Спектрофотометрия в фармакопейном анализе
Содержание
Вступление
Глава
1. Оптические методы анализа
.1
Понятие оптических методов анализа
.2
Классификация оптических методов
.3
Некоторые элементы теории поглощения света
Глава
2. Спектрофотометрия в ультрафиолетовой и видимой областях
.1
Электронные спектры
.2
Спектрофотометры
.3
Методика спектрофотометрических измерений
.4
Кривые поглощения
.5
Калибровка спектрофотометров
.6
Отклонение от закона Ламберта - Бера
Глава
3. Инфракрасная спектрофотометрия
.1
Основа метода
.2
Инфракрасные спектрофотометры
.3
Методика измерений
Глава
4. Применение спектрофотометрии в фармакопейном анализе
.1
Испытание на подлинность органических лекарственных веществ спектометрией в
ультрафиолетовом спектре
.2
Испытание на чистоту спектометрией в ультрафиолетовом спектре
.3
Количественное определение спектометрией в ультрафиолетовом спектре
.4
Инфракрасные спектры поглощения и их применение для идентификации лекарственных
веществ
.5
Количественное определение по поглощению в инфракрасной области
.
Экспериментальная часть
.1
Методика исследования
.2
Результаты исследования
Выводы
Список
использованной литературы
Вступление
Одна из наиболее важных задач фармацевтической химии - это разработка и
совершенствование методов оценки качества лекарственных средств.
Для установления чистоты лекарственных веществ используют различные
физические, физико-химические, химические методы анализа или их сочетание.
К физическим и физико-химическим методам относятся: определение
температур плавления и затвердевания, а также температурных пределов перегонки;
определение плотности, показателей преломления (рефрактометрия), оптического
вращения (поляриметрия); спектрофотометрия - ультрафиолетовая, инфракрасная;
фотоколориметрия, эмиссионная и атомно-абсорбционная спектрометрия,
флуориметрия, спектроскопия ядерного магнитного резонанса, масс-спектрометрия;
хроматография - адсорбционная, распределительная, ионообменная, газовая,
высокоэффективная жидкостная; электрофорез (фронтальный, зональный,
капиллярный); электрометрические методы (потенциометрическое определение рН,
потенциометрическое титрование, амперометрическое титрование,
вольтамперометрия).
Спектрофотометрия (абсорбционная) - физико-химический метод исследования
растворов и твёрдых веществ, основанный на изучении спектров поглощения в ультрафиолетовой
(200-400 нм), видимой (400-760 нм) и инфракрасной (>760 нм) областях
спектра. Основная зависимость, изучаемая в спектрофотометрии зависимость
интенсивности поглощения падающего света от длины волны.
Спектрофотометрия широко применяется при изучении строения и состава
различных соединений (комплексов, красителей, аналитических реагентов и др.),
для качественного и количественного определения веществ (определения следов
элементов в металлах, сплавах, технических объектах, лекарственных препаратах).
Электронные спектры молекул являются весьма сложными.
Зависимость между строением вещества и его электронным спектром
продолжает оставаться предметом изучения многих исследователей.
Именно поэтому анализ качества лекарственных препаратов с помощью спектрофотометрии
является весьма актуальной проблемой.
Цель данной работы - осветить вопросы идентификации, методик анализа и
количественного определения препаратов с помощью спектрофотометрии .
В экспериментальной части работы проведен анализ раствора ретинола
ацетата в масле 3,44% - 10 мл, методом спектрофотометрии.
Глава 1.
Оптические методы анализа
.1 Понятие
оптических методов анализа
Оптические методы анализа основаны на использовании оптических свойств
исследуемых соединений.
Свет, как известно, представляет собой формулу лучевой энергии,
испускаемой в виде электромагнитных волн. Эти волны характеризуются длиной
волны или их частотой. Зависимость между длиной волны и ее частотой выражается
следующим уравнением:
λ * ν = С
где λ - длина волны,
ν - частота колебаний волны в циклах в
секунду,
С - скорость света в секунду в вакууме.
Световая энергия, применяющаяся в аналитических целях, ультрафиолетовая
видимая, инфракрасная, является определенной частью электромагнитного спектра
(табл. 1).
Таблица 1
Электромагнитные
спектры
|
Молекулярные вращения
|
Молекулярные колебания
|
Переходы валентных
электронов
|
|
Инфракрасная область
далекая ближняя
|
Видимая
|
Ультрафиолетовая область
ближняя далекая
|
|
150 мкм 40 мкм, 3 мкм
|
780 нм
|
380 нм, 200 нм 100 нм
|
За пределами 150 мкм находится область, близкая к микроволнам, а выше 100
нм - близкая к лучам Рентгена.
1.2
Классификация оптических методов
К оптическим относятся следующие методы:
Эмиссионный спектральный анализ - основан на наблюдении линейчатых
спектров, излучаемых парами веществ при их нагревании в пламени газовой
горелки, искры или электрической дуге. Метод дает возможность определять
элементный состав веществ.
Абсорбционный спектральный анализ в ультрафиолетовой, видимой и
инфракрасной областях спектра.
Различают спектрофотометрический и фотоколориметрический методы.
Спектрофотометрический метод анализа основан на измерении поглощения
света (монохроматического излучения) определенной длины волны, которая
соответствует максимуму кривой поглощения вещества.
Фотоколориметрический метод анализа основан на измерении светопоглощения
или определения спектра поглощения в приборах - фотоколориметрах в видимом
участке спектра.
Рефрактометрия - основана на измерении коэффициента преломления, по
которому следует судить о природе вещества, чистоте и содержании в растворах.
Поляриметрия - основана на измерении вращения плоскости поляризации.
Вещества, обладающие свойством изменять направление колебаний при прохождении
через них поляризованного света, называются оптически активными. У
поляризованного луча, пропущенного через слой раствора оптически активного
вещества, меняется направление колебаний, а плоскость поляризации оказывается
повернутой на некоторый угол, называемый углом поворота плоскости поляризации,
который зависит от поворота плоскости поляризации, концентрации и толщины слоя
раствора, длины волны поляризованного луча и температуры.
Нефелометрия - основана на использовании явлений отражения или
рассеивания света неокрашенными частицами, взвешенными в растворе. Метод дает
возможность определять очень малые количества вещества, находящиеся в растворе
в виде взвеси.
Турбидиметрия - основанная на использовании явлений отражения или
рассеивания света окрашенными частицами, которые находятся во взвешенном
состоянии в растворе. Свет, поглощенный раствором или прошедший через него,
измеряют так же, как и при фотоколориметрии окрашенных растворов.
Люминесцентный или флуоресцентный анализ - основан на флуоресценции
веществ, которые подвергаются облучению ультрафиолетовым светом. При этом
измеряется интенсивность излучаемого или видимого света.
Пламенная фотометрия (фотометрия пламени) - основана на распылении
раствора исследуемых веществ в пламени, выделении характерного для
анализируемого элемента излучения и измерении его интенсивности. Метод
используют для анализа щелочных, щелочноземельных и некоторых других элементов.
.3 Некоторые
элементы теории поглощения света
Применение оптических методов основано на свойстве веществ поглощать
световую энергию. При этом используются следующие характеристики свойств света:
длина волны (или частота) и интенсивность света.
Длина волны определяет тот предел, до которого луч света способен
взаимодействовать с любым веществом, а путем измерения интенсивности света
можно количественно определять взаимодействие между веществом и энергией луча
света.
При рассмотрении способа взаимодействия вещества и света энергию света
представляют разделенной на отдельные единицы, носящие название фотонов, или
квантов. Энергия фотона зависит от частоты излучения и определяется уравнением:
Е = ν * h,
где Е - энергия фотона в эргах;
ν - частота колебания волны в циклах в
секунду; - постоянная Планка, равная 6,624*10-27 эргов в секунду.
Следовательно, излучение при определенной длине волны состоит из фотонов,
имеющих абсолютно равное количество энергии. Интенсивность, или световая
энергия, пропорциональна числу фотонов, которые в единицу времени проходят
через единицу площади, перпендикулярной к направлению луча света.
Общая энергия молекулы для любого ее состояния может быть выражена
следующим уравнением:
Еобщ = Еэлектр + Еколеб + Евращ
Каждый из компонентов общей энергии может иметь только определенную
величину, называемую энергетическим уровнем. Молекула, у которой электронная,
колебательная и вращательная энергии имеют их наименьшее значение, находится в
так называемом основном состоянии. В этом состоянии молекула может поглощать
энергию, однако лишь в определенных количествах. Если молекула подверглась
воздействию фотонов, чья энергия соответствует разности энергии между основным
и возбужденным состояниями молекулы, то происходит поглощение молекулой энергии
и вследствие этого молекула переходит на более высокий энергетический уровень.
Более высокие уровни называют первым, вторым и т. д. возбужденными состояниями.
Каждому электронному уровню соответствует одно основное и несколько
возбужденных колебательных состояний, аналогично каждому колебательному уровню
соответствует один основной и несколько возбужденных вращательных уровней.
С другой стороны, если существует значительная разница в энергии фотонов
и разности энергий двух состояний, может не быть никакого поглощения.
Таким образом, электронные, колебательные и вращательные энергии молекулы
могут иметь только определенные, дискретные значения, иначе говоря, энергии в
молекуле квантизированы.
Поглощение молекулой излучения может привести в зависимости от энергии
фотона к следующим изменениям:
1. увеличению электронной энергии вследствие перераспределения
электронов и перехода их на более высокий уровень;
2. увеличению колебательной энергии (распределение энергии между
двумя ядрами);
. увеличению вращательной энергии (уокорение вращения диполя).
Если молекула поглощает (небольшое количество энергии, излучаемой
источником в далекой инфракрасной или микроволновой области, то изменяется
только ее вращательная энергия, а электронная и колебательная энергия остаются
прежними. Бели же источник излучения характеризуется более высокой энергией,
соответствующей близкой инфракрасной области, то возрастает как вращательная,
так и колебательная энергия молекулы. Излучение более высокой энергии,
соответствующей ультрафиолетовой и видимой областям, приводит к изменениям всех
трех видов энергии - вращательной, колебательной и электронной.
Молекулы вещества очень недолго находятся в возбужденном состоянии,
продолжительность их существования порядка 10-8 сек. Следовательно, энергия не
аккумулируется в системе, а .вещество немедленно растрачивает избыточную
энергию несколькими путями, которые могут быть физическими или химическим.
Энергия может выделиться в виде тепла или флюоресцентного излучения.
Повторное излучение энергии в виде флюоресценции происходит ;в молекулах,
у которых процессы деактивации протекают несколько иначе и полная деактивация
путем столкновения или химической реакции затруднена. Такие молекулы могут
иметь более высокую колебательную энергию в возбужденном состоянии, чем в
основном состоянии. Эта колебательная энергия теряется путем столкновения на
высшем электронном уровне, после чего молекула флюоресцирует, т. е.
возвращается в основное состояние с выделением энергии в виде излучения.
Флюоресцентная энершя меньше по величине, чем энергия падающего света, т. е.
имеет большую длину волны. Флюоресценция немедленно прекращается при устранении
источника радиации, что и отличает это свойство от фосфоресценции, которая
продолжается некоторое время после устранения источника излучения.
Вещество может подвергнуться гомолитической диссоциации или ионизации.
Выше уже отмечалось, что излучения разнятся по содержанию энергии в зависимости
от длин волн. Для разрыва межатомной связи в молекуле требуется энергия порядка
50-100 ккал/моль; следовательно, для разрыва связи необходимо поглощение
квантов видимого света (от 55 до 70 ккал/моль) или ультрафиолетового (около 140
ккал/моль).
Изучением химических реакций, возникающих при воздействии
электромагнитного излучения, занимается фотохимия.
Определения, связанные с измерением поглощения света, основаны на двух
физических законах.
Когда свет проходит через вещество, интенсивность излучения уменьшается
по сравнению с интенсивностью излучения, падающего на вещество (рис. 1).
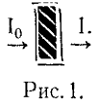
Закон Бугера-Ламберта связывает поглощение с толщиной слоя поглощающего
вещества и выражается соотношением:
(I0 / I) = k1 * b,
где I0 - интенсивность излучения,
падающего на вещество; - интенсивность излучения, прошедшего через вещество;
b -
толщина слоя вещества в сантиметрах;
Второй закон поглощения Бера связывает интенсивность падающего света и
света, прошедшего через раствор определенной толщины, с концентрацией раствора.
