Молекулярно-динамическое моделирование систем заряженных частиц с использованием графических ускорителей
Введение
Использованию компьютерных технологий в
современной физике отводится важное место, вычислительные методы широко
применяются в самых разных задачах и научных областях. В данной работе
исследуется возможность применения программируемых логических интегральных схем
(ПЛИС, FPGA- FieldProgrammableGateArray) для методов молекулярной динамики(МД)
и Монте-Карло(МК), являющихся одними из самых популярных методов, используемых
для моделирования систем, состоящих из многих частиц. Для этих методов используется
также термин «атомистическое моделирование», указывающий на то, что информация
о свойствах веществ получается на основе решения уравнений движения для
большого числа частиц - атомов, молекул, молекулярных фрагментов, электронов и
ионов (в зависимости от рассматриваемой системы и ее математической модели).
Алгоритмы МД и МКдают возможность исследовать
свойства различных объектов на малых масштабах, а также строить модели
происходящих в них сложных динамических процессов. Данные методытребуют высокой
производительности компьютерных систем, в связи с чем появляется острая
необходимость в изучении современных аппаратных и программных технологий,
которые позволяют производить сложные расчеты.На протяжении многих лет
единственной возможностью ускорения МД/МК расчетов было использование
суперкомпьютерных кластеров, состоящих из большого числа одно- или
многопроцессорных модулей, объединенных высокопроизводительной сетью. Несмотря
на то, что алгоритмы МД/МК моделирования показывают сравнительно неплохую
эффективность распараллеливания на кластерах (по сравнению, например, с
решением уравнений в частных производных на сетке), проведение масштабных
расчетов всегда ограничивается пропускной способностью коммуникационной сети.
В последние годы большое распространение получили
расчеты с применением графических ускорителей (ГУ, GPU -
GraphicsProcessingUnit), с помощью которых удается значительно повысить
скорость работы программы на одном узле вычислительного кластера и, таким
образом, снизить количество необходимых узлов и требования к коммуникационной
сети. Алгоритмы МД/МК позволяют использовать значительную часть ресурсов ГУ,
особенно для систем с большим количеством частиц, и получать высокую
эффективность распараллеливания по сравнению с теми же сеточными алгоритмами.
Однако, при сравнительно небольшом числе частиц на вычислительный узел
эффективность ГУ существенно падает, и его применение становится неэффективным.
Это связано с внутренней архитектурой ГУ, которая не может быть изменена на
программном уровне.
С другой стороны, для решения многих
технологических задач успешно применяются ПЛИС, которые представляют собой
интегральные схемы с программируемой архитектурой, т.е. связи и
функциональность вычислительных блоков не являются фиксированными, а задаются в
процессе специальной процедуры программирования ПЛИС, которая может быть
повторена неограниченное количество раз. Такие системы нашли широкое применение
в задачах, связанных с обработкой больших потоков информации, криптографии,
задачах распознавания образов, компьютерного зрения и т.п. вычислительный молекулярный динамика ускоритель
В последнее время наблюдается интерес к
применению ПЛИС и для решения научных задач, в частности методами компьютерного
моделирования. Очевидно, это связано с развитием средств разработки программ
для ПЛИС. Долгое время программирование ПЛИС осуществлялось только с помощью
специализированных низкоуровневых языков программирования, таких какVerilog,
VHDL и др. Однако, с появлением новой аппаратной платформы AlteraSDK создавать
программы для ПЛИС стало возможным с помощью высокоуровневого языка OpenCL
(Open Computing Language), уже хорошо зарекомендовавшего себя в задачах
параллельного программирования для систем с общей памятью. Использование OpenCL
существенно упрощает процесс разработки, отладки и модификации программы, и,
тем самым, открывает возможность применения ПЛИС для задач компьютерного
моделирования, в частности, основанных на методах МД и МК.
Цель данной работы - реализация алгоритмов МД и
МК на ПЛИС, с использованием OpenCL, проведение сравнительного анализа
производительности центральных процессоров (ЦП,CPU), ГУи ПЛИСв задачах МД/МК
моделирования с различными потенциалами взаимодействия. В качестве примера
рассмотрены широко известный короткодействующий потенциал Леннарда-Джонса, а
также специализированный потенциал, применяемый для моделирования
электронионной плазмы, который имеет дальнодействующую кулоновскую асимптотику
на больших расстояниях.
1.Обзор методов атомистического моделирования
.1 Методы молекулярной динамики и Монте-Карло
Методы компьютерного моделирования стали мощным
инструментом для исследования систем, состоящих из множества частиц, и получили
широкое распространение в задачах статистической физики, физической химии и
биофизике[1,2]. Несмотря на то, что теоретическое описание сложных систем в
статистической физике развито достаточно хорошо и использование вычислительных
методов достаточно трудоемко, часто только с помощью компьютерного
моделирования возможно изучить специфические аспекты систем, такие как,динамика
дефектов в твердых телах, фазовые переходы, релаксация и колебательные процессы
в неидеальной жидкости и неидеальной плазме, динамика макромолекул и полимеров.
С другой стороны, моделирование требует специфичных входных параметров, которые
характеризуют систему и находятся либо с помощью эксперимента, либо из
теоретических соображений.Часто моделирование используется в качестве
альтернативы или дополнения как экспериментальных, так и теоретических
исследований.
Традиционные методы компьютерного моделирования
многочастичных систем можно разделить на два класса: стохастические и
детерминированные. Представителями этих классов являются методы Монте-Карло и
молекулярной динамики, соответственно. Метод МК основан на случайных смещениях
частиц. Согласно алгоритму Метрополиса, показателем для принятия или отвержения
очередной новой конфигурации является изменение энергии системы между шагами nи
n + 1. Свойства системы могут быть вычислены с помощью усреднения по всем
итерациям в методе МК.
Метод МД основан на численном решении уравнений
движения частиц, в основе которыхлежит гамильтонова динамика. На основе
разностных методов на каждом шаге по времени определяются новые координаты и
скорости частиц. По сравнению с методом МК данный метод дает больше информации
о системе. Несмотря на то, что существует множество технологий для
моделирования многочастичных систем, для всех них необходима модель
взаимодействия составляющих систему частиц (атомов, ионов, молекул и т.д.). Эта
модель должна соответствовать экспериментально полученным данным или известным
теоретическим сведениям. Программа моделирования методом МД состоит из
следующих компонент:
)алгоритмы расчета взаимодействия между
частицами(атомами, молекулами, ионами и т.д.), часто полагается, что частицы
взаимодействуют посредством парного потенциала, что сильно упрощает расчёт;
) интегратора, который решает уравнения движения
для определения координат и скоростей частиц в разные моменты времени;
) алгоритма реализации выбранных граничных
условий;
) термостата или баростата, контролирующих такие
величины как температура или давление;
) модуля имитации внешнего воздействия на
систему;
) модулей для расчета требуемых параметров
системы (термодинамических и структурных параметров, корреляционных функций, и
др.);
) модуля статистического усреднения по времени
или по ансамблю независимых МД траекторий;
) модуля ввода-вывода данных, преобразования
форматов и визуализации.
В случае значительного расхождения результатов
моделирования с экспериментальными или теоретическими данными, модель
взаимодействия частиц должна быть пересмотрена.
