ребиндер
П. А., Поверхностные явления в дисперсных системах.
2.
Коллоидная химия, Избр. труды, М., 1978; Дерягин Б. В., "Успехи
химии", 1979, т. 48, в. 4, с. 675-721
3. Урьев Н.
Б., Высококонцентрированные дисперсные системы, М., 1980
4.
Коагуляционные контакты в дисперсных системах, М., 1982
5.
Капиллярная химия, под ред. К. Тамару, пер. с япон., М., 1983
6. Щукин Е.
Д., Перцов А. В., Амелина Е. А., Коллоидная химия, М., 1982
7. Также
лит. при статьях Коллоидная химия. Поверхностные явления. Физико-химическая
механика. Л. А. Шиц. Е. Д. Щукин.