При этом предполагается, что растворитель не поглощает в данной области спектра:
(I0 / I) = k2 *
С,
где k2 - константа, зависящая от способа
выражения концентрации раствора;
С - концентрация раствора.
Оба закона могут быть сведены в одно уравнение, которое известно под
названием закона Бугера - Ламберта - Бера, закона Ламберта - Бера или просто
закона Бера:
(I0 / I) = k * b * С,
Раздел терминологии, относящейся к оптическим методам анализа, остается
унифицированным, описывается согласно Государственной фармакопеи X издания с
некоторыми изменениями согласно Второму изданию Международной фармакопеи.
Соотношение lg (I0 / I) известно как поглощение (А), оптическая плотность (D), или как экстинкция (Е).
Значение k зависит от
единиц, в которых выражают концентрацию вещества и толщину слоя. Если выразить
С в грамм-молях на 1 л раствора, а b в сантиметрах, то коэффициент поглощения будет равен молярному
коэффициенту поглощения. Последний изображается греческой буквой эпсилон - ε.
Если
концентрация выражается в граммах вещества на 100 мл раствора, то эта величина
называется удельным показателем поглощения и обозначается символом 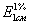 или Е (1 %, 1 см).
или Е (1 %, 1 см).
Известно
также выражение поглощения при концентрации в граммах вещества на 1 л раствора
- поглощаемость - а. Эта величина в 10 раз меньше, чем удельный показатель
поглощения.
Приведенные
ниже формулы определяют зависимость между величиной поглощения, Е (1 %, 1см), и
молярным коэффициентом поглощения.
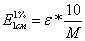
где
М - молекулярный вес и соответственно
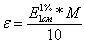
Глава
2. Спектрофотометрия в
ультрафиолетовой и видимой областях
.1
Электронные спектры
Ультрафиолетовый спектр включает область длин волн от 200 до 380 нм,
видимая часть спектра находится в пределах от 380 до 780 нм.
Электронные спектры молекул являются весьма сложными. Возникающие полосы
поглощения являются результатом взаимодействий различных видов энергий,
сопровождающих электронные изменения. Ультрафиолетовые спектры многоатомных
молекул даже в газовой фазе состоят из широких полос поглощения или
перекрывающихся полос, так как наблюдается большое количество близко
расположенных подуровней. При переходе из основного состояния в возбужденное
перемещение электронов в ультрафиолетовой области является наиболее вероятным и
значительным по величине. Изменение других видов энергии, связанное с
различными количествами колебательной энергии, также возможно, но проявляется
менее заметно. Изменения энергии вращения также сопровождают электронные
изменения, но они имеют еще меньшую величину и обусловливают тонкую структуру,
налагаемую на электронно-колебательное изменение.
В случае спектров жидких веществ и растворов вращательная и
вращательно-колебательная структуры могут отсутствовать вследствие
взаимодействия между соседними молекулами .растворенного вещества и влияния
растворителя. Большинство химических исследований относится именно к этим
условиям.
Зависимость между строением вещества и его электронным спектром
продолжает оставаться предметом изучения многих исследователей.
.2
Спектрофотометры
Измерение поглощения в ультрафиолетовой и видимой областях производится
на фотоэлектрических спектрофотометрах. В Советском Союзе выпускались
однолучевые, призменные, нерегистрирующие приборы СФ-4 и СФ-4А для измерений в
ультрафиолетовой, видимой и ближней инфракрасной областях спектра (от 220 до 1100 нм), нерегистрирующий
прибор с дифракционной решеткой СФД-2 для измерений от 220 до 1100 нм,
однолучевой, призмепной, нерегистрирующей спектрофотометр СФ-5М для измерений
от 380 до 1100 нм, и двухлучевые, призменные, регистрирующие приборы СФ-2М и
СФ-10 для измерений в видимой части спектра от 400 до 750 нм.
За рубежом и в современной Украине используются также нерегистрирующие и
регистрирующие спектрофотометры типа Бекман (США), Перкин-Элмер (США), Уникам
(Англия), Хилгер-Увиспек (Англия), Цейс (ГДР) и другие серийные приборы.
Основными частями любого спектрофотометра являются источник непрерывного
излучения, монохроматор,
кювета для
анализируемого раствора, детектор и регистрирующее устройство.
Оптическая схема простейшего спектрофотометра приведена на рис. 2.
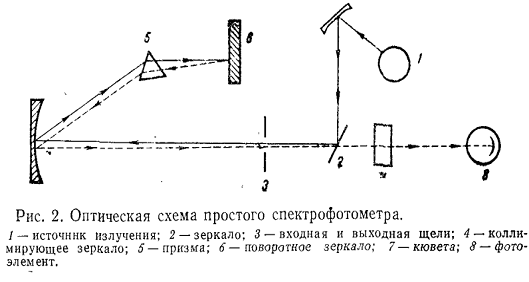
В качестве источников излучения в приборах наиболее широко используются газоразрядная
водородная лампа и вольфрамовая лампа накаливания.
Газоразрядная водородная лампа обеспечивает сплошной спектр в
ультрафиолетовой области и особенно удобна для измерений от 200 до 350 нм.
Вольфрамовая лампа накаливания используется для работы в ближней
ультрафиолетовой области, видимой и ближней инфракрасной области, т. е. в
пределах от 320 до 3000 нм. Ртутные лампы обеспечивают очень высокую
интенсивность в ультрафиолетовой и видимой областях, давая интенсивную линию
спектра ртути и сплошное излучение. Ртутные лампы необходимо нагревать в
течение 15 минут, прежде чем они начнут давать постоянное излучение.
Недостатком является высокая температура, которую ртутная лампа
приобретает при работе.
Ксеноновые разрядные лампы применяются в ряде приборов для измерений в
области от 200 до 900 нм.
Монохроматор - приспособление для изолирования очень узкой полосы
излучения ;из источника света. Смешанное излучение проходит через щель в
монохроматор, в котором луч разделяется на спектр при помощи призмы или дифракционной
решетки. Этот спектр фокусируется на выход щели. Путем вращения призмы или
дифракционной решетки можно выделить определенную часть спектра, которая через
щель направляется в кюветное отделение, где находится раствор исследуемого
вещества.
Угол отклонения между первоначальным направлением луча и направлением, в
котором он проходит через призму, зависит от показателя преломления материала,
из которого сделана призма. Показатель преломления любого материала изменяется
в зависимости от длины волны, что определяется следующим уравнением:
= n0 + C / (λ - λ0),
где n - показатель преломления при
определенной длине волны;
λ - длина волны;
C; n0; - константы.
Следовательно, когда луч немонохроматической радиации входит в призму,
составляющие его длины волн отклоняются под разными углами. Тот же процесс
повторяется при выходе луча из призмы. Таким образом, получается спектр, в
котором короткие волны отклоняются от их начального направления больше, чем
длинные.
Угловая дисперсия - это изменение угла диспергированного луча с
изменением длины волны. Дисперсия не изменяется линейно в зависимости от длины
волны.
Разрешающая сила призмы определяется способностью инструмента разделять
две спектральные линии, отличающиеся на длину волны dλ.
= λ / dλ
= t * dn / dλ
где λ- средняя длина волны двух линий, незначительно отличающихся
друг от друга;
dλ- различие в длинах воли двух линий;
t -
толщина основания призмы;
n -
показатель преломления призмы.
Материал, из которого изготавливаются призмы, выбирается с расчетом
получения максимальной дисперсии и хорошей пропускаемости в определенной
области спектра. Призмы из стекла используются в видимой области, из кварца - в
ультрафиолетовой и ближней инфракрасной области. Призмы по сравнению с
дифракционными решетками обеспечивают более чистый спектр.
Дифракционные решетки дешевле, чем призмы, и могут быть использованы для
всех областей спектра, так как пропускаемость в данном случае не имеет
определяющего значения. Дифракционная решетка состоит из большого числа
параллельных линий, нанесенных на стекло или на поверхность металла. Спектры,
получаемые с дифракционной решеткой, не так чисты, как призменные, потому, что
образуется спектр более чем одного «порядка».
Когда свет отражается от дифракционной поверхности, спектры образуются на
обеих сторонах перпендикуляра в соответствии со следующим уравнением:
* λ
= d * (sin i + sin Θ),
где n - порядок спектра;
λ - длина волны; d - расстояние между линиями дифракционной решетки;
i -
угол падения;
Θ - угол дифракции.
Дисперсия от дифракции остается практически постоянной при изменении
длины волны, и разрешающая сила решетки определяется порядком спектра и числом
линий на освещенной части дифракционной решетки.
= n * N,
где R - разрешающая сила решетки;
n -
порядок спектра; - число линий.
Разрешающая сила также зависит от качества дифракционной решетки. Любые
недостатки в точности нанесения линий могут привести к появлению несколько
смещенного изображения линий. Обычно получают спектр несколько более высокого
порядка, чем ожидаемый.
Для обеих систем диспергирования света необходимы коллимирующие и
фокусирующие линзы или зеркала, обычно комбинируемые с диспергирующим
устройством.
В абсорбционной спектроскопии применяются кюветы разных размеров,
изготовленные из кварца или стекла. Как и призмы, кюветы сделаны из материала,
обладающего высокой пропускной способностью в определенной части спектра.
Кварцевые кюветы пригодны для измерений как ультрафиолетовой, так и в видимой
области; стеклянные же могут быть использованы только в видимой области.
Толщина слоя в кюветах колеблется от 0,1 до 10 см. Чаще всего измерения
проводят в кюветах с толщиной слоя 1 см. Трудно производить кюветы, абсолютно
идентичные по пропускаемости, поэтому одну и ту же кювету обычно используют
только для растворителя. Поправка на различное поглощение кювет определяется
путем сравнения поглощения обеих кювет, наполненных чистым растворителем.
Следует обращать внимание на чистоту кювет и состояние их оптической
поверхности, так как оба этих фактора влияют на показания поглощения.
Для измерения поглощения света необходимо фотометрическое устройство.
Применяемые для этих целей фотоэлементы, фотоэмиссионные лампы и фотоумножители
основаны на известном эффекте перехода световой энергии в электрическую.
Фотоэлементы дают относительно сильный ток, который может быть измерен
при помощи гальванометра. Фотоэлементы чаще всего применяются в
фотоэлектроколориметрах.
Фотоэмиссионные лампы - это разреженные трубки, содержащие два электрода,
один из которых при облучении испускает электроны, так как покрыт
светочувствительным материалом (щелочной металл, нанесенный на слой окиси
серебра или сурьмы). Возникающий при этом ток очень слабый, поэтому необходимо
применять усилительные устройства.
Эмиссионные лампы применяют по следующим основным причинам. Вследствие
низкого внутреннего сопротивления усиление тока в фотоэлементе затруднено. В
спектрофотометре используется более узкий луч света, чем в колориметре,
благодаря чему ток в фотоэлементе был бы слишком слаб для измерения. Ток фотоэлемента,
подвергаемого постоянному освещению, медленно снижается во времени. Наконец,
спектральный ответ фотоэлементов ограничивается видимой частью спектра,
фотоэлементы почти бесполезны в ультрафиолетовой области.
Природа покрытия определяет область волн, в которой эмиссионная лампа
может быть использована (от 300 до 500 нм для слоя металлического натрия и от
200 до 700 нм для слоя калия).
Фотоумножительные устройства являются дальнейшим развитием
фотоэмиссионных ламп. Первичные электроны, испускаемые фоточувствительным
электродом, направляются на следующий электрод, который в свою очередь
испускает несколько электронов на каждый падающий на него электрон и т. д.
После ряда таких этапов удается значительно усилить ток при сохранении очень
небольшой величины начального тока.
.3 Методика
спектрофотометрических измерений
Существует два типа спектрофотометров: однолучевые и двухлучевые.
В однолучевом приборе луч света, выходящий из монохроматора, проходит
через одну кювету и затем попадает в детектор. Определение поглощения
производят следующим образом. Вначале прибор устанавливают на нуль
пропускаемости (бесконечная величина поглощения) с детектором в темноте, что
делается для компенсации слабого тока, который имеется даже при отсутствии
излучения и возникает вследствие эмиссии тепловых электронов. Затем в луч
помещают кювету, содержащую растворитель, и прибор устанавливается для
измерения в единицах пропускаемости (нуль поглощения) при определенной длине
волны. Наконец, кювету с растворителем заменяют кюветой с раствором
исследуемого вещества и производят измерение.