Метод молекулярной динамики - это метод, в
котором временная эволюция системы взаимодействующих частиц или атомов
определяется интегрированием их уравнений движения[3,4]:
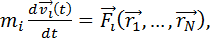 (1.1)
(1.1)
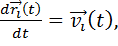 (1.2)
(1.2)
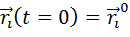 ,
,
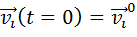 ,i
= 1,…,N.(1.3)
,i
= 1,…,N.(1.3)
Прежде всего необходимо указать начальные
условие, такие как, координаты частиц и их скорости. После этого с заданным
шагом по времени интегрируются уравнения движения и производится расчёт сил,
действующих на частицы.Свойства системы зависят от выбранного потенциала
взаимодействия 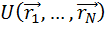 , который описывает
зависимость энергии взаимодействия частиц от расстояния между ними
, который описывает
зависимость энергии взаимодействия частиц от расстояния между ними
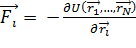 .(1.4)
.(1.4)
Важным аспектом, который необходимо учитывать
при разработке моделей, является характерное время рассчитываемого процесса и
возможный размер системы (см. рис. 1).
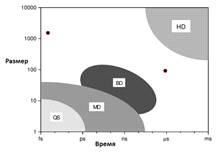
Рис. 1. Сравнение допустимых времен и размеров
для разных техник моделирования:QS-квантовая молекулярная динамика,
MD-классическая молекулярная динамика, BD-броуновская динамика, HD -
гидродинамика
Метод классической молекулярной динамики дает
возможность, используя передовые информационные технологии,производить расчёт
систем, состоящих из миллионов атомов на временах порядка наносекунд. Благодаря
использованию разнообразных средств оптимизации возможно увеличить шаг
интегрирования, с помощью чего увеличить доступное для наблюдения время до
нескольких микросекунд.
В наше время метод классической молекулярной
динамики применяется для решения большого класса задач, таких как, исследование
биомолекул, изучение свойств жидкостей, определения свойств поверхностей,
молекулярных кластеров, дефектов в кристаллах, динамических процессов в плотной
плазме и т.д.
Прямое молекулярно-динамическое моделирование
макроскопических объектов неосуществима и требует многомасштабного приближения,
в котором МД используется для получения свойств маленьких фрагментов системы.
Вычислительное время в классической МД без применения специальных
оптимизационных алгоритмов растет как t ~ N2 ~ L6. Где N- это число частиц, а L
- это параметр, определяющий размер системы[5,6]. В связи с этим размер МД
системы должен быть минимальным и регулироваться характерным масштабом
изучаемой системы. Нет необходимости в увеличении размера системы для лучшего
усреднения, так как это может быть заменено усреднением по времени на
равновесной траектории.
Типичный прием МД моделирования, используемый
для эффективного увеличения размеров системы - это периодические граничные
условия, где каждая частица взаимодействует с ближайшими образами других частиц
(метод ближайшего образа) или со всеми периодическими образами, как в методе
Эвальда. Метод Эвальда больше подходит для упорядоченных систем, таких как
кристаллы, а также иногда используется для моделирования неидеальной плазмы. В
некоторых работах авторы используют отражающие стенки, это означает
незамедлительное отражение частицы, как только она пересекла границу. Это может
повлечь за собой нарушение закона сохранения энергии и формирование
приповерхностного слоя со свойствами, отличными от свойств основной части
системы, поэтому данный метод используется сравнительно редко. Все типы
граничных условий используют неявное отсечение потенциала, поэтому в системах с
дальними взаимодействиями этот эффект требует особого внимания.
Для моделирования равновесных систем в начале
расчета используется термостат для вывода системы на нужную температуру.
Выполнение основной части моделирования без использования термостата позволяет
избежать нефизического искажения функции распределения скоростей. Недостаток
состоит в том, что фактическая средняя температура получается путем усреднения
по траекториии может быть отличной от необходимой.
.2 Потенциалы взаимодействия частиц
Для каждой задачи требуется индивидуальный выбор
потенциала взаимодействия. Одним из наиболее простых и хорошо изученных
является потенциал Леннард-Джонса, применяемый для моделирования взаимодействия
инертных газов, других слабовзаимодействующих незаряженных атомов или молекул,
а также в качестве модельного потенциала для исследования фундаментальных
свойств молекулярно-динамических систем. Потенциал Леннард-Джонса - это
короткодействующий потенциал, для которого на сравнительно небольшом расстоянии
можно сделать «отсечку», то есть, считать, что на расстоянии больше этого
частицы не взаимодействуют.
В общем случае с ростом числа частиц сложность
вычислений растёт как O(N2). Однако, для короткодействующих потенциалов
существует много методов оптимизации вычислений, например, вычисление списков
ближайших соседей. Суть метода в том, что для каждой частицы создаётся список,
в который включаются частицы, находящиеся в определённом расстоянии вокруг неё.
Считается, что с другими частицами она не взаимодействует. На каждом шаге
проводятся расчёты взаимодействий только внутри списка, а сами списки
пересчитываются через определённые интервалы по времени. Применение такого
метода (часто в совокупности с методом связанных списков) позволяет добиться
роста сложности как О(N).
В случае короткодействующего потенциала
достаточно просто использовать параллельные вычисления, так как наличие отсечки
позволяет разделить область вычислений на несколько более мелких областей, и
расчёт на каждом из участков можно проводить отдельным процессором. Грамотно
подобранное разбиение на области и составление списков ближайших соседей
позволяет сократить время передачи информации между процессорами и
оптимизировать вычисления.
Тем не менее, большое количество задач требует
применения дальнодействующих потенциалов. Например, расчёт электростатических
или гравитационных взаимодействий. В этом случае отсечку нужно устанавливать на
достаточно большом расстоянии, и разбиение на области становится неэффективным
в силу того, что тратится много времени на коммуникацию между процессорами.
Сложность вычисления сильно возрастает с ростом числа частиц, и для таких задач
необходимы иные методы оптимизации [1], которые, однако, не являются столь же
эффективными, как в случае короткодействующих потенциалов, поэтому размеры
моделируемых системы обычно ограничиваются тысячами или миллионами частиц.
Задание потенциала взаимодействия между
частицами является одним из ключевых этапов моделирования МД, определяющим
эволюцию системы. В программах МД существует несколько способов задания
потенциалов: в виде таблиц и в виде аналитических зависимостей.
При моделировании крупных органических молекул
(белков, полимеров) потенциальная энергия задается следующей формулой:
=UBMC +Uугл+Uторс+Uэлектростат+Uэксп,
где UВМС - потенциал внутримолекулярной связи,
Uугл - потенциал угловой связи, Uторс - торсионный потенциал, Uэлектростат -
электростатический потенциал, Uэксп - эмпирическая часть полного потенциала[7].