По этой методике измеряют два фототока - один пропорциональный
интенсивности луча, прошедшего через растворитель, и второй - пропорциональный
интенсивности луча, прошедшего через раствор вещества. Чтобы соотношения этих
токов были эквивалентны пропускаемости, надо источник излучения и детектор
оставлять постоянными в пределах, когда пропускаемость установлена на единицу и
когда пропускаемость уменьшается при измерении поглощения вещества. Следовательно,
особое внимание необходимо обращать на постоянное напряжение, подающееся на
лампу.
В двухлучевом спектрофотометре эта проблема разрешена следующим образом.
Излучение, выходящее из монохроматора, разделяется на два луча, имеющие
одинаковые интенсивности и спектральные распределения. Один из лучей проходит
через кювету с растворителем, другой - через кювету с исследуемым веществом. На
отношение излучений, выходящих из обеих кювет, величина источника света не
оказывает никакого влияния. Отношение излучений может быть измерено двумя
способами.
Лучи, вышедшие из кювет, направляются на катоды двух фотоэмиссионных ламп
или фотоумножителей. Выходы этих детекторов связаны серией сопротивлений,
усиливают разницу между двумя фототоками и регистрируют величину поглощения.
Удобством двухлучевых приборов является возможность регистрации показаний.
Обычно для получения спектров требуется от 0,1 до 100 мг вещества в
зависимости оу молярного коэффициента поглощения. Растворы, применяемые в
спектрофотометрии, являются очень разведенными, что обусловливает необходимость
точного взвешивания образца и точного отмеривания раствора при последующих
разведениях. Подходящей для измерений концентрацией является та, которая обеспечивает
показания поглощения между 0,2 и 0,7, что соответствует пропускаемости от 65 до
20%.
Определенное повышение точности спектрофотометрического анализа
достигается путем, так называемой дифференциальной фотометрии. Этот метод
основан на сравнении поглощения неизвестного раствора и поглощения стандартного
раствора, последний подобран таким образом, что разница в поглощениях будет
находиться в пределах, в которых измерение может быть выполнено с достаточной
точностью. Дифференциальный способ принят Скандинавской фармакопеей в качестве
основного метода для спектрофотометрических определений.
Согласно одному из вариантов дифференциального метода, называемого иногда
ΔЕ или Δε методом, измерение проводится путем
сравнения .неизвестного вещества при определенной величине рН к той же
концентрации вещества, но при другой, отличной от первой, величине рН.
Например, для определения фенолов разводят часть раствора щелочным, буфером, а
другую часть кислотным буфером. Измеряют поглощение более щелочного раствора,
используя для сравнения кислотный раствор. Рассчитывают содержание фенолов,
применяя ΔЕ значение, найденное для чистого вещества в той же паре
буферных растворов.
Таким образом определяется содержание гексахлорофена в жидком мыле по
Фармакопее США XVII. Так как ряд веществ не показывает изменений в спектральных
характеристиках при изменении рН, дифференциальный метод для фенолов можно
считать более специфичным, чем химический метод.
.4 Кривые
поглощения
Электронные спектры молекул графически изображаются |В виде кривых
поглощения. Для этого наносят по оси ординат единицы поглощения, а по оси
абсцисс - единицы длин волн. Единицы поглощения обычно выражают в виде
абсолютного поглощения А, удельного показателя поглощения Е (1%, 1 см) или
молярного показателя поглощения, или в процентах пропускания Т. Значения волн
выражают в нанометрах (нм), иногда в волновых числах (см-1) и редко в
ангстремах (Å).
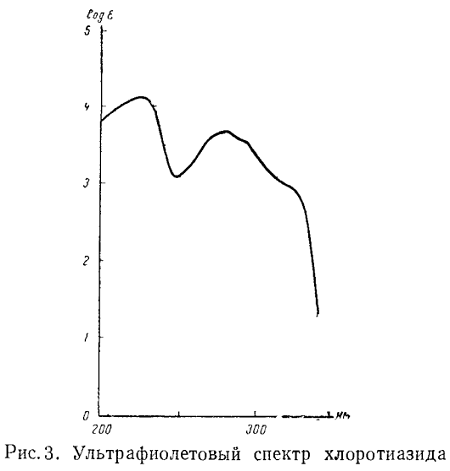
Так как величина поглощения зависит от концентрации раствора и толщины
слоя, то при построении кривых поглощения рекомендуют переводить ее в молярный
показатель поглощения, который является характерным для вещества (рис. 3).
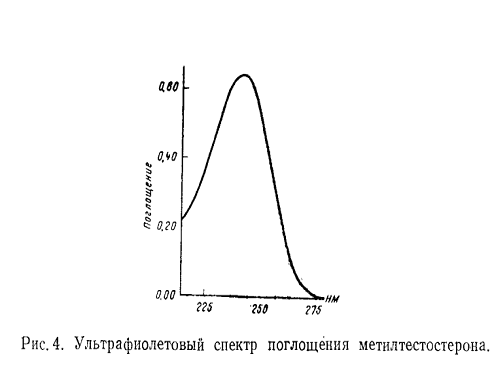
Однако при условии постоянной толщины слоя и одинаковой концентрации
построение графика зависимости поглощение - длина волны является наиболее
удобным для быстрого установления характера спектра и количественных
определений. Последний способ изображения принят большинством фармакопей (рис.
4).
Электронные спектры поглощения неорганических и органических веществ
издаются в разных странах периодически в виде отдельных сборников. Число таких
спектров достигает обычно нескольких сотен, что затрудняет работу аналитиков,
интересующихся спектрами лекарственных веществ и близких к ним соединений.
Данное обстоятельство говорит о целесообразности выпуска атласов
ультрафиолетовых спектров лекарственных веществ.
.5 Калибровка
спектрофотометров
Приборы следует проверять время от времени в отношении шкалы длин волн и
точности фотометрической шкалы поглощения (разрешающая способность прибора).
Наилучшим источником для калибровки шкалы длин волн является
ртутно-кварцевая лампа, линии которой при 239,95, 248,30, 253,65, 280,4,
302,25, 313,16, 334,15, 365,48, 404,66, 435,83, 546,1, 578,0, 623,44, 671,52 и
690,72 нм рекомендуются большинством фармакопей. Линии спектра газоразрядной
водородной лампы при 486,13 и 656,28 нм могут быть также использованы. Шкалу
длин волн проверяют при помощи подходящих стеклянных фильтров, которые имеют
характерные полосы поглощения в видимой и ультрафиолетовой областях.
Рекомендованы стандартные стекла, содержащие дидимий (смесь редкоземельных
элементов празеодимия и неодимия). Известно также применение для этих целей
стекла, содержащего холмий.
Шкалу поглощения проверяют, по цветным фильтрам, имеющим стандартное
поглощение, или по стандартным растворам щелочного хромата калия или
подкисленного бихромата калия. Рекомендуются также растворы сульфата меди и
сульфата кобальт-аммония. Наиболее удобен раствор бихромата калия, так как он
поглощает не только в ближней, ультрафиолетовой, но и в видимой области.
.6 Отклонение
от закона Ламберта - Бера
Отклонения от закона Ламберта - Бера могут быть обусловлены различными
изменениями с анализируемым веществом или относятся к недостаткам прибора.
Нарушение закона иногда является результатом изменения концентрации
растворенного вещества вследствие ассоциации между молекулами растворенного
вещества или между молекулами растворенного вещества и растворителя, или
вследствие ионизации, что часто зависит от природы растворителя.
Ширина щели является одной из наиболее важных переменных величин в
спектрофотометрии. Чистота (монохроматизм) излучения частично зависит от ширины
щели. Относительная интенсивность полосы при любой длине волны будет
неодинаковой; наивысшая интенсивность приходится на центр полосы и уменьшается
к ее краям. Если входную щель уменьшить, относительная интенсивность становится
более однородной. Однако нельзя уменьшать ширину щели бесконечно, существует
предельная величина (оптимальная ширина щели), за которой любое уменьшение щели
снижает интенсивность излучения. Центр полосы, имеющий максимальную
интенсивность, называется средней длиной волны и является величиной, отмечаемой
на шкале длин волн.
Ширина щели имеет большое значение при анализе веществ с узкими
адсорбционными полосами. Если используется слишком широкая щель, то существует
возможность, что измерение будет выполнено при длинах волн на каждой стороне
острого максимума («пика»), в связи с чем показание (величина) поглощения
окажется низким.
Имея в виду этот факт, Британская фармакопея указывает, что при измерении
поглощения три максимуме адсорбции ширина спектральной щели должна быть меньше
половины ширины полосы поглощения, иначе может быть ошибочно определена низкая
величина поглощения. Особенно внимательно нужно относиться к измерениям таких
веществ, как апоморфина гидрохлорид, хингамин, дийодгидроксихинолин, нафазолина
нитрат, папаверина гидрохлорид и проциклидина гидрохлорид, которые имеют очень
узкую полосу поглощения.
При обычных рабочих условиях ширина полосы составляет от 1 до 5 нм на
щель, что является удовлетворительным, за исключением узких максимумов.
Возможные ошибки независимо от ширины щели вероятны в случае, если
излучение источника и чувствительность фотоэлемента низкие, пропускаемость
растворителя, кювет или призм неудовлетворительная вследствие старения,
загрязнения, повреждения или если определение проводится на пределе возможной
области.
С призменными инструментами ширина полосы, соответствующая установленной
ширине щели, не является постоянной по всей шкале волн, так как разрешение
спектра изменяется с длиной волны и увеличивается с ее уменьшением. Настройка
ширины щели не является необходимой для ряда приборов, в которых всегда
используются условия максимальной чувствительности.
Большинство спектрофотометрических измерений проводится в растворах. В
качестве растворителей для области от 220 до 780 нм используются
дистиллированная вода, метиловый, этиловый и другие спирты, диоксан, эфиры и низшие
углеводороды. При выборе растворителя важно, чтобы он не поглощал в той же
части спектра, что и растворенное вещество. Для получения спектроскопически
чистых растворителей последние подвергают дополнительной очистке. Проба
растворителей на отсутствие бензола может быть выполнена путем измерения
поглощения в области от 230 до 265 нм, образцом для сравнения служит
дистиллированная вода.
Британская фармакопея определяет, что «при измерении поглощения раствора
при определенной длине волны поглощение контрольной кюветы и растворителя не
должно превышать 0,4 и в общем должно быть меньше чем 0,2, когда измеряется по
отношению к воздуху при той же длине волны». Растворители должны быть свободны
от флюоресценции, что может привести к возникновению эффекта рассеяния света и
последующей ошибке при измерении.
Структура и положение полос поглощения для ряда веществ зависят от
природы растворителя. Полярные растворители, как правило, изменяют положение
полос и упрощают их колебательную структуру. На рис. 5 и 6 приведены спектры
адреналина и сульфафуразола в зависимости от реакции среды.
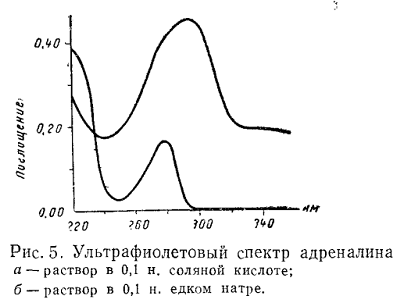
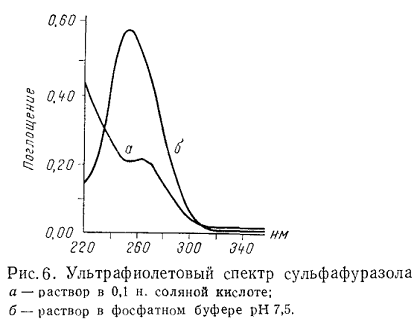
При сравнении двух кривых поглощения необходимо использовать один и тот
же растворитель, поэтому в фармакопейном анализе указывают рН среды или
растворитель, который используют в опыте.
Отклонения от закона Ламберта - Бера могут быть также вызваны рассеянием
света. Рассеяние света - это любое излучение, которое попадает в фотоэлемент,
минуя раствор. Причинами возникновения рассеяния света могут быть неправильный
выбор фильтров, слишком широкая полоса поглощения, установленная на приборе, а
также сильное поглощение и флюоресценция растворителя.
Вследствие недостатков оптической системы прибора небольшое количество рассеянного
излучения может обычно присутствовать, однако оно возрастает по мере старения
зеркал и источника излучения, что иногда приводит к получению ложного
максимума. Такие максимумы обычно для предела ультрафиолетовой области около и
ниже 220 нм, следовательно, показания ниже 220 нм нельзя считать достоверными,
если не доказано отсутствие рассеянного света или его количеством можно
пренебречь.