Ниже приведены наиболее часто употребляемые
потенциалы[5,6], описывающие взаимодействие атомов, не связанных между собой
химическими связями:
Потенциал Ленарда-Джонса
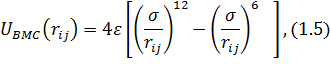
где 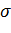 -
эффективный диаметр атома,
-
эффективный диаметр атома, 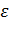 - глубина
потенциальной ямы
- глубина
потенциальной ямы
Потенциал Букингема
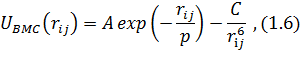
где А - амплитуда, а р - эффективный радиус
взаимодействия
Потенциал Борна-Хаггинса-Мейера
 , (1.7)
, (1.7)
Энергия взаимодействия внутримолекулярных
связей, торсионная энергия вращения задаются в виде:
Потенциал гармонической связи
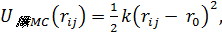 (1.8)
(1.8)
где k- жесткость связи
Потенциал Морса
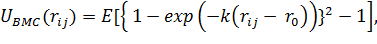 (1.9)
(1.9)
где Е - амплитуда потенциала, а k-постоянный
параметр
Потенциал валентной гармонической связи
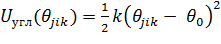 ,(1.10)
,(1.10)
где 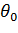 -
равновесное значение валентного угла
-
равновесное значение валентного угла
Потенциал гармонической косинусоидальной связи
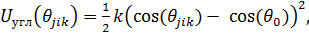 (1.11)
(1.11)
Потенциал торсионной гармонической связи
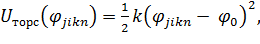 (1.12)
(1.12)
где 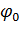 -
равновесное значение торсионного угла.
-
равновесное значение торсионного угла.
Классический кулоновский потенциал, описывающий
взаимодействие заряженных частиц, не всегда применяем для моделирования
электрон-ионного взаимодействия на малых расстояниях. Из-за резкого убывания в
нуле на короткой дистанции при расчётах получаются очень большие по модулю
отрицательные значения энергии. Это противоречит квантово-механическим
представлениям о наличии минимального уровня энергии (основного состояния) в
системе электрона и иона. Для приближенного учета этого явления куловновский
потенциал взаимодействия принято заменять псевдопотенциалом, который при
нулевом расстоянии между атомами имеет определённое конечное значение. Примером
является потенциал вида:
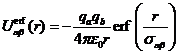 , (1.13)
, (1.13)
2. Применение гетерогенных вычислительных систем
в задачах молекулярной динамики
.1 Методы ускорения молекулярно-динамического моделирования
на ЦП
Для метода МД, особенно для многочастичных
систем с дальнодействующим потенциалом, необходима высокая производительность
вычислительных систем, в связи с чем, молекулярно-динамическое моделирование
часто возможно исключительно на передовых суперкомпьютерах с параллельной
архитектурой. Из-за этого значительную роль в моделировании играет
эффективность распараллеливания программы. Скорость работы, как и эффективность
распараллеливания, зависят от потенциала взаимодействия частиц и их количества
в рассматриваемой системе.
В задачах молекулярной динамики для
распараллеливания вычислений на многоядерныхCPUактивно используется OpenMP
(Open Multi-Processing) - это набор директив компилятора, библиотечных процедур
и переменных окружения, предназначенных для разработки многопоточных приложений
на многопроцессорных системах с общей памятью (SMP-системах).
Первый стандарт OpenMP был разработан в 1997 г.
как API, ориентированный на программирование легко переносимых многопоточных
приложений. Изначально он был основан на языке Fortran, однако в последствии
включил в себяязыки программирования СииСи++. На данный момент интерфейс OpenMP
является одной из самых популярных технологий параллельного программирования
систем с общей памятью,онуспешно применяется как при разработки
суперкомпьютерных систем с большим количеством процессоров, так и в настольных
пользовательских системах или, например, в Xbox 360.
Разработку спецификации OpenMP ведут несколько
крупных производителей вычислительной техники и программного обеспечения, чья
работа регулируется некоммерческой организацией «OpenMP Architecture Review
Board» [15].
В OpenMP используется модель параллельного
выполнения «ветвление-слияние». Программа OpenMP начинается как единственный
поток выполнения, называемый начальным потоком. Когда начальный поток
выполнения встречает параллельную конструкцию, он создает новую группу потоков,
состоящую из себя и некоторого числа дополнительных потоков, и становится
главным в новой группе. Все потоки новой группы (включая главный) выполняют код
внутри параллельной конструкции. В конце параллельной конструкции существует
неявный барьер. После окончания параллельной конструкции исполнение
пользовательского кода продолжает лишь главный поток. В параллельный регион
могут быть вложены другие параллельные регионы, в которых каждый поток
первоначального региона становится основным для своей группы потоков. Вложенные
регионы могут в свою очередь включать регионы более глубокого уровня
вложенности.
Существует несколько способов контролировать
число потоков выполняющихся параллельно. Один из них - использование переменной
окружения OMP_NUM_THREADS. Другой способ - вызов процедуры
omp_set_num_threads(). Еще один способ - использование выражения num_threads в
сочетании с директивой parallel.
Однако, современные ЦПв задачах молекулярной
динамики не могут обеспечить скорость работы сравнимую с ГУ, что обусловлено их
архитектурными отличиями.
.2 Архитектура ГУ и их использование в задачах
молекулярной динамики
Особое распространение в молекулярной
динамике получили гетерогенные вычислительные системы, в которые кроме обычных
процессоров могут входить графические ускорители (ГУ)[8,9], сигнальный цифровой
процессор (DSP), со-процессор, специализированная интегральная схема (ASIC -
Applicationspecificintegratedcircuit) или программируемая пользователем
вентильная матрица
<https://ru.wikipedia.org/wiki/%D0%9F%D1%80%D0%BE%D0%B3%D1%80%D0%B0%D0%BC%D0%BC%D0%B8%D1%80%D1%83%D0%B5%D0%BC%D0%B0%D1%8F_%D0%BF%D0%BE%D0%BB%D1%8C%D0%B7%D0%BE%D0%B2%D0%B0%D1%82%D0%B5%D0%BB%D0%B5%D0%BC_%D0%B2%D0%B5%D0%BD%D1%82%D0%B8%D0%BB%D1%8C%D0%BD%D0%B0%D1%8F_%D0%BC%D0%B0%D1%82%D1%80%D0%B8%D1%86%D0%B0>
(ПЛИС).Наиболее востребованы в задачах МД остаютсяГУ, в данной работе
проводится исследование возможности применения ПЛИС в задачах МД и
сравнительный анализ производительности ЦП, ГУи ПЛИС.Первоначально ГУ
разрабатывались как часть видеокарты, но в последствии продемонстрировали
высокую эффективность в научных вычислениях.В отличие от современных ЦП,
которые содержат маленькое число физических ядер (в основном, от 2 до 24), ГУ
разрабатывалась с использованием многоядерной архитектуры, c количеством ядер,
исчисляющимися тысячами (рис. 3).

Рис. 3Архитектура ЦПи ГУNvidia[9]
Архитектурные отличия определяют и
разницу в принципах функционирования. ГПУ изначально разрабатывались для
обработки компьютерной графики, в следствии чего ориентированы на параллелизм
на уровне данных (SIMD), в то время как архитектура ЦП изначально
разрабатывалась для последовательной обработки данных и лишь в процессе
развития получила поддержу SIMD-инструкций, внеочередное исполнение инструкций
и конвеерный параллелизм.
ГУ компании Intel (см. рис. 4-5) в
отличии от ГУ производства Nvidia содержат порядка 40 исполнительных устройств,
из-за чего их производительность в задачах молекулярной динамики значительно
уступает ГУ производства Nvidia.