Поправку на рассеяние света можно определить путем измерения поглощения
для двух или более различающихся слоев, где поглощения должны быть в прямой
зависимости от толщины слоя.
Глава 3.
Инфракрасная спектрофотометрия
.1 Основа
метода
Спектрофотометрия в инфракрасной области сравнительно недавно стала
применяться в фармакопейном анализе. Она была впервые введена в ГФХ, Фармакопею
США XVI, Британскую фармакопею 1963 г. и во Второе издание Международной
фармакопеи.
Известно, что молекулы вещества, независимо от его физического состояния
и его природы, находятся в динамическом состоянии. Кроме переходов с одного уровня
на другой, электроны колеблются вокруг и между двух или более положительно
заряженных атомных ядер. Ядра в свою очередь сами движутся не только как целая
единица, но и колеблются по отношению друг к другу, а также вращаются вокруг
центра тяжести в молекуле. Все эти движения происходят с установленными
частотами и характеризуются определенной величиной энергии.
Выше отмечалось, что молекулы вещества могут поглощать различные виды
световой энергии. В частности, энергия, необходимая для получения эффекта колебания,
приходится на область от 0,5 до 25 мкм.
Частота колебания молекулы или отдельной функциональной группы
определяется геометрическим расположением всех атомов в молекуле, массой
атомов, участвующих в движении, и прочностью в положении равновесия связи или
связей, на которые оказано воздействие. Следовательно, любое вещество поглощает
в инфракрасной области и лишь немногие вещества имеют поглощение в виде одной
или нескольких полос.
Для двухатомной молекулы волновое число поглощения, выраженное в см-1,
определяется следующей зависимостью:
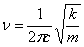 ,
,
где
k - прочность связи; с -скорость света; m -
уменьшение массы двух атомов, которое в свою очередь выражается уравнением:
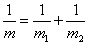
В
целом спектры инфракрасного поглощения являются чрезвычайно сложными, так как
определяются большой группой взаимозависимых факторов.
.2
Инфракрасные спектрофотометры
Приборы, применяемые в инфракрасной области, отличаются от
ультрафиолетовых в отношении источников излучения, оптических материалов и
детекторов.
Источниками излучения в большинстве приборов являются лампы накаливания.
Они представляют собой палочки карбида кремния (силитовый стержень - глобар)
или очищенных окисей редкоземельных элементов - циркония, тория, иттрия (штифт
Нернста). Оба элемента нагреваются электрически и при температуре 1200-2000°
излучают радиацию с максимумом между 1,5 и 2,5 мкм, по типу радиации черного
тела.
Кварцевые и стеклянные призмы вследствие их сильного поглощения
непригодны для работы в основной инфракрасной области, т. е. ниже 3,6 мкм.
Здесь необходимо использовать ионные кристаллы, имеющие колебания низкой
частоты.
Невозможно использовать одну и ту же призму для всей инфракрасной
области. Выбор призмы представляет собой компромисс между диспергирующей
способностью и пропускаемостью призмы. Как, правило, призма становится
наилучшим диспергирующим материалом при условиях наихудшей пропускаемости.
На практике в большинстве приборов применяют призмы из хлорида натрия
(каменная соль). Если необходима большая точность или избирательность,
используют призмы из фторида лития или фторида кальция. Иногда призменное
устройство комбинируют с дифракционной решеткой.
Галоиды щелочных металлов должны быть защищены от высоких концентраций
паров воды, в связи с чем следует проводить контроль температуры и влажности в
лабораторном помещении.
Как и в ультрафиолетовой области, здесь применяют систему зеркал. Чаще
всего основной частью монохроматора является оптическое устройство
(автоколлимационная схема Литтрова), в котором луч дважды проходит через призму
и таким образом достигается двойное диспергирование.
Исключительная сложность инфракрасных спектров обусловила создание регистрирующих
автоматических приборов, являющихся в большинстве своем двухлучевыми.
Схематическое изображение одного из таких приборов (типа Бекман) приведено на
рис. 7.
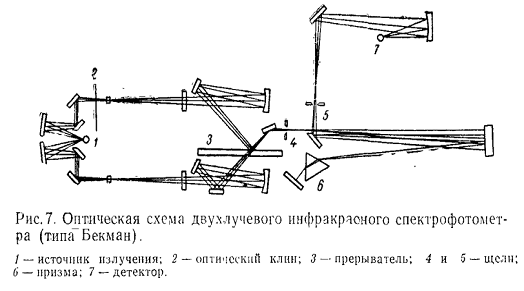
Инфракрасное излучение источника разделяется зеркалами на два луча, один
из которых проходит через образец, а другой через контроль. Луч, выходящий из
контрольного отделения, отражается на посеребренную часть прерывателя, а затем
на другое зеркало. Прерыватель последовательно направляет энергию от образца, а
затем от контроля через оптическую систему (монохроматор) к детектору для
измерения. После прохождения щели энергия через сферическое зеркало
натравляется на систему, состоящую из призмы и зеркала, которые медленно
вращаются для обеспечения необходимой спектральной области. Луч дважды проходит
через призму и затем через выходную щель попадает в детектор, который таким
образом последовательно получает импульсы энергии, прошедшей через образец, и
энергии, прошедшей через контроль.
Так как невозможно получить постоянное количество энергии излучения из
инфракрасного источника, две щели автоматически расширяются и сужаются для
получения постоянного уровня энергии в нужной спектральной области н
обеспечения чистоты спектрального излучения.
Вращающееся призменное устройство синхронизировано с движением диаграммы,
на которой регистрируется спектр.
Интенсивность радиации, прошедшей через контрольную кювету, обычно выше,
чем прошедшей через образец, так как последний поглощает часть энергии. Для
компенсации энергии детектор связывают с оптическим клином, который механически
входит в контрольный луч и снижает его интенсивность. Перо регистрирующего
устройства связано с компенсатором и следует его движению, отмечая по оси
ординат на бумаге проценты пропускаемости или поглощение при соответствующей
длине волны или волновом числе, указываемых по оси абсцисс. В результате
получают спектр вещества.
В качестве детекторов применяют термопары, болометры, термисторы или
пневматический детектор Голэя.
Термопара, как известно, представляет собой два различных металла,
соединенные по концам. Если одно соединение находится при температуре, отличной
от другого соединения, то возникает разность напряжений и протекает слабый ток.
Болометры и термисторы состоят из веществ, сопротивление которых или
возрастает с увеличением температуры, или уменьшается.
Пневматический детектор Голэя, который сейчас все чаще применяется для
инфракрасных измерений, представляет собой камеру, содержащую газ низкой
теплопроводности и закрытую с одной стороны окном из бромида калия, через
которое излучение попадает на тонкую адсорбирующую пленку. Так как пленка имеет
низкую теплоемкость, она реагирует на инфракрасную радиацию и нагревает газ,
находящийся в камере. Увеличение температуры газа вызывает изменение давления в
камере и изменение зеркальной мембраны, которая закрывает камеру с другой
стороны. Эти изменения направляются через оптическую систему в фотоэлемент, где
превращаются в соответствующее напряжение, которое подвергают дальнейшему
усилению и измерению.
Для калибрования приборов к ним обычно прилагаются стандартные пленки
полистирена, по которым устанавливается воспроизводимость прибора по длине
волны. Известны также таблицы волновых номеров, предложенные Международным
союзом теоретической и прикладной химии.
.3 Методика
измерений
Подготовка образца для анализа является наиболее важным моментом при
определениях в инфракрасной области спектра. Тот факт, что все растворители
имеют характерное инфракрасное поглощение, несколько усложняет методику. Однако
твердые вещества, которые вследствие рассеяния света непригодны для
ультрафиолетовой и видимой области, широко применяются для инфракрасной
спектрофотометрии.
Энергия инфракрасных фотонов в отличие от ультрафиолетовой и видимой
энергии незначительна и поэтому структурные изменения под их воздействием
встречаются очень редко.
Измерения поглощения в инфракрасной области обычно проводят с растворами,
взвесями в парафиновом масле или с твердыми веществами в виде дисперсии со
щелочными галоидами.
Жидкости вносят в прибор в виде капиллярной пленки между двумя
пластинками каменной соли или в чистом виде в кювете толщиной 1 мм или меньше.
Твердые вещества и жидкости могут также измеряться в растворах.
Сероуглерод и четыреххлористый углерод являются наиболее подходящими
веществами. Эти растворители также имеют свои характерные полосы поглощения,
однако, если комбинировать различные растворители, можно получить спектр
вещества по всей его области. Из других растворителей могут быть рекомендованы
хлороформ, тетрахлорэтилен, метиленхлорид.
Для уменьшения поглощения растворителя предпочитают использовать
концентрированные растворы и кюветы от 1 до 0,05 мм.
Растворители, кроме хорошей пропускаемости, естественно, не должны
взаимодействовать с растворенным веществом и с материалом, из которого сделана
кювета.
Если вещество имеет низкую температуру плавления, несколько кристаллов
помещают на пластинку соли и нагревают в шкафу. Когда вещество расплавится, его
накрывают второй пластинкой и, таким образом, получают тонкую пленку вещества.
В зависимости от температуры, при которой поддерживается кюветное отделение
прибора, можно получить спектр вещества в жидком или твердом состоянии.
Иногда концентрированный раствор вещества в летучем растворителе, эфир,
хлороформ, спирт и т. п. наносят по каплям на пластинку каменной соли. После
испарения растворителя на пластинке остается пленка твердого вещества.
Чаще всего готовят взвесь вещества в жидком парафине или дисперсию в виде
дисков (таблеток) в щелочном галоиде.
Твердые вещества растирают с небольшим количеством парафина
(спектроскопического качества) до получения гомогенной смеси, которую затем
помещают между пластинками хлорида натрия и производят измерение. Рассеивание
света при этом зависит от размера частиц вещества и различия в коэффициенте
преломления исследуемого вещества и парафинового масла. Повышение остроты и
высоты полос поглощения возрастает с уменьшением размера частиц. В области выше
7 мкм для частиц размером 3 мкм влиянием этого фактора можно пренебречь.
Наибольшему воздействию рассеивания света подвергаются сильно выраженные полосы
поглощения.
Составные компоненты взвеси должны иметь .близкие изменения коэффициента
преломления в инфракрасной области, иначе около полосы поглощения может
возникнуть асимметрическая полоса поглощения со смещением.
Недостатком измерений в жидком парафине является тот факт, что поглощение
углеводородов маскирует полосы связей С-Н исследуемого вещества. Если
необходимо изучать специфически эту область, рекомендуют использовать
перфтор-керосин или гексахлорбутадиен. Кроме того, требуется определенный навык
для получения взвесей с низкой степенью рассеивания и четкостью полос
поглощения.
Для получения дисперсии вещество растирают с сухим мелко измельченным
бромидом калия или хлоридом калия (спектроскопического качества) в отношении к
щелочному галоиду 1:200. Часть смеси переносят в специальную матрицу,
подвергают воздействию вакуума (для удаления воздуха) и прессуют. Полученный
прозрачный диск-таблетку помещают в специальный держатель и проводят измерение.
Трудности, связанные с полиморфизмом, могут быть преодолены
приготовлением растворов стандарта и испытуемого вещества в подходящем
растворителе. Затем растворитель удаляют выпариванием, готовят для каждого
остатка новые диски, которые затем подвергают вторичному исследованию.
Методика определений со взвесями или дисперсиями в щелочном галоиде чаще
всего используется для текущего фармацевтического анализа.
При изучении структур, однако, рекомендуют проводить измерения в
растворах.
Инфракрасные спектрофотометры регистрируют на бумаге процент пропускания
или поглощение по отношению к волновому числу или длине волны. Техника
измерений практически не отличается от подобной в ультрафиолетовой области.
Двухлучевые приборы обеспечивают показания, свободные от влияния паров
воды и поглощения углекислого газа, и дают возможность компенсации поглощения
растворителя при работе с растворами.
При дифференциальных измерениях можно получить спектр отдельных полос
поглощения, менее заметных при обычных определениях. Для этого основной
компонент по аналогии с измерениями в ультрафиолетовой области помещают в
контрольную кювету и в результате компенсации основных показателей спектра
получают спектр отдельных вторичных функциональных групп.
Методика дифференциальных определений может быть применена для испытания
на чистоту, так как при компенсации основных компонентов любая примесь при
условии, что в контрольной кювете находится более очищенное вещество, будет
заметна на спектре. Таким образом, можно обнаружить и количественно измерить до
0,05% примесей.
Тот же способ можно применить для количественного определения
лекарственных форм, из которых трудно извлечь действующее вещество. Например,
при анализе масляных растворов, применяя в качестве контроля масло, можно
получить и измерить полосу поглощения, характерную для исследуемого вещества.
Однако этот метод имеет ограниченную ценность для фармакопейного анализа
вследствие необходимости иметь вещество определенного качества для сравнения.
Трудности, возникающие при дифференциальных измерениях, обусловлены тем,
что при сильном поглощении в обоих лучах (изучение структурных особенностей)
этот метод может быть исключительно полезным.
Глава 4.
Применение спектрофотометрии в фармакопейном анализе
оптический
анализ спектрофотометрия измерение
4.1 Испытание
на подлинность органических лекарственных веществ спектометрией в
ультрафиолетовом спектре
Спектрофотометрия в ультрафиолетовой области является одним из основных
общих методов анализа лекарственных веществ и их препаратов, включенных в любую
современную фармакопею.
Установление зависимости между строением веществ и их электронными
спектрами является сложной проблемой, рассмотрение которой находится вне
пределов данной работы. Следует, однако, остановиться на некоторых
закономерностях, определяющих характер спектров.
Поглощение в ультрафиолетовой и видимой частях спектра обычно связывают с
наличием в молекуле вещества определенных групп - хромофоров. К ним относятся
двойные и тройные углеродные связи, карбонильная, карбоксильная, азо-, нитро- и
другие группы. Известно также, что некоторые группы, не являясь хромофорными,
увеличивают интенсивность окраски вещества - такие группы называют ауксохромными,
или ауксохромами. Типичными примерами ауксохромов могут быть гидроксильная и
аминогруппы.
Влияние различных заместителей на характер поглощения удобно наблюдать
при рассмотрении спектров простых молекул. Интерпретация спектров органических
веществ, имеющих сложное строение, затруднена ввиду присутствия в молекуле
более чем одной хромофорной и ауксохромной группы. Часто определенные группы
хромофоров проявляются спектрально как единый комплекс. Так, например,
структура бензола имеет характерные полосы поглощения при 200 и при 255 нм.
Если введение новой группы в молекулу вещества не затрагивает имеющиеся
хромофорные структуры, то не произойдет какого-либо значительного изменения
спектра. Поэтому ряд фармакопейных веществ (фенамин, эфедрин и лидол) имеют
типичную полосу поглощения бензола при 257 нм.
При введении в бензольное ядро фенольной группы полоса поглощения
смещается к области 280 нм, где особенно возрастает влияние растворителя, так
как имеется возможность образования ауксохрома в виде - ОН (кислая и
нейтральная среда) или О- (щелочная среда). Типичными примерами одноатомных
фенолов являются морфин и эстрадиол, и двухатомных фенолов - адреналин и
изопреналин.
Интенсивное поглощение многих кетостероидов полностью зависит от
сопряженной системы колца А. Остальные группы в молекуле не оказывают влияния
на характер спектра. Поэтому ряд стероидов (преднизон, преднизолон,
прогестерон, дезоксикортикостерон, гидрокортизон, кортизон и метилтестостерон)
имеет подобный спектр с максимумом около 240 им.
Производные барбитуровой кислоты проявляют максимум при рН 7 вследствие
возникновения хромофорной группы
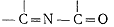
Для идентификации неизвестного вещества в органической аналитической
химии спектр исследуемого вещества обычно сравнивают с полученным при тех же
условиях спектром вещества, строение которого известно.
Установление подлинности вещества по ультрафиолетовому спектру является
ценным дополнением к химическим и физико-химическим методам фармакопейного
анализа. Далее мы рассматриваем некоторые случаи поглощения в ультрафиолетовой
области, используемые рядом фармакопей для определения подлинности органических
лекарственных веществ.
Указание длин волн при максимумах поглощения является лишь
ориентировочной характеристикой, так как не дает возможности судить о виде
спектра.
,002% раствор препарата в 0,01Н растворе соляной кислоты в области от 220
до 350 нм имеет максимум поглощения около 251 нм (Имизин, ГФХ).
В данном случае, очевидно, следует или целиком промерить указанную область,
что при отсутствии регистрирующего прибора требует значительного времени, или
сделать ряд измерений в области, близкой к 251 нм, например ±10 нм, и
установить максимум.
Чаще приводят максимумы и минимумы при определенных длинах волн и
соответствующие величины поглощения.
Ультрафиолетовый спектр раствора в 0,1Н соляной кислоте имеет два
максимума - при 241 нм и при 291 нм, поглощение 0,001% раствора при толщине
слоя 1 см при 241 нм, около 0,50 и при 291 нм, около 0,67. (Антазолина
гидрохлорид, Международная фармакопея, Второе издание).
Иногда величину поглощения выражают в виде удельного показателя
поглощения, Е (1%, 1 см).
Удельный показатель поглощения, Е (1 %, 1 см), при длине волны 278 нм
290-305 (0,002% водный раствор) (Левомицетин, ГФХ).
Если кривая поглощения изменяется в зависимости от pH раствора, указывают значение pH, при котором проводится измерение.
Ультрафиолетовый спектр раствора в фосфатном буфере, pH 7,5, имеет максимум только при 252
нм, поглощение 0,0005 раствора при толщине слоя 1 см около 0,4 (Сульфафуразол,
Международная фармакопея, Второе издание).
Раствор в 0,01 н. едком натре имеет максимум при 326 нм, Е (1%, 1 см) -
около 65, и минимум при 294 нм, Е (1%, 1 см) - около 35.
Раствор в смеси 10 объемов 0,1Н соляной кислоты и 40 объемов 95% спирта,
доведенных водой до 100 мл, имеет максимум при 300 нм, Е (1%, 1 см) - около 50,
и минимум при 275 нм, Е (1 %, 1 см) - около 40 (Левотироксин-натрий,
Скандинавская фармакопея).
Некоторые фармакопеи вместо абсолютных величин поглощения принимают
отношение адсорбции при максимуме к абсорбции при минимуме, что дает
возможность до некоторой степени судить о наличии поглощающих примесей.
,001 % раствор препарата в 0,1Н растворе едкого натра имеет максимумы
поглощения при 256, 283 и 365 нм. Отношение D при 256 нм к D при
365 нм составляет 2,8-3,0 (Фолиевая кислота, ГФХ).
Для феноксиметилпеннцииллина по той же фармакопее проводят измерение
следующим образом.
Около 0,1 г препарата (точная навеска) растворяют в 4 мл 5% раствора
гидрокарбоната натрня, разводят водой до 500 мл и определяют оптическую
плотность (D) при длине волны 268 нм и при длине
волны 274 нм в кювете с толщиной слоя 1 см. Контрольным раствором служат 4 мл
5% раствора гидрокарбоната иатрия, разведенные водой до 500 мл.
Отношение D
при длине волны 268 нм к
D при длине волны 274 им должно быть не
менее 1,21 и не более 1,24.
В случае стандарта идентификация веществ сводится к сравнению двух
спектров поглощения, полученных при одних и тех же условиях.
Ультрафиолетовый спектр 0,0005% раствора в спирте имеет максимумы и
минимумы яри тех же длинах волн, что н раствор стандарта, одинаковой
концентрации и одновременно измеренный; соответствующие величины поглощения,
рассчитанные на сухое вещество, при максимуме поглощения около 281 нм не отличаются
более чем на 3% (Эгинилэстрадиол, Фармакопея США XVII).
Последний метод обеспечивает наиболее достоверные результаты и должен
быть рекомендован для фармакопейных испытаний на подлинность.
.2 Испытание
на чистоту спектометрией в ультрафиолетовом спектре
Метод спектрофотометрии в ультрафиолетовой области успешно используется
как для идентификации и количественного определения, так и в испытаниях на
чистоту.
Применение ультрафиолетовой спектрофотометрии для проверки
доброкачественности фармакопейных препаратов является наиболее ценным в
случаях, когда примеси или продукты разложения поглощают в области, отличной от
исследуемого вещества. Таким образом определяют содержание адреналона, имеющего
максимум при 310 им, в адреналине, максимум поглощения при 278 нм.
Указываемые в фармакопейных статьях величины поглощения, определяющие
предельное содержание примеси как «не более...», устанавливаются эмпирическим
путем.
-оксифенилтриметиламмоний бромид, сопутствующий йеостигмину бромиду,
имеет максимум при 294 нм.
Поглощение 0,05% раствора в 0,1М карбонате натрия при 294 нм не более
0,03 - предел содержания 3 мг на 1 г (Неостнгмнн бромид, Скандинавская
фармакопея).
Аналогично определяется содержание гитоксина в дигитоксине по
Международной фармакопее Второго издания.
Растворяют около 5 мг препарата (точная навеска) в 1 мл метилового спирта
и доводят глицерином или концентрированной соляной кислотой до объема 25 мл.
Поглощение этого раствора после часа стояния при 352 нм около 0,28 (не более
5%).
Предел содержания поглощающих примесей может быть установлен по величине
отношений абсорбций при различных максимумах. При анализе циаиокобаламина по
Государственной фармакопее X издания определяют оптическую плотность (D) раствора, приготовленного для
количественного определения в кювете с толщиной слоя 1 см при длинах волн 278,
361 и 548 нм.
Отношение D при 361 нм к D при 548 нм должно быть от 3,0 до
3,4.
Отношение D при 361 нм к D при 278 нм должно быть от 1,7 до
1,88.
Изучение ультрафиолетовых спектров является ценным при испытаниях на
чистоту и при исследованиях стабильности лекарственных средств, если изменения
в характере спектра позволяют судить об изменениях и превращениях вещества.
4.3
Количественное определение спектометрией в ультрафиолетовом спектре
Спектрофотометрия в ультрафиолетовой области широко используется для
количественного определения лекарственных веществ и их препаратов и включена во
все современные фармакопеи.
Идеальное вещество для таких измерений должно иметь четко выраженную
полосу поглощения с широким максимумом (для уменьшения ошибки за счет ширины
щели) и со значительной величиной поглощения при максимуме.
Чувствительность анализа определяется в основном способностью вещества
поглощать свет и выражается, как было указано выше, молярным коэффициентом
поглощения. Предельные концентрации веществ, анализируемые при помощи
спектрофотометрии, как правило, меньше, чем при обычных и потенциометрических
титрованиях или весовых измерениях, что и объясняет тот факт, что
спектрофотометрия используется при определении небольших количеств веществ.
Основным условием для количественного анализа является соблюдение закона
Ламберта - Бера в пределах указанных концентраций. Для проверки соответствия
закону строят график зависимости: поглощение - длина волны, если все точки
лежат на прямой - закон выполняется. Точность метода будет зависеть от наклона
прямой: чем больше наклон, тем выше точность.
По другому способу рассчитывают фактор для каждого стандартного раствора
и определяют область концентраций, в пределах которой величина D/С остается постоянной.
Известны два принципиально различных способа спект- рофотометрических
количественных определений. По одному из них содержание вещества рассчитывают
на основании предварительно вычисленной величины поглощения. Чаще всего в таких
.случаях используется значение Е (1%, 1 см) и закон Ламберта - Бера принимает
следующую форму:
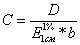
Британская
фармакопея 1968 г. приняла этот метод определения как для лекарственных веществ,
так и для лекарственных форм.
Растворяют
около 10 мг (точная навеска) в безводном спирте и доводят спиртом до 100 мл,
разводят 5 мл полученного раствора до 50 мл безводным спиртом и измеряют
поглощение полученного раствора в кювете с толщиной слоя 1 см при 240 им Е (1%,
1 см) для С20Н30О2 при максимуме около 240 нм - 535 (Метилтестостерэн).
Государственная
фармакопея СССР X издания следующим образом рекомендует определять кортизона
ацетат в таблетках.
Около
0,1 г (точная навеска) порошка растертых таблеток помещают в мерную колбу
емкостью 100 мл с притертой пробкой, прибавляют 60-70 мл 95% спирта и
растворяют при легком подогревании на водяной бане прн помешивании в течение
10-15 минут. По охлаждении раствор доводят до метки 95% спиртом, хорошо перемешивают
и дают отстояться в течение часа. Не взмучивая осадка, 5 мл раствора переносят
в другую мерную колбу емкостью 100 мл и доводят тем же спиртом до метки.
Измеряют оптическую плотность полученного раствора на спектрофотометре при
длине волны 238 нм в кювете с толщиной слоя в 1 см. В контрольную кювету
помещают спирт.