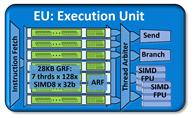
Рис.4Архитектура исполнительного
устройства в IntelHDgraphics [25]
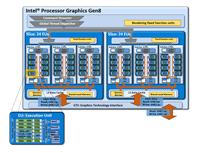
Рис.5Архитектура графического
ускорителя IntelHDgraphics [25]
Широкое распространение графических
ускорителей в задачах, несвязанных с компьютерной графикой (GPGPU), обусловлено
разработкой компаниями языков программирования и технологий, облегчающих
использование ускорителей в научных расчетах и прикладных программах, таких как
CUDA и OpenCL(рис. 6).
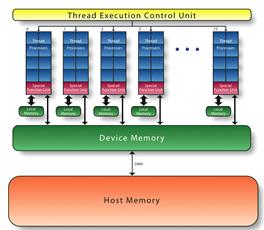
Рис.6Структура гетерогенной вычислительной
системы(ГУ)
Использование ГУ в задачах МД зачастую
обеспечивает быстродействие, во много раз превышающее быстродействие систем,
использующих только ЦП.При всех плюсах использования ГУ, нужно учитывать, что
разработка программ, эффективно использующих все вычислительные возможности
современных ГУ-довольно трудоемкий процесс, который требует рассмотрения
особенностей архитектуры отдельно для каждой задачи моделирования, в противном
случае производительность может быть не столь высокой. Максимальная
производительностьГУ в сравнении с использованием ЦП при моделировании систем
методом МД достигается при большом количестве частиц в системе, что позволяет
загрузить большое количество ядер ГУ.
.3 Архитектура ПЛИС и их использование в задачах
молекулярной динамики
ПЛИС представляют собой интегральные схемы,
предназначенные для конфигурации пользователем или разработчиком после
производства.ПЛИСдает возможность управлять обработкой данных на самом низком
уровне: создавать кэши нужного размера в нужном месте, организовывать
конвейризацию, описывать явный параллелизм. До недавнего времени
программирование ПЛИС осуществлялось только посредством hardwaredes
criptionlanguage (HDL), таких как Verilogи VHDL.Разработка на HDL, отличается
высокой сложностью, из-за чего применение ПЛИСв прикладных программах и научных
расчетах было крайне трудоемким процессом.В ноябре 2012ого года компания
Altera, на данный момент являющаяся частью корпорации Intel, выпустила первую
версию AlteraOpenCLCompiler, позволяющего программировать ПЛИСс использованием
стандарта OpenCL, аналогичная технология - SDAccelсуществует у компании Xilinx,
одного из ведущего производителя ПЛИС.
ПЛИСсостоит из массива программируемых
логических блоков и перенастраиваемых интерконнектов, позволяющих соединять
блоки вместе. Логические блоки могут быть настроены для выполнения сложных
функций или сконфигурированы как простые логические вентили ANDили OR.
[10,16,17] В большинстве ПЛИС логические блоки также включают в себя элементы
памяти. (рис. 7)
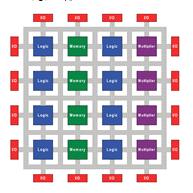
Рис.7Архитектура ПЛИС
С использованием ПЛИС можно реализовать все те
же логические функции, что и на ASIC, главное отличие состоит в том, что
пользователь может самостоятельно перенастроить ПЛИС, на реализацию другой
функции.
Чип с FPGAможет быть размещен на PCIeкарте,
подключенной к материнской плате через соответствующий разъем. Посредством DMAи
PCIeFPGAможет обращаться к DDR памяти основного процессора (ЦП). Наряду с ней
на плате можно разместить внешнюю память, доступную только для ПЛИС(рис. 8).
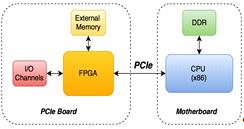
Рис.8Схема подключения FPGA посредством PCI
Внешняя память необходима для хранения
промежуточных результатов вычислений: обращение к ней происходит быстрее,
нежели доступ через DMAв память ЦП.
Данные для обработки возможно передавать в ядро
не только через глобальную память, но также и с I/O каналов, таких как
Ethernet-порты.
Второй способ размещения ПЛИС используется в
системах типаSoС(SystemonChip), в которых на одном кристалле находится
ARM-процессор и программируемая логика (рис.9).
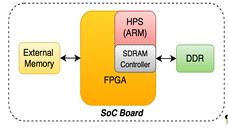
Рис.9Использование ПЛИСв SoC
память, закрашенная на рис. 9 зеленым цветом,
является разделяемым ресурсом: с одной стороны, им пользуется ЦП, а с другой,
ПЛИС может «напрямую» читать/писать в эту память через SDRAM-контроллер с
минимальными накладными расходами. Как и в случае с размещением на PCIe, к ПЛИС
может быть подключена внешняя память, но необходимость в этом меньше, из-за
наличия DDR памяти, подключенной к ПЛИС напрямую.
Процесс разработки с использованием Altera Open CL
Compiler состоит из следующих этапов (рис. 10):
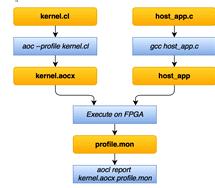
Рис.10Процесс разработки с использование
AlteraOpenCLCompiler
Код, исполняемый на ПЛИС,(ядро) описывается в
файле *.cl.
Приложение для ЦП разрабатывается на С/C++ и отвечает
за выделение и передачу необходимых объемов памяти.
С помощью утилиты aoc, которая входит в Altera
OpenCL SDK, «компилируется» ядро в aocx-файл. С помощью компилятора (например,
gcc) собирается приложение для ЦП.
При запуске приложения произойдет загрузка
прошивки ПЛИС, в ядро загрузятся подготовленные данные и начнется их обработка.
Счетчики для профилирования собирают данные,
которые поместятся в файлprofile.mon.
С помощью утилиты aocl можно посмотреть этот
отчет и сделать вывод: удовлетворяет ли по времени
выполнения/производительности текущая реализация.
Процесс сборки aocx-файла состоит из следующих
этапов (рис. 11):

Рис.11Процесс сборки aocx-файла
.cl подается на вход clang, который переводит
описание в IR, а также проводит различные оптимизации.
Генерируется RTL Verilog IP-ядро.
Сгенерированные файлы доступны для чтения и могут быть просимулированы в
обычном симуляторе (например, ModelSim).
Полученное IP «присоединяется» к базовому
проекту для платы и получается обычный проект дляСАПР Quartus.
Проходит сборка проекта. Именно этот пункт
занимает наибольшее время (от десяти минут до нескольких часов): поиск
оптимальных мест расположения примитивов требует много вычислений.
Результат сборки, информация о плате и прочее
размещаются в aocx, который является просто ELF-файлом.
Таким образом, мы видим, что создание и
выполнение приложений для ПЛИС с использованием OpenCL представляет собой
довольно сложный процесс, однако его большая часть автоматизирована, а спецификация
OpemCL хорошо документирована, благодаря этому разработка на ПЛИС с
использованием OpenCL значительно легче написания HDL кода. В тоже время
применение ПЛИС для многих задач обеспечивает производительность лучшую, чем
ГУ.