Содержание
кортизона ацетата в граммах вычисляют по формуле:
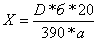
где
D-оптическая плотность испытуемого раствора;
-
удельный показатель поглощения кортизона ацетата Е (1%, 1 см) при 238 нм;
а
- навеска препарата в граммах;
б
- средний вес таблетки в граммах.
Количественное
определение по описанному выше методу подвергается постоянной критике. Основной
причиной для этого является общеизвестный факт, что различные спектрофотометры
(даже различные приборы одной и той же модели и одного производства) дают
значительные отклонения по величине поглощения для одного и того же
стандартного раствора.
Более
правильным способом является сравнение поглощения испытуемого вещества с
поглощением стандартного образца, определенного в тех же условиях. Таким
образом достигается наибольшая точность, так как при этом учитываются
многочисленные факторы, влияющие на спектрофотометрические измерения, как,
например, установка длины волны, ширина щели, поглощение кюветы, поправки на
поглощение растворителя и т. д.
Многие
методы физико-химического анализа лекарственных средств связаны со
сравнительной оценкой испытуемого препарата по отношению к веществам, химический
состав которых отличается постоянством и может быть установлен с необходимой
точностью. Такие вещества называют стандартными образцами. Выражение «стандарт»
особенно часто применяется в биологических методах анализа. Однако в связи с
тем, что термин одновременно применяют и для обозначения технической
документации (ГОСТ), представляется более правильным называть все вещества,
используемые для сравнения при определении качества, стандартными образцами.
В
частности, при количественном спектрофотометрическом определении индивидуальных
веществ эти фармакопеи рекомендуют применять специально приготовленные
стандартные образцы. При расчете количественного содержания определяемого
вещества по ГФХ и Международной фармакопее, если нет специального указания,
стандартный образец принимают за 100%; по Скандинавской фармакопее учитывают
фактическое содержание чистого вещества в стандартном образце, что,
по-видимому, следует делать лишь при содержании чистого вещества ниже 97,
например при анализе некоторых витаминных препаратов.
Следующий
текст используется в статьях X издания Государственной фармакопеи при анализе
индивидуальных веществ.
Измеряют
оптическую плотность 0,001 % раствора препарата в 95% спирте на
спектрофотометре при длине волны 241 нм в кювете с толщиной слоя 1 см. Повторяют
такое же измерение с 0,001% раствором стандартного образца прегнина.
Содержание
прегнина в процентах (X) вычисляют по формуле:
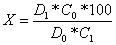
где
D1 - оптическая плотность испытуемого раствора; D0 -
оптическая плотность раствора стандартного образца; С1 - концетрация
испытуемого раствора; С0 - концентрация раствора стандартного образца.
Содержание
С21Н28О2 в препарате должно быть не менее 97% и не более 103,0% (Прегнин).
При
количественном определении лекарственных форм, если нет специальных указаний,
стандартным образцом служит вещество, отвечающее всем требованиям фармакопеи.
При расчетах стандартный образец принимают за 100%. По Международной фармакопее
при спектрофотометрическом анализе лекарственных форм в качестве стандартного
вещества применяют вещество, определяемое по фармакопее объемным, весовым или
другими, но не спектрофотометрическими методами. Так как в ряде случаев этого
требования может быть недостаточно, во Второе издание Международной фармакопеи
включена оговорка о возможном применении для анализа лекарственных форм
специального стандартного образца.
Методика
количественного определения дипрофиллина в таблетках дипрофиллина по ГФХ.
Около
0,12 г (точная навеска) порошка растертых таблеток помещают в мерную колбу
емкостью 100 мл, прибавляют небольшое количество воды, доводят объем раствора
водой до метки, перемешивают и фильтруют, 1 мл фильтрата переносят в мерную
колбу емкостью 100 мл, доводят объем раствора водой до метки и измеряют оптическую
плотность полученного раствора на спектрофотометре при длине волны 273 нм в
кювете с толщиной слоя 1 см.
Параллельно
проводят измерение оптической плотности раствора стандартного образца
дипрофиллина.
Содержание
дипрофиллина в одной таблетке в граммах (X) вычисляют по формуле:

где
D1 - оптическая плотность испытуемого раствора;
D0 - оптическая
плотность раствора стандартного образна;
а
- навеска в граммах;
б
- средний вес таблетки в граммах.
Содержание
С10Н14О4 должно быть 0,19-0,21, считая на средний вес одной таблетки.
Примечание.
Приготовление раствора стандартного образца дипрофиллина. 0,1000 г стандартного
образца дипрофиллина растворяют в воде в мерной колбе емкостью 100 мл и доводят
объем раствора водой до метки. 1 мл полученного раствора переносят в мерную
колбу емкостью 100 мл, доводят объем раствора водой до метки и перемешивают.
мл
раствора стандартного образца содержит 0,01 мг дипрофиллина.
Расчет
содержания вещества по стандартам не представляет особых трудностей. Как
правило, построение калибровочного графика не является необходимым, так как
ультрафиолетовые измерения, выполненные на современных приборах, практически
всегда следуют закону Ламберта - Бера и одно измерение стандартного и
исследуемого вещества обеспечивает точные результаты. При текущем анализе
желательно время от времени проверять поглощения стандартных растворов для
предотвращения ошибки вследствие неожиданных изменений характеристики
спектрофотометра.
Необходимо
упомянуть способ расчета, принятый Скандинавской фармакопеей. Содержание
вещества в испытуемом препарате определяют по формуле:
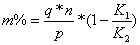 ,
,
где
q - концентрация стандарта в миллиграммах на 100мл раствора;
b - процентное
содержание чистого вещества в стандарте;
р
- концентрация испытуемого вещества в милиграммах на 100 мл;
К1
- разница в поглощении стандартного раствора, измеренного к контрольному
раствору;
К2
- разница в поглощении стандартного раствора, измеренного к испытуемому
раствору.
Второе
издание Международной фармакопеи не указывает способа расчета содержания
вещества.
Рассчитывают
содержание вещества по величине поглощения, полученной со стандартным образцом,
определенной при условиях опыта.
Рассмотренные
до сих пор методы спектрофотометрических количественных определений относились
к анализу одного компонента при условии отсутствия поглощающих примесей в
области максимума и при условии, когда расчет поправки на дополнительное поглощение
не представляет специальной проблемы.
Однако
поправку на примеси следует определять, если спектрофотометрически
анализируются природные извлечения или сложные смеси. Существует несколько
способов установления поправок в зависимости от характера кривой поглощения и
примесей.
Один
из (них, известный под названием способа Мортон - Стаббса, используется в
фармакопейном определении ретинола (витамина А). Линейная зависимость
содержания поглощающих примесей по отношению к длине волны является основным
условием для расчета поправки по способу Мортон - Стаббса. Поглощение измеряют
в трех точках спектра таким образом, что второе измерение приходится на
максимум поглощения. Остальные величины поглощения при двух длинах волн равны
между собой и составляют определенную часть поглощения при максимуме; эта
последняя рассчитывается по результатам, полученным для чистого вещества.
Метод
спектрофотометрического определения витамина А рекомендован Международным
союзом теоретической и прикладной химии в 1956 г. и принят большинством
фармакопей.
Метод.
Все процедуры должны проводиться быстро, насколько это возможно, следует
принимать меры во избежание воздействия света и окислительных агентов.
Так
как положение максимума поглощения является важным критерием и поправочные
уравнения требуют измерений при точных длинах волн, необходимо, чтобы шкала
длин волн спектрофотометра была проверена немедленно перед определением.
Ртутные линии при 313,16 и 334,15 нм являются подходящими точками и для
удобства настройка прибора по этим линиям может быть связана с его настройкой
по водородным линиям при 379,7 и 486,1 нм. Точность исправленного поглощения
значительно ниже, чем три непосредственно измеренных поглощения, из которых оно
рассчитывается; следовательно, измерения требуют особой осторожности и следует
проводить не менее двух количественных определений.
Витамин
А в форме эфира. Образец, нерастворимый непосредственно В циклогексане, может
быть обработан экстракцией или другими средствами, не связанными с омылением.
Если это невозможно, поступают так, как описано в разделе «Другие формы
витамина А».
Растворяют
точно взвешенное количество образца или приготовленного извлечения в
достаточном количестве цик- логексана для получения раствора с содержанием от 9
до 15 Международных единиц в миллилитрах. Определяют длину волны максимума
поглощения. Измеряют поглощения при длинах волн, приведенных в табл. 2, и
рассчитывают их как отношение к поглощению при 328 нм.
Таблица
2
Относительное поглощение при разных длинах волн
|
Длина волны (в нм)
|
Относительное поглощение
|
|
300
|
0,555
|
|
316
|
0,907
|
|
328
|
1,000
|
|
340
|
0,811
|
|
360
|
0,299
|
Рассчитывают также поглощение при 328 нм в виде Е(1%, 1 см) или в виде
«а» - поглощаемость.
Если максимум поглощения лежит между 326 и 329 нм и наблюдаемые
относительные поглощения находятся в пределах 0,02 от указанных в таблице,
определяют активность образца в Международных единицах и в граммах по формуле:
Е(1%, 1 см)328 * 1900.
Если максимум поглощения лежит между 326 и 329 нм, но относительные
поглощения не находятся в пределах 0,02 от указанных в табл. 2, рассчитывают
исправленное поглощение при 328 нм, применяя найденные показания в уравнении:
А328 (испр.) = 3,52 (2A328 -
А316 - А340).
Если исправленное поглощение лежит в пределах -15% или +3%
неисправленного поглощения, активность рассчитывают по исправленному
поглощению.
Если исправленное поглощение лежит вне -15% или +3% неисправленного
поглощения или максимум поглощения не приходится на область 326 и 329 нм,
образец подвергают анализу, как это описано в разделе «Другие формы витамина
А».
Другие формы витамина А. Смешивают точно взвешенное количество образца,
содержащего не менее 500 Международных единиц витамина А и не более 1 г жира с
30 мл безводного спирта и 3 мл 50% раствора едкого кали, кипятят с обратным
холодильником в течение 30 минут, под током азота, свободного от кислорода,
быстро охлаждают и прибавляют 30 мл воды. Переносят гидролизат в делительную
воронку с помощью трех количеств, каждое 50 мл, эфира и извлекают витамин А
встряхиванием в течение минуты. После полного разделения слоев отбрасывают
водный слой и промывают извлечение четырьмя порциями, каждая 50 мл, воды
смешивая осторожно во избежание образования эмульсии во время первых двух
промываний. Упаривают определенный экстракт до 5 мл и удаляют оставшийся
растворитель в токе азота, свободного от кислорода, без применения нагревания.
Растворяют остаток в достаточном количестве изопропилового спирта для получения
раствора с содержанием от 9 до 15 Международных единиц в 1 миллилитре и
измеряют поглощения при 300, 310, 325 и 334 нм. Определяют длину волны
максимума поглощения.
Если максимум поглощения лежит между 323 и 327 нм и отношение поглощения
при 300 им к поглощению при 325 нм не превышает 0,73, получают исправленное
поглощение по уравнению:
A325 (испр.)
=6,815 А325 - 2,555 А310 - 4,260 А334.
Активность образца в Международных единицах в граммах рассчитывают по
формуле:
Е325 (испр.) * 18300,
за исключением случая, когда исправленное поглощение лежит в пределах
±3,0% неисправленного поглощения, исправленным поглощением можно пренебречь, и
активность рассчитывают по неисправленному поглощению.
Если максимум поглощения лежит вне области 323 до 327 им или отношение
поглощения при 300 им к поглощению при 325 им превышает 0,73, неомыленную часть
образца нужно подвергать хроматографированию.
При выполнении спектрофото метрического анализа витамина А следует
принимать во внимание, что растворы ретинола исключительно чувствительны по
отношению к свету.
В качестве растворителей применяются петролейный эфир, циклогексан,
изопропиловый спирт и реже хлороформ. Чаще всего предпочитают циклогексан, так
как спектральные различия в области 310 до 315 нм между нео- и транс-витамином
А менее проявляются в этом растворителе. Растворители подвергают дополнительной
очистке путем перегонки; рекомендуется также проверять их спектральную чистоту
в области от 300 до 350 нм.
Согласно ГФХ, содержание ретинола определяют в индивидуальном веществе
спектрофотометрически при 326 нм в растворе в абсолютном спирте. Предварительно
проводят испытание на поглощающие примеси. Расчет производят принимая 1550 за
величину Е (1%, 1 см) для 100% ретинола при длине волны 326 нм.