.4 Сравнение архитектур ГУ и ПЛИС
Пиковая производительность ПЛИС значительно
превышает производительность ГУ, и достигает 10 TFLOPS, что обусловлено
уникальной архитектурой ПЛИС. Ключевое отличие между исполнением ядра на ГУи
ПЛИСзаключается в типе параллелизма. Архитектура ГУадаптирована под параллелизм
типа SIMD (SingleInstruction, MultipleData), когда группа вычислительных
элементов выполняет одну операцию в своих собственных потоках исполнения. С
другой стороны, ПЛИС поддерживает конвеерный параллелизм - разные стадии
инструкций применяются к разным потокам параллельно.
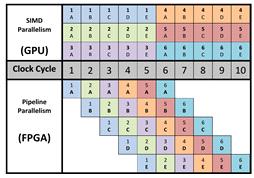
Рис. 12SIMD и конвеерный параллелизм [19]
На рисунке 12 изображено различие между этими
двумя подходами. В показанном примере используется шесть потоков выполнения,
исполняющих ядро, и пять стадий, причем оба метода распараллеливания
обеспечивают одинаковое время исполнения. Различия в стратегии
распараллеливания в ГУи ПЛИС основываются на аппаратной архитектуре устройств.
ГУ состоит из сотен ядер, исполняющих одну и ту же инструкцию, в то время как в
ПЛИС каждое ядро компилируется в отдельную цепь. (см. рис. 13)
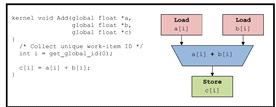
Рис. 13Упрощенный пример FPGAсхемы для ядра
OpenCL [19]
Ключевое отличие в этих подходах заключается в
способе обработки ветвей исполнения из-за того, что в ГУ используется
SIMDпараллелизм, при использовании условного перехода с N возможными опциями,
итоговое время исполнения может быть равно времени исполнения всех N опций. На
ПЛИС различные ветви выполнения программы не являются столь большой проблемой,
из-за того, что все пути исполнения устанавливаются аппаратно, во время
компиляции. Все ветви могут быть исполнены параллельно или даже вычислены
спекулятивно. (см. рис. 14)
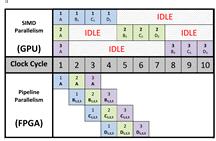
Рис.14Сравнение схем обработки ветвей исполнения
[19]
Наряду с конвеерным параллелизмом, ПЛИСтак же
поддерживает векторизацию ядра т.е. SIMD инструкции, причем у разработчика есть
возможность самостоятельно задать число SIMD потоков. Помимо SIMDинструкций
ПЛИС поддерживает множественную репликацию ядра. Разница между двумя этими
подходами заключается в эффективности передачи данных ядру, количестве
обращений к памяти. (см. рис. 15)
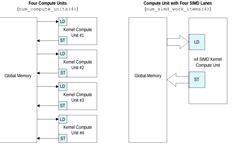
Рис.15Сравнение нескольких репликаций ядра и
SIMD [18]
является стандартом для программирования
гетерогенных вычислительных систем, который в настоящее время поддерживается
устройствами, такими как графические процессоры, процессоры общего назначения и
ПЛИС. OpenCL код полностью переносим между разными платформами в функциональном
плане, но не является переносимым с точки зрения производительности, это
означает, что один и тот же код будет выполняться на различных устройствах,
однако для достижения максимальной производительности необходимо учитывать
архитектурные особенности различных устройств. Понимание различий между SIMD
параллелизмом и pipeliningпараллелизмом, и использование таких преимуществ
ПЛИС, как I/Oканалы, анролинг циклов и поддержка гетерогенной памяти является
ключом к достижению максимальной производительности на ватт.
.1 Используемая аппаратура
В данной работе расчеты проводились
с использованием различного оборудования. В качестве ПЛИСбыла выбранаTerasicDE1-SOC,
в основе которой лежит кристалл Cyclone V
<https://www.altera.com/products/soc/portfolio/cyclone-v-soc/overview.html>(рис.
16).

Рис.14 TerasicDE1-SoC [21]
Данная плата, весьма ограниченна в
своих вычислительных возможностях, относится к низшей ценовой категории и
обладает следующими характеристиками:
Cyclone V
SoC 5CSEMA5F31C6 Devicecore ARM Cortex-A9 (HPS)
K
Programmable Logic Elements
,450 Kbits
embedded memory
6 Fractional PLLs
Hard Memory Controllers
На рисунке 17 представлена
блок-схема данной FPGA.
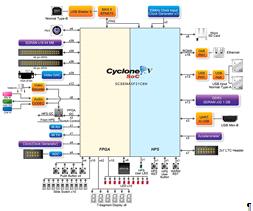
Рис.17БлокдиаграммаTerasic DE1-SoC [21]
В данной ПЛИС, всего 32 тысячи логических
элементов, в то время как наиболее производительные ПЛИСсодержат миллионы
макроячеек.
В качестве ЦП/ГУ использовались следующие
устройства:
ГУNVIDIAGeForce 960M, содержащую 640 CUDA ядер
(см. рис. 18);

Рис. 18Архитектура NVIDIAGeforce 960M [9]
ГУIntelHDGraphics 4600, содержащий 20
исполнительных устройств;
ЦПIntelCorei7 c ядром haswell, 4-мя физическими
и 8-ю логическими ядрами.
Указанные устройства являются одними из наиболее
простых и недорогих устройств, которые удалось получить для выполнения данной
работы. Однако,их архитектура, принцип функционирования и средства разработки
программ полностью соответствуют более мощным аналогам, которые и могут
составить в дальнейшем конкуренцию современным высокопроизводительным системам
на базе традиционных процессоров. Выполнение тестов на данном оборудовании позволяет:
а) определить принципиальную возможность и трудоемкость реализации на них
программ МД/МК моделирования; б) выяснить требовательность к ресурсам и
ограничение на число частиц, которые могут рассчитываться одновременно одним
устройством; в) провести оценку производительности и экстраполировать
полученные результаты на аналогичные устройства, обладающие большим числом
вычислительных блоков, тактовой частотой и пропускной способностью.
.2 Реализация алгоритмов молекулярной динамики и
Монте-Карло на ПЛИС
Для проведения тестов были созданы оригинальные
программы молекулярной динамики и Монте-Карло, адаптированные для расчетов на
ГУ, ЦП иПЛИС, с использованием средств распараллеливания OpenCLи OpenMP. В
качестве потенциалов взаимодействия рассматривался короткодействующий потенциал
- Ленард-Джонса(1.5)и дальнодействующий кулоновский потенциал
(1.13)Принципиальное отличие между ними состоит в невозможности введения
радиуса отсечки в дальнодействующих потенциалах.
.2.1 Реализация алгоритма молекулярной динамики
на ПЛИС
Алгоритм молекулярной динамики является
итеративным по времени. Используется массив трехмерных координат молекул, и
наряду с ним массивы скоростей и сил взаимодействий. На каждой временной
итерации последовательно выполняются два вложенных цикла по молекулам,
осуществляющих расчет сил и энергий взаимодействия. После чего происходит
обновление значений скоростей и координат согласно методу Эйлера. (рис.19)
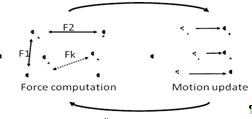
Рис.19метод молекулярной динамики
Расчет сил и энергии требует наибольших
вычислительных ресурсов, и именно он был реализован с использованием FPGA. Было
разработано NDRangeOpenCLядро, которое вычисляет энергии и силы взаимодействия
выбранной частицы со всеми остальными, наряду с ним рассматривалась реализация
OpenCLядра вычисляющего только попарные взаимодействия на ПЛИС\ГУ, однако
данный метод оказался менее эффективным с точки зрения производительности из-за
неэффективного использования пропускной способности памяти(см. рис. 20,
приложение 1,2)
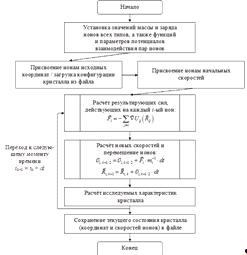
Рис.20Реализация алгоритма МД на ПЛИС
Результаты молекулярно-динамического
моделирования для кулоновского и Ленард-Джонсовского потенциалов приведены на
рисунках 21-24.
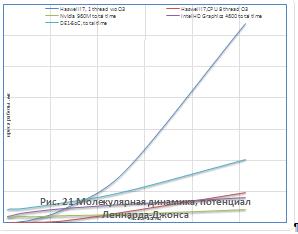

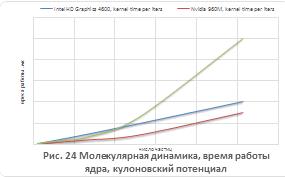
Как видно из рисунков 21 - 24, для систем,
состоящих из более чем 500 незаряженных частиц применение графических
ускорителей обеспечивает лучшую производительность в сравнении с ЦП. В то
время, как для кулоновского потенциала выигрыш в производительности
наблюдается, начиная с 50 частиц, отчасти это обусловлено разной реализацией
функции ошибок в OpenCLи math.h. Также, стоит отметить, что вплоть до 250
частиц ПЛИСCyclone 5 обеспечивала лучшую производительность, чем IntelГУ.
.2.2 Реализация метода Монте-Карло на ПЛИС
Так же, как и в методе молекулярной динамики,
расчет энергий взаимодействия был перенесен на FPGA (рис 25, приложение 3,4),
что позволило повысить производительность. Было разработано NDRangeOpenCLядро,
которое вычисляет энергии взаимодействия выбранной частицы со всеми остальными,
наряду с ним рассматривалась реализация OpenCLядра вычисляющего только попарные
взаимодействия на ПЛИС\ГУ, однако данный метод оказался менее эффективным с
точки зрения производительности из-за неэффективного использования пропускной
способности памяти, также стоит отметить, что в отличии от метода МД в методе
МК не удалось полностью использовать ресурсы ПЛИС.
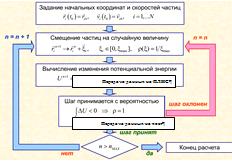
Рис. 25Алгоритм Метрополиса
Результаты моделирования методом Монте-Карло для
кулоновского и Ленард-Джонсовского потенциалов приведены на рисунках 26-29.
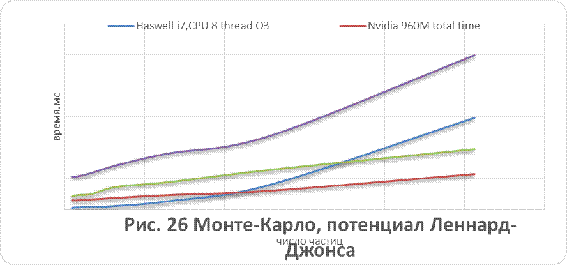
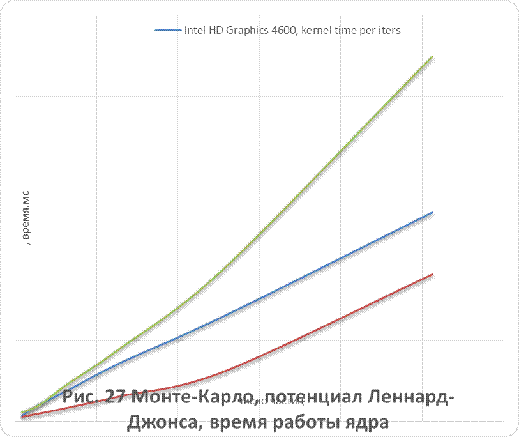
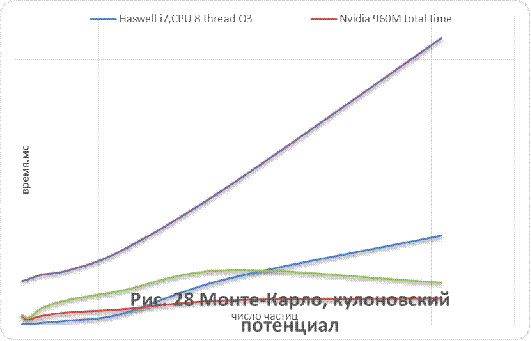
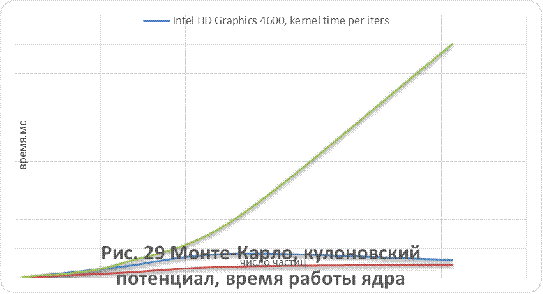
Как видно из рисунков 25 - 29, для систем,
состоящих из более чем 500 незаряженных частиц, применение графических
ускорителей обеспечивает лучшую производительность в сравнении с CPUверсией.
На данный момент наиболее
высокопроизводительными ПЛИС фирмы Intel являются ПЛИС семейства Stratix 5 и
Aria 10, содержащие порядка миллиона логических элементов и тысячи DSP
элементов, в то время как Cyclone 5, использующийся в данной работе, содержит
только 86000 логических элементов и 70 DSP блоков. Для задач молекулярной
динамики необходимы интенсивные вычисления чисел с плавающей точкой, которые
осуществляются DSPблоками. В качестве простейшей оценки можно провести линейную
экстраполяцию производительности ПЛИС в зависимости от числа логических и DSP
блоков, вплоть до ограничений связанных с пропускной способностью памяти. В
этом случае мы ожидаем 10 кратный прирост производительности на ПЛИС семейства
Stratix 5 и Aria 10в сравнении с используемой в данной работе ПЛИС Cyclone 5,
что позволит получить скорость работы лучшую чем на ГУ Nvidia, вплоть до
нескольких тысяч частиц.
Заключение
В работе была исследована возможность реализации
алгоритмов молекулярной динамики и Монте-Карло на программируемых логических
интегральных схемам (ПЛИС, FPGA) с различными типами потенциалов
взаимодействия: короткодействующим потенциалом Леннарда-Джонса, описывающим
диполь-дипольное взаимодействие незаряженных атомов, и дальнодействующим
кулоновским потенциалом для электронов и ионов в неидеальной плазме. При этом
были протестированные ПЛИС последнего поколения, поддерживающие
программирование на языкеOpenCL, что, как показала данная работа, значительно
упрощает разработку программ.