Фармакопея США XVII указывает, что при условиях определения (поглощение
измеряют после омыления) следует ожидать расхождения в данных, полученных
различными лабораториями при Р = 0,05 около ±8%.
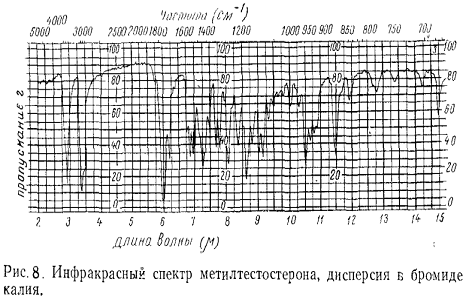
Для интерпретации спектров обычно используют имеющиеся данные о связи
между инфракрасными полосами поглощения и структурными элементами молекул.
Такие данные сводятся, как правило, в виде корреляционных таблиц-диаграмм.
В области от 2,5 до 6,5 мкм с помощью таблиц по сильному и среднему
поглощению можно с определенной точностью идентифицировать определенную
функциональную группу. Инфракрасные полосы поглощения, соответствующие
валентным колебаниям С-Н, находятся в области 3,3 мкм, для алифатических
соединений характерные полосы лежат от 3,3 до 3,7 мкм; для ароматических
веществ - от 3,2 до 3,3 мкм. Свободная гидроксильная группа будет иметь слабую,
но четко выраженную полосу при 2,75 мкм, водородная связь значительно увеличит
ее интенсивность и вызовет смещение полости до 2,85 или до 3,0 мкм.
Наличие полос валентных колебаний =С - Н при 3,3 мкм и колебаний в
области от 6,1 до 6,25 мкм указывает на присутствие вещества, имеющего
структуру ароматического типа. Положение полос поглощения определенной
функциональной группы не является постоянным в применении к разным молекулам
вследствие влияния соседних атомов и групп. Например, положение карбонила, С =
0, при 5,75-5,8 мкм, характерно для альдегидов и кетонов; при 6,0 - 6,1 мкм -
для кислот; при 5,75 - 5,85 мкм - для эфиров; ангидриды имеют две полосы
поглощения: при 4,9 - 5,05 мкм и при 5,60 - 5,75 мкм.
Ниже 6,25 мкм, однако, соотношение между характерными частотами диаграммы
и наблюдаемыми при опыте частотами не соблюдается, так как в этой области
проявляются характеристики не отдельных функциональных групп, а всей молекулы.
Это явление объясняется частично тем, что здесь существует много полос для
большинства молекул и что нет простого способа установления соответствия полос
поглощения определенным колебаниям и положение полос в большей мере
подвергается непредусмотренным изменениям. Прежде всего зависимость характера
спектра поглощения от строения определенной молекулы может быть связана с
фактом, что существуют многие колебания, имеющие приблизительно одинаковую
частоту, и что эти колебания взаимодействуют друг с другом.
Область от 8 до 15 мкм носит иногда название области «отпечатков пальцев»
(finger print), так как именно эта часть спектра обусловливает
уникальность спектра отдельного вещества.
Существует несколько возможностей описания фармакопейных испытаний на
подлинность посредством инфракрасных спектрофотометрических измерений.
Прежде всего это издание сборников инфракрасных спектров для веществ,
включенных в фармакопею, с тем, чтобы впоследствии эти спектры использовались
для сравнения с соответствующим спектром исследуемого вещества. Коллекции
инфракрасных спектров постоянно публикуются и расширяются. Описаны спектры
стандартных веществ, включенных в Фармакопею США XVI.
Другую возможность представляет указание длин волн, при которых
наблюдаются полосы поглощения, и выражение их интенсивности как сильной,
средней, слабой и переменной.
Инфракрасные спектры, являясь уникальным средством идентификации
вещества, в то же время, как было показано выше, подвержены влиянию многих
факторов. Спектры могут быть разными у двух различных спектроскопистов.
Качество растворителей, условия приготовления образца для анализа,
кристаллические формы вещества составляют краткий перечень факторов, влияющих
на спектр. В связи с этим в фармакопейном анализе основным правилом для
инфракрасной идентификации является получение спектра стандартного вещества в
то же время и при тех же условиях, что и спектр образца, с последующим
сравнением двух спектров.
Британская фармакопея 1968 г. и Международная фармакопея Второго издания
описывают определение инфракрасного поглощения для установления подлинности стероидов,
гликозидов и полусинтетических пенициллинов. В разделе общих методов анализа
излагаются методики измерений в виде взвеси и в виде дисперсии со щелочным
галоидом.
В Государственную фармакопею X издания впервые включена инфракрасная
спектрофотометрия для анализа натриевых солей метициллина и оксациллина и
фторотана (галотана).
Инфракрасный спектр препарата имеет те же максимумы поглощения, что и
стандартный образец натриевой соли метициллина (Метициллина натриевая соль,
ГФХ).
Идентификация органических оснований по инфракрасному спектру в растворе
сероуглерода является общим методом Фармакопеи США XVII.
Растворяют 50 мг вещества в 25 мл воды или взбалтывают эквивалентное
количество порошка таблеток или содержимого капсул с 25 мл 0,01Н соляной
кислоты в течение 10 минут. Переносят жидкость в делительную воронку, фильтруя,
если необходимо, и промывая фильтр и остаток небольшими порциями воды. Во
второй делительной воронке растворяют 50 мг стандарта в 25 мл воды. Затем
каждый раствор обрабатывают следующим образом: прибавляют 2 мл 10% раствора
едкого натра и 4 мл сероуглерода и встряхивают в течение 2 минут.
Центрифугируют, если необходимо сделать прозрачной нижнюю фазу, и фильтруют ее
через сухой фильтр, собирая фильтрат в небольшую колбу с притертой пробкой.
Определяют спектры поглощения обоих отфильтрованных растворов без задержки, в
кювете 1 мм в области от 7 до 15 мкм в подходящем спектрофотометре, применяя в
качестве контрольного раствора сероуглерод. Спектр раствора, полученный из
образца, имеет все важнейшие полосы поглощения, что и раствор стандарта. Если
спектр образца имеет полосы, отличающиеся от стандарта, образец может быть
подвергнут очистке и опыт повторен.
Для остальных веществ в Фармакопее США XVII описывается определение с
дисперсией препарата в бромиде калия.
4.5
Количественное определение по поглощению в инфракрасной области
Единственной фармакопеей, рекомендующей количественное определение по
поглощению в инфракрасной области, остается Фармакопея США XVII. Принципы
количественного анализа в инфракрасной области те же, что и в ультрафиолетовой
или в видимой области.
Любое поглощение, не связанное с поглощением основного компонента, носит
название постороннего поглощения, или поглощения фона. При инфракрасных
измерениях такое поглощение может быть значительным, и поэтому установлению
нулевой линии и линии 100% пропускаемости придается особое значение.
Для большинства приборов неопределенность нулевой линии обычно небольшая
и она уменьшается по мере увеличения чувствительности прибора к сигналам низкой
интенсивности.
При установлении линии 100% пропускаемости некоторые отклонения могут
быть вызваны влиянием паров воды, которые могут конденсироваться на стенках
кюветы при испарении растворителей. Толщина кюветы при количественных
определениях приобретает особое значение, так как работают с концентрированными
растворами в слое от 1 мм и менее. Даже при тщательном подборе кювет не всегда
удается обеспечить одинаковую толщину слоя. Поэтому на практике линия отсчета
100% пропускаемости устанавливается несколько произвольно (рис. 9).
Для измерения пропускаемости определяют интенсивность лучей, прошедших
через образец и через контроль. На спектре эта зависимость изображается
линейно, поэтому измеряют расстояние на диаграмме от 0% пропускаемости до кривой
поглощения образца, которое относят к расстоянию от 0 до 100% пропускаемости,
откуда при измерении абсолютных величин получают: А = lg (100/I).
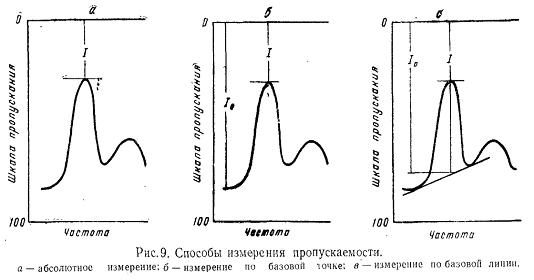
При условии, что любое постороннее поглощение изменяется линейно в зависимости
от концентрации анализируемого вещества, условно принимают за линию 100%
пропускаемости расстояние от 0% до минимума кривой. В этом случае зависимость
имеет вид: А = lg (I0/I).
Неточный выбор 100% линии не нарушает линейной зависимости, а только
изменяет величину отрезка на графике поглощение - концентрация.
При измерениях часто используют метод определения так называемой основной
базовой линии. Для этого на отрезке кривой спектра выбирают три точки, которые
обычно, но не обязательно, соответствуют максимуму и двум минимумам. Проводят
между двумя точками, лежащими вне кривой, прямую линию и измеряют высоту
спектра между максимумом и произвольно проведенной основной линией. В таком
случае А = lg (I0/I).
Следует учитывать, что при определениях по данному методу все измерения
должны быть выполнены точно при одних и тех же частотах, если даже имеются
отклонения в спектре исследуемого вещества. Ошибки, вызванные небольшим
изменением в положении измерений, будут наименьшими, если точки действительно соответствуют
максимумам и минимумам. Если другие вещества поглощают в измеряемой области и
характер этого поглощения неизвестен, применение метода может дать неправильные
результаты.
Вместо того чтобы полагаться на линейную зависимость закона Бера и
применять соответствующие уравнения, часто более точным и удобным является
сравнение поглощения образца с поглощением стандартного образца, определенного
в одно и то же время, при одних и тех же условиях. Идентичность условий, при
которых производятся измерения, делает возможным достигать точность определения
более чем 1%.
Наиболее разработанными являются методики количественного определения в
растворах. Для уменьшения взаимодействия между растворенным веществом и
растворителем рекомендуют применять неполярные жидкости: сероуглерод,
хлороформ, четыреххлористый углерод. Реже используют пиридин, диметилформамид.
Так как каждый из этих растворителей имеет характерные полосы поглощения,
количественные измерения проводят в узкой части спектра, где растворитель имеет
наилучшую пропускаемость.
Растворяют около 200 мг диакарба (точная навеска) в небольшом объеме
пиридина в 10 мл мерной колбе, доводят раствор пиридином до метки и
перемешивают. Таким же образом растворяют точную навеску стандартного образца
диакарба в пиридине для получения концентрации около 20 мг в 1 мл. Одновременно
определяют поглощения обоих растворов в кювете толщиной 0,1 мм при длине волны
максимума поглощения около 7,38 мкм на подходящем инфракрасном
спектрофотометре, используя пиридин в качестве контроля. Рассчитывают
содержание в миллиграммах, C4H6N403S2 в части взятого на анализ диакарба по формуле
*С*А/Ас,
в которой С - концентрация в мг/мл стандартного образца,
А и Ас - соответственно поглощения испытуемого раствора и стандартного
образца.
5. Экспериментальная
часть
.1 Методика
исследования
Retinoli acetatis oleosa 3,44%
Раствор ретинола ацетата в масле 3,44%
Состав:
· Ретинола ацетата - 34,4 г
· Масла соевого рафинированного, полученного путем прессования,
или гидратированного, первого сорта (ГОСТ 7825-55) - до 1 л
Описание. Прозрачная маслянистая жидкость от светло-желтого до
темно-желтого цвета, без прогорклого запаха и вкуса.
Подлинность. 1 каплю препарата растворяют в 1 мл хлороформа. К
полученному раствору прибавляют 5 мл раствора хлорида сурьмы; появляется
нестойкое синее окрашивание.
Приготовленный для количественного определения раствор препарата в
абсолютном спирте для спектрофотометрии имеет максимум поглощения при 326±1 нм,
а раствор в циклогексане - при 327,5±1 нм.
Кислотное число не более 1,0.
Поглощающие примеси. Измеряют оптическую плотность раствора препарата в
абсолютном спирте, приготовленного для количественного определения, при длинах
волн 311,5 нм, 326 нм и 337 нм.
Отношения значений оптической плотности при 311,5 нм и 337 нм к
оптической плотности при 326 нм должны быть равны 0,857±0,03.