Для TerasicDE1-SOCна основе ПЛИС
AlteraCyclone V <https://www.altera.com/products/soc/portfolio/cyclone-v-soc/overview.html>
были проведены тесты производительности, результаты которых сопоставлены с
аналогичеными тестами, проведенными наЦПHaswelli7, а также с применением
графических ускорителей ____Nvidia 960Mи IntelHDGraphics 4600_____. Было показано,
что для систем, состоящих из более чем 500 незаряженных частиц применение
графических ускорителей обеспечивает лучшую производительность в сравнении
сверсией для ЦП. В то время, как для кулоновского потенциала выигрыш в
производительности наблюдается, начиная с меньшего частиц. Было показано, что
ПЛИСCyclone 5 обеспечивает производительность, сравнимую с Intel HDG raphics
4600 в задачах молекулярной динамики. Экстраполяция результатов на более мощные
модели ПЛИС показала, что ПЛИС семейства Stratix 5 и Altera 10 обеспечат
производительность в 10 раз превышающую производительность Cyclone 5,
используемого в данной работе.
Полученные результаты говорят о том,
что применение ПЛИС целесообразно в том случае, если планируется проведение
большого объема вычислений для систем со сравнительно небольшим числом частиц,
например, в задачах, связанных с усреднением по траекториями или поиском
оптимальных параметров.
Список цитированной литературы
AllenM.P.,
Tildesley
D.J.
Computer
Simulation
of Liquids.
Oxford : Clarendon Press, 1989.D., Smit B.
Understanding Molecular Simulation: From Algorithms to Applications. San Diego:
Academic Press, 2002. L. Computer "Experiments" on Classical Fluids.
Phys. Rev., v. 159, pp. 98-103, 1967; v. 165, pp. 201-214, 1968;Phys. Rev A.,
v. 2, pp. 2514-2528, 1970; v. 7, pp. 1690-1700, 1973.G., Classical molecular
dynamics. In: Quantum Simulations of Complex Many-Body Systems: From Theory to
Algorithms (eds. J. Grotendorst, et al), Julich: NIC, Vol. 10, pp. 211-254,
2002.A. Correlations in the Motion fo Atoms in Liquid Argon. Phys. Rev., v.
136, pp. A405-411, 1964.. Erkoc, Empirical many-body potential energy functions
used in computer simulations of condensed mater properties. Physics Reports.,
v. 278, pp. 79-105, 1997.P., Sutmann G. Long-Range Interactions in
Many-Particle Simulation. In: Quantum Simulations of Complex Many-Body Systems:
From Theory to Algorithms (eds. J. Grotendorst, et
al), Julich: NIC, Vol. 10, pp. 467-506, 2002.
Боресков,
А. В. Параллельные вычисления на GPU. М. Изд-во Моск. ун-та, 2012. - 333 с.
Официальный
сайт NVIDIACUDA http://www.nvidia.ru/object/cuda_home_new_ru.html
Официальный
сайт Altera https://www.altera.com/support/literature/lit-index.html
T.
Raitza, H. Reinholz, G. Roepke, I.V. Morozov. Collision frequency of electrons
in laser excited small clusters // J. Phys. A. 2009. V. 42. P. 214048.
T.
Raitza, G. Röpke, H. Reinholz, I. Morozov. Spatially resolved dynamic
structure factor of finite systems from molecular dynamics simulations //
Phys. Rev.
E. 2011. V. 84. P. 036406.
И.В.
Морозов. Моделирование кластерной наноплазмы методом молекулярной динамики //
Наноструктуры. Математическая физика и моделирование. 2011. Т. 5, № 1/2. С.
39-56..G. Bystryi, I.V. Morozov. Electronic
oscillations in ionized sodium nanoclusters // J. Phys. B. 2015. V.
48. P. 015401.
Официальный
сайт OpenMPhttp://www.openmp.org/
Стешенко
В.Б. ПЛИС фирмы ALTERA, система проектирования и языки описания аппаратуры //
ДМК Пресс, 2015.
Наваби
З. Проектирование встраиваемых систем на ПЛИС // ДМК Пресс, 2016.
Intel
FPGA SDK for OpenCL Best practice guide
https://www.altera.com/en_US/pdfs/literature/hb/opencl-sdk/aocl-best-practices-guide.pdf
Hasitha Muthumala Waidyasooriya
<http://ieeexplore.ieee.org/search/searchresult.jsp?searchWithin=%22Authors%22:.QT.Hasitha%20Muthumala%20Waidyasooriya.QT.&newsearch=true>,
Masanori Hariyama
<http://ieeexplore.ieee.org/search/searchresult.jsp?searchWithin=%22Authors%22:.QT.Masanori%20Hariyama.QT.&newsearch=true>,Kota
Kasahara <http://ieeexplore.ieee.org/search/searchresult.jsp?searchWithin=%22Authors%22:.QT.Kota%20Kasahara.QT.&newsearch=true>Architecture
of an FPGA accelerator for molecular dynamics simulation using OpenCL.
Приложение 1
Расчет энергии и сил взаимодействия на OpenCL
для кулоновского потенциала
|
/**
|
|
|
*
@file md_coulomb.cl
|
|
* @brief OpenCL kernel which calculate energy
and force
|
|
*/
|
|
|
|
/**
|
|
* @brief OpenCL kernel for coulomb potential
|
|
*
@param particles Position array
|
|
*
@param charge Charge array
|
|
* @param out_energy Energy which describe how
one particles iteract which all others
|
|
* @param out_force Force acting on the
particle from all others
|
|
*
@return void
|
|
*/
|
|
__attribute__((reqd_work_group_size(particles_count,
1, 1)))
|
|
__kernel void md(__global const float3
*restrict particles,
|
|
__global constint
*restrict charge,
|
|
__global float *restrict out_energy,
|
|
__global float3 *restrict out_force) {
|
|
intindex
= get_global_id(0);
|
|
float
energy = 0;
|
|
float3
force = (float3)(0, 0, 0);
|
|
#pragma
unroll 2
|
|
for (int i = 0; i < particles_count; i++) {
|
|
float x = particles[i].x - particles[index].x;
|
|
float y = particles[i].y - particles[index].y;
|
|
float z = particles[i].z - particles[index].z;
|
|
/* second part of implementation periodic
boundary conditions */
|
|
if
(x > half_box)
|
|
x
-= box_size;
|
|
else
{
|
|
if
(x < -half_box)
|
|
x
+= box_size;
|
|
}
|
|
if
(y > half_box)
|
|
y
-= box_size;
|
|
else
{
|
|
if
(y < -half_box)
|
|
y
+= box_size;
|
|
}
|
|
if
(z > half_box)
|
|
z
-= box_size;
|
|
else
{
|
|
if
(z < -half_box)
|
|
z
+= box_size;
|
|
}
|
|
if
(i != index) {
|
|
float3 r = (float3)(x, y, z);
|