Количественное определение. Около 0,1 г препарата (точная навеска)
растворяют в абсолютном спирте для спектрофотометрии в мерной колбе емкостью
100 мл, доводят объем раствора тем же спиртом до метки и перемешивают. Отбирают
точное количество полученного раствора и разводят тем же спиртом до получения
концентрации ретинола ацетата около 3 мкг (8-10 ME) в 1 мл. Измеряют оптическую плотность полученного раствора
на спектрофотометре при длине волны 326 нм в кювете с толщиной слоя 1 см. В
качестве контрольного раствора применяют абсолютный спирт для
спектрофотометрии.
Содержание ретинола ацетата в 1 мл препарата в граммах (X) вычисляют по
формуле:
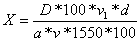
где
D- оптическая плотность испытуемого раствора;
v-объем раствора
первого разведения, взятого для приготовления раствора второго разведения, в
миллилитрах;
v1-объем
раствора второго разведения в миллилитрах;
d- плотность
препарата;
а-
навеска препарата в граммах;
-
удельный показатель поглощения Е (1%, см) при длине волны 326 нм для раствора
100% ретинола ацетата в абсолютном спирте.
Содержание
С22Н32О2 в 1 мл препарата соответственно должно быть 0,0310-1,0378 г
(90000-110000 ME), 0,0619-0,0757 г (180000-220000 ME),
0,0774-0,0946 г (225000-275000 ME).
Примечание.
Определение может быть проведено с использованием циклогексана вместо
абсолютного спирта, в этом случае готовят раствор препарата в циклогексане для
спектрофотометрии с содержанием в 1 мл около 3 мкг ретинола ацетата и измеряют
оптическую плотность этого раствора при длине волны 327,5 нм. В расчетную
формулу вместо 1550 подставляют 1515 - Е (1%, см) при длине волны 327,5 нм для
раствора 100% ретинола ацетата в циклогексане .
Хранение.
Список Б. В доверху заполненных, хорошо укупоренных склянках оранжевого стекла,
в защищенном от света месте, при температуре не выше +10°.
5.2
Результаты исследования
|
Название и лекарственная форма препарата
|
Пока-затель
|
№ п/п
|
Методика исследования
|
Результат
|
|
Раствор ретинола ацетата в
масле 3,44%
|
Подлин-ность
|
1
|
1 каплю препарата
растворяют в 1 мл хлороформа. К полученному раствору прибавляют 5 мл раствора
хлорида сурьмы; появляется нестойкое синее окрашивание.
|
Соответ-ствует
|
|
Раствор ретинола ацетата в
масле 3,44%
|
Подлин-ность
|
2
|
Около 0,1 г препарата
(точная навеска) растворяют в абсолютном спирте для спектрофотометрии в
мерной колбе емкостью 100 мл, доводят объем раствора тем же спиртом до метки
и перемешивают. Отбирают точное количество полученного раствора и разводят
тем же спиртом до получения концентрации ретинола ацетата около 3 мкг (8-10 ME)
в 1 мл. Раствор препарата имеет максимум поглощения при 326±1 нм, а раствор
в циклогексане - при 327,5±1 нм.
|
Соответ-ствует
|
Примеси
|
3
|
Измеряют оптическую
плотность раствора препарата из п.№2 при длинах волн 311,5 нм, 326 нм и 337
нм. Отношения значений оптической плотности при 311,5 нм и 337 нм к
оптической плотности при 326 нм должны быть равны 0,857±0,03.
|
Соответ-ствует
|
|
Раствор ретинола ацетата в
масле 3,44%
|
Количес-твенное
опреде-ление
|
4
|
Измеряют оптическую
плотность раствора препарата из п.№2 на спектрофотометре при длине волны 326
нм в кювете с толщиной слоя 1 см. В качестве контрольного раствора применяют
абсолютный спирт для спектрофотометрии. Содержание ретинола ацетата в 1 мл
препарата в граммах (X) вычисляют по формуле. Содержание С22Н32О2 в 1 мл
препарата соответственно должно быть 0,0310-1,0378 г (90000-110000 ME),
0,0619-0,0757 г (180000-220000 ME), 0,0774-0,0946 г (225000-275000 ME).
|
Соответ-ствует
|
Выводы
Спектрофотометрия - оптический метод исследования газообразных, жидких и
твердых веществ, основанный на определении интенсивности поглощения света
веществом (абсорбционная спектрофотометрия) или интенсивности излучения им
света (эмиссионная спектрофотометрия) в зависимости от длины волны.
Получаемые при этом (при помощи специальных приборов - спектрофотометров)
абсорбционные и эмиссионные спектры являются характерными для каждого данного
вещества.
Различают спектрофотометрию в ультрафиолетовой (УФ), видимой и
инфракрасной (ИК) областях спектра.
Спектрофотометрия широко применяется в клинических, биохимических,
санитарно-гигиенических, судебно-медицинских и фармацевтических лабораториях
для качественного и количественного анализа различного рода объектов
биологического происхождения (сыворотка крови, спинномозговая жидкость, моча и
др.), лекарственных средств, продуктов питания и т. д.
Спектрофотометрия в ультрафиолетовой области является одним из основных
общих методов анализа лекарственных веществ и их препаратов, включенных в любую
современную фармакопею.
Поглощение в ультрафиолетовой и видимой частях спектра обычно связывают с
наличием в молекуле вещества определенных групп - хромофоров. К ним относятся
двойные и тройные углеродные связи, карбонильная, карбоксильная, азо-, нитро- и
другие группы. Известно также, что некоторые группы, не являясь хромофорными,
увеличивают интенсивность окраски вещества - такие группы называют
ауксохромными, или ауксохромами. Типичными примерами ауксохромов могут быть
гидроксильная и аминогруппы.
Для идентификации неизвестного вещества в органической аналитической
химии спектр исследуемого вещества обычно сравнивают с полученным при тех же
условиях спектром вещества, строение которого известно.
Установление подлинности вещества по ультрафиолетовому спектру является
ценным дополнением к химическим и физико-химическим методам фармакопейного
анализа.
Метод спектрофотометрии в ультрафиолетовой области успешно используется как
для идентификации и количественного определения, так и в испытаниях на чистоту.
Применение ультрафиолетовой спектрофотометрии для проверки
доброкачественности фармакопейных препаратов является наиболее ценным в
случаях, когда примеси или продукты разложения поглощают в области, отличной от
исследуемого вещества.
Изучение ультрафиолетовых спектров является ценным при испытаниях на
чистоту и при исследованиях стабильности лекарственных средств, если изменения
в характере спектра позволяют судить об изменениях и превращениях вещества.
Спектрофотометрия в ультрафиолетовой области широко используется для
количественного определения лекарственных веществ и их препаратов и включена во
все современные фармакопеи.
Идеальное вещество для таких измерений должно иметь четко выраженную
полосу поглощения с широким максимумом (для уменьшения ошибки за счет ширины
щели) и со значительной величиной поглощения при максимуме.
Чувствительность анализа определяется в основном способностью вещества
поглощать свет и выражается, как было указано выше, молярным коэффициентом
поглощения. Предельные концентрации веществ, анализируемые при помощи
спектрофотометрии, как правило, меньше, чем при обычных и потенциометрических
титрованиях или весовых измерениях, что и объясняет тот факт, что
спектрофотометрия используется при определении небольших количеств веществ.
Основным условием для количественного анализа является соблюдение закона
Ламберта - Бера в пределах указанных концентраций. Для проверки соответствия
закону строят график зависимости: поглощение - длина волны, если все точки
лежат на прямой - закон выполняется. Точность метода будет зависеть от наклона
прямой: чем больше наклон, тем выше точность.
По другому способу рассчитывают фактор для каждого стандартного раствора
и определяют область концентраций, в пределах которой величина D/C остается
постоянной.
Более правильным способом является сравнение поглощения испытуемого
вещества с поглощением стандартного образца, определенного в тех же условиях.
Таким образом достигается наибольшая точность, так как при этом учитываются
многочисленные факторы, влияющие на спектрофотометрические измерения, как,
например, установка длины волны, ширина щели, поглощение кюветы, поправки на
поглощение растворителя и т. д.
Оценка инфракрасных спектров поглощения является более сложной, чем
ультрафиолетовых, вследствие большого числа полос, что в то же время составляет
основу идентификации вещества
Для интерпретации спектров обычно используют имеющиеся данные о связи
между инфракрасными полосами поглощения и структурными элементами молекул.
Такие данные сводятся, как правило, в виде корреляционных таблиц-диаграмм.
Инфракрасные спектры, являясь уникальным средством идентификации
вещества, в то же время подвержены влиянию многих факторов. Спектры могут быть
разными у двух различных спектроскопистов. Качество растворителей, условия
приготовления образца для анализа, кристаллические формы вещества составляют
краткий перечень факторов, влияющих на спектр. В связи с этим в фармакопейном
анализе основным правилом для инфракрасной идентификации является получение
спектра стандартного вещества в то же время и при тех же условиях, что и спектр
образца, с последующим сравнением двух спектров.
Принципы количественного анализа в инфракрасной области те же, что и в ультрафиолетовой
или в видимой области.
Следует учитывать, что при определениях по данному методу все измерения
должны быть выполнены точно при одних и тех же частотах, если даже имеются
отклонения в спектре исследуемого вещества. Ошибки, вызванные небольшим
изменением в положении измерений, будут наименьшими, если точки действительно
соответствуют максимумам и минимумам. Если другие вещества поглощают в
измеряемой области и характер этого поглощения неизвестен, применение метода
может дать неправильные результаты.
Вместо того чтобы полагаться на линейную зависимость закона Бера и
применять соответствующие уравнения, часто более точным и удобным является
сравнение поглощения образца с поглощением стандартного образца, определенного
в одно и то же время, при одних и тех же условиях. Идентичность условий, при
которых производятся измерения, делает возможным достигать точность определения
более чем 1%.
Наиболее разработанными являются методики количественного определения в
растворах.
Проанализированный раствор ретинола ацетата в масле 3,44% по показателям
идентификации соответствуют требованиям нормативно-технической документации.
Список
использованной литературы
1. Фармацевтична хімія. Підручник для студентів вищ. фармац. начальних
закладів і фарм. фак. вищих мед. навчальних закладів III-IV рівня акредитації / За заг. ред. П.О. Безуглого. - Вінниця: Нова книга,
2008. - 560 с.
2. Арзамасцев А.П. Фармакопейный анализ - М.: Медицина, 1971.
. Беликов В.Г. Фармацевтическая химия. В 2 частях. Часть 1. Общая
фармацевтическая химия: Учеб. для фармац. ин-тов и фак. мед. ин-тов. - М.:
Высш. шк., 1993. - 432 с.
. Глущенко Н. Н. Фармацевтическая химия: Учебник для студ. сред.
проф. учеб. заведений / Н. Н. Глущенко, Т. В. Плетенева, В. А. Попков; Под ред.
Т. В. Плетеневой. - М.: Издательский центр «Академия», 2004. - 384 с.
. Драго Р. Физические методы в химии - М.: Мир, 1981
. Кольтгоф И.М., Стенгер В.А. Объемный анализ В 2 томах - М.:
Государственное научно-техническое издательство химической литературы, 1950
. Коренман И.М. Фотометрический анализ - М.: Химия, 1970
8. Коростелев
П. П, Фотометрический и комплексометрический анализ в металлургии - М.:
Металлургия, 1984, 272 с.
9. Логинова Н. В., Полозов Г. И. Введение в фармацевтическую химию:
Учеб. пособие - Мн.: БГУ, 2003.-250 с.
. Мелентьева Г. А., Антонова Л. А. Фармацевтическая химия. - М.:
Медицина, 1985.- 480 с.
. Мискнджьян С.П. Кравченюк Л.П. Полярография лекарственных препаратов. - К.: Вища школа, 1976. 232 с
12. Фармацевтическая
химия: Учеб. пособие / Под ред. Л.П.Арзамасцева. - М.: ГЭОТАР-МЕД, 2004. - 640
с.
13. Фармацевтический
анализ лекарственных средств / Под общей редакцией В.А.Шаповаловой - Харьков:
ИМП «Рубикон», 1995
14. Фармацевтичний аналіз: Навч. посіб. для студ. вищ. фармац. навч.
закл. III-IV рівнів акредитації/П.О. Безуглий,
В. О. Грудько, С. Г. Леонова та ін.; За ред. П.О. Безуглого,- X.: Вид-во НФАУ;
Золоті сторінки, 2001.- 240 с.
15. Халецкий A.M. Фармацевтическая химия - Ленинград:
Медицина, 1966
. Эшворт М.Р. Титриметрические методы анализа органических
соединений кн.1,2 - М.: Химия, 1972