|
float dist = fast_length(r);
|
|
float inv_dist = native_divide(1, dist);
|
|
float inv_dist_cub = native_divide(1, dist *
dist * dist);
|
|
if ((charge[index] == -1) || (charge[i] ==
-1)){
|
|
float erf_arg = native_divide(dist, SIGMA);
|
|
float multiplier = erf(erf_arg);
|
|
float inv_dist_square = inv_dist * inv_dist;
|
|
energy += charge[i] * charge[index] *
native_divide(multiplier, dist);
|
|
force += r * charge[i] * charge[index] *
((-DERIVATIVE_ERF * native_exp(-erf_arg * erf_arg) * inv_dist_square) +
multiplier * inv_dist_cub);
|
|
}
|
|
else{
|
|
energy += charge[i] * charge[index] *
inv_dist;
|
|
force += r * (charge[i] * charge[index] *
inv_dist_cub);
|
|
}
|
|
}
|
|
}
|
|
out_force[index]
= force;
|
|
out_energy[index]
= energy;
|
|
}
|
Приложение 2
Расчет энергии и сил взаимодействия на OpenCL
для потенциала Ленарда-Джонса
|
/**
|
|
|
*
@file md_lj.cl
|
|
* @brief OpenCL kernel which calculate energy
and force
|
|
*/
|
|
#include"parameters.h"
|
|
/**
|
|
* @brief OpenCL kernel for LJ
|
|
* @param out_energy Energy which describe how
one particles iteract which all others
|
|
* @param out_force Force acting on the
particle from all others
|
|
*
@return void
|
|
*/
|
|
__attribute__((reqd_work_group_size(particles_count,
1, 1)))
|
|
__kernel void md(__global const float3
*restrict particles,
|
|
__global float *restrict out_energy,
|
|
__global float3 *restrict out_force) {
|
|
intindex
= get_global_id(0);
|
|
float
energy = 0;
|
|
float3
force = (float3)(0, 0, 0);
|
|
#pragma
unroll 4
|
|
for (int i = 0; i < particles_count; i++) {
|
|
float x = particles[i].x - particles[index].x;
|
|
float y = particles[i].y - particles[index].y;
|
|
float z = particles[i].z - particles[index].z;
|
|
/* second part of implementation periodic boundary
conditions */
|
|
if
(x > half_box)
|
|
x
-= box_size;
|
|
else
{
|
|
if
(x < -half_box)
|
|
x
+= box_size;
|
|
}
|
|
if
(y > half_box)
|
|
y
-= box_size;
|
|
else
{
|
|
if
(y < -half_box)
|
|
y
+= box_size;
|
|
}
|
|
if
(z > half_box)
|
|
z
-= box_size;
|
|
else
{
|
|
if
(z < -half_box)
|
|
z
+= box_size;
|
|
}
|
|
float3 r = (float3)(x, y, z);
|
|
float sq_dist = x * x + y * y + z * z;
|
|
if ((sq_dist < (rc * rc)) && (i !=
index)) {
|
|
float r6 = sq_dist * sq_dist * sq_dist;
|
|
float
r12 = r6 * r6;
|
|
float r8 = r6 * sq_dist;
|
|
float r14 = r12 * sq_dist;
|
|
force
+= r * (24 * (2 / r14 - 1 / r8));
|
|
energy
+= 4 * (1 / r12 - 1 / r6);
|
|
}
|
|
}
|
|
out_force[index]
= force;
|
|
out_energy[index]
= energy;
|
|
}
|
Приложение 3
Расчет энергии взаимодействия на OpenCL для
кулоновского потенциала
|
/**
|
|
|
*
@file mc_coulomb.cl
|
|
* @brief OpenCL kernel which calculate energy
|
|
*/
|
|
#include"parameters.h"
|
|
/**
|
|
* @brief OpenCL kernel for coulomb potential
|
|
*
@param particles Position array
|
|
*
@param charge Charge array
|
|
* @param out_energy Energy which describe how
one particles iteract which all others
|
|
*
@return void
|
|
*/
|
|
__attribute__((reqd_work_group_size(particles_count,
1, 1)))
|
|
__kernel void mc(__global const float3
*restrict particles,
|
|
__global constint
*restrict charge,
|
|
__global float *restrict out_energy) {
|
|
intindex
= get_global_id(0);
|
|
float
energy = 0;
|
|
#pragma
unroll 4
|
|
float x = particles[i].x - particles[index].x;
|
|
float y = particles[i].y - particles[index].y;
|
|
float z = particles[i].z - particles[index].z;
|
|
if
(x > half_box)
|
|
x
-= box_size;
|
|
else
{
|
|
if
(x < -half_box)
|
|
x
+= box_size;
|
|
}
|
|
if
(y > half_box)
|
|
y
-= box_size;
|
|
else
{
|
|
if
(y < -half_box)
|
|
y
+= box_size;
|
|
}
|
|
if
(z > half_box)
|
|
z
-= box_size;
|
|
else
{
|
|
if
(z < -half_box)
|
|
z
+= box_size;
|
|
}
|
|
if
(i != index) {
|
|
float3 r = (float3)(x, y, z);
|
|
float dist = fast_length(r);
|
|
float inv_dist = native_divide(1, dist);
|
|
if ((charge[index] == -1) || (charge[i] ==
-1)){
|
|
float erf_arg = native_divide(dist, SIGMA);
|
|
float multiplier = erf(erf_arg);
|
|
energy += charge[i] * charge[index] *
multiplier * inv_dist;
|
|
}
|
|
else{
|
|
energy += charge[i] * charge[index] *
inv_dist;
|
|
}
|
|
}
|
|
}
|
|
out_energy[index]
= energy;
|
|
}
|
Приложение 4
Расчет энергии взаимодействия на OpenCL для
потенциала Ленарда-Джонса
|
/**
|
|
|
*
@file mc_lj.cl
|
|
* @brief OpenCL kernel which calculate energy
|
|
*/
|
|
#include"parameters.h"
|
|
/**
|
|
* @brief OpenCL kernel for LJ
|
|
*
@param particles Position array
|
|
* @param out_energy Energy which describe how
one particles iteract which all others
|
|
*
@return void
|
|
*/
|
|
__attribute__((reqd_work_group_size(particles_count,
1, 1)))
|
|
__kernel void mc(__global const float3
*restrict particles,
|
|
__global float *restrict out_energy) {
|
|
intindex
= get_global_id(0);
|
|
float
energy = 0;
|
|
#pragma
unroll 8
|
|
for (int i = 0; i < particles_count; i++) {
|
|
float x = particles[i].x - particles[index].x;
|
|
float y = particles[i].y - particles[index].y;
|
|
float z = particles[i].z - particles[index].z;
|
|
/* second part of implementation periodic
boundary conditions */
|
|
if
(x > half_box)
|
|
x
-= box_size;
|
|
else
{
|
|
if
(x < -half_box)
|
|
x
+= box_size;
|
|
}
|
|
if
(y > half_box)
|
|
y
-= box_size;
|
|
else
{
|
|
y
+= box_size;
|
|
}
|
|
if
(z > half_box)
|
|
z
-= box_size;
|
|
else
{
|
|
if
(z < -half_box)
|
|
z
+= box_size;
|
|
}
|
|
float sq_dist = x * x + y * y + z * z;
|
|
if ((sq_dist < rc * rc) && (i !=
index)) {
|
|
float r6 = sq_dist * sq_dist * sq_dist;
|
|
float
r12 = r6 * r6;
|
|
energy
+= 4 * (1 / r12 - 1 / r6);
|
|
}
|
|
}
|
|
out_energy[index]
= energy;
|
|
}